Yong Won Choi1, In Kyoung Lim *
1 Dipartimento di Biochimica e Biologia Molecolare, BK21 Cell Transformation and Restoration Project, Ajou University School of Medicine, Suwon 443-721, Repubblica di Corea
* Dipartimento di Biochimica e Biologia Molecolare, Ajou University School of Medicine, San 5 Woncheon-dong, Yeongtonggu, Suwon 443-721, Repubblica di Corea; Tel.: +82 31 219 5051; fax: +82 31 219 5059. Indirizzo e-mail: [email protected] (I.K. Lim).
Ricevuto: 29 ottobre 2013
Accettato: 20 gennaio 2014
Revisionato: in forma rivista 8 gennaio 2014
Pubblicato/disponibile online: 27 gennaio 2014
Abstract
Per studiare la sensibilizzazione alla citotossicità della metformina, le cellule tumorali sono state trattate con dicloroacetato (DCA), un inibitore della piruvato deidrogenasi chinasi (PDK). La citotossicità della metformina dipendeva principalmente dalla disponibilità di glucosio e dal potere riducente generato dalla via del pentoso fosfato, mentre il cotrattamento con DCA ha potenziato la citotossicità della metformina attraverso la riprogrammazione del metabolismo del glucosio, inibendo la PDK e aumentando la respirazione mitocondriale . Il cotrattamento con DCA ha provocato la morte cellulare piuttosto che la sopravvivenza delle cellule, nonostante le condizioni di alto glucosio e alto GSH. In conclusione, il DCA ha sensibilizzato la citotossicità della metformina riprogrammando il metabolismo del glucosio in parte dalla glicolisi aerobica all’ossidazione mitocondriale, come dimostrato dalle misurazioni del consumo di glucosio, del rilascio di lattato e del rapporto tra tasso di consumo di ossigeno e tasso di acidificazione extracellulare.
Parole chiave: Metformina dicloroacetato (DCA), Stress ossidativo, Privazione di glucosio, Contenuto di glutatione
© 2014 Pubblicato da Elsevier Ireland Ltd.
INTRODUZIONE
Le crescenti evidenze indicano che le perturbazioni metaboliche delle cellule tumorali, come la glicolisi aerobica o la dipendenza da glutammina, sono inevitabili per la carcinogenesi, al di là dell’epifenomeno [13], [31], e sono stati compiuti enormi sforzi per trovare bersagli farmacologici e sostanze chimiche candidate sul metabolismo del cancro [5], [30]. Sulla base dei dati epidemiologici, preclinici e clinici, la metformina, una biguanide utilizzata per il trattamento del diabete mellito, è diventata uno dei farmaci più interessanti e promettenti per il metabolismo del cancro. Sebbene il meccanismo della metformina non sia del tutto chiarito, è noto che la sua funzione intracellulare inibisce il complesso I della catena respiratoria [8], [21]. Attualmente, molte evidenze indicano che l’attivazione di AMPK è un nodo principale degli effetti antitumorali della metformina [3], [16],[23], pertanto le cellule tumorali LKB1-/- sono più resistenti alla citotossicità indotta dalla metformina nel sistema di coltura in vitro [22], [34]. Al contrario, è stato dimostrato che l’attivazione dell’AMPK protegge le cellule tumorali dallo stress energetico attraverso la regolazione dell’omeostasi del NADPH [12], pertanto le cellule tumorali LKB1-/- sono più sensibili allo stress metabolico indotto dalla fenformina nel modello di tumore polmonare del topo [24]. In contesti confusi, è stato riportato che gli effetti antitumorali della metformina dipendono dalla concentrazione di glucosio nel terreno di coltura [11], [17], [25]; la privazione di glucosio aumenta significativamente la citotossicità della metformina [17], mentre l’attivazione dell’AMPK indotta dalla metformina è inefficace in condizioni di glucosio elevato (25 mM) [25]. Pertanto, prima di utilizzare la metformina come agente antitumorale incentrato sul metabolismo del glucosio, è necessario chiarire il meccanismo biochimico della citotossicità della metformina.
L’effetto Warburg, frequentemente osservato nelle cellule tumorali, è importante non solo per la produzione di energia, ma anche per mantenere uno stato ridotto in un microambiente tumorale ostile. Il glucosio è la principale fonte di potere riducente, NADPH, attraverso la via del pentoso fosfato [1], [9], [22]. In effetti, la morte delle cellule tumorali mediata dal 2-deossiglucosio(2DG) dipende principalmente dall’intollerabile stress ossidativo oltre che dalla crisi energetica [15]. Sulla base del concetto che l’aumento della glicolisi protegge le cellule tumorali dallo stress ossidativo, si è pensato che la glicolisi aumentata dalla metformina sia in grado di proteggere le cellule dallo stress ossidativo mitocondriale derivante dall’inibizione del complesso I della catena respiratoria [35]. Pertanto, abbiamo studiato se lo stress mitocondriale indotto dalla metformina possa essere aumentato dallo stress ossidativo dovuto alla deplezione di GSH in condizioni di deprivazione di glucosio o al trattamento con H2O2 in condizioni di glucosio sufficiente. Sulla base di quanto riportato, l’inibizione della glicolisi da parte del 2-DG potenzia gli effetti citotossici della metformina [2], [6], [14] e che il dicloroacetato (DCA) riduce la glicolisi attivando la piruvato deidrogenasi (PDH) [32] insieme al metabolismo ossidativo e all’attività antitumorale [20].
Nel presente studio abbiamo analizzato gli effetti del DCA sulla citotossicità della metformina e sulla regolazione del metabolismo del glucosio. A differenza degli effetti della sola metformina, il trattamento combinato delle cellule tumorali con metformina e DCA ha aumentato significativamente l’attività della PDH, ma non la glicolisi, recuperando così la respirazione mitocondriale nelle cellule tumorali. La riprogrammazione del metabolismo del glucosio era strettamente associata a un grave stress ossidativo. In sintesi, la riprogrammazione mediata dal DCA della glicolisi aerobica verso l’ossidazione mitocondriale ha aumentato lo stress redox mitocondriale e cellulare indotto dalla metformina, sufficiente a indurre una morte cellulare massiva nonostante l’alto livello di glucosio.
Materiali e metodi
Cellule e reagenti
Le cellule HeLa, MCF7 e MDA-MB-231 sono state coltivate in DMEM (Gibco) contenente glucosio (0-25 mM) integrato con 10% di siero fetale bovino e 100 U/mL di gentamicina a 37 °C e 5% diCO2. Metformina, dicloroacetato (DCA), l-butionina-sulfossimina (BSO), H2O2, glutatione monoetil estere (GSH-MEE) e N-acetil-l-cisteina (NAC) sono stati ottenuti da Sigma Chemical Co. (St. Louis, MO). Il trolox è stato prodotto da Biomol International, L.P., mentre il 2′,7′-diclorodiidrofluoresceina diacetato (H2DCFDA), il MitoSOX Red, lo ioduro di propidio (PI) e la calceina AM sono stati ottenuti da Molecular Probes (Eugene, OR).
