Yong Won Choi1, In Kyoung Lim *
1 Abteilung für Biochemie und Molekularbiologie, BK21 Cell Transformation and Restoration Project, Ajou University School of Medicine, Suwon 443-721, Republik Korea*
Abteilung für Biochemie und Molekularbiologie, Ajou University School of Medicine, San 5 Woncheon-dong, Yeongtonggu, Suwon 443-721, Republik Korea; Tel
.:
+82 31 219 5051; Fax: +82 31 219 5059. E-Mail-Adresse: [email protected] (I.K. Lim).
Received: 29. Oktober 2013Angenommen
: 20. Januar 2014Geändert
: in überarbeiteter Form 8. Januar
2014Veröffentlicht/online
verfügbar
: 27. Januar 2014
Zusammenfassung
Um die Sensibilisierung der Metformin-Zytotoxizität zu untersuchen, wurden Krebszellen mit Dichloracetat (DCA), einem Inhibitor der Pyruvat-Dehydrogenase-Kinase (PDK), behandelt. Die Metformin-Zytotoxizität hing hauptsächlich von der Verfügbarkeit von Glukose und der durch den Pentosephosphatweg erzeugten Reduktionskraft ab, während die DCA-Kombinationsbehandlung die Metformin-Zytotoxizität über eine Umprogrammierung des Glukosestoffwechsels durch Hemmung der PDK und Erhöhung der mitochondrialen Atmung verstärkte. Die DCA-Kombinationsbehandlung löste trotz hoher Glukose- und GSH-Konzentration eher den Zelltod als das Überleben der Zellen aus. Zusammenfassend lässt sich sagen, dass DCA die Metformin-Zytotoxizität sensibilisiert, indem es den Glukosestoffwechsel teilweise von der aeroben Glykolyse auf die mitochondriale Oxidation umprogrammiert, was durch Messungen des Glukoseverbrauchs, der Laktatfreisetzung und des Verhältnisses zwischen Sauerstoffverbrauchsrate und extrazellulärer Übersäuerungsrate belegt wird.
Schlüsselwörter: Metformin-Dichloracetat (DCA), Oxidativer Stress, Glukoseentzug, Glutathiongehalt
© 2014 Veröffentlicht von Elsevier Ireland Ltd.
EINLEITUNG
Es gibt immer mehr Hinweise darauf, dass Stoffwechselstörungen von Krebszellen wie aerobe Glykolyse oder Glutaminsucht für die Krebsentstehung unvermeidlich sind und nicht nur ein Epiphänomen darstellen [13], [31], und es wurden enorme Anstrengungen unternommen, um arzneimittelwirksame Ziele und chemische Kandidaten für den Krebsstoffwechsel zu finden [5], [30]. Auf der Grundlage epidemiologischer, präklinischer und klinischer Daten wurde Metformin, ein Biguanid, das zur Behandlung von Diabetes mellitus eingesetzt wird, zu einem der attraktivsten und vielversprechendsten Medikamente, die auf den Krebsstoffwechsel abzielen. Obwohl der Mechanismus von Metformin noch nicht vollständig geklärt ist, ist bekannt, dass Metformin die intrazelluläre Funktion des Atmungskettenkomplexes I hemmt [8], [21]. Derzeit deuten viele Hinweise darauf hin, dass die AMPK-Aktivierung ein Hauptknotenpunkt der Anti-Tumor-Wirkungen von Metformin ist [3], [16],[23], weshalb LKB1-/–Krebszellen im In-vitro-Kultursystem resistenter gegen die Metformin-induzierte Zytotoxizität sind [22], [34]. Im Gegensatz dazu hat sich gezeigt, dass die AMPK-Aktivierung Krebszellen über die Regulierung der NADPH-Homöostase vor Energiestress schützt [12], weshalb LKB1-/–Krebszellen im Maus-Lungenkrebsmodell empfindlicher auf Phenformin-induzierten metabolischen Stress reagieren [24]. In den verwirrenden Zusammenhängen gibt es Berichte, dass die Anti-Tumor-Wirkung von Metformin von der Glukosekonzentration im Kulturmedium abhängt [11], [17], [25]; Glukose-Entzug verstärkt die Metformin-Zytotoxizität erheblich [17], während die Metformin-induzierte AMPK-Aktivierung unter hohen Glukosebedingungen (25 mM) ineffizient ist [25]. Daher muss das Verständnis des biochemischen Mechanismus der Metformin-Zytotoxizität noch vertieft werden, bevor Metformin als Krebsmittel mit Schwerpunkt auf dem Glukosestoffwechsel eingesetzt wird.
Der in Krebszellen häufig beobachtete Warburg-Effekt ist nicht nur für die Energiegewinnung wichtig, sondern auch für die Aufrechterhaltung eines reduzierten Status unter der feindlichen Mikroumgebung des Tumors. Glukose ist die Hauptquelle für reduzierende Energie, NADPH, über den Pentosephosphatweg [1], [9], [22]. Der durch 2-Desoxyglukose(2DG) vermittelte Tod von Krebszellen hängt hauptsächlich von einem unerträglichen oxidativen Stress und einer Energiekrise ab [15]. Auf der Grundlage des Konzepts, dass eine erhöhte Glykolyse Krebszellen vor oxidativem Stress schützt, wurde angenommen, dass eine durch Metformin gesteigerte Glykolyse in der Lage ist, Zellen vor mitochondrialem oxidativem Stress zu schützen, der aus der Hemmung des Atmungskettenkomplexes I resultiert [35]. Daher untersuchten wir, ob Metformin-induzierter mitochondrialer Stress durch oxidativen Stress aufgrund von GSH-Verlust unter Glukose-Entzug oderH2O2-Behandlungunter Glukose-ausreichenden Bedingungen verstärkt werden kann. Basierend auf den Berichten, dass die Hemmung der Glykolyse durch 2-DG die zytotoxische Wirkung von Metformin verstärkt [2], [6], [14] und dass Dichloracetat (DCA) die Glykolyse durch Aktivierung der Pryruvat-Dehydrogenase (PDH) [32] zusammen mit dem oxidativen Stoffwechsel und der Antitumoraktivität [20] reduziert.
In der vorliegenden Studie untersuchten wir die Auswirkungen von DCA auf die Metformin-Zytotoxizität und die Regulierung des Glukosestoffwechsels. Im Gegensatz zu den Wirkungen von Metformin allein erhöhte die kombinierte Behandlung von Krebszellen mit Metformin und DCA deutlich die PDH-Aktivität, nicht aber die Glykolyse, wodurch die mitochondriale Atmung in Krebszellen wiederhergestellt wurde. Die Umprogrammierung des Glukosestoffwechsels stand in engem Zusammenhang mit schwerem oxidativem Stress. Zusammenfassend lässt sich sagen, dass die DCA-vermittelte Umprogrammierung der aeroben Glykolyse auf mitochondriale Oxidation den Metformin-induzierten mitochondrialen und zellulären Redoxstress verstärkte, der ausreichte, um trotz eines hohen Glukosespiegels einen massiven Zelltod auszulösen.
Materialien und Methoden
Zellen und Reagenzien
HeLa-, MCF7- und MDA-MB-231-Zellen wurden in DMEM (Gibco) mit Glukose (0-25 mM), ergänzt mit 10 % fötalem Rinderserum und 100 U/mL Gentamycin bei 37 °C und 5 %CO2 gezüchtet. Metformin, Dichloracetat (DCA), l-Buthionin-Sulfoximin (BSO),H2O2, Glutathion-Monoethylester (GSH-MEE) und N-Acetyl-l-Cystein (NAC) wurden von Sigma Chemical Co. (St. Louis, MO) bezogen. Trolox stammte von Biomol International, L.P., und 2′,7′-Dichlordihydrofluorescein-Diacetat (H2DCFDA), MitoSOX Red, Propidiumiodid (PI) und Calcein AM wurden von Molecular Probes (Eugene, OR) bezogen.
