Yong Won Choi1, In Kyoung Lim *
1 Afdeling Biochemie en Moleculaire Biologie, BK21 Cell Transformation and Restoration Project, Ajou University School of Medicine, Suwon 443-721, Republiek Korea
* Afdeling Biochemie & Moleculaire Biologie, Ajou University School of Medicine, San 5 Woncheon-dong, Yeongtonggu, Suwon 443-721, Republiek Korea; Tel.: +82 31 219 5051; fax: +82 31 219 5059. E-mailadres: [email protected] (I.K. Lim).
Ontvangen: 29 oktober 2013
Aanvaard: 20 januari 2014
Herzien: in herziene vorm 8 januari 2014
Gepubliceerd/online beschikbaar: 27 januari 2014
Abstract
Om sensitisatie van metformine-cytotoxiciteit te onderzoeken, werden kankercellen behandeld met dichlooracetaat (DCA), een remmer van pyruvaat dehydrogenase kinase (PDK). Metformine-cytotoxiciteit was voornamelijk afhankelijk van de beschikbaarheid van glucose en het reductievermogen gegenereerd door de pentosefosfaatroute, terwijl DCA co-behandeling de metformine-cytotoxiciteit verhoogde via herprogrammering van het glucosemetabolisme door remming van PDK en verhoging van de mitochondriale ademhaling. Gelijktijdige behandeling met DCA leidde eerder tot celdood dan tot celoverleving, ondanks hoge glucose- en hoge GSH-conditie. De conclusie is dat DCA metformine-cytotoxiciteit heeft gesensibiliseerd door het glucosemetabolisme gedeeltelijk te herprogrammeren van aërobe glycolyse naar mitochondriale oxidatie, zoals blijkt uit metingen van glucoseverbruik, lactaatafgifte en de verhouding zuurstofverbruik/extracellulaire verzuring.
Trefwoorden: Metformine Dichlooracetaat (DCA), Oxidatieve stress, Glucose-deprivatie, Glutathion-gehalte
© 2014 Gepubliceerd door Elsevier Ireland Ltd.
INLEIDING
Er zijn steeds meer aanwijzingen dat metabole verstoringen van kankercellen, zoals aërobe glycolyse of glutamineverslaving, onvermijdelijk zijn voor carcinogenese die verder gaat dan het epifenomeen [13], [31], en er zijn enorme inspanningen geleverd om geneesbare doelen en kandidaatchemicaliën voor het kankermetabolisme te vinden [5], [30]. Op basis van de epidemiologische, preklinische en klinische gegevens werd metformine, een biguanide die wordt gebruikt voor de behandeling van diabetes mellitus, een van de meest aantrekkelijke en veelbelovende geneesmiddelen die gericht zijn op het kankermetabolisme. Hoewel het mechanisme van metformine niet volledig is opgehelderd, is bekend dat de intracellulaire functie van metformine het ademhalingsketencomplex I remt [8], [21]. Momenteel wijzen veel aanwijzingen erop dat AMPK-activering een hoofdknooppunt is van antitumoreffecten van metformine [3], [16],[23], daarom zijn LKB1-/- kankercellen in het in vitro kweeksysteem resistenter tegen metformine-geïnduceerde cytotoxiciteit [22], [34]. Daarentegen is aangetoond dat AMPK-activering kankercellen beschermt tegen energiestress via regulering van de NADPH-homeostase [12], daarom zijn LKB1-/- kankercellen gevoeliger voor door fenformine veroorzaakte metabole stress in het longkankermodel van de muis [24]. In de verwarrende context zijn er rapporten dat de anti-tumor effecten van metformine afhankelijk zijn van de glucoseconcentratie in het kweekmedium [11], [17], [25]; Glucose-deprivatie versterkt de metformine-cytotoxiciteit aanzienlijk [17], terwijl de metformine-geïnduceerde AMPK-activering inefficiënt is onder hoge glucose (25 mM) conditie [25]. Daarom moet een diepgaand inzicht in het biochemische mechanisme van metformine-cytotoxiciteit worden verkregen alvorens metformine te gebruiken als antikankermiddel dat zich richt op het glucosemetabolisme.
Het vaak in kankercellen waargenomen Warburg-effect is niet alleen belangrijk voor de opwekking van energie, maar ook voor de handhaving van de verminderde status in een vijandige tumormicro-omgeving. Glucose is de belangrijkste bron van reductievermogen, NADPH, via de pentosefosfaatroute [1], [9], [22]. Inderdaad, 2-deoxyglucose(2DG)-gemedieerde kanker celdood is voornamelijk afhankelijk van de ondraaglijke oxidatieve stress naast energie crisis [15]. Op basis van het concept dat een verhoogde glycolyse kankercellen beschermt tegen oxidatieve stress, werd gedacht dat metformine-verhoogde glycolyse cellen kan beschermen tegen mitochondriale oxidatieve stress als gevolg van de remming van ademhalingsketen complex I [35]. Daarom onderzochten wij of metformine-geïnduceerde mitochondriale stress kan worden vergroot door oxidatieve stress als gevolg van GSH-depletie bij glucosedeprivatie of H2O2-behandeling bij glucose-voldoende conditie. Gebaseerd op de rapporten dat remming van de glycolyse door 2-DG de cytotoxische effecten van metformine versterkt [2], [6], [14] en dat dichlooracetaat (DCA) de glycolyse vermindert door activering van pryruvaat dehydrogenase (PDH) [32] samen met oxidatief metabolisme en antitumoractiviteit [20].
Wij onderzochten in de huidige studie de effecten van DCA op de metformine-cytotoxiciteit en de regulering van het glucosemetabolisme. In tegenstelling tot de effecten van metformine alleen, verhoogde de gecombineerde behandeling van kankercellen met metformine en DCA aanzienlijk de PDH-activiteit, maar niet de glycolyse, waardoor de mitochondriale ademhaling in kankercellen werd hersteld. De herprogrammering van het glucosemetabolisme was nauw verbonden met ernstige oxidatieve stress. Kortom, DCA-gemedieerde herprogrammering van aerobe glycolyse naar mitochondriale oxidatie versterkte metformine-geïnduceerde mitochondriale en cellulaire redox stress die voldoende was om massale celdood te induceren ondanks een hoog glucoseniveau.
Materialen en methoden
Cellen en reagentia
HeLa, MCF7 en MDA-MB-231 cellen werden gekweekt in DMEM (Gibco) met glucose (0-25 mM) aangevuld met 10% foetaal runderserum en 100 U/mL gentamycine bij 37 °C en5%CO2. Metformine, dichlooracetaat (DCA), l-buthionine-sulfoximine (BSO), H2O2, glutathionmonoëthylester (GSH-MEE) en N-acetyl-l-cysteïne (NAC) werden verkregen van Sigma Chemical Co. (St. Louis, MO). Trolox was afkomstig van Biomol International, L.P., en 2′,7′-dichlorodihydrofluoresceïne-diacetaat (H2DCFDA), MitoSOX Red, propidiumjodide (PI) en Calcein AM werden verkregen van Molecular Probes (Eugene, OR).