Misura della vitalità cellulare e saggio di formazione di colonie in soft agar
La vitalità cellulare dopo i vari trattamenti per i tempi indicati è stata valutata mediante esclusione del colorante blu di tripan (Sigma-Aldrich). Per il test, 4 ×104 cellule sono state piastrate in piastre a 12 pozzetti e trattate con sostanze chimiche il giorno successivo. Le cellule sono state tripsinizzate e mescolate con lo 0,4% di tripan blu (1:1). La percentuale di cellule vitali rappresenta il numero di cellule non macchiate/numero di cellule totali × 100. La morte cellulare è stata misurata anche con il kit per il test del rilascio di lattatodeidrogenasi (LDH) secondo il protocollo della ditta produttrice (Takara, Giappone). Per il saggio di formazione di colonie in soft agar, le cellule HeLa (1,5 ×103) sono state piastrate in una soluzione di agarosio allo 0,6% e stratificate su un letto di agarosio all’1,0% prima del trattamento con diverse concentrazioni di metformina, DCA o entrambi in combinazione. Le colonie di dimensioni superiori a 125 μm in 2 settimane sono state contate come positive.
Misurazione dei livelli di ROS cellulari
I ROS intracellulari e mitocondriali sono stati misurati utilizzando la sonda fluorescente sensibile all’ossidazione diclorodiidroflourescina diacetato (H2DCF-DA) e MitoSOX Red (Invitrogen), rispettivamente. Le cellule HeLa trattate con metformina, DCA o entrambi per i tempi indicati sono state incubate con DCF-DA (20 μM) o MitoSOX (5 μM) per 10 min a 37 °C. Quindi, le cellule sono state lavate due volte con tampone fosfato (PBS) e sottoposte a citometria a flusso (BD FACSCanto II, BD Biosciences, San Jose, CA) per l’acquisizione e l’analisi. L’intensità della fluorescenza è stata determinata anche mediante microscopia a fluorescenza.
Misurazione dei livelli di glutatione cellulare
Per determinare lo stato redox del glutatione intracellulare, i livelli di glutatione ridotto e ossidato (GSH, GSSG) sono stati analizzati secondo il metodo precedentemente descritto [29]. Per determinare il glutatione totale, le cellule sono state raccolte e sospese in acido solfosalicilico all’1%. Dopo aver rimosso gli aggregati mediante centrifugazione a 8000g per 10 minuti, i surnatanti sono stati utilizzati per il dosaggio. La reazione è stata avviata aggiungendo il surnatante (1 μg) a una piastra di microtitolazione a 96 pozzetti contenente 200 μl di miscela di dosaggio [125 mM NaPO4, pH 7,5, 0,3 mM NADPH, 6 mM 5,50-ditiobis(acido 2-nitrobenzoico) (DNTB; Sigma, St. Louis, MO), 0,12 U glutatione reduttasi] e il tasso massimo di riduzione del DNTB da parte del GSH è stato ottenuto misurando l’assorbanza a 405 nm. La quantità di glutatione totale è stata calcolata dalla curva di calibrazione utilizzando il GSH standard (Sigma). Per ottenere il livello di GSSG, le cellule sono state pretrattate con 2-vinilpiridina per privare il GSH esistente e il surnatante (50 μg) è stato trattato secondo lo stesso protocollo utilizzato per il glutatione totale.
Dosaggio dell’attività della piruvato deidrogenasi (PDH)
L’attività della PDH è stata misurata utilizzando il PDH Enzyme Activity Microplate Assay Kit (Abcam, Cambridge, MA) secondo il protocollo. Le cellule sono state lisate nel tampone di campionamento fornito con il kit e la proteina cellulare totale a una concentrazione di 5 mg/ml è stata caricata in triplicato sulla micropiastra precoatata con anticorpo anti-PDH e incubata per 3 ore a temperatura ambiente. Dopo aver lavato i campioni, è stata aggiunta la miscela di reazione e l’assorbanza del prodotto di reazione della riduzione accoppiata al colorante reporter di NAD+ a NADH è stata misurata a 450 nm per 50 minuti a intervalli di 0,5 minuti utilizzando lo spettrofotometro per micropiastre Eon (BioStack, Winooski, VT). L’attività della PDH è stata calcolata dalla pendenza della curva mOD per tempo (min) (ΔmOD/min) e normalizzata rispetto al gruppo di controllo.
Analisi del consumo di glucosio e della produzione di lattato
Le celluleHela(1 ×104/ml) coltivate in DMEM con 25 mM di glucosio sono state trattate con metformina, DCA o entrambi. Dopo 48 ore, le concentrazioni di glucosio e lattato nei mezzi di coltura sono state misurate con il sistema bioanalitico multiparametrico YSI 7100. Per la misurazione della concentrazione iniziale di glucosio e lattato sono stati utilizzati terreni senza coltura cellulare. Consumo di glucosio = (concentrazione di glucosio nei terreni senza coltura cellulare) – (concentrazione di glucosio nei terreni del gruppo trattato). Produzione di lattato = (concentrazione di lattato nei terreni del gruppo trattato) – (concentrazione di lattato nei terreni senza coltura cellulare).
Misurazione dei tassi di fosforilazione ossidativa e glicolisi
Il tasso di consumo diossigeno(OCR) e il tasso di acidificazione extracellulare (ECAR) sono stati misurati utilizzando un analizzatore di flusso extracellulare Seahorse Bioscience (North Billerica, MA) XF24 [33]. Pertanto, le cellule HeLa sono state seminate in micropiastre XF a 24 pozzetti (1 ×104) e incubate a 37 °C per 24 ore prima del trattamento con metformina, DCA o entrambi per 12 ore. Successivamente, le cellule sono state lavate ed equilibrate con DMEM integrato con GlutaMax-1 (200 mM), glucosio (25 mM), cloruro di sodio (32 mM) e rosso fenolo senza bicarbonato a 37 °C per 1 ora in un incubatore senzaCO2. L’OCR (pmol/min) e l’ECAR (mpH/min) sono stati misurati 3 volte per 3 minuti ciascuno. Dopo l’esperimento, è stato contato il numero di cellule per la normalizzazione.
Analisi immunoblot
I lisati cellulari (40 μg) preparati in tampone RIPA con inibitori della fosfatasi sono stati separati su SDS-PAGE prima di essere trasferiti su membrana PVDF (Millipore Corp). I blot sono stati ibridati con gli anticorpi primari e visualizzati con il sistema ECL (Amersham Biosciences, Buckinghamshire, Regno Unito). Gli anticorpi sono stati acquistati, anti-p-PDH (S293) da Calbiochem, anti-PARP e anti-PDH da Abcam, anti-Caspasi 3 da Cell signaling, anti-ciclina D1, anti-ciclina E e anti-β-actina da Santa Cruz Biotechnology.