Messung der Zelllebensfähigkeit und Soft-Agar-Koloniebildungstest
Die Zelllebensfähigkeit wurde nach verschiedenen Behandlungen für die angegebenen Zeiten durch Ausschluss des Farbstoffs Trypanblau (Sigma-Aldrich) bestimmt. Für den Test wurden 4 ×104 Zellen in 12-Well-Platten plattiert und am nächsten Tag mit Chemikalien behandelt. Die Zellen wurden trypsinisiert und mit 0,4 % Trypanblau (1:1) gemischt. Der Prozentsatz der lebensfähigen Zellen entspricht der Anzahl der ungefärbten Zellen/Anzahl der Gesamtzellen × 100. Der Zelltod wurde auch mit dem Laktatdehydrogenase(LDH)-Freisetzungs-Assay-Kit nach dem Protokoll des Herstellers (Takara, Japan) gemessen. Für den Soft-Agar-Koloniebildungstest wurden HeLa-Zellen (1,5 ×103) in 0,6 %iger Agaroselösung ausplattiert und auf ein 1,0 %iges Agarosebett geschichtet, bevor sie mit verschiedenen Konzentrationen von Metformin, DCA oder beiden in Kombination behandelt wurden. Kolonien, die nach 2 Wochen größer als 125 μm waren, wurden als positiv gezählt.
Messung der zellulären ROS-Werte
Intrazelluläre und mitochondriale ROS wurden mit der oxidationsempfindlichen Fluoreszenzsonde Dichlordihydroflourescin-Diacetat (H2DCF-DA) bzw. MitoSOX Red (Invitrogen) gemessen. HeLa-Zellen, die für die angegebenen Zeiten mit Metformin, DCA oder beiden behandelt wurden, wurden 10 Minuten lang bei 37 °C mit DCF-DA (20 μM) oder MitoSOX (5 μM) inkubiert. Anschließend wurden die Zellen zweimal mit phosphatgepufferter Kochsalzlösung (PBS) gewaschen und zur Erfassung und Analyse einer Durchflusszytometrie (BD FACSCanto II, BD Biosciences, San Jose, CA) unterzogen. Die Fluoreszenzintensität wurde auch durch Fluoreszenzmikroskopie bestimmt.
Messung des zellulären Glutathionspiegels
Zur Bestimmung des Redox-Status des intrazellulären Glutathions wurde der Gehalt an reduziertem und oxidiertem Glutathion (GSH, GSSG) nach der zuvor beschriebenen Methode analysiert [29]. Zur Bestimmung des Gesamtglutathions wurden die Zellen geerntet und in 1%iger Sulfosalicylsäure suspendiert. Nach der Entfernung der Aggregate durch 10-minütiges Zentrifugieren bei 8000 g wurden die Überstände für den Test verwendet. Die Reaktion wurde gestartet, indem der Überstand (1 μg) in eine 96-Well-Mikrotiterplatte gegeben wurde, die 200 μl der Testmischung [125 mM NaPO4, pH 7,5, 0,3 mM NADPH, 6 mM 5,50-Dithiobis(2-nitrobenzoesäure) (DNTB; Sigma, St. Louis, MO), 0,12 U Glutathionreduktase] enthielt. Die maximale Reduktionsrate von DNTB durch GSH wurde durch Messung der Absorption bei 405 nm ermittelt. Die Menge des Gesamtglutathions wurde anhand der Kalibrierungskurve unter Verwendung von Standard-GSH (Sigma) berechnet. Um den GSSG-Gehalt zu bestimmen, wurden die Zellen mit 2-Vinylpyridin vorbehandelt, um vorhandenes GSH zu entfernen, und der Überstand (50 μg) wurde nach demselben Protokoll wie für das Gesamtglutathion verarbeitet.
Bestimmung der Pyruvatdehydrogenase (PDH)-Aktivität
Die PDH-Aktivität wurde mit dem PDH Enzyme Activity Microplate Assay Kit (Abcam, Cambridge, MA) gemäß dem Protokoll gemessen. Die Zellen wurden in dem mit dem Kit gelieferten Probenpuffer lysiert, und das gesamte Zellprotein in einer Konzentration von 5 mg/ml wurde in dreifacher Ausfertigung auf die mit einem Anti-PDH-Antikörper beschichtete Mikroplatte gegeben und 3 Stunden lang bei Raumtemperatur inkubiert. Nach dem Waschen der Proben wurde die Reaktionsmischung zugegeben, und die Absorption des Reaktionsprodukts der mit dem Reporterfarbstoff gekoppelten Reduktion von NAD+ zu NADH wurde 50 Minuten lang in 0,5-minütigen Abständen bei 450 nm mit dem Eon-Mikroplatten-Spektrophotometer (BioStack, Winooski, VT) gemessen. Die PDH-Aktivität wurde aus der Steigung der mOD pro Zeit (min) Kurve (ΔmOD/min) berechnet und auf die Kontrollgruppe normiert.
Analyse des Glukoseverbrauchs und der Laktatproduktion
Hela-Zellen (1 ×104/ml), die in DMEM mit 25 mM Glukose kultiviert wurden, wurden mit Metformin, DCA oder beiden behandelt. Nach 48 Stunden wurden die Glukose- und Laktatkonzentrationen in den Medien mit dem YSI 7100 Multiparameter-Bioanalytiksystem gemessen. Medien ohne Zellkultur wurden für die Messung der ursprünglichen Glukose- und Laktatkonzentration verwendet. Glucoseverbrauch = (Glucosekonzentration der Medien ohne Zellkultur) – (Glucosekonzentration der Medien der behandelten Gruppe). Laktatproduktion = (Laktatkonzentration der Medien der behandelten Gruppe) – (Laktatkonzentration der Medien ohne Zellkultur).
Messung der Raten der oxidativen Phosphorylierung und der Glykolyse
Die Sauerstoffverbrauchsrate(OCR) und die extrazelluläre Übersäuerungsrate (ECAR) wurden mit einem XF24 Extracellular Flux Analyzer von Seahorse Bioscience (North Billerica, MA) gemessen [33]. Dazu wurden HeLa-Zellen in XF-Mikroplatten mit 24 Vertiefungen (1 ×104) ausgesät und 24 Stunden lang bei 37 °C inkubiert, bevor sie 12 Stunden lang mit Metformin, DCA oder beidem behandelt wurden. Anschließend wurden die Zellen gewaschen und 1 Stunde lang bei 37 °C in einem Inkubator ohneCO2 mit DMEM, ergänzt mit GlutaMax-1 (200 mM), Glukose (25 mM), Natriumchlorid (32 mM) und Phenolrot ohne Bikarbonat, äquilibriert. Die OCR (pmol/min) und ECAR (mpH/min) wurden dreimal für jeweils 3 Minuten gemessen. Nach dem Experiment wurde die Anzahl der Zellen zur Normalisierung gezählt.