Meting van de levensvatbaarheid van de cellen en soft agar colony forming assay
De levensvatbaarheid van de cellen na verschillende behandelingen gedurende aangegeven tijden werd beoordeeld met behulp van trypan blue dye exclusion (Sigma-Aldrich). Voor de test werden 4×104 cellen uitgezet in platen met 12 putjes en de volgende dag behandeld met chemicaliën. De cellen werden getrypsiniseerd en gemengd met 0,4% trypanblauw (1:1). Het percentage levensvatbare cellen is het aantal niet-bevlekte cellen/aantal totale cellen × 100. Celdood werd ook gemeten met de lactaat-dehydrogenase(LDH) release assay kit volgens het protocol van de fabrikant (Takara, Japan). Voor soft agar kolonievormingstest werden HeLa-cellen (1,5×103) uitgezet in 0,6% agaroseoplossing en gelaagd op een 1,0% agarosebed vóór behandeling met verschillende concentraties metformine, DCA of beide in combinatie. Kolonies groter dan 125 μm in 2 weken werden als positief geteld.
Meting van cellulaire ROS-niveaus
Intracellulaire en mitochondriale ROS werden gemeten met respectievelijk de oxidatiegevoelige fluorescentiesonde dichloordihydroflourescinediacetaat (H2DCF-DA) en MitoSOX Red (Invitrogen). HeLa-cellen behandeld met metformine, DCA of beide gedurende aangegeven tijden werden geïncubeerd met DCF-DA (20 μM) of MitoSOX (5 μM) gedurende 10 minuten bij 37 °C. Vervolgens werden de cellen tweemaal gewassen met fosfaatgebufferde zoutoplossing (PBS) en onderworpen aan flowcytometrie (BD FACSCanto II, BD Biosciences, San Jose, CA) voor acquisitie en analyse. De fluorescentie-intensiteit werd ook bepaald door fluorescentiemicroscopie.
Meting van cellulaire glutathionniveaus
Om de redoxstatus van intracellulair glutathion te bepalen, werden gereduceerde en geoxideerde glutathionniveaus (GSH, GSSG) geanalyseerd volgens de eerder beschreven methode [29]. Om het totale glutathion te bepalen werden de cellen geoogst en gesuspendeerd in 1% sulfosalicylzuur. Na verwijdering van de aggregaten door centrifugeren bij 8000g gedurende 10 min, werden de supernatanten gebruikt voor de analyse. De reactie werd gestart door toevoeging van het supernatant (1 μg) aan een 96-well microtiterplaat met 200 μl testmengsel [125 mM NaPO4, pH 7,5, 0,3 mM NADPH, 6 mM 5,50-dithiobis(2-nitrobenzoëzuur) (DNTB; Sigma, St. Louis, MO), 0,12 U glutathionreductase], en de maximale reductiesnelheid van DNTB door GSH werd verkregen door meting van de absorptie bij 405 nm. De hoeveelheid totaal glutathion werd berekend aan de hand van de ijklijn met standaard GSH (Sigma). Om het niveau van GSSG te verkrijgen, werden de cellen voorbehandeld met 2-vinylpyridine om bestaand GSH te verwijderen, en het supernatant (50 μg) werd verwerkt volgens hetzelfde protocol als dat voor totaal glutathion.
Analyse van de pyruvaatdehydrogenase (PDH)-activiteit
De PDH-activiteit werd gemeten met behulp van de PDH Enzyme Activity Microplate Assay Kit (Abcam, Cambridge, MA) volgens het protocol. De cellen werden gelyseerd in de bij de kit geleverde monsterbuffer, en het totale cellulaire eiwit in een concentratie van 5 mg/ml werd in drievoud geladen op de vooraf gecoate microtiterplaat met anti-PDH-antilichaam en gedurende 3 uur bij kamertemperatuur geïncubeerd. Na het wassen van de monsters werd reactiemengsel toegevoegd, en de absorptie van het reactieproduct van de reporter-kleurstofgekoppelde reductie van NAD+ tot NADH werd gemeten bij 450 nm gedurende 50 min met tussenpozen van 0,5 min met behulp van de Eon Microplate Spectrophotometer (BioStack, Winooski, VT). De PDH-activiteit werd berekend uit de helling van de mOD per tijd (min) curve (ΔmOD/min) en genormaliseerd ten opzichte van de controlegroep.
Analyse van glucoseverbruik en lactaatproductie
Hela-cellen (1×104/ml) gekweekt in DMEM met 25 mM glucose werden behandeld met metformine, DCA of beide. Na 48 uur werden de glucose- en lactaatconcentraties in de media gemeten met het YSI 7100 multiparameter bioanalysesysteem. Media zonder celcultuur werden gebruikt voor het meten van de initiële glucose- en lactaatconcentratie. Glucoseverbruik = (glucoseconcentratie van media zonder celcultuur) – (glucoseconcentratie van media van behandelde groep). Lactaatproductie = (lactaatconcentratie van media van behandelde groep) – (lactaatconcentratie van media zonder celcultuur).
Meting van de snelheid van oxidatieve fosforylering en glycolyse
De zuurstofverbruikssnelheid(OCR) en de extracellulaire verzuringssnelheid (ECAR) werden gemeten met een Seahorse Bioscience (North Billerica, MA) XF24 extracellulaire fluxanalysator [33]. Zo werden HeLa-cellen gezaaid in XF 24-wells microtiterplaten (1×104) en geïncubeerd bij 37 °C gedurende 24 uur voor behandeling met metformine, DCA of beide gedurende 12 uur. Vervolgens werden de cellen gewassen en geëquilibreerd met DMEM aangevuld met GlutaMax-1 (200 mM), glucose (25 mM), natriumchloride (32 mM) en fenolrood zonder bicarbonaat bij 37 °C gedurende 1 uur in een incubatorzonderCO2. De OCR (pmol/min) en ECAR (mpH/min) werden 3 keer gemeten gedurende telkens 3 minuten. Na het experiment werd het aantal cellen geteld voor normalisatie.
Immunoblotanalyse
Cellysaten (40 μg), bereid in RIPA-buffer met fosfataseremmers, werden gescheiden op SDS-PAGE alvorens te worden overgebracht op PVDF-membraan (Millipore Corp). De blots werden gehybridiseerd met primaire antilichamen en gevisualiseerd met het ECL-systeem (Amersham Biosciences, Buckinghamshire, UK). De antilichamen werden gekocht, anti-p-PDH (S293) van Calbiochem, anti-PARP en anti-PDH van Abcam, anti-Caspase 3 van Cell signaling, anti-cycline D1, anti-cycline E en anti-β-actine van Santa Cruz Biotechnology.