Analisi statistica
I dati numerici sono stati presentati come media ± SD delle determinazioni indipendenti. È stato applicato il test tindipendente o l’ANOVA e i confronti multipli sono stati valutati mediante Tukey HSD. I valori di P inferiori a 0,05 sono stati considerati significativi.
Risultati
Lacitotossicità della metfromina è recuperata dalla disponibilità di glucosio
Per comprendere le vie biochimiche che regolano la morte delle cellule tumorali indotta dalla metformina, le cellule HeLa mantenute in DMEM con 5,5 mM di glucosio sono state trattate con metformina (5 mM) insieme all’aggiunta giornaliera di glucosio (0-2 mM) per 72 ore. L’integrazione di glucosio ha aumentato significativamente la vitalità delle cellule trattate con metformina (Fig. 1A, #p < 0,05 e *p < 0,001 rispetto a nessuna aggiunta), in buon accordo con i rapporti precedenti [11], [17], [25]. Quando le cellule HeLa mantenute in mezzi di coltura contenenti 5-25 mM di glucosio sono state incubate con 0-10 mM di metformina per 72 ore, la morte cellulare indotta dalla metformina dipendeva dalla concentrazione di glucosio nei mezzi di coltura. In particolare, la condizione di glucosio elevato (25 mM) ha protetto significativamente la citotossicità da metformina, indipendentemente dalla concentrazione di metformina (Fig. 1B). La morte cellulare indotta da 10 mM di metformina è stata protetta da 25 mM di glucosio nel terreno di coltura non solo nelle cellule HeLa, ma anche nelle cellule di cancro al seno MCF7 e MDA-MB231, mentre 5,5 mM di glucosio non sono riusciti a proteggere le cellule dall’effetto tossico (Fig. 1C), osservato al microscopio a contrasto di fase. I risultati indicano che l’equilibrio tra le concentrazioni di metformina e glucosio nei terreni di coltura è importante per la regolazione della citotossicità della metformina.
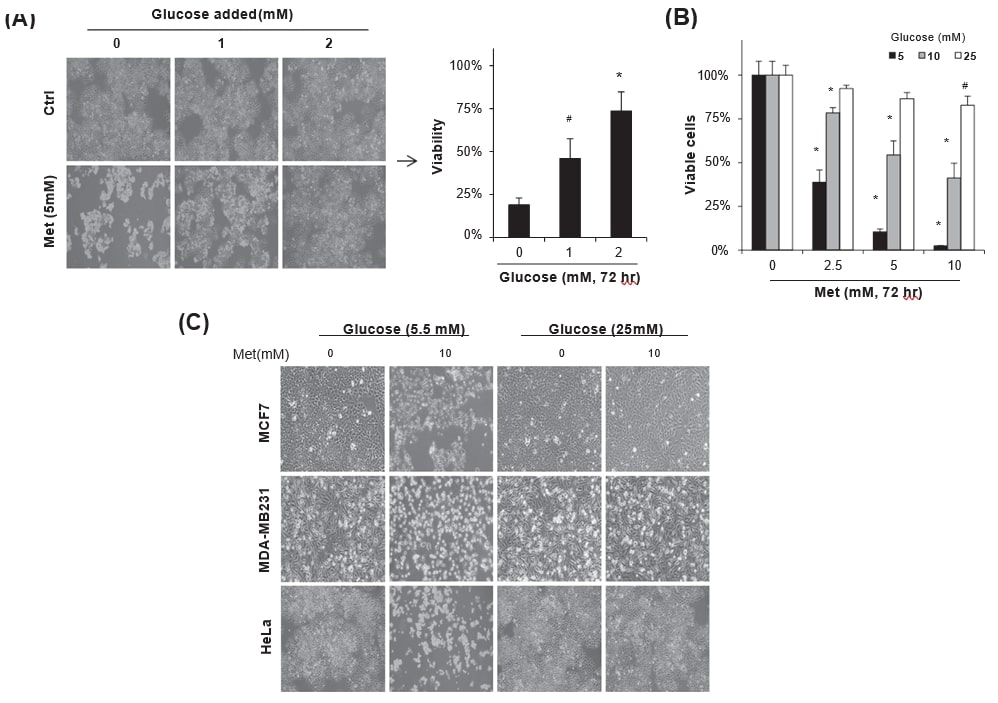
Lacitotossicità della metformina è regolata da cambiamenti redox mediati dal GSH
Poiché la metformina aumenta il livello di ROS mitocondriali attraverso l’inibizione del complesso I della catena respiratoria [8], [21], [36], abbiamo quindi esaminato se lo stress ossidativo mitocondriale indotto dalla metformina fosse abbastanza grave da indurre la morte delle cellule tumorali in condizioni di glucosio insufficiente. Le cellule HeLa mantenute in terreno contenente glucosio sono state trattate con 5 mM di metformina, quindi i livelli di superossido mitocondriale e di ROS cellulare totale sono stati valutati utilizzando rispettivamente MitoSox Red e DCFDA (Fig. 2A). In effetti, la metformina ha aumentato i livelli di ROS mitocondriale e totale cellulare in modo dipendente dalla concentrazione di metoformina, tuttavia la fluorescenza del MitoSox Red è stata per lo più superiore a quella del livello di ROS cellulare totale nelle cellule HeLa (Fig. 2B). Come previsto, il livello di ROS cellulare indotto dalla metformina era significativamente più alto in condizioni di assenza di glucosio rispetto ai mezzi contenenti glucosio (Fig. 2C). Questi dati supportano fortemente la relazione [15] secondo cui la privazione di glucosio riduce il pool di glutatione cellulare (GSH + GSSG). Pertanto, quando è stato misurato il pool di glutatione, la riduzione del GSH rispetto al GSSG è stata più significativa nella condizione senza glucosio (p = 0,000) in risposta alla metformina; tuttavia, il contenuto totale di glutatione è stato mantenuto senza un aumento significativo del GSSG nei terreni contenenti glucosio (5,5 mM) (Fig. 2D), indicando che il glucosio nei terreni di coltura ha svolto un ruolo importante nella generazione di GSH. Per indagare la regolazione della citotossicità della metformina da parte dello stress redox in condizioni di deprivazione di glucosio, sono stati impiegati gli antiossidanti. Il trattamento delle cellule HeLa con 10 mM di glutammina o N-acetil-l-cisteina (NAC) da solo ha ridotto significativamente la morte cellulare osservata nei mezzi privi di glucosio senza aggiunta di metformina (Fig. 2E, rispettivamente p = 0,000 e p = 0,004 rispetto all’ assenza di trattamento), ma lo stesso trattamento non è riuscito a migliorare la citotossicità della metformina (5 mM) nei mezzi privi di glucosio. D’altra parte, il trattamento combinato di glutammina e NAC ha protetto le cellule HeLa dalla citotossicità della metformina (5 mM) (Fig. 2E, p = 0,000). Per confermare direttamente il ruolo del glutatione ridotto nell’inibizione della citotossicità della metformina, è stata utilizzata una forma di GSH permeabile alle cellule (GSH-MEE). L’aggiunta di GSH-MEE ha ridotto significativamente la morte delle cellule HeLa, rispetto al controllo con veicolo, anche in condizioni di assenza di glucosio (Fig. 2F). I dati rivelano l’effetto sinergico della privazione di glucosio nella citotossicità della metformina attraverso lo stress ossidativo per deplezione di GSH.