Immunoblot-Analyse
Zelllysate (40 μg), die in RIPA-Puffer mit Phosphatase-Inhibitoren hergestellt wurden, wurden auf SDS-PAGE getrennt, bevor sie auf eine PVDF-Membran (Millipore Corp) übertragen wurden. Die Blots wurden mit primären Antikörpern hybridisiert und mit dem ECL-System (Amersham Biosciences, Buckinghamshire, UK) visualisiert. Die Antikörper wurden gekauft: anti-p-PDH (S293) von Calbiochem, anti-PARP und anti-PDH von Abcam, anti-Caspase 3 von Cell Signaling, anti-cyclin D1, anti-cyclin E und anti-β-actin von Santa Cruz Biotechnology.
Statistische Analyse
Die numerischen Daten wurden als Mittelwert ± SD der unabhängigen Bestimmungen angegeben. Es wurde ein unabhängiger t-Testoder eine ANOVA durchgeführt, und Mehrfachvergleiche wurden mit Tukey HSD ausgewertet. P-Werte von weniger als 0,05 wurden als signifikant angesehen.
Ergebnisse
DieMetfromin-Zytotoxizität wird durch verfügbare Glukose wiederhergestellt
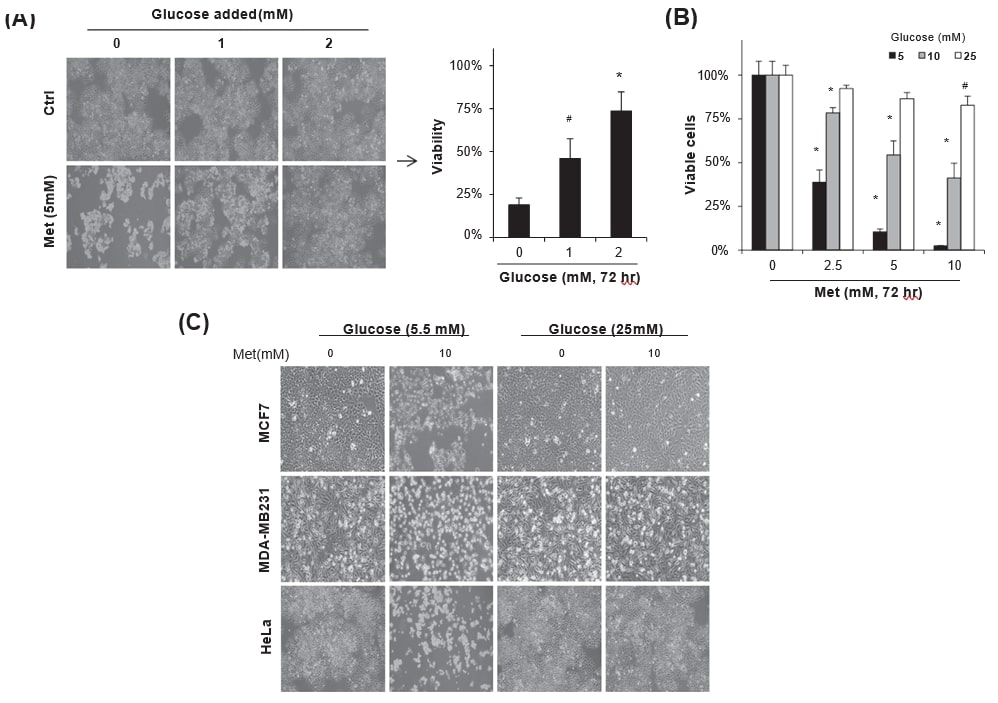
Um die biochemischen Wege zu verstehen, die den Metformin-induzierten Krebszelltod regulieren, wurden HeLa-Zellen, die in DMEM mit 5,5 mM Glukose aufbewahrt wurden, 72 Stunden lang mit Metformin (5 mM) und täglicher Zugabe von Glukose (0-2 mM) behandelt. Die Zugabe von Glukose erhöhte die Lebensfähigkeit der mit Metformin behandelten Zellen signifikant (Abb. 1A, #p < 0,05 und *p < 0,001 im Vergleich zu keiner Zugabe), was gut mit früheren Berichten übereinstimmt [11], [17], [25]. Wenn HeLa-Zellen, die in Medien mit 5-25 mM Glukose gehalten wurden, 72 Stunden lang mit 0-10 mM Metformin inkubiert wurden, war der Metformin-induzierte Zelltod abhängig von der Glukosekonzentration im Kulturmedium. Insbesondere ein hoher Glukosegehalt (25 mM) schützte die Metformin-Zytotoxizität unabhängig von der Metformin-Konzentration signifikant (Abb. 1B). Der durch 10 mM Metformin induzierte Zelltod wurde durch 25 mM Glukose im Kulturmedium nicht nur in HeLa-Zellen, sondern auch in MCF7- und MDA-MB231-Brustkrebszellen geschützt, während 5,5 mM Glukose die Zellen nicht vor der toxischen Wirkung schützte (Abb. 1C), was unter dem Phasenkontrastmikroskop beobachtet wurde. Die Ergebnisse deuten darauf hin, dass das Gleichgewicht zwischen den Konzentrationen von Metformin und Glukose in den Kulturmedien wichtig für die Regulierung der Metformin-Zytotoxizität ist.
DieMetformin-Zytotoxizität wird durch GSH-vermittelte Redox-Veränderungen reguliert
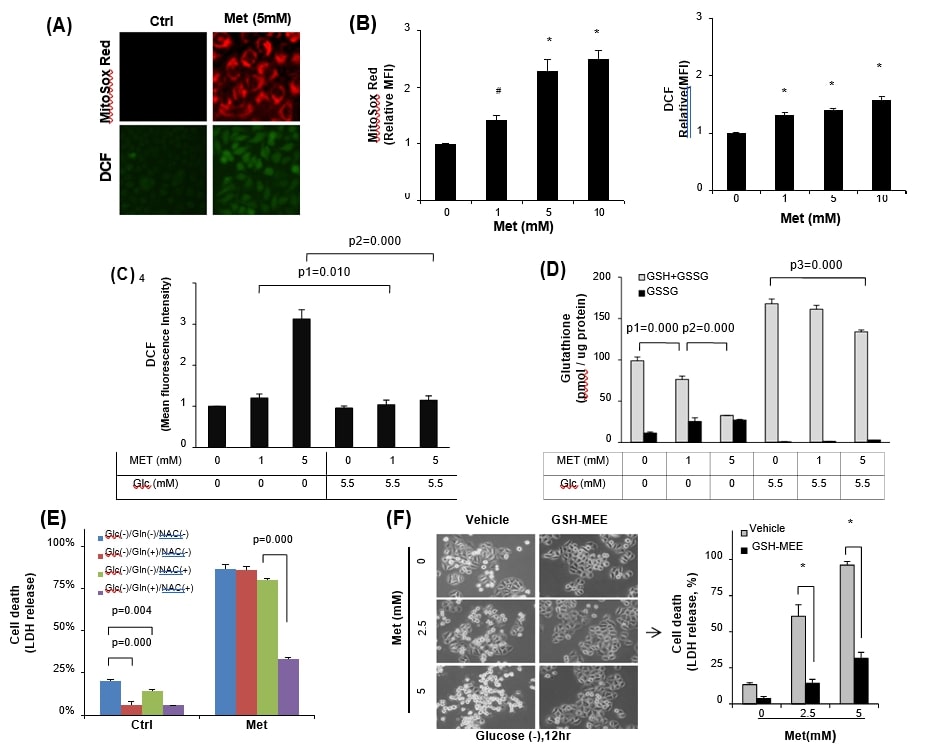
Da Metformin den mitochondrialen ROS-Spiegel durch Hemmung des Atmungskettenkomplexes I erhöht [8], [21], [36], haben wir daher untersucht, ob der Metformin-induzierte mitochondriale oxidative Stress schwerwiegend genug war, um den Tod von Krebszellen unter Glukose-insuffizienten Bedingungen zu induzieren. HeLa-Zellen, die in glukosehaltigen Medien gehalten wurden, wurden mit 5 mM Metformin behandelt, und anschließend wurden die mitochondriale Superoxid- und die gesamte zelluläre ROS-Konzentration mithilfe von MitoSox Red bzw. DCFDA bestimmt (Abb. 2A). Tatsächlich erhöhte Metformin die mitochondriale und die gesamte zelluläre ROS-Konzentration in Abhängigkeit von der Metformin-Konzentration, jedoch war die Fluoreszenz von MitoSox Red meist höher als die der gesamten zellulären ROS-Konzentration in HeLa-Zellen (Abb. 2B). Wie erwartet war der Metformin-induzierte