Statistische analyse
Numeriekegegevenswerden gepresenteerd als gemiddelde ± SD van de onafhankelijke bepalingen. Er werd een onafhankelijke t-toetsofANOVA toegepast, en meervoudige vergelijkingen werden geëvalueerd door Tukey HSD. P-waarden kleiner dan 0,05 werden als significant beschouwd.
Resultaten
Metfromine-cytotoxiciteit wordt hersteld door glucose beschikbaar
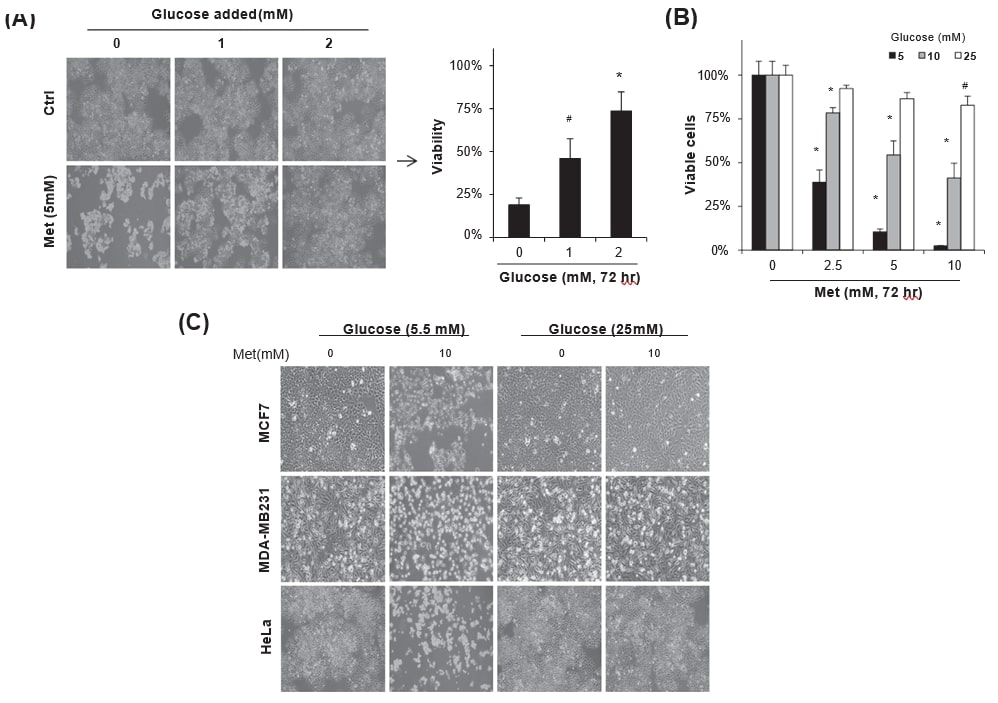
Om de biochemische routes te begrijpen die de metformine-geïnduceerde kanker celdood regelen, werden HeLa cellen gehandhaafd in DMEM met 5,5 mM glucose behandeld met metformine (5 mM) samen met dagelijkse toevoeging van glucose (0-2 mM) gedurende 72 uur. Glucosetoevoeging verhoogde de levensvatbaarheid van de met metformine behandelde cellen aanzienlijk (Fig. 1A, #p < 0,05 en *p < 0,001 vs. geen toevoeging) in goede overeenstemming met eerdere rapporten [11], [17], [25]. Wanneer HeLa-cellen in media met 5-25 mM glucose gedurende 72 uur werden geïncubeerd met 0-10 mM metformine, was de metformine-geïnduceerde celdood afhankelijk van de glucoseconcentratie in de kweekmedia. Vooral een hoge glucoseconcentratie (25 mM) beschermde de metformine-cytotoxiciteit aanzienlijk, ongeacht de metformineconcentratie (fig. 1B). De celdood als gevolg van 10 mM metformine werd beschermd door 25 mM glucose in de kweekmedia, niet alleen bij HeLa-cellen, maar ook bij MCF7- en MDA-MB231-borstkankercellen, terwijl 5,5 mM glucose de cellen niet tegen het toxische effect beschermde (fig. 1C), waargenomen onder een fasecontrastmicroscoop. De resultaten wijzen erop dat het evenwicht tussen de concentraties van metformine en glucose in de kweekmedia belangrijk is voor de regulering van de metformine-cytotoxiciteit.
Metformine-cytotoxiciteit wordt gereguleerd door GSH-gemedieerde redox-veranderingen
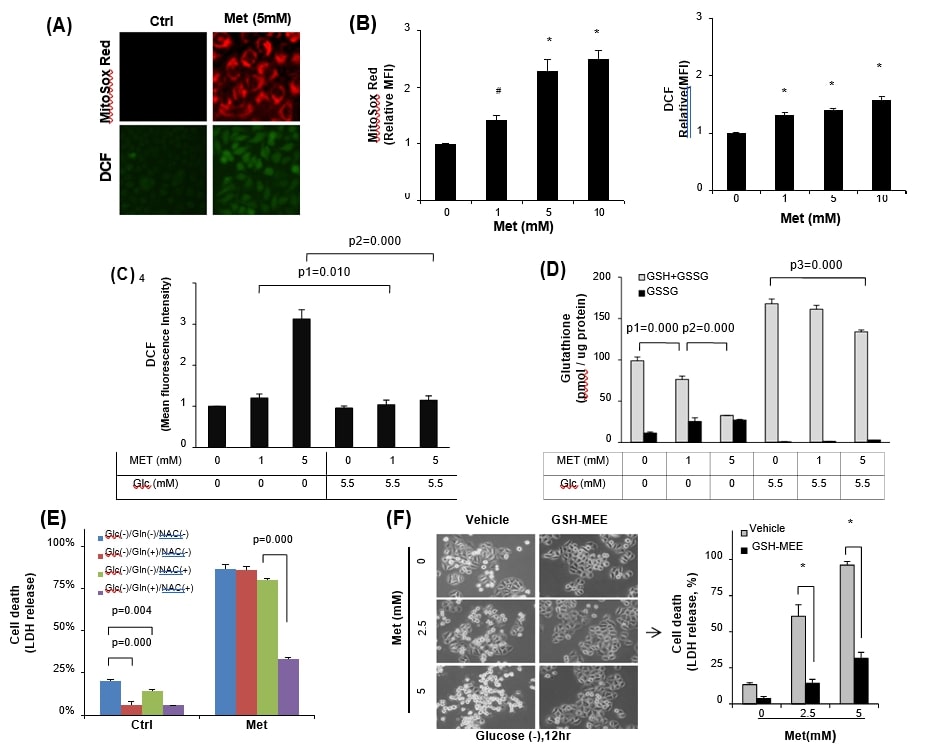
Aangezien metformine het mitochondriale ROS-niveau verhoogt door remming van het respiratoire ketencomplex I [8], [21], [36], onderzochten wij daarom of de door metformine veroorzaakte mitochondriale oxidatieve stress ernstig genoeg was om de dood van kankercellen te induceren onder glucose-arme omstandigheden. HeLa-cellen in glucosehoudende media werden behandeld met 5 mM metformine, en vervolgens werden de mitochondriale superoxide- en totale cellulaire ROS-niveaus geëvalueerd met behulp van respectievelijk MitoSox Red en DCFDA (fig. 2A). Inderdaad verhoogde metformine de mitochondriale en totale cellulaire ROS-niveaus op metformine-concentratie-afhankelijke wijze, maar de fluorescentie van MitoSox Red was meestal hoger dan die van het totale cellulaire ROS-niveau in HeLa-cellen (fig. 2B). Zoals verwacht was het metformine-geïnduceerde cellulaire ROS-niveau significant hoger in de glucosevrije toestand dan in de glucose bevattende media (fig. 2C). Deze gegevens ondersteunen het verslag [15] dat glucosedeprivatie de cellulaire glutathionpool (GSH + GSSG) vermindert. Toen de glutathionpool werd gemeten, was de