Jong-Lyel Roh a, *, Jin Young Park a, Eun Hye Kim a, Hye Jin Jang a, Minsu Kwon b
a Afdeling Otolaryngologie, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republiek Korea
b Afdeling Otolaryngologie, Gyeongsang National University Hospital School of Medicine, Jinju, Republiek Korea
Correspondentie auteur: Tel: +82 2 3010 3965; fax: +82 2 489 2773. E-mailadres: [email protected] (J.-L. Roh).
Ontvangen: 9 september 2015
Herzien: herzien 13 november 2015
Aanvaard: 14 november 2015
Abstract
Dichlooracetaat (DCA), een weesgeneesmiddel dat een verschuiving van glycolyse naar oxidatieve fosforylering bevordert, is opnieuw gebruikt voor kankertherapie. De huidige studie onderzocht of DCA cisplatineresistentie in hoofd- en halskanker (HNC) kan overwinnen. Twee cisplatineresistente HNC-cellijnen (AMC-HN4R en -HN9R), hun ouderlijnen en andere menselijke HNC-lijnen werden gebruikt. Het effect van DCA, alleen en in combinatie met cisplatine, werd beoordeeld door het meten van celcyclus, levensvatbaarheid, sterfte, productie van reactieve zuurstofsoorten (ROS), mitochondriaal membraanpotentieel (ΔΨm) en eiwitexpressie in preklinische muis xenograft modellen. Verhoogde glycolyse correleerde met verminderde gevoeligheid voor cisplatine en werd verminderd door DCA. Cisplatine-resistente cellen brachten pyruvaat dehydrogenase kinase 2 (PDK2) tot overexpressie. DCA induceerde HNC-celdood door verlaging van ΔΨm en bevordering van de mitochondriale ROS-productie. Dit effect werd verminderd door het antioxidant N-acetyl-l-cysteïne of door remming van de door caspase gemedieerde apoptose. Activering van mitochondriale glucose-oxidatie door DCA activeerde uiteindelijk downstream mitochondriale apoptotische signalering, wat leidde tot de dood van chemoresistente kankercellen. Daarom maakte DCA resistente HNC-cellen significant gevoelig voor cisplatine in vitro en in vivo. Hoge glycolyse en PDK2-overexpressie zijn nauw verbonden met cisplatineresistentie in HNC-cellen; dit laatste kan worden ondervangen door DCA.
Trefwoorden: Hoofd- en halskanker, Cisplatineresistentie, PDK2, Dichlooracetaat, Mitochondriale remodellering
Afkortingen: HNC, hoofd-halskanker; DCA, dichlooracetaat; CDDP, cisplatine; OXPHOS, oxidatieve fosforylering; PDK2, pyruvaat dehydrogenase kinase 2; PDHE1α, pyruvaat dehydrogenase isoform E1α; ROS, reactieve zuurstofsoorten; ΔΨm, mitochondriaal membraanpotentiaal; NAC, N-acetyl-l-cysteïne; DCF-DA, 2′,7′- dichloorfluoresceïne-diacetaat; PARP, poly(ADP-ribose) polymerase; siRNA, short interfering RNA; 18F-FDG, 18F-fluorodeoxyglucose; PET, positron emissie tomografie; SUV, standardized uptake value; MTV, metabool tumorvolume; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
© 2015 Elsevier Ireland Ltd. Alle rechten voorbehouden.
INLEIDING
Hoofd-halskanker (HNC) is wereldwijd de achtste meest voorkomende vorm van kanker, met meer dan een half miljoen nieuwe gevallen per jaar [1]. De tumoren ontstaan in de bovenste luchtwegen, waaronder de mond- of neusholte, de keelholte en het strottenhoofd, en metastaseren naar regionale lymfeklieren en afgelegen plaatsen. De huidige zorgstandaard bij HNC omvat een multimodale aanpak met chirurgie, chemotherapie en radiotherapie, met name bij gevorderde HNC. Samen met de recente belangstelling voor strategieën voor orgaanbehoud worden niet-chirurgische modaliteiten, zoals radiotherapie in combinatie met chemotherapie, steeds vaker gebruikt als eerstelijnstherapie bij HNC [2]. Bij de behandeling van patiënten met gevorderd HNC is systemische chemotherapie nu een centraal onderdeel van verschillende curatieve benaderingen, waaronder combinatie met definitieve radiotherapie of inductiebehandeling, en meer dan alleen voor palliatie [3]. Cisplatine, het platinederivaat cis-diaminedichloroplatinum (II) (CDDP), blijft een eerstelijns chemotherapeutisch middel in niet-chirurgische modaliteiten tegen HNC, ondanks recente vooruitgang in gerichte therapie [4]. In de afgelopen drie decennia is de totale overleving van HNC, ondanks de vooruitgang op diagnostisch en therapeutisch gebied, echter niet wezenlijk veranderd, een gevolg van de hardnekkige resistentie van kankercellen tegen therapie, waaronder cisplatine en bestraling [2].
Metabolische veranderingen zijn een gemeenschappelijk kenmerk van kankercellen: een verschuiving in de cellulaire energieopwekking van mitochondriale oxidatieve fosforylering naar aërobe glycolyse biedt kankercellen een biosynthetisch voordeel [5]. Verhoogde glycolyse in kankercellen, het Warburg-effect genoemd, wordt gewoonlijk in verband gebracht met fenotypische veranderingen, waaronder aanpassing aan hypoxie en lage voedingsstoffen, weerstand tegen oxidatieve stress en apoptotische prikkels, en verhoogde biomassasynthese [6]. Een ontregeld metabolisme is gekoppeld aan resistentie tegen behandelingen bij kankertherapie [7]. In de glycolytische route wordt de upregulatie van onder meer de glucose transporter (GLUT), hexokinase (HK), pyruvaat kinase M2 (PKM2), pyruvaat dehydrogenase kinase (PDK), lactaat dehydrogenase-A (LDHA), vetzuursynthese (FASN) en glutaminase in verband gebracht met resistentie tegen kankermedicijnen [7]. Er zijn aanwijzingen dat het reguleren van het metabolisme van kankercellen de therapie kan verbeteren en de resistentie tegen chemotherapie of radiotherapie kan overwinnen [8,9].
Dichlooracetaat (DCA), een weesgeneesmiddel tegen melkziekte, verschuift het kankermetabolisme van glycolyse naar oxidatieve fosforylering [10]. DCA remt selectief PDK, dat in veel kankers wordt geactiveerd, wat leidt tot activering van pyruvaatdehydrogenase (PDH), een complex van enzymen dat cytosolisch pyruvaat omzet in mitochondriaal acetyl-CoA. Inhibitie van PDK met small interfering RNA (siRNA) of behandeling met DCA verandert de bio-energetica van kanker en herstelt mitochondria-afhankelijke apoptose in kankercellen [11]. DCA is effectief bij de behandeling van kankers met agressieve fenotypes in vitro en in vivo[12,13], en overwint resistentie tegen sorafenib in hepatocellulair carcinoom door mitochondriale oxidatieve fosforylering te activeren [ 14]. Daarom kan DCA een geschikt antikankergeneesmiddel zijn om de werkzaamheid van chemotherapie te verhogen en om resistentie tegen chemotherapeutische middelen bij menselijke kanker te overwinnen; hoewel DCA uitgebreid is onderzocht bij kanker, is het zelden getest bij HNC-cellen [15], met name in de setting van resistentie tegen chemotherapeutische middelen. Verder onderzoek naar de mechanismen van DCA en de synergie met conventionele chemotherapeutische middelen is nodig. Hier tonen we aan dat DCA de bioenergetica van HNC verschuift naar mitochondriale glucose-oxidatie en leidt tot cellulaire accumulatie van mitochondriale reactieve zuurstofspecies (mROS), waardoor chemoresistente HNC-cellen in vitro en in vivo gevoelig worden voor cisplatine.
Materialen en methoden
Celkweek en vorming van cisplatineresistente HNC-cellen
De menselijke hoofd-halskankercellen (HNC) AMC-HN2, -HN3, -HN4, -HN5, -HN9 en -HN10 werden gekweekt in Eagle’s minimum essential medium (Life TechnologiesTM, Carlsbad, CA, USA); SNU-1041, -1066, en -1076 cellen werden gekweekt in Roswell Park Memorial Institute medium (Life TechnologiesTM ); en UMSCC1 cellen werden gekweekt in Dulbecco’s modified Eagle medium (Life TechnologiesTM ). De media werden aangevuld met 10% foetaal runderserum. Alle kankercellijnen werden geverifieerd aan de hand van DNA-profielen (short-tandem-repeat, STR) van de Korean Cell Line Bank. De cellen werden geïncubeerd bij 37 °C in een vochtige atmosfeer met 5% CO2.
Cisplatine-resistente AMC-HN4