zelluläre ROS-Spiegel unter glukosefreien Bedingungen deutlich höher als in glukosehaltigen Medien (Abb. 2C). Diese Daten stützen den Bericht [15], dass Glukoseentzug den zellulären Glutathionpool (GSH + GSSG) reduziert. Als der Glutathion-Pool gemessen wurde, war die Verringerung von GSH im Verhältnis zu GSSG unter den glukosefreien Bedingungen als Reaktion auf Metformin signifikanter (p = 0,000). Der Gesamtglutathion-Gehalt wurde jedoch ohne signifikanten Anstieg von GSSG in den glukosehaltigen (5,5 mM) Medien aufrechterhalten (Abb. 2D), was darauf hindeutet, dass Glukose in den Kulturmedien eine wichtige Quelle für die GSH-Bildung ist. Um die Regulierung der Metformin-Zytotoxizität durch Redox-Stress unter Glukose-Entzug zu untersuchen, wurden Antioxidantien eingesetzt. Die Behandlung von HeLa-Zellen mit entweder 10 mM Glutamin oder N-Acetyl-l-Cystein (NAC) allein reduzierte den Zelltod, der in den glukosefreien Medien ohne Metformin-Zusatz beobachtet wurde, signifikant (Abb. 2E, p = 0,000 bzw. p = 0,004 im Vergleich zu keiner Behandlung), jedoch konnte dieselbe Behandlung die Metformin (5 mM)-Zytotoxizität in den glukosefreien Medien nicht verbessern. Andererseits konnte eine kombinierte Behandlung von Glutamin mit NAC die HeLa-Zellen vor der Metformin (5 mM)-Zytotoxizität schützen (Abb. 2E, p = 0,000). Um direkt zu bestätigen, ob reduziertes Glutathion bei der Hemmung der Metformin-Zytotoxizität eine Rolle spielt, wurde die zellpermeable Form von GSH (GSH-MEE) verwendet. Die Zugabe von GSH-MEE verringerte den Zelltod der HeLa-Zellen im Vergleich zur Vehikelkontrolle auch unter glukosefreien Bedingungen erheblich (Abb. 2F). Die Daten zeigen einen synergistischen Effekt des Glukoseentzugs bei der Metformin-Zytotoxizität über oxidativen Stress durch Verarmung von GSH.
Verstärkung des oxidativen Stresses verschlimmert die Metformin-Zytotoxizität trotz hohem Glukosespiegel
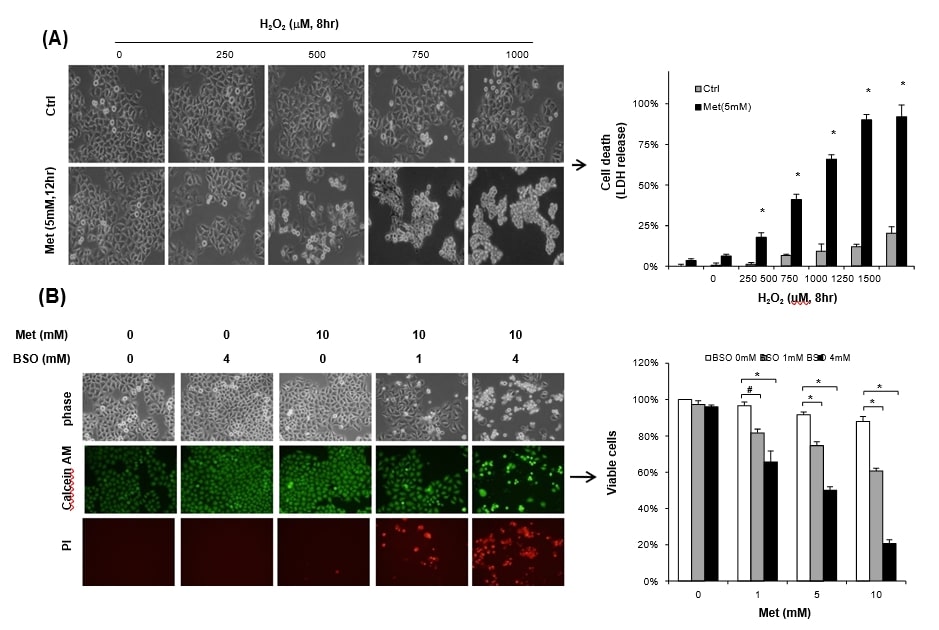
Um sicherzustellen, dass die Zellen, die Metformin ausgesetzt sind, anfälliger für Stress werden, wurden HeLa-Zellen, die mit 25 mM Glukose versorgt wurden, 12 Stunden lang mit 5 mM Metformin zusammen mitH2O2behandelt. Die alleinige Behandlung mitH2O2oder 5 mM Metformin führte nicht zu einem signifikanten Zelltod, während die kombinierte Behandlung von Metformin mitH2O2(über 500 μM) den Zelltod deutlich erhöhte, gemessen an der LDH-Freisetzung (Abb. 3A). Nicht nur exogenesH2O2, sondern auch die Verarmung des Glutathion-Pools durch BSO, einen Inhibitor der GSH-Synthese, führte ebenfalls zu einem signifikanten Grad an Metformin-Zytotoxizität. Eine 48-stündige Inkubation mit Metformin (10 mM) oder 4 mM BSO allein führte unter hohen Glukosebedingungen (25 mM) nicht zu einem signifikanten Zelltod, wie die Calcein AM-Färbung zeigt (Abb. 3B links). Die kombinierte Behandlung von Metformin mit BSO führte jedoch zu einem signifikanten Anstieg des Propidiumjodid-positiven Zelltods, der durch Trypanblau-Ausschluss gemessen wurde. BSO (1 mM) sensibilisierte signifikant die 1 mM Metformin-Zytotoxizität unter den Bedingungen der hohen Glukose (25 mM) (Abb. 3B rechts). Unsere Ergebnisse werden durch frühere Studien bestätigt, wonach GSH das am häufigsten vorkommende und wichtigste biologische Antioxidans gegen oxidativen Stress in Krebszellen ist [7], [10].