vermindering van GSH ten opzichte van GSSG significanter in de glucosevrije conditie (p = 0,000) in reactie op metformine, maar het totale glutathiongehalte werd gehandhaafd zonder significante toename van GSSG in de glucose (5,5 mM) bevattende media (Fig. 2D), wat erop wijst dat glucose in de kweekmedia speelde als een belangrijke bron van GSH-generatie. Om de regulering van metformine-cytotoxiciteit door redoxstress onder glucosedeprivatie te onderzoeken, werden antioxidanten gebruikt. Behandeling van HeLa-cellen met 10 mM glutamine of N-acetyl-l-cysteïne (NAC) alleen verminderde de celdood in de glucosevrije media zonder toevoeging van metformine aanzienlijk (Fig. 2E, p = 0,000 en p = 0,004 vs. geen behandeling, respectievelijk), maar dezelfde behandeling kon de metformine (5 mM)-cytotoxiciteit in de glucosevrije media niet verbeteren. Anderzijds kon de gecombineerde behandeling van glutamine met NAC HeLa-cellen beschermen tegen de metformine (5 mM)-cytotoxiciteit (fig. 2E, p = 0,000). Om rechtstreeks te bevestigen of gereduceerd glutathion een rol speelde bij de remming van metformine-cytotoxiciteit, werd gebruik gemaakt van de celdoorlaatbare vorm van GSH (GSH-MEE). Toevoeging van GSH-MEE verminderde de celdood bij HeLa aanzienlijk, vergeleken met die van de voertuigcontrole, zelfs in de glucosevrije toestand (fig. 2F). De gegevens wijzen op een synergetisch effect van glucosedeprivatie op de metformine-cytotoxiciteit via oxidatieve stress door depletie van GSH.
Toename van oxidatieve stress verergert metformine-cytotoxiciteit ondanks hoge glucosespiegel
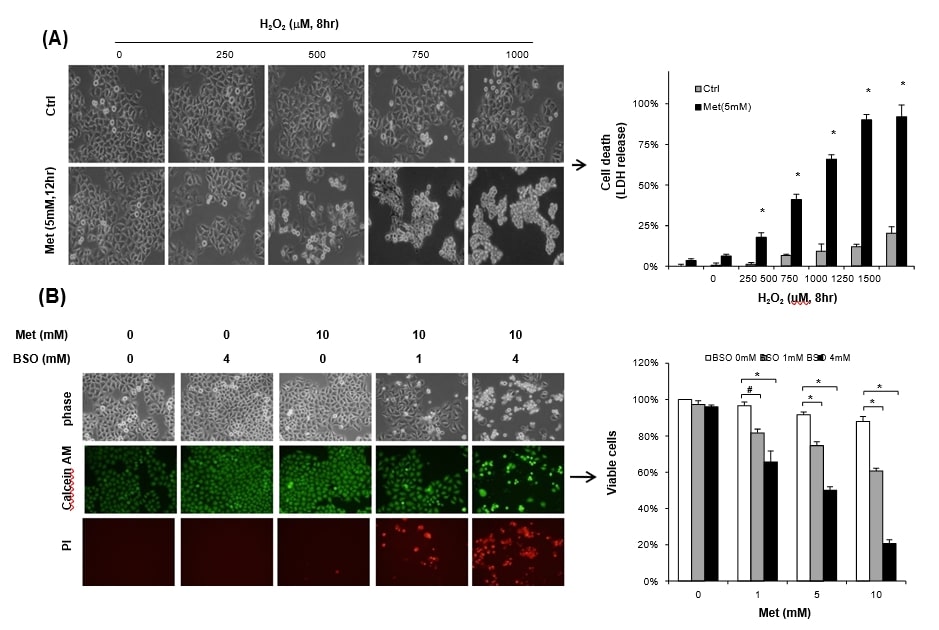
Om na te gaan of de aan metformine blootgestelde cellen kwetsbaarder worden voor stress, werden HeLa-cellen die werden onderhouden met 25 mM glucose gedurende 12 uur behandeld met 5 mM metformine samen met H2O2. Enkelvoudige behandeling met H2O2 of 5 mM metformine leidde niet tot significante celdood, maar gecombineerde behandeling van metformine met H2O2 (meer dan 500 μM) verhoogde duidelijk de celdood, gemeten aan de hand van de afgifte van LDH (fig. 3A). Niet alleen exogene H2O2, maar ook uitputting van de glutathionpool door BSO, een remmer van de GSH-synthese, resulteerde in een aanzienlijke mate van metformine-cytotoxiciteit. Incubatie met hetzij metformine (10 mM) of 4 mM BSO alleen gedurende 48 uur gaf geen significant teken van celdood onder hoge glucosewaarde (25 mM) bij evaluatie met Calceïne AM-kleuring (Fig. 3B, linkerpaneel). Echter, gecombineerde behandeling van metformine met BSO aanzienlijk verhoogd propidiumjodide positieve celdood, toen het werd gecontroleerd door trypan blauw uitsluiting. BSO (1 mM) sensibiliseerde significant 1 mM metformine-cytotoxiciteit onder de hoge glucose (25 mM) conditie (Fig. 3B rechter paneel). Onze bevindingen worden goed ondersteund door eerdere studies dat GSH de meest overvloedige en belangrijke biologische antioxidant is tegen oxidatieve stress in kankercellen [7], [10].