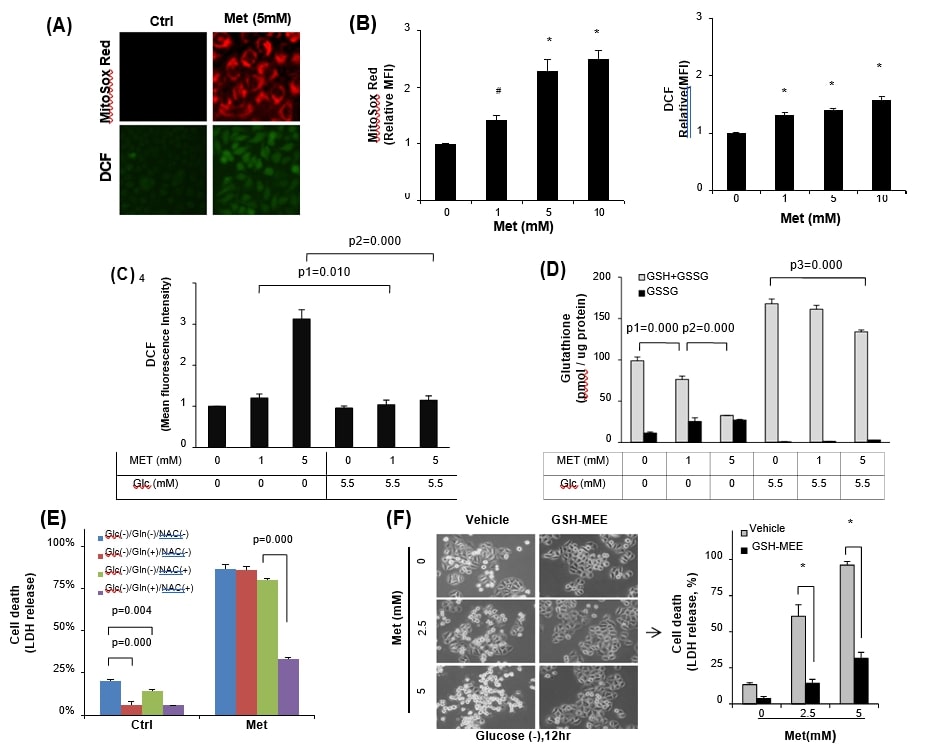
L’aumento dello stress ossidativo aggrava la citotossicità della metformina nonostante l’alto livello di glucosio
Per verificare se le cellule esposte alla metformina diventano più vulnerabili allo stress, le cellule HeLa mantenute con 25 mM di glucosio sono state trattate con 5 mM di metformina e H2O2 per 12 ore. Il trattamento singolo con H2O2 o 5 mM di metformina non ha indotto una morte cellulare significativa, ma il trattamento combinato di metformina e H2O2 (oltre 500 μM) ha aumentato notevolmente la morte cellulare, misurata dal rilascio di LDH (Fig. 3A). Non solo l’H2O2 esogeno, ma anche la deplezione del pool di glutatione mediante BSO, inibitore della sintesi di GSH, ha determinato un livello significativo di citotossicità da metformina. L’incubazione con metformina (10 mM) o BSO 4 mM da sola per 48 ore non ha mostrato segni significativi di morte cellulare in condizioni di glucosio elevato (25 mM), valutati mediante colorazione con Calceina AM (Fig. 3B pannello sinistro). Tuttavia, il trattamento combinato di metformina e BSO ha aumentato significativamente la morte cellulare positiva allo ioduro di propidio, quando è stata monitorata mediante esclusione del blu di tripan. La BSO (1 mM) ha sensibilizzato in modo significativo la citotossicità della metformina 1 mM in condizioni di glucosio elevato (25 mM) (Fig. 3B pannello destro). I nostri risultati sono ben supportati da studi precedenti secondo cui il GSH è l’antiossidante biologico più abbondante e importante contro lo stress ossidativo nelle cellule tumorali [7], [10].
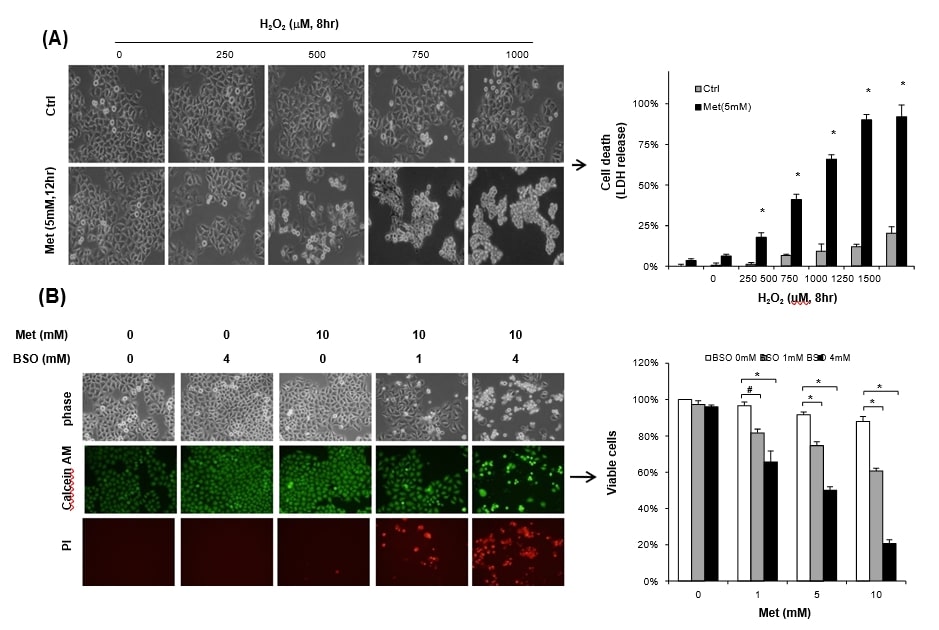
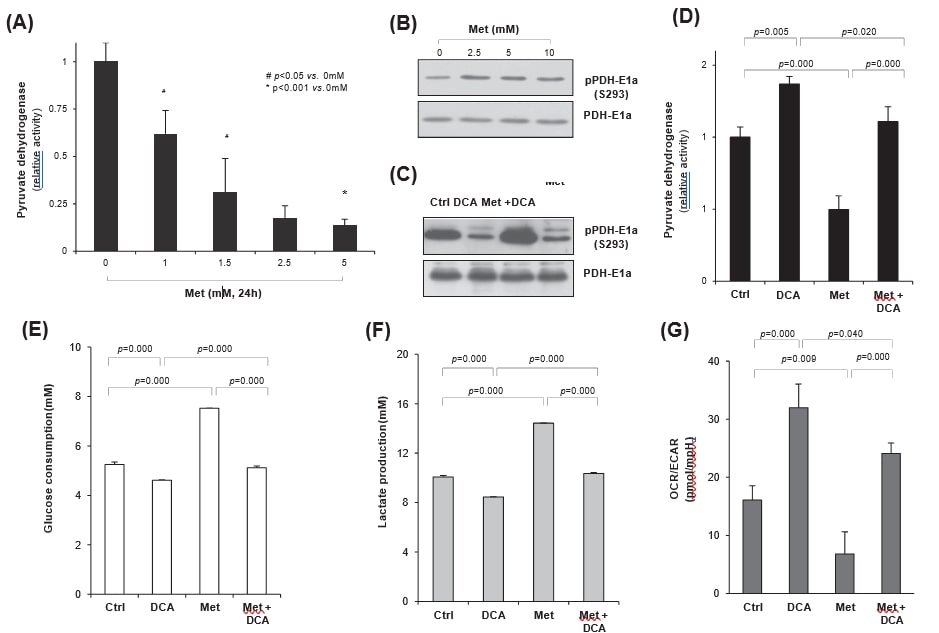
Ildicloroacetato aumenta la respirazione mitocondriale attraverso l’attivazione della piruvato deidrogenasi
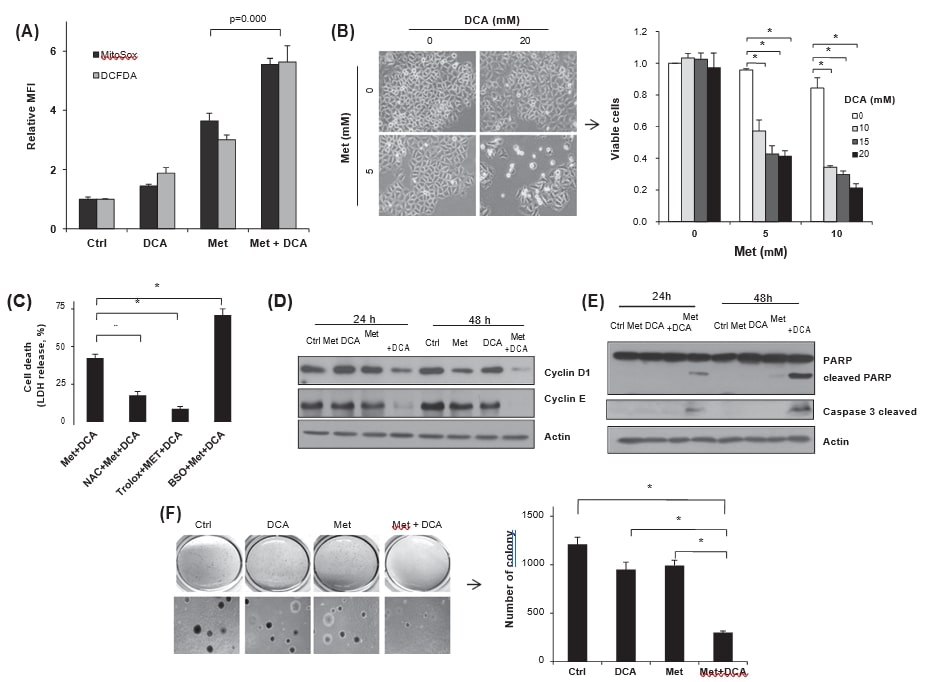
Poiché la PDH determina il destino del metabolismo del glucosio nei mitocondi o nel citosol, è molto probabile che il potenziamento dell’attività della PDH possa aumentare la respirazione mitocondriale piuttosto che la glicolisi aerobica nelle cellule tumorali. Pertanto, abbiamo studiato se la glicolisi indotta dalla metformina fosse regolata dal DCA, un inibitore della PDH chinasi (PDK). Quando le cellule HeLa sono state trattate con metformina, l’attività della PDH è diminuita significativamente in modo dipendente dalla dose (Fig. 4A). Il trattamento con metformina ha aumentato chiaramente la fosforilazione della