Dichloracetat steigert die mitochondriale Atmung durch Aktivierung der Pyruvatdehydrogenase
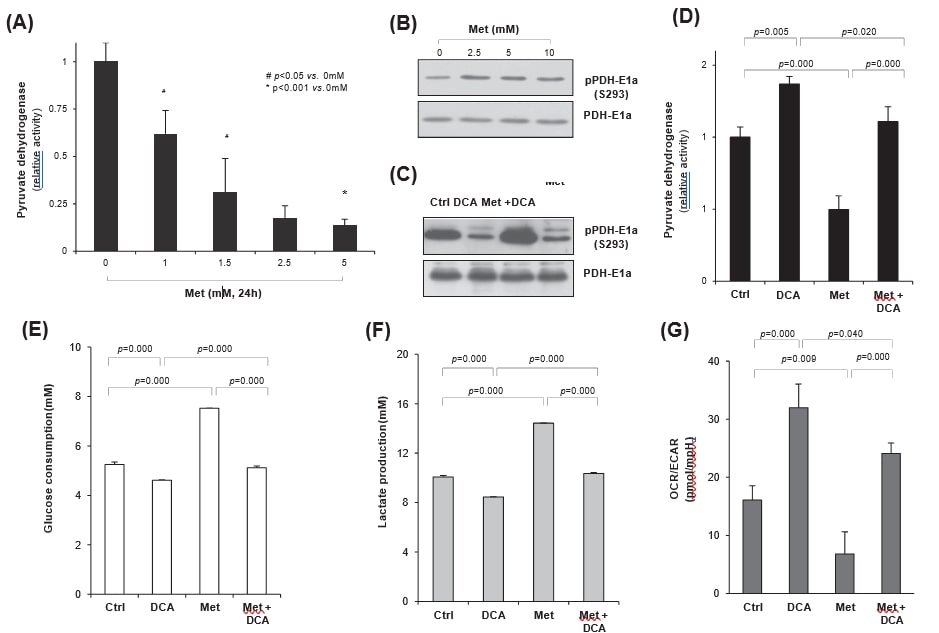
Da die PDH das Schicksal des Glukosestoffwechsels entweder in der Mitochondrie oder im Zytosol bestimmt, ist es sehr wahrscheinlich, dass eine Steigerung der PDH-Aktivität die mitochondriale Atmung und nicht die aerobe Glykolyse in Krebszellen erhöht. Daher untersuchten wir, ob die Metformin-induzierte Glykolyse durch DCA, einen Inhibitor der PDH-Kinase (PDK), reguliert wird. Wenn HeLa-Zellen mit Metformin behandelt wurden, war die PDH-Aktivität dosisabhängig deutlich verringert (Abb. 4A). Die Behandlung mit Meformin erhöhte die PDH-E1a-Phosphorylierung deutlich, was auf eine PDK-Aktivierung im Gegensatz zur PDH-Hemmung hinweist (Abb. 4B). Andererseits reduzierte die DCA-Behandlung die Metformin-induzierte PDH-E1a-Phosphorylierung am S293-Rest unabhängig von der Metformin-Behandlung signifikant, was auf eine Erhöhung oder Aufrechterhaltung der PDH-Aktivität durch DCA hindeutet (Abb. 4C). Tatsächlich erhöhte DCA die PDH-Aktivität signifikant (p = 0,005), im Gegensatz zur signifikanten Hemmung durch Metformin (p = 0,000), im Vergleich zur Kontrolle. Darüber hinaus wurde durch die gleichzeitige Behandlung von DCA und Metformin die durch Metformin gehemmte PDH-Aktivität signifikant wiederhergestellt (p = 0,000), und zwar bis auf das Kontrollniveau (Abb. 4D); DCA wirkte der Wirkung von Metformin auf die PDH-Aktivität entgegen. Um die Regulierung der metabolischen Veränderungen durch DCA oder Metformin zu bewerten, wurden die Raten des Glukoseverbrauchs und der Laktatproduktion in den Kulturmedien gemessen; die DCA-Behandlung führte zu einer signifikanten Verringerung des Glukoseverbrauchs (p = 0,000), im Gegensatz zu einem signifikanten Anstieg durch Metformin (p = 0,000, Abb. 4E). Allerdings war die Veränderung durch die gleichzeitige Behandlung mit Metformin und DCA unbedeutend (p = 0,125 gegenüber der Kontrolle), was wiederum die antagonistische Beziehung zwischen DCA und Metformin bestätigt. Darüber hinaus zeigte die Laktatfreisetzung (Abb. 4F) sowohl bei DCA als auch bei Metformin allein das gleiche Muster wie die Glukoseverbrauchsrate (Abb. 4E), was auf einen Wechsel des Glukosestoffwechsels von der Metformin-induzierten aeroben Glykolyse zur mitochondrialen Oxidation durch die DCA-Kombehandlung hindeutet. Bei der Messung der Sauerstoffverbrauchsrate (OCR) und der extrazellulären Übersäuerungsrate (ECAR) mit dem XF24-Analysegerät wurde das Verhältnis von OCR/ECAR durch die DCA-Behandlung signifikant erhöht (p = 0,000), was auf eine erhöhte mitochondriale Atmung hindeutet, während es durch Metformin (p = 0,009) im Vergleich zur Kontrolle verringert wurde. In der Tat erholte sich die mitochondriale Atmung durch die gleichzeitige Behandlung von Metformin und DCA im Vergleich zu Metformin allein signifikant (p = 0,000), auch wenn sie etwas niedriger war als bei DCA allein (p = 0,04). Insgesamt programmierte die gemeinsame Behandlung von Metformin und DCA den Glukosestoffwechsel teilweise von der aeroben Glykolyse zur mitochondrialen Oxidation um (Abb. 4G).
Dichloracetat erhöht den oxidativen Stress und die Metformin-Zytotoxizität unter hohen Glukosebedingungen
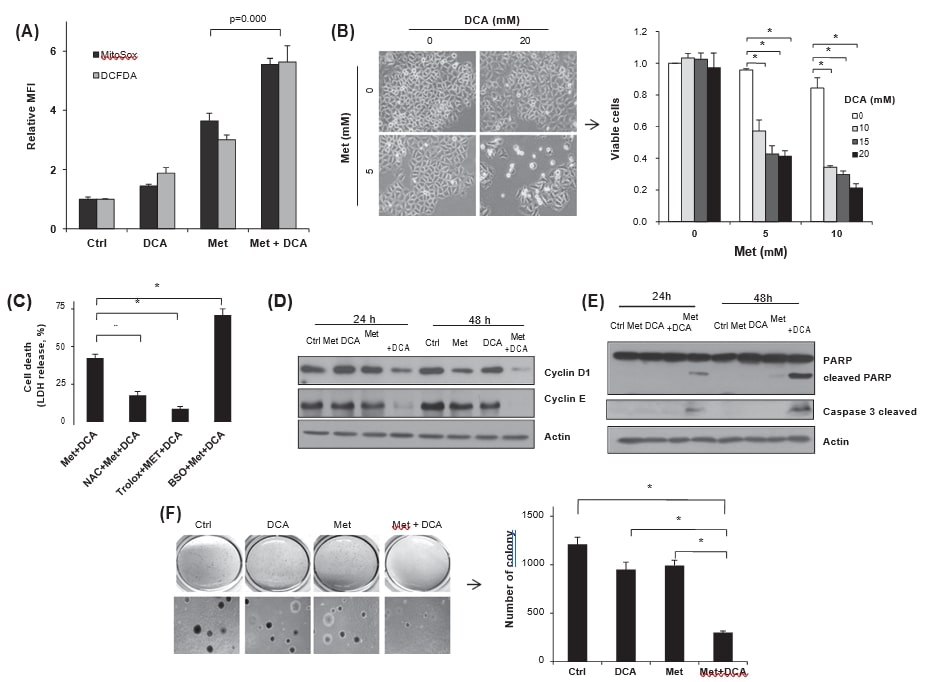
Um die Auswirkungen der DCA-vermittelten metabolischen Umprogrammierung auf den zellulären ROS-Spiegel zu untersuchen, wurden FACS-Analysen mit MitoSox Red und DCFDA durchgeführt. Die gemeinsame Behandlung von HeLa-Zellen mit Metformin und DCA erhöhte sowohl den mitochondrialen Superoxid- als auch den gesamten zellulären ROS-Spiegel (Abb. 5A), was darauf hindeutet, dass die metabolische Umprogrammierung den oxidativen Stress durch DCA verschlimmern könnte. Um festzustellen, ob die durch DCA verstärkte ROS-Bildung die Metformin-Zytotoxizität erhöht oder nicht, wurde die Lebensfähigkeit der Zellen nach der Behandlung mit Metformin, DCA oder beidem gezählt. Im Gegensatz zum vernachlässigbaren Zelltod durch DCA allein (∼20 mM) reduzierte die gleichzeitige Behandlung von DCA und Metformin die Lebensfähigkeit trotz hoher Glukose deutlich (Abb. 5B). Um die Auswirkungen der erhöhten ROS auf den Zelltod zu untersuchen, wurden die Antioxidantien NAC oder Trolox oder das Prooxidans BSO vor der gemeinsamen Behandlung mit Metformin und DCA vorbehandelt. Tatsächlich reduzierten die Antioxidantien die LDH-Freisetzung in das Medium signifikant, während sie durch BSO vor der Behandlung erhöht wurde (Abb. 5C), was auf eine signifikante Regulierung der Metformin-Zytotoxizität durch die Modulation des oxidativen Stresses durch DCA hinweist. Ein solcher synergistischer Effekt der Zytotoxizität wurde auch bei anderen Krebszellen, MDA-MB-231, EJ und MCF7, beobachtet (Daten nicht gezeigt). Darüber hinaus hemmte die Co-Behandlung von Metformin und DCA signifikant die Synthese von Cyclin D1 und Cyclin E sowie die PARP-Spaltung (Abb. 5D und E), was auf eine verringerte Zellproliferation und Apoptose-Induktion hindeutet. Schließlich reduzierte die gemeinsame Behandlung das verankerungsunabhängige Wachstum von HeLa-Zellen signifikant im Vergleich zu den Einzelbehandlungen und der Kontrolle (Abb. 5F).