Dichlooracetaat verhoogt de mitochondriale respiratie via activering van pyruvaatdehydrogenase
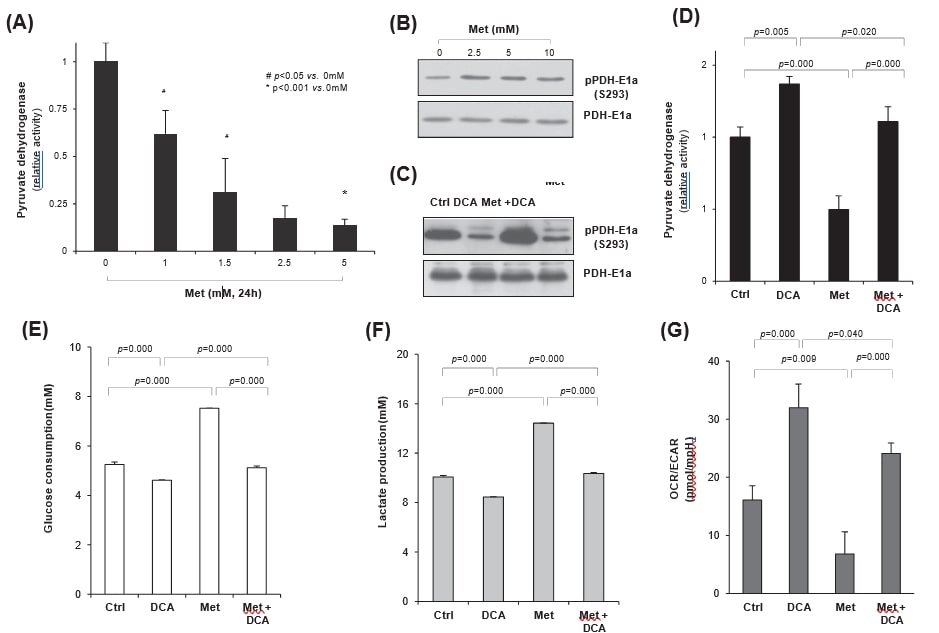
Aangezien PDH het lot van het glucosemetabolisme bepaalt, hetzij in de mitochondriën, hetzij in het cytosol, is het zeer waarschijnlijk dat het verhogen van de PDH-activiteit de mitochondriale respiratie kan verhogen in plaats van de aërobe glycolyse in kankercellen. Daarom onderzochten wij of metformine-geïnduceerde glycolyse werd gereguleerd door DCA, een remmer van PDH-kinase (PDK). Wanneer HeLa-cellen werden behandeld met metformine, werd de PDH-activiteit significant verminderd op dosisafhankelijke wijze (fig. 4A). Behandeling met meformine verhoogde duidelijk de fosforylering van PDH-E1a, wat wijst op activering van PDK in tegenstelling tot remming van PDH (fig. 4B). Aan de andere kant verminderde DCA-behandeling aanzienlijk de metformine-geïnduceerde PDH-E1a-fosforylering op S293-residu, onafhankelijk van de metformine-behandeling, wat resulteert in de toename of het behoud van de PDH-activiteit door DCA (fig. 4C). Inderdaad verhoogde DCA de PDH-activiteit aanzienlijk (p = 0,005), in tegenstelling tot de significante remming door metformine (p = 0,000), vergeleken met de controle. Bovendien herstelde co-behandeling van DCA met metformine de door metformine geremde PDH-activiteit aanzienlijk (p = 0,000) tot het controleniveau (fig. 4D); DCA antagoneerde het effect van metformine op de PDH-activiteit. Om de regulering van metabole veranderingen door DCA of metformine te evalueren, werden de snelheid van glucoseverbruik en lactaatproductie gemeten in kweekmedia; DCA-behandeling verminderde het glucoseverbruik significant (p = 0,000), in tegenstelling tot een significante toename door metformine (p = 0,000, fig. 4E). De verandering was echter niet significant door metformine plus DCA co-behandeling (p = 0,125 vs. controle), wat opnieuw de antagonistische relatie tussen DCA en metformine bevestigt. Bovendien vertoonde de afgifte van lactaat (fig. 4F) door zowel DCA als metformine alleen hetzelfde patroon als dat van het glucoseverbruik (fig. 4E), wat wijst op de verandering van het glucosemetabolisme van door metformine geïnduceerde aërobe glycolyse naar mitochondriale oxidatie door DCA medebehandeling. Toen de zuurstofverbruikssnelheid (OCR) en de extracellulaire verzuringssnelheid (ECAR) werden gemeten met de XF24-analysator, werd de verhouding OCR/ECAR significant verhoogd door DCA-behandeling (p = 0,000), wat wijst op een verhoogde mitochondriale ademhaling, maar verlaagd door metformine (p = 0,009) in vergelijking met de controle. Inderdaad herstelde de co-behandeling van metformine met DCA de mitochondriale ademhaling aanzienlijk in vergelijking met die van metformine alleen (p = 0,000), hoewel deze iets lager was dan die van DCA alleen (p = 0,04). Al met al herprogrammeerde co-behandeling van metformine met DCA het glucosemetabolisme gedeeltelijk van aërobe glycolyse naar mitochondriale oxidatie (fig. 4G).
Dichlooracetaat verhoogt oxidatieve stress en metformine-cytotoxiciteit onder hoge glucoseconditie
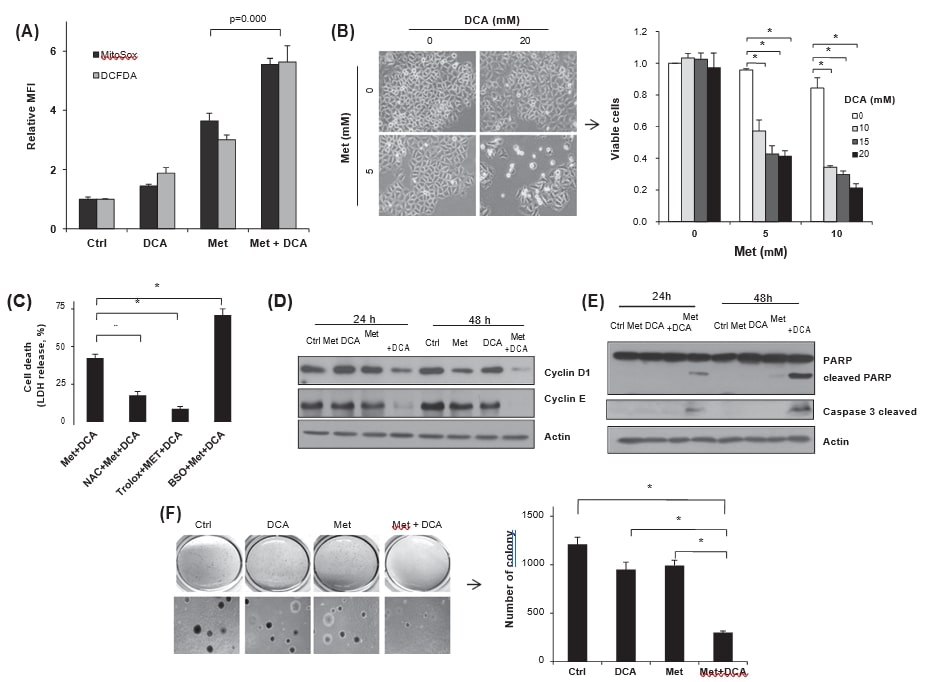
Om het effect van DCA-gemedieerde metabole herprogrammering op het cellulaire ROS-niveau te onderzoeken, werden FACS-analyses met MitoSox Red en DCFDA gebruikt. De gelijktijdige behandeling van HeLa-cellen met metformine en DCA verhoogde zowel het mitochondriale superoxide als het totale cellulaire ROS-niveau dan elke behandeling afzonderlijk (fig. 5A), wat suggereert dat metabole herprogrammering de oxidatieve stress door DCA zou kunnen verergeren. Om na te gaan of de door DCA verhoogde ROS-generatie de metformine-cytotoxiciteit al dan niet verhoogt, werd de levensvatbaarheid van de cellen na behandeling met metformine, DCA of beide geteld. In tegenstelling tot een verwaarloosbaar niveau van celdood door DCA alleen (∼20 mM), verminderde co-behandeling van DCA met metformine de levensvatbaarheid aanzienlijk, ondanks de hoge glucosewaarde (fig. 5B). Om het effect van de verhoogde ROS op de celdood te evalueren, werden anti-oxidanten, NAC of trolox, of een pro-oxidant, BSO, voorbehandeld vóór de co-behandeling met metformine en DCA. De antioxidanten verminderden inderdaad significant de LDH-afgifte in de media, terwijl deze werd verhoogd door BSO voorbehandeling (fig. 5C), hetgeen wijst op een significante regulering van de metformine-cytotoxiciteit via modulatie van de oxidatieve stress door DCA. Een dergelijk synergetisch effect van de cytotoxiciteit werd ook waargenomen in andere kankercellen, MDA-MB-231, EJ en MCF7 (gegevens niet getoond). Bovendien remde de cotreatment van metformine en DCA de synthese van cycline D1 en cycline E aanzienlijk, samen met PARP-splitsing (Fig. 5D en E), wat wijst op verminderde cel proliferatie en inductie van apoptose. Ten slotte verminderde de medebehandeling de ankerplaatsonafhankelijke groei van HeLa-cellen aanzienlijk in vergelijking met de afzonderlijke behandelingen en de controle (fig. 5F).
Bespreking
Wij toonden aan dat herprogrammering van het glucosemetabolisme door behandeling van HeLa-cellen met meformine en dicholoroacetaat de celdood bevorderde door verhoging van het ROS-niveau door verhoging van de mitochondriale oxidatie in plaats van de aërobe glycolyse (fig. 6). DCA werd gebruikt als een middel om oxidatieve fosforylering te bevorderen in plaats van de glycolyse en was bedoeld om het metforminegemedieerde glucosemetabolisme te herprogrammeren. DCA activeert PDH via remming van PDK, waardoor pyruvaat kan worden omgezet in acetyl-CoA in plaats van lactaat. Als biochemisch mechanisme van de door glucosetekort veroorzaakte gevoeligheid van kankercellen voor metformine-cytotoxiciteit toonden wij aan dat de door metformine geïnduceerde oxidatieve stress onder glucosetekort werd versterkt (fig. 2C). Metformine verbruikte inderdaad veel meer glutathion dan het basale gebruik (fig. 2D), terwijl dit veel minder was in aanwezigheid van glucose (5,5 mM). Hoewel voorbehandeling van HeLa-cellen met NAC de celdood onder glucosevrije omstandigheden aanzienlijk blokkeerde, was aanvulling van cysteïne door NAC zonder glutamine onvoldoende om het glutathionniveau op peil te houden om metformine-geïnduceerde oxidatieve stress te doorstaan (fig. 2E). Dit fenomeen kan worden ondersteund door de studie dat glutamine belangrijk is voor reductieve carboxylering in tumorcellen met defecte mitochondriën [19]. Daarom verminderde aanvulling van NAC met glutamine aanzienlijk de metformine-cytotoxiciteit onder de glucosevrije conditie (Fig. 2E).
Onder hoge glucose (25 mM) conditie vertoonde 10 mM metformine slechts een mild tumoristatisch effect, maar geen significante celdood (Fig. 3B). Daarentegen leidde 2,5 mM metformine tot significante celdood onder 5,5 mM glucoseconcentratie (fig. 1B), hetgeen erop wijst dat glucose een belangrijke bron van reductievermogen is. Daarom was suppletie met 1-2 mM glucose voldoende om de metformine-cytotoxiciteit te blokkeren onder het gebruikelijke glucoseniveau (fig. 1A), wat suggereert dat de metformine-cytotoxiciteit onvoldoende zou kunnen zijn om celdood te induceren onder fysiologische glucoseconditie. Behandeling met metformine verhoogde echter aanzienlijk de mitochondriale ROS in de toestand zonder glucosebeperking. Er is nog geen rapport over de mitochondriale ROS-generatie in de context van metformine-cytotoxiciteit. Hier constateerden wij dat 1 mM metformine reeds een aanzienlijke toename van mitochondriaal superoxide tezamen met cellulaire ROS teweegbracht (Fig. 2B), hetgeen mitochondriale zwelling in kankercellen induceerde en de zwelling was niet onomkeerbaar (Aanvullende Figs. S1A en S1B). Toch lijkt het verhoogde mitochondriale ROS niet voldoende om het totale cellulaire ROS en de oxidatieve schade te verhogen. Kankercellen die aan metformine worden blootgesteld, proberen de oxidatieve schade te minimaliseren om de levensvatbaarheid van de cellen te handhaven door de inductie van aërobe glycolyse en NADPH-productie. Deze opvatting kan gedeeltelijk worden gestaafd door het feit dat uitputting van het reductievermogen door BSO of exogeen H2O2 de metformine-cytotoxiciteit verhoogde ondanks de hoge glucoseconditie (fig. 3). Kankercellen kunnen oxidatieve stress verdragen door aërobe glycolyse en de hoeveelheid GSH kan voornamelijk in stand worden gehouden door NADPH dat via de pentosefosfaatroute wordt geproduceerd [12].
Anti-tumoractiviteit van DCA is aangetoond in vitro [4] en in vivo muismodellen [27], [28], en bij glioblastomapatiënten [18], maar de IC50-waarde van DCA is relatief hoog (>17 mM, 48 uur) om celdood te induceren. Bovendien is er geen tumorcelspecificiteit in de geteste kankercellijnen [26], en is DCA selectief actief tegen cellen met een defect in mitochondriën zoals rho(0) cellen of HCT116 p53 null cellen met complex IV deficiëntie. Wij constateerden ook dat 20 mM DCA alleen geen effectieve celdood induceerde, maar in combinatie met metformine synergetiseerde de anti-tumoractiviteit van DCA met 10 mM (Fig. 5B en F).
Wanneer cellen werden behandeld met metformine, werd de PDH-activiteit aanzienlijk geremd door fosfolytatie op Ser293-residu (Fig. 4C) zonder significante verandering van PDK-mRNA-expressie (gegevens niet getoond), wat wijst op posttranscriptionele modificatie. Hoewel we het mechanisme van de metformine-geïnduceerde remming van de PDH-activiteit niet konden definiëren, versterkte de afname van de PDK-activiteit door DCA de metformine-geïnduceerde oxidatieve stress door onderdrukking van de PDH-fosforylering (fig. 4), wat duidt op remming van de PDK-activiteit door DCA, maar op toename van de oxidatieve fosforylering samen met redox-stress. In overeenstemming met bovenstaande bevinding herstelde DCA de door metformine geremde mitochondriale ademhaling aanzienlijk (fig. 4F) en verhoogde het de door metformine geïnduceerde mitochondriale ROS-generatie in hoge mate (fig. 5A). De beschermende rol van anti-oxidanten tegen de gecombineerde behandeling van metformine en DCA bracht ons tot de conclusie dat DCA de metformine-cytotoxiciteit kan versterken door herprogrammering en verhoging van de oxidatieve stress (fig. 5C).
Kortom, de metformine-geïnduceerde cytotoxiciteit werd versterkt door glucosedeprivatie. Zelfs in een hoge glucoseconditie verhoogde extra ROS-stress door uitputting van GSH of exogene H2O2-belasting de metformine-cytotoxiciteit, samen met een verhoogde mitochondriale ademhaling. Wij concluderen dat DCA-gemedieerde herprogrammering van het glucosemetabolisme van aërobe glycolyse naar oxidatieve ademhaling metformine-geïnduceerde oxidatieve stress verergert en metformine-cytotoxiciteit in kankercellen gevoelig maakt (fig. 6).

Belangenverstrengeling
Er is geen concurrerend financieel belang met betrekking tot dit werk.
Erkenningen
Dit werk werd ondersteund door een subsidie van het National R&D Program for Cancer Control, Ministry for Health and Welfare, Republic of Korea (131280).
Bijlage A. Aanvullend materiaal
Aanvullende gegevens bij dit artikel zijn, in de online versie, te vinden op http://dx.doi.org/10.1016/j.canlet.2014.01.015.
VERWIJZINGEN
1 I.M. Ahmad, N. Aykin-Burns, J.E. Sim, S.A. Walsh, R. Higashikubo, G.R. Buettner, S. Venkataraman, M.A. Mackey, S.W. Flanagan, L.W. Oberley, D.R. Spitz Mitochondrial O2*- en H2O2 mediate glucose deprivation-induced stress in human cancer cells J. Biol. Chem., 280 (2005), pp. 4254-42632 I. Ben Sahra, K. Laurent, S. Giuliano, F. Larbret, G. Ponzio, P. Gounon, Y. Le Marchand-Brustel, S. Giorgetti-Peraldi, M. Cormont, C. Bertolotto, M. Deckert, P. Auberger, J.F. Tanti, F. Bost Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells Cancer Res., 70 (2010), pp. 2465-2475
3 I. Ben Sahra, Y. Le Marchand-Brustel, J.F. Tanti, F. Bost Metformine in kankertherapie: een nieuw perspectief voor een oud antidiabeticum? Mol. Cancer Ther., 9 (2010), pp. 1092-1099
4 S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C.T. Lee, G.D. Lopaschuk, L. Puttagunta, S. Bonnet, G. Harry, K. Hashimoto, C.J. Porter, M.A. Andrade, B. Thebaud, E.D. Michelakis Een mitochondria-K+ kanaal as wordt onderdrukt in kanker en de normalisatie ervan bevordert apoptose en remt kanker groei Cancer Cell, 11 (2007), pp. 37-51
5 J.R. Cantor, D.M. Sabatini Kankercelmetabolisme: één kenmerk, vele gezichten Cancer Discov., 2 (2012), pp. 881-898
6 J.H. Cheong, E.S. Park, J. Liang, J.B. Dennison, D. Tsavachidou, C. Nguyen-Charles, K. Wa Cheng, H. Hall, D. Zhang, Y. Lu, M. Ravoori, V. Kundra, J. Ajani, J.S. Lee, W. Ki Hong, G.B. Mills Dual inhibition of tumor energy pathway by 2-deoxyglucose and metformin is effective against a broad spectrum of preclinical cancer models Mol. Cancer Ther., 10 (2011), pp. 2350-2362
7 E.P. Clark, E.R. Epp, J.E. Biaglow, M. Morse-Gaudio, E. Zachgo Glutathion depletion, radiosensitization, and misonidazole potentiation in hypoxic Chinese hamster ovary cells by buthionine sulfoximine Radiat. Res., 98 (1984), blz. 370-380
8 M.Y. El-Mir, V. Nogueira, E. Fontaine, N. Averet, M. Rigoulet, X. Leverve Dimethylbiguanide remt de celademhaling via een indirect effect gericht op het ademhalingsketencomplex I J. Biol. Chem., 275 (2000), pp. 223-228
9 R.B. Hamanaka, N.S. Chandel Celbiologie. Warburg-effect en redoxbalans Science, 334 (2011), pp. 1219-1220
10 T. Ishimoto, O. Nagano, T. Yae, M. Tamada, T. Motohara, H. Oshima, M. Oshima, T. Ikeda, R. Asaba, H. Yagi, T. Masuko, T. Shimizu, T. Ishikawa, K. Kai, E. Takahashi, Y. Imamura, Y. Baba, M. Ohmura, M. Suematsu, H. Baba, H. Saya CD44-variant regelt redoxstatus in kankercellen door de xCT-subeenheid van systeem xc(-) te stabiliseren en bevordert daardoor tumorgroei Cancer Cell, 19 (2011), pp. 387-400
11 S. Javeshghani, M. Zakikhani, S. Austin, M. Bazile, M.J. Blouin, I. Topisirovic, J. St-Pierre, M.N. Pollak Koolstofbron en myc-expressie beïnvloeden de antiproliferatieve werking van metformine Cancer Res., 72 (2012), pp. 6257-6267
12 S.M. Jeon, N.S. Chandel, N. Hay AMPK reguleert NADPH-homeostase om overleving van tumorcellen tijdens energiestress te bevorderen Nature, 485 (2012), pp. 661-665
13 G. Kroemer, J. Pouyssegur Tumorcelmetabolisme: de achilleshiel van kanker Cancer Cell, 13 (2008), pp. 472-482
14 M.A. Lea, J. Chacko, S. Bolikal, J.Y. Hong, R. Chung, A. Ortega, C. Desbordes Toevoeging van 2-deoxyglucose versterkt de groeiremming maar keert de verzuring in darmkankercellen behandeld met fenformine om Anticancer Res., 31 (2011), pp. 421-426
15 X. Lin, F. Zhang, C.M. Bradbury, A. Kaushal, L. Li, D.R. Spitz, R.L. Aft, D. Gius 2-Deoxy-d-glucose-geïnduceerde cytotoxiciteit en radiosensitisatie in tumorcellen wordt gemedieerd via verstoringen in het thiolmetabolisme Cancer Res., 63 (2003), pp. 3413-3417
16 L. Luo, W. Huang, R. Tao, N. Hu, Z.X. Xiao, Z. Luo ATM- en LKB1-afhankelijke activering van AMPK sensibiliseert kankercellen voor etoposide-geïnduceerde apoptose Cancer Lett., 328 (2013), pp. 114-119
17 J.A. Menendez, C. Oliveras-Ferraros, S. Cufi, B. Corominas-Faja, J. Joven, B. Martin-Castillo, A. Vazquez-Martin Metformine is synthetisch dodelijk bij glucoseonttrekking in kankercellen Cell Cycle, 11 (2012), pp. 2782-2792
18 E.D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, C. Maguire, T.L. Gammer, J.R. Mackey, D. Fulton, B. Abdulkarim, M.S. McMurtry, K.C. Petruk Metabole modulatie van glioblastoom met dichlooracetaat Sci. Translat. Med., 2 (2010), p. 31ra34
19 A.R. Mullen, W.W. Wheaton, E.S. Jin, P.H. Chen, L.B. Sullivan, T. Cheng, Y. Yang, W.M. Linehan, N.S. Chandel, R.J. DeBerardinis Reductieve carboxylering ondersteunt groei in tumorcellen met defecte mitochondriën Nature, 481 (2012), pp. 385-388
20 M.R. Niewisch, Z. Kuci, H. Wolburg, M. Sautter, L. Krampen, B. Deubzer, R. Handgretinger, G. Bruchelt Invloed van dichlooracetaat (DCA) op lactaatproductie en zuurstofverbruik in neuroblastoomcellen: is DCA een geschikt geneesmiddel voor neuroblastoomtherapie? Cel. Physiol. Biochem: Int. J. Exp. Cell. Physiol., Biochem., Pharmacol., 29 (2012), pp. 373-380
21 M.R. Owen, E. Doran, A.P. Halestrap Evidence that metformin exert its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain Biochem. J., 348 (Pt 3) (2000), pp. 607-614
22 C. Riganti, E. Gazzano, M. Polimeni, E. Aldieri, D. Ghigo De pentosefosfaatroute: een antioxidante verdediging en een kruispunt in het lot van tumorcellen Free Radical Biol. Med., 53 (2012), pp. 421-436
23 G.Z. Rocha, M.M. Dias, E.R. Ropelle, F. Osorio-Costa, F.A. Rossato, A.E. Vercesi, M.J. Saad, J.B. Carvalheira Metformine versterkt chemotherapie-geïnduceerde AMPK-activering en antitumorale groei Clin. Cancer Res: Official J. Am. Assoc. Cancer Res., 17 (2011), pp. 3993-4005
24 D.B. Shackelford, E. Abt, L. Gerken, D.S. Vasquez, A. Seki, M. Leblanc, L. Wei, M.C. Fishbein, J. Czernin, P.S. Mischel, R.J. Shaw LKB1-inactivatie dicteert therapeutische respons van niet-kleincellige longkanker op het stofwisselingsmedicijn fenformine Cancer Cell, 23 (2013), pp. 143-158
25 J. Sinnett-Smith, K. Kisfalvi, R. Kui, E. RozengurtMetformine remming van mTORC1 activering, DNA-synthese en proliferatie in pancreaskankercellen: afhankelijkheid van glucoseconcentratie en rol van AMPK Biochem. Biophys. Res. Commun., 430 (2013), pp. 352-357
26 L.H. Stockwin, S.X. Yu, S. Borgel, C. Hancock, T.L. Wolfe, L.R. Phillips, M.G. Hollingshead, D.L. Newton Natriumdichlooracetaat richt zich selectief op cellen met defecten in het mitochondriale ETC Int. J. Cancer J. Int. Cancer, 127 (2010), pp. 2510-2519
27 R.C. Sun, M. Fadia, J.E. Dahlstrom, C.R. Parish, P.G. Board, A.C. Blackburn Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo Breast Cancer Res. Treat., 120 (2010), pp. 253-260
28 G. Sutendra, P. Dromparis, A. Kinnaird, T.H. Stenson, A. Haromy, J.M. Parker, M.S. McMurtry, E.D. Michelakis iMitochondriale activering door remming van PDKII onderdrukt HIF1a signalering en angiogenese in kanker Oncogene, 32 (2013), pp. 1638-1650
29 F. Tietze Enzymische methode voor de kwantitatieve bepaling van nanogramhoeveelheden totaal en geoxideerd glutathion: toepassingen op zoogdierbloed en andere weefsels m8 Anal. Biochem., 27 (1969), pp. 502-522
30 M.G. Vander Heiden Targeting cancer metabolism: a therapeutic window opens Nat. Rev. Drug Discovery, 10 (2011), pp. 671-684
31 P.S. Ward, C.B. Thompson Metabolic reprogramming: a cancer hallmark even warburg did not anticipate Cancer Cell, 21 (2012), pp. 297-308
32 S. Whitehouse, R.H. Cooper, P.J. Randle Mechanisme van activering van pyruvaatdehydrogenase door dichlooracetaat en andere gehalogeneerde carbonzuren Biochem. J., 141 (1974), pp. 761-774
33 M. Wu, A. Neilson, A.L. Swift, R. Moran, J. Tamagnine, D. Parslow, S. Armistead, K. Lemire, J. Orrell, J. Teich, S. Chomicz, D.A. Ferrick Multiparameter metabole analyse onthult een nauw verband tussen verminderde mitochondriale bio-energetische functie en verhoogde glycolyse afhankelijkheid in menselijke tumorcellen Am. J. Physiol. Cell Physiol., 292 (2007), pp. C125-136
34 X. Xiao, Q. He, C. Lu, K.D. Werle, R.X. Zhao, J. Chen, B.C. Davis, R. Cui, J. Liang, Z.X. Xu Metformine vermindert de groei van leverkinase B1-intacte baarmoederhalskankercellen Gynecol. Oncol., 127 (2012), pp. 249-255
35 Y. Zhuang, W.K. Miskimins Metformine induceert zowel caspase-afhankelijke als poly(ADP-ribose) polymerase-afhankelijke celdood in borstkankercellen Mol. Cancer Res: MCR, 9 (2011), pp. 603-615
36 J.W. Zmijewski, E. Lorne, X. Zhao, Y. Tsuruta, Y. Sha, G. Liu, G.P. Siegal, E. Abraham Mitochondrial respiratory complex I regulates neutrophilation and severity of lung injury Am. J. Respir. Crit. Care Med., 178 (2008), pp. 168-179