PDH-E1a, indicando l’attivazione della PDK rispetto all’inibizione della PDH (Fig. 4B). D’altra parte, il trattamento con DCA ha ridotto significativamente la fosforilazione della PDH-E1a indotta dalla metformina sul residuo S293, indipendentemente dal trattamento con metformina, con conseguente aumento o mantenimento dell’attività della PDH da parte del DCA (Fig. 4C). Infatti, il DCA ha aumentato significativamente l’attività della PDH (p = 0,005), in contrasto con la significativa inibizione da parte della metformina (p = 0,000), rispetto al controllo. Inoltre, il co-trattamento di DCA con metformina ha recuperato significativamente l’attività PDH inibita dalla metformina (p = 0,000) fino al livello di controllo (Fig. 4D); il DCA ha antagonizzato l’effetto della metformina sull’attività PDH. Per valutare la regolazione dei cambiamenti metabolici da parte del DCA o della metformina, sono stati misurati i tassi di consumo di glucosio e di produzione di lattato nei terreni di coltura; il trattamento con DCA ha ridotto significativamente il consumo di glucosio (p = 0,000), in contrasto con l’aumento significativo della metformina (p = 0,000, Fig. 4E). Tuttavia, la variazione è stata insignificante con il cotrattamento metformina più DCA (p = 0,125 rispetto al controllo), confermando ancora una volta la relazione antagonista tra DCA e metformina. Inoltre, il rilascio di lattato (Fig. 4F) da parte del DCA o della metformina da soli ha rivelato lo stesso andamento del tasso di consumo di glucosio (Fig. 4E), suggerendo il cambiamento del metabolismo del glucosio dalla glicolisi aerobica indotta dalla metformina all’ossidazione mitocondriale con il cotrattamento con DCA. Quando il tasso di consumo di ossigeno (OCR) e il tasso di acidificazione extracellulare (ECAR) sono stati misurati con l’analizzatore XF24, il rapporto OCR/ECAR è risultato significativamente aumentato con il trattamento con DCA (p = 0,000), che rappresenta un aumento della respirazione mitocondriale, ma è diminuito con la metformina (p = 0,009) rispetto al controllo. In effetti, il cotrattamento di metformina con DCA ha recuperato significativamente la respirazione mitocondriale rispetto alla sola metformina (p = 0,000), anche se era leggermente inferiore rispetto al solo DCA (p = 0,04). Nel complesso, il cotrattamento di metformina con DCA ha riprogrammato il metabolismo del glucosio in parte dalla glicolisi aerobica all’ossidazione mitocondriale (Fig. 4G).
Ildicloroacetato aumenta lo stress ossidativo e la citotossicità della metformina in condizioni di glucosio elevato
Per indagare l’effetto della riprogrammazione metabolica mediata dal DCA sul livello dei ROS cellulari, sono state effettuate analisi FACS con MitoSox Red e DCFDA. Il cotrattamento delle cellule HeLa con metformina e DCA ha aumentato i livelli di superossido mitocondriale e di ROS cellulare totale rispetto a quelli di ciascun trattamento (Fig. 5A), suggerendo che la riprogrammazione metabolica potrebbe aggravare lo stress ossidativo da parte del DCA. Per valutare se la generazione di ROS potenziata dal DCA aumenti o meno la citotossicità della metformina, è stata contata la vitalità cellulare dopo il trattamento con metformina, DCA o entrambi. A differenza del livello trascurabile di morte cellulare causato dal solo DCA (∼20 mM), il co-trattamento di DCA con metformina ha ridotto notevolmente la vitalità nonostante la condizione di glucosio elevato (Fig. 5B). Per valutare l’effetto dell’aumento dei ROS sulla morte cellulare, prima del cotrattamento con metformina e DCA sono stati pretrattati antiossidanti, NAC o trolox, o un pro-ossidante, BSO. In effetti, gli antiossidanti hanno ridotto significativamente il rilascio di LDH nel terreno, mentre è aumentato con il pretrattamento con BSO (Fig.5C), indicando una significativa regolazione della citotossicità della metformina attraverso la modulazione dello stress ossidativo da parte del DCA. Tale effetto sinergico della citotossicità è stato osservato anche in altre cellule tumorali, MDA-MB-231, EJ e MCF7 (dati non mostrati). Inoltre, il cotrattamento di metformina e DCA ha inibito significativamente la sintesi della ciclina D1 e della ciclina E insieme alla scissione di PARP (Fig. 5D ed E), indicando una riduzione della proliferazione cellulare e l’induzione dell’apoptosi. Infine, il co-trattamento ha ridotto significativamente la crescita ancoraggio-indipendente delle cellule HeLa rispetto ai trattamenti singoli e al controllo (Fig. 5F).
Discussione
Abbiamo dimostrato che la riprogrammazione del metabolismo del glucosio mediante trattamento delle cellule HeLa con meformina e dicloracetato ha aumentato la morte cellulare attraverso l’aumento del livello di ROS, incrementando l’ossidazione mitocondriale piuttosto che la glicolisi aerobica (Fig. 6). Il DCA è stato impiegato come agente per promuovere la fosforilazione ossidativa rispetto alla glicolisi e per riprogrammare il metabolismo del glucosio mediato dalla metformina. Il DCA attiva la PDH attraverso l’inibizione della PDK, per cui il piruvato può essere convertito in acetil-CoA anziché in lattato. Come meccanismo biochimico della sensibilizzazione delle cellule tumorali alla citotossicità indotta dalla deprivazione di glucosio, abbiamo dimostrato che lo stress ossidativo indotto dalla metformina era aggravato in condizioni di deprivazione di glucosio (Fig. 2C). In effetti, la metformina ha consumato glutatione in misura molto maggiore rispetto all’uso basale (Fig. 2D), mentre era molto minore in presenza di glucosio (5,5 mM). Sebbene il pretrattamento delle cellule HeLa con NAC abbia bloccato in modo significativo la morte cellulare in assenza di glucosio, l’integrazione di cisteina con NAC senza glutammina non è stata sufficiente a mantenere il livello di glutatione per sopportare lo stress ossidativo indotto dalla metformina (Fig. 2E). Il fenomeno può essere supportato dallo studio secondo cui la glutammina è importante per la carbossilazione riduttiva nelle cellule tumorali con mitocondri difettosi [19]. Pertanto, l’integrazione di NAC con glutammina ha ridotto significativamente la citotossicità della metformina in condizioni di assenza di glucosio (Fig. 2E).
In condizioni di glucosio elevato (25 mM), la metformina 10 mM ha mostrato solo un lieve effetto tumorale, ma non una morte cellulare significativa (Fig. 3B). Al contrario, 2,5 mM di metformina hanno indotto una morte cellulare significativa in presenza di una concentrazione di glucosio pari a 5,5 mM (Fig. 1B), indicando nel glucosio un’importante fonte di potere riducente. Pertanto, l’integrazione con 1-2 mM di glucosio è stata sufficiente a bloccare la citotossicità della metformina al livello abituale di glucosio (Fig. 1A), suggerendo che la citotossicità della metformina potrebbe essere insufficiente a indurre la morte cellulare in condizioni fisiologiche di glucosio. Tuttavia, il trattamento con metformina ha aumentato significativamente i ROS mitocondriali nella condizione di assenza di limitazione del glucosio. Non esiste ancora un rapporto incentrato sulla generazione di ROS mitocondriali nel contesto della citotossicità della metformina. Qui abbiamo osservato che già 1 mM di metformina aumentava significativamente il superossido mitocondriale insieme ai ROS cellulari (Fig. 2B), inducendo un rigonfiamento mitocondriale nelle cellule tumorali che non era irreversibile (Figg. S1A e S1B supplementari). Tuttavia, l’aumento dei ROS mitocondriali non sembra essere sufficiente per aumentare i ROS cellulari totali e il danno ossidativo. Le cellule tumorali esposte alla metformina cercano di minimizzare il danno ossidativo per mantenere la vitalità cellulare attraverso l’induzione della glicolisi aerobica e la produzione di NADPH. Questa idea può essere in parte supportata dal fatto che la deplezione del potere riducente mediante BSO o H2O2 esogeno ha aumentato la citotossicità della metformina nonostante la condizione di glucosio elevato (Fig. 3). Le cellule tumorali possono sopportare lo stress ossidativo attraverso la glicolisi aerobica e la quantità di GSH può essere mantenuta principalmente dal NADPH prodotto attraverso la via del pentoso fosfato [12].
L’attività antitumorale del DCA è stata dimostrata in vitro [4] e in modelli murini in vivo [27], [28] e in pazienti affetti da glioblastoma [18], tuttavia il valore IC50 del DCA è relativamente alto (>17 mM, 48 h) per indurre la morte cellulare. Inoltre, non esiste una specificità delle cellule tumorali nelle linee cellulari testate [26], e il DCA è selettivamente attivo contro le cellule con difetti nei mitocondri, come le cellule rho(0) o le cellule HCT116 p53 null con deficit del complesso IV. Abbiamo anche osservato che il DCA 20 mM da solo non è riuscito a indurre una morte cellulare efficace, tuttavia, combinato con la metformina, ha sinergizzato l’attività antitumorale del DCA con 10 mM (Fig. 5B e F).
Quando le cellule sono state trattate con metformina, l’attività della PDH è stata significativamente inibita attraverso la sua fosfoliazione sul residuo Ser293 (Fig. 4C) senza alcun cambiamento significativo dell’espressione dell’mRNA della PDK (dati non mostrati), indicando una modifica post-trascrizionale. Sebbene non siamo stati in grado di definire il meccanismo dell’inibizione dell’attività della PDH indotta dalla metformina, la diminuzione dell’attività della PDK da parte del DCA ha aumentato lo stress ossidativo indotto dalla metformina reprimendo la fosforilazione della PDH (Fig. 4), indicando l’inibizione dell’attività della PDK da parte del DCA, ma l’aumento della fosforilazione ossidativa insieme allo stress redox. In accordo con quanto sopra, il DCA ha ripristinato in modo significativo la respirazione mitocondriale inibita dalla metformina (Fig. 4F) e ha aumentato notevolmente la generazione di ROS mitocondriali indotta dalla metformina (Fig. 5A). Il ruolo protettivo degli antiossidanti nei confronti del trattamento combinato di metformina e DCA ci ha portato a concludere che il DCA può potenziare la citotossicità della metformina attraverso la riprogrammazione e il potenziamento dello stress ossidativo (Fig. 5C).
In sintesi, la citotossicità indotta dalla metformina è stata potenziata dalla privazione di glucosio. Anche in condizioni di glucosio elevato, un ulteriore stress da ROS, dovuto alla deplezione di GSH o all’insulto esogeno di H2O2, ha aumentato la citotossicità della metformina e l’aumento della respirazione mitocondriale. Concludiamo che la riprogrammazione mediata dal DCA del metabolismo del glucosio dalla glicolisi aerobica alla respirazione ossidativa aggrava lo stress ossidativo indotto dalla metformina e sensibilizza la citotossicità della metformina nelle cellule tumorali (Fig. 6).

Conflitto di interessi
Non ci sono interessi finanziari concorrenti legati a questo lavoro.
Riconoscimenti
Questo lavoro è stato sostenuto da una sovvenzione del National R&D Program for Cancer Control, Ministero della Salute e del Benessere, Repubblica di Corea (131280).
Appendice A. Materiale supplementare
I dati supplementari associati a questo articolo sono disponibili, nella versione online, all’indirizzo http://dx.doi.org/10.1016/j.canlet.2014.01.015.
RIFERIMENTI
1 I.M. Ahmad, N. Aykin-Burns, J.E. Sim, S.A. Walsh, R. Higashikubo, G.R. Buettner, S. Venkataraman, M.A. Mackey, S.W. Flanagan, L.W. Oberley, D.R. Spitz L’O2*- e l’H2O2 mitocondriali mediano lo stress indotto dalla deprivazione di glucosio nelle cellule tumorali umane J. Biol. Chem., 280 (2005), pp. 4254-42632 I. Ben Sahra, K. Laurent, S. Giuliano, F. Larbret, G. Ponzio, P. Gounon, Y. Le Marchand-Brustel, S. Giorgetti-Peraldi, M. Cormont, C. Bertolotto, M. Deckert, P. Auberger, J.F. Tanti, F. Bost Mirando al metabolismo delle cellule tumorali: la combinazione di metformina e 2-deossiglucosio induce l’apoptosi p53-dipendente nelle cellule di cancro alla prostata Cancer Res., 70 (2010), pp. 2465-2475
3 I. Ben Sahra, Y. Le Marchand-Brustel, J.F. Tanti, F. Bost Metformina nella terapia del cancro: una nuova prospettiva per un vecchio farmaco antidiabetico? Mol. Cancer Ther., 9 (2010), pp. 1092-1099
4 S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C.T. Lee, G.D. Lopaschuk, L. Puttagunta, S. Bonnet, G. Harry, K. Hashimoto, C.J. Porter, M.A. Andrade, B. Thebaud, E.D. Michelakis L’asse mitocondrio-canale K+ è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita tumorale Cancer Cell, 11 (2007), pp. 37-51
5 J.R. Cantor, D.M. Sabatini Il metabolismo delle cellule tumorali: un segno distintivo, molte facce Cancer Discov., 2 (2012), pp. 881-898
6 J.H. Cheong, E.S. Park, J. Liang, J.B. Dennison, D. Tsavachidou, C. Nguyen-Charles, K. Wa Cheng, H. Hall, D. Zhang, Y. Lu, M. Ravoori, V. Kundra, J. Ajani, J.S. Lee, W. Ki Hong, G.B. Mills La doppia inibizione della via energetica tumorale da parte di 2-deossiglucosio e metformina è efficace contro un ampio spettro di modelli tumorali preclinici Mol. Cancer Ther., 10 (2011), pp. 2350-2362
7 E.P. Clark, E.R. Epp, J.E. Biaglow, M. Morse-Gaudio, E. Zachgo Deplezione del glutatione, radiosensibilizzazione e potenziamento del misonidazolo in cellule ovariche di criceto cinese ipossiche mediante la butionina sulfoximina Radiat. Res., 98 (1984), pp. 370-380
8 M.Y. El-Mir, V. Nogueira, E. Fontaine, N. Averet, M. Rigoulet, X. Leverve Il dimetilbiguanide inibisce la respirazione cellulare attraverso un effetto indiretto mirato al complesso della catena respiratoria I J. Biol. Chem., 275 (2000), pp. 223-228
9 R.B. Hamanaka, N.S. Chandel Biologia cellulare. Effetto Warburg ed equilibrio redox Science, 334 (2011), pp. 1219-1220
10 T. Ishimoto, O. Nagano, T. Yae, M. Tamada, T. Motohara, H. Oshima, M. Oshima, T. Ikeda, R. Asaba, H. Yagi, T. Masuko, T. Shimizu, T. Ishikawa, K. Kai, E. Takahashi, Y. Imamura, Y. Baba, M. Ohmura, M. Suematsu, H. Baba, H. Saya La variante CD44 regola lo stato redox nelle cellule tumorali stabilizzando la subunità xCT del sistema xc(-) e promuovendo così la crescita tumorale Cancer Cell, 19 (2011), pp. 387-400
11 S. Javeshghani, M. Zakikhani, S. Austin, M. Bazile, M.J. Blouin, I. Topisirovic, J. St-Pierre, M.N. Pollak La fonte di carbonio e l’espressione di myc influenzano le azioni antiproliferative della metformina Cancer Res., 72 (2012), pp. 6257-6267
12 S.M. Jeon, N.S. Chandel, N. Hay L’AMPK regola l’omeostasi del NADPH per promuovere la sopravvivenza delle cellule tumorali durante lo stress energetico Nature, 485 (2012), pp. 661-665
13 G. Kroemer, J. Pouyssegur Il metabolismo delle cellule tumorali: il tallone d’Achille del cancro Cancer Cell, 13 (2008), pp. 472-482
14 M.A. Lea, J. Chacko, S. Bolikal, J.Y. Hong, R. Chung, A. Ortega, C. Desbordes L’aggiunta di 2-deossiglucosio aumenta l’inibizione della crescita ma inverte l’acidificazione in cellule di cancro del colon trattate con fenformina Anticancer Res., 31 (2011), pp. 421-426
15 X. Lin, F. Zhang, C.M. Bradbury, A. Kaushal, L. Li, D.R. Spitz, R.L. Aft, D. Gius la citotossicità e la radiosensibilizzazione indotta dal 2-deossi-d-glucosio nelle cellule tumorali è mediata da alterazioni del metabolismo tiolico Cancer Res., 63 (2003), pp. 3413-3417
16 L. Luo, W. Huang, R. Tao, N. Hu, Z.X. Xiao, Z. Luo L’attivazione ATM e LKB1 dipendente di AMPK sensibilizza le cellule tumorali all’apoptosi indotta dall’etoposide Cancer Lett., 328 (2013), pp. 114-119
17 J.A. Menendez, C. Oliveras-Ferraros, S. Cufi, B. Corominas-Faja, J. Joven, B. Martin-Castillo, A. Vazquez-Martin La metformina è sinteticamente letale con l’astinenza da glucosio nelle cellule tumorali Ciclo cellulare, 11 (2012), pp. 2782-2792
18 E.D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, C. Maguire, T.L. Gammer, J.R. Mackey, D. Fulton, B. Abdulkarim, M.S. McMurtry, K.C. Petruk Metabolic modulation of glioblastoma with dichloroacetate Sci. Translat. Med., 2 (2010), p. 31ra34
19 A.R. Mullen, W.W. Wheaton, E.S. Jin, P.H. Chen, L.B. Sullivan, T. Cheng, Y. Yang, W.M. Linehan, N.S. Chandel, R.J. DeBerardinis La carbossilazione riduttiva sostiene la crescita di cellule tumorali con mitocondri difettosi Nature, 481 (2012), pp. 385-388
20 M.R. Niewisch, Z. Kuci, H. Wolburg, M. Sautter, L. Krampen, B. Deubzer, R. Handgretinger, G. Bruchelt Influenza del dicloroacetato (DCA) sulla produzione di lattato e sul consumo di ossigeno nelle cellule di neuroblastoma: il DCA è un farmaco adatto alla terapia del neuroblastoma? Cell. Physiol. Biochem: Int. J. Exp. Cell. Physiol., Biochem., Pharmacol., 29 (2012), pp. 373-380
21 M.R. Owen, E. Doran, A.P. Halestrap Prova che la metformina esercita i suoi effetti antidiabetici attraverso l’inibizione del complesso 1 della catena respiratoria mitocondriale Biochem. J., 348 (Pt 3) (2000), pp. 607-614
22 C. Riganti, E. Gazzano, M. Polimeni, E. Aldieri, D. Ghigo La via del pentoso fosfato: una difesa antiossidante e un crocevia nel destino delle cellule tumorali Free Radical Biol. Med., 53 (2012), pp. 421-436
23 G.Z. Rocha, M.M. Dias, E.R. Ropelle, F. Osorio-Costa, F.A. Rossato, A.E. Vercesi, M.J. Saad, J.B. Carvalheira La metformina amplifica l’attivazione dell’AMPK indotta dalla chemioterapia e la crescita antitumorale Clin. Cancer Res: Official J. Am. Assoc. Cancer Res., 17 (2011), pp. 3993-4005
24 D.B. Shackelford, E. Abt, L. Gerken, D.S. Vasquez, A. Seki, M. Leblanc, L. Wei, M.C. Fishbein, J. Czernin, P.S. Mischel, R.J. Shaw L’inattivazione di LKB1 detta la risposta terapeutica del carcinoma polmonare non a piccole cellule al farmaco metabolico fenformina Cancer Cell, 23 (2013), pp. 143-158
25 J. Sinnett-Smith, K. Kisfalvi, R. Kui, E. RozengurtL’inibizione da parte della metformina dell’attivazione di mTORC1 dell’attivazione di mTORC1, della sintesi di DNA e della proliferazione in cellule di cancro del pancreas: dipendenza dalla concentrazione di glucosio e ruolo dell’AMPK Biochem. Biophys. Res. Commun., 430 (2013), pp. 352-357
26 L.H. Stockwin, S.X. Yu, S. Borgel, C. Hancock, T.L. Wolfe, L.R. Phillips, M.G. Hollingshead, D.L. Newton Il dicloroacetato di sodio colpisce selettivamente le cellule con difetti nell’ETC mitocondriale Int. J. Cancer J. Int. Cancer, 127 (2010), pp. 2510-2519
27 R.C. Sun, M. Fadia, J.E. Dahlstrom, C.R. Parish, P.G. Board, A.C. Blackburn L’inversione del fenotipo glicolitico da parte del dicloroacetato inibisce la crescita delle cellule metastatiche del cancro al seno in vitro e in vivo Breast Cancer Res. Treat., 120 (2010), pp. 253-260
28 G. Sutendra, P. Dromparis, A. Kinnaird, T.H. Stenson, A. Haromy, J.M. Parker, M.S. McMurtry, E.D. Michelakis L’attivazione mitocondriale mediante l’inibizione di PDKII sopprime la segnalazione di HIF1a e l’angiogenesi nel cancro Oncogene, 32 (2013), pp. 1638-1650
29 F. Tietze Metodo enzimatico per la determinazione quantitativa di quantità nanometriche di glutatione totale e ossidato: applicazioni al sangue di mammiferi e ad altri tessuti m8 Anal. Biochem., 27 (1969), pp. 502-522
30 M.G. Vander Heiden Puntare sul metabolismo del cancro: si apre una finestra terapeutica Nat. Rev. Drug Discovery, 10 (2011), pp. 671-684
31 P.S. Ward, C.B. Thompson Riprogrammazione metabolica: un segno distintivo del cancro che nemmeno Warburg aveva previsto Cancer Cell, 21 (2012), pp. 297-308
32 S. Whitehouse, R.H. Cooper, P.J. Randle Meccanismo di attivazione della piruvato deidrogenasi da parte del dicloroacetato e di altri acidi carbossilici alogenati Biochem. J., 141 (1974), pp. 761-774
33 M. Wu, A. Neilson, A.L. Swift, R. Moran, J. Tamagnine, D. Parslow, S. Armistead, K. Lemire, J. Orrell, J. Teich, S. Chomicz, D.A. Ferrick L’analisi metabolica multiparametrica rivela uno stretto legame tra l’attenuazione della funzione bioenergetica mitocondriale e la maggiore dipendenza dalla glicolisi nelle cellule tumorali umane Am. J. Physiol. Cell Physiol., 292 (2007), pp. C125-136
34 X. Xiao, Q. He, C. Lu, K.D. Werle, R.X. Zhao, J. Chen, B.C. Davis, R. Cui, J. Liang, Z.X. Xu La metformina ostacola la crescita delle cellule di carcinoma cervicale intatte alla chinasi B1 del fegato Gynecol. Oncol., 127 (2012), pp. 249-255
35 Y. Zhuang, W.K. Miskimins La metformina induce la morte cellulare sia caspasi-dipendente che polimerasi-dipendente in cellule di cancro al seno Mol. Cancer Res: MCR, 9 (2011), pp. 603-615
36 J.W. Zmijewski, E. Lorne, X. Zhao, Y. Tsuruta, Y. Sha, G. Liu, G.P. Siegal, E. Abraham Il complesso respiratorio mitocondriale I regola l’attivazione dei neutrofili e la gravità del danno polmonare Am. J. Respir. Crit. Care Med., 178 (2008), pp. 168-179