Diskussion
Wir konnten nachweisen, dass die Umprogrammierung des Glukosestoffwechsels durch die Behandlung von HeLa-Zellen mit Meformin und Dicholoroacetat den Zelltod durch eine Hochregulierung des ROS-Niveaus fördert, indem die mitochondriale Oxidation anstelle der aeroben Glykolyse gesteigert wird (Abb. 6). DCA wurde als Mittel zur Förderung der oxidativen Phosphorylierung gegenüber der Glykolyse eingesetzt und sollte den Metformin-vermittelten Glukosestoffwechsel umprogrammieren. DCA aktiviert die PDH durch Hemmung der PDK, so dass Pyruvat anstelle von Laktat in Acetyl-CoA umgewandelt werden kann. Als biochemischen Mechanismus der durch Glukoseentzug induzierten Sensibilisierung von Krebszellen für die Metformin-Zytotoxizität konnten wir nachweisen, dass der durch Metformin induzierte oxidative Stress unter Glukoseentzug verstärkt wurde (Abb. 2C). Tatsächlich verbrauchte Metformin viel mehr Glutathion als im Grundzustand (Abb. 2D), während der Verbrauch in Gegenwart von Glukose (5,5 mM) viel geringer war. Obwohl die Vorbehandlung der HeLa-Zellen mit NAC den Zelltod unter glukosefreien Bedingungen signifikant blockierte, reichte die Cysteinergänzung durch NAC ohne Glutamin nicht aus, um den Glutathionspiegel aufrechtzuerhalten, um den durch Metformin verursachten oxidativen Stress zu überstehen (Abb. 2E). Dieses Phänomen kann durch die Studie unterstützt werden, dass Glutamin für die reduktive Carboxylierung in Tumorzellen mit defekten Mitochondrien wichtig ist [19]. Daher reduzierte die Ergänzung von NAC mit Glutamin die Metformin-Zytotoxizität unter glukosefreien Bedingungen deutlich (Abb. 2E).
Unter Bedingungen mit hohem Glukosegehalt (25 mM) zeigte 10 mM Metformin nur eine leichte tumoristische Wirkung, aber keinen signifikanten Zelltod (Abb. 3B). Im Gegensatz dazu löste 2,5 mM Metformin bei einer Glukosekonzentration von 5,5 mM einen signifikanten Zelltod aus (Abb. 1B), was darauf hindeutet, dass Glukose eine wichtige Quelle für reduzierende Kraft ist. Daher reichte eine Supplementierung mit 1-2 mM Glukose aus, um die Metformin-Zytotoxizität unter der üblichen Glukosekonzentration zu blockieren (Abb. 1A), was darauf hindeutet, dass die Metformin-Zytotoxizität nicht ausreicht, um den Zelltod unter physiologischen Glukosebedingungen auszulösen. Die Behandlung mit Metformin führte jedoch zu einem signifikanten Anstieg der mitochondrialen ROS unter den Bedingungen ohne Glukose-Limitierung. Es gibt bisher keinen Bericht, der sich auf die mitochondriale ROS-Bildung im Zusammenhang mit der Metformin-Zytotoxizität konzentriert. Hier beobachteten wir, dass bereits 1 mM Metformin das mitochondriale Superoxid zusammen mit den zellulären ROS signifikant erhöhte (Abb. 2B), was eine mitochondriale Schwellung in Krebszellen induzierte, die nicht irreversibel war (ergänzende Abb. S1A und S1B). Dennoch scheint die erhöhte mitochondriale ROS nicht ausreichend zu sein, um die gesamte zelluläre ROS und die oxidative Schädigung zu erhöhen. Krebszellen, die Metformin ausgesetzt sind, versuchen, oxidative Schäden zu minimieren, um die Lebensfähigkeit der Zellen zu erhalten, indem sie die aerobe Glykolyse und die NADPH-Produktion anregen. Dieser Gedanke wird teilweise durch die Tatsache gestützt, dass die Verringerung der reduzierenden Kraft durch BSO oder exogenesH2O2die Metformin-Zytotoxizität trotz der hohen Glukosebedingungen verstärkte (Abb. 3). Krebszellen können oxidativem Stress durch aerobe Glykolyse standhalten, und der GSH-Gehalt kann hauptsächlich durch NADPH aufrechterhalten werden, das über den Pentosephosphatweg produziert wird [12].
Die Anti-Tumor-Aktivität von DCA wurde in vitro [4] und in vivo in Mausmodellen [27], [28] sowie bei Glioblastom-Patienten [18] nachgewiesen, allerdings ist der IC50-Wert von DCA relativ hoch (>17 mM, 48 h), um den Zelltod auszulösen. Darüber hinaus gibt es in den getesteten Krebszelllinien keine Tumorzellspezifität [26], und DCA ist selektiv gegen Zellen mit Defekten in den Mitochondrien wie rho(0)-Zellen oder HCT116 p53-Null-Zellen mit Komplex-IV-Mangel aktiv. Wir beobachteten auch, dass 20 mM DCA allein keinen wirksamen Zelltod auslöste, die Kombination mit Metformin jedoch die Anti-Tumor-Aktivität von DCA mit 10 mM synergierte (Abb. 5B und F).
Wurden die Zellen mit Metformin behandelt, wurde die PDH-Aktivität durch Phospholytierung am Ser293-Rest signifikant gehemmt (Abb. 4C), ohne dass sich die PDK-mRNA-Expression signifikant veränderte (Daten nicht gezeigt), was auf eine posttranskriptionelle Modifikation hinweist. Obwohl wir nicht in der Lage waren, den Mechanismus der Metformin-induzierten Hemmung der PDH-Aktivität zu definieren, verstärkte die Verringerung der PDK-Aktivität durch DCA den Metformin-induzierten oxidativen Stress durch die Unterdrückung der PDH-Phosphorylierung (Abb. 4), was auf die Hemmung der PDK-Aktivität durch DCA, aber die Erhöhung der oxidativen Phosphorylierung zusammen mit dem Redox-Stress hinweist. In Übereinstimmung mit dem obigen Befund stellte DCA die durch Metformin gehemmte mitochondriale Atmung teilweise wieder her (Abb. 4F) und verstärkte die durch Metformin induzierte mitochondriale ROS-Erzeugung erheblich (Abb. 5A). Die schützende Rolle der Antioxidantien gegen die kombinierte Behandlung von Metformin und DCA führte uns zu dem Schluss, dass DCA die Metformin-Zytotoxizität durch Reprogrammierung und Verstärkung des oxidativen Stresses verstärken kann (Abb. 5C).
Zusammenfassend lässt sich sagen, dass die Metformin-induzierte Zytotoxizität durch Glukoseentzug verstärkt wurde. Selbst bei hohem Glukosegehalt wurde die Metformin-Zytotoxizität durch zusätzlichen ROS-Stress aufgrund von GSH-Verarmung oder exogenemH2O2-Insultverstärkt und die mitochondriale Atmung erhöht. Wir schließen daraus, dass die DCA-vermittelte Umprogrammierung des Glukosestoffwechsels von aerober Glykolyse zu oxidativer Atmung den Metformin-induzierten oxidativen Stress verschlimmert und die Metformin-Zytotoxizität in Krebszellen sensibilisiert (Abb. 6).

Interessenkonflikt
Es gibt keine konkurrierenden finanziellen Interessen im Zusammenhang mit dieser Arbeit.
Danksagung
Diese Arbeit wurde durch einen Zuschuss des Nationalen Forschungs- und Entwicklungsprogramms zur Krebsbekämpfung, Ministerium für Gesundheit und Wohlfahrt, Republik Korea (131280), unterstützt.
Anhang A. Ergänzendes Material
Ergänzende Daten zu diesem Artikel sind in der Online-Version unter http://dx.doi.org/10.1016/j.canlet.2014.01.015 zu finden.
REFERENZEN
1 I.M. Ahmad, N. Aykin-Burns, J.E. Sim, S.A. Walsh, R. Higashikubo, G.R. Buettner, S. Venkataraman, M.A. Mackey, S.W. Flanagan, L.W. Oberley, D.R. Spitz Mitochondriales O2*- und H2O2 vermitteln den durch Glukoseentzug ausgelösten Stress in menschlichen Krebszellen J. Biol. Chem., 280 (2005), S. 4254-42632 I. Ben Sahra, K. Laurent, S. Giuliano, F. Larbret, G. Ponzio, P. Gounon, Y. Le Marchand-Brustel, S. Giorgetti-Peraldi, M. Cormont, C. Bertolotto, M. Deckert, P. Auberger, J.F. Tanti, F. Bost Targeting cancer cell metabolism: the combination of metformin and 2-deoxyglucose induces p53-dependent apoptosis in prostate cancer cells Krebsforschung, 70 (2010), S. 2465-2475
3 I. Ben Sahra, Y. Le Marchand-Brustel, J.F. Tanti, F. Bost Metformin in der Krebstherapie: eine neue Perspektive für ein altes Antidiabetikum? Mol. Cancer Ther., 9 (2010), S. 1092-1099
4 S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C.T. Lee, G.D. Lopaschuk, L. Puttagunta, S. Bonnet, G. Harry, K. Hashimoto, C.J. Porter, M.A. Andrade, B. Thebaud, E.D. Michelakis Eine Mitochondrien-K+-Kanal-Achse ist bei Krebs unterdrückt und ihre Normalisierung fördert die Apoptose und hemmt das Krebswachstum Krebszelle, 11 (2007), S. 37-51
5 J.R. Cantor, D.M. Sabatini Krebszellstoffwechsel: ein Merkmal, viele Gesichter Cancer Discov. 2 (2012), S. 881-898
6 J.H. Cheong, E.S. Park, J. Liang, J.B. Dennison, D. Tsavachidou, C. Nguyen-Charles, K. Wa Cheng, H. Hall, D. Zhang, Y. Lu, M. Ravoori, V. Kundra, J. Ajani, J.S. Lee, W. Ki Hong, G.B. Mills Die duale Hemmung des Tumorenergiestoffwechsels durch 2-Desoxyglukose und Metformin ist gegen ein breites Spektrum präklinischer Krebsmodelle wirksam Mol. Cancer Ther., 10 (2011), S. 2350-2362
7 E.P. Clark, E.R. Epp, J.E. Biaglow, M. Morse-Gaudio, E. Zachgo Glutathionverarmung, Radiosensibilisierung und Misonidazol-Potenzierung in hypoxischen Ovarialzellen des chinesischen Hamsters durch Buthioninsulfoximin Radiat. Res., 98 (1984), S. 370-380
8 M.Y. El-Mir, V. Nogueira, E. Fontaine, N. Averet, M. Rigoulet, X. Leverve Dimethylbiguanid hemmt die Zellatmung über eine indirekte Wirkung, die auf den Atmungskettenkomplex I abzielt J. Biol. Chem., 275 (2000), S. 223-228
9 R.B. Hamanaka, N.S. Chandel Zellbiologie. Warburg-Effekt und Redox-Gleichgewicht Wissenschaft, 334 (2011), S. 1219-1220
10 T. Ishimoto, O. Nagano, T. Yae, M. Tamada, T. Motohara, H. Oshima, M. Oshima, T. Ikeda, R. Asaba, H. Yagi, T. Masuko, T. Shimizu, T. Ishikawa, K. Kai, E. Takahashi, Y. Imamura, Y. Baba, M. Ohmura, M. Suematsu, H. Baba, H. Saya CD44-Variante reguliert Redox-Status in Krebszellen durch Stabilisierung der xCT-Untereinheit des Systems xc(-) und fördert dadurch Tumorwachstum Cancer Cell, 19 (2011), S. 387-400
11 S. Javeshghani, M. Zakikhani, S. Austin, M. Bazile, M.J. Blouin, I. Topisirovic, J. St-Pierre, M.N. Pollak Kohlenstoffquelle und Myc-Expression beeinflussen die antiproliferative Wirkung von Metformin Cancer Res. 72 (2012), S. 6257-6267
12 S.M. Jeon, N.S. Chandel, N. Hay AMPK reguliert die NADPH-Homöostase, um das Überleben von Tumorzellen bei Energiestress zu fördern Nature, 485 (2012), S. 661-665
13 G. Kroemer, J. Pouyssegur Tumorzellstoffwechsel: die Achillesferse des Krebses Cancer Cell, 13 (2008), S. 472-482
14 M.A. Lea, J. Chacko, S. Bolikal, J.Y. Hong, R. Chung, A. Ortega, C. Desbordes Die Zugabe von 2-Desoxyglucose verstärkt die Wachstumshemmung, kehrt aber die Übersäuerung von mit Phenformin behandelten Dickdarmkrebszellen um Anticancer Res. 31 (2011), S. 421-426
15 X. Lin, F. Zhang, C.M. Bradbury, A. Kaushal, L. Li, D.R. Spitz, R.L. Aft, D. Gius 2-Desoxy-d-Glucose-induzierte Zytotoxizität und Radiosensibilisierung in Tumorzellen wird durch Störungen des Thiol-Stoffwechsels vermittelt Cancer Res. 63 (2003), S. 3413-3417
16 L. Luo, W. Huang, R. Tao, N. Hu, Z.X. Xiao, Z. Luo ATM- und LKB1-abhängige Aktivierung von AMPK sensibilisiert Krebszellen für Etoposid-induzierte Apoptose Cancer Lett. 328 (2013), S. 114-119
17 J.A. Menendez, C. Oliveras-Ferraros, S. Cufi, B. Corominas-Faja, J. Joven, B. Martin-Castillo, A. Vazquez-Martin Metformin ist synthetisch tödlich bei Glukoseentzug in Krebszellen Zellzyklus, 11 (2012), S. 2782-2792
18 E.D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, C. Maguire, T.L. Gammer, J.R. Mackey, D. Fulton, B. Abdulkarim, M.S. McMurtry, K.C. Petruk Metabolische Modulation des Glioblastoms mit Dichloracetat Sci. Translat. Med., 2 (2010), S. 31ra34
19 A.R. Mullen, W.W. Wheaton, E.S. Jin, P.H. Chen, L.B. Sullivan, T. Cheng, Y. Yang, W.M. Linehan, N.S. Chandel, R.J. DeBerardinis Reduktive Carboxylierung unterstützt Wachstum in Tumorzellen mit defekten Mitochondrien Nature, 481 (2012), S. 385-388
20 M.R. Niewisch, Z. Kuci, H. Wolburg, M. Sautter, L. Krampen, B. Deubzer, R. Handgretinger, G. Bruchelt Einfluss von Dichloracetat (DCA) auf die Laktatproduktion und den Sauerstoffverbrauch in Neuroblastomzellen: Ist DCA ein geeignetes Medikament für die Neuroblastomtherapie? Cell. Physiol. Biochem: Int. J. Exp. Cell. Physiol. Biochem. Pharmacol., 29 (2012), S. 373-380
21 M.R. Owen, E. Doran, A.P. Halestrap Nachweis, dass Metformin seine antidiabetischen Wirkungen durch Hemmung von Komplex 1 der mitochondrialen Atmungskette ausübt Biochem. J., 348 (Pt 3) (2000), S. 607-614
22 C. Riganti, E. Gazzano, M. Polimeni, E. Aldieri, D. Ghigo Der Pentosephosphatweg: eine antioxidative Verteidigung und ein Scheideweg im Tumorzellschicksal Free Radical Biol. Med., 53 (2012), S. 421-436
23 G.Z. Rocha, M.M. Dias, E.R. Ropelle, F. Osorio-Costa, F.A. Rossato, A.E. Vercesi, M.J. Saad, J.B. Carvalheira Metformin verstärkt die Chemotherapie-induzierte AMPK-Aktivierung und das antitumorale Wachstum Clin. Cancer Res: Official J. Am. Assoc. Cancer Res., 17 (2011), S. 3993-4005
24 D.B. Shackelford, E. Abt, L. Gerken, D.S. Vasquez, A. Seki, M. Leblanc, L. Wei, M.C. Fishbein, J. Czernin, P.S. Mischel, R.J. Shaw LKB1-Inaktivierung bestimmt das therapeutische Ansprechen von nicht-kleinzelligem Lungenkrebs auf das Stoffwechselmedikament Phenformin Cancer Cell, 23 (2013), S. 143-158
25 J. Sinnett-Smith, K. Kisfalvi, R. Kui, E. RozengurtMetforminhemmung der mTORC1 aktivierung, DNA-Synthese und Proliferation in Bauchspeicheldrüsenkrebszellen: Abhängigkeit von der Glukosekonzentration und Rolle von AMPK Biochem. Biophys. Res. Commun. 430 (2013), S. 352-357
26 L.H. Stockwin, S.X. Yu, S. Borgel, C. Hancock, T.L. Wolfe, L.R. Phillips, M.G. Hollingshead, D.L. Newton Natriumdichloracetat wirkt selektiv auf Zellen mit Defekten im mitochondrialen ETC Int. J. Cancer J. Int. Cancer, 127 (2010), S. 2510-2519
27 R.C. Sun, M. Fadia, J.E. Dahlstrom, C.R. Parish, P.G. Board, A.C. Blackburn Umkehrung des glykolytischen Phänotyps durch Dichloracetat hemmt das Wachstum metastasierender Brustkrebszellen in vitro und in vivo Breast Cancer Res. Treat, 120 (2010), S. 253-260
28 G. Sutendra, P. Dromparis, A. Kinnaird, T.H. Stenson, A. Haromy, J.M. Parker, M.S. McMurtry, E.D. Michelakis iMitochondriale Aktivierung durch Hemmung von PDKII unterdrückt HIF1a-Signalisierung und Angiogenese bei Krebs Oncogene, 32 (2013), S. 1638-1650
29 F. Tietze Enzymatische Methode zur quantitativen Bestimmung von Nanogramm-Mengen an Gesamtglutathion und oxidiertem Glutathion: Anwendungen bei Säugetierblut und anderen Geweben m8 Anal. Biochem, 27 (1969), S. 502-522
30 M.G. Vander Heiden Zielgerichteter Krebsstoffwechsel: ein therapeutisches Fenster öffnet sich Nat. Rev. Drug Discovery, 10 (2011), S. 671-684
31 P.S. Ward, C.B. Thompson Metabolische Umprogrammierung: ein Krebsmerkmal, das selbst Warburg nicht vorausgesehen hat Cancer Cell, 21 (2012), S. 297-308
32 S. Whitehouse, R.H. Cooper, P.J. Randle Mechanismus der Aktivierung von Pyruvatdehydrogenase durch Dichloracetat und andere halogenierte Carbonsäuren Biochem. J., 141 (1974), S. 761-774
33 M. Wu, A. Neilson, A.L. Swift, R. Moran, J. Tamagnine, D. Parslow, S. Armistead, K. Lemire, J. Orrell, J. Teich, S. Chomicz, D.A. Ferrick Die Multiparameter-Stoffwechselanalyse zeigt eine enge Verbindung zwischen einer abgeschwächten mitochondrialen bioenergetischen Funktion und einer verstärkten Glykolyse-Abhängigkeit in menschlichen Tumorzellen Am. J. Physiol. Zellphysiol. 292 (2007), S. C125-136
34 X. Xiao, Q. He, C. Lu, K.D. Werle, R.X. Zhao, J. Chen, B.C. Davis, R. Cui, J. Liang, Z.X. Xu Metformin beeinträchtigt das Wachstum von Leberkinase B1-intakten Gebärmutterhalskrebszellen Gynecol. Oncol., 127 (2012), S. 249-255
35 Y. Zhuang, W.K. Miskimins Metformin induziert sowohl den Caspase-abhängigen als auch den Poly(ADP-ribose)-Polymerase-abhängigen Zelltod in Brustkrebszellen Mol. Cancer Res: MCR, 9 (2011), S. 603-615
36 J.W. Zmijewski, E. Lorne, X. Zhao, Y. Tsuruta, Y. Sha, G. Liu, G.P. Siegal, E. Abraham Mitochondrialer Atmungskomplex I reguliert die neutrophile Aktivierung und die Schwere der Lungenverletzung Am. J. Respir. Crit. Care Med., 178 (2008), S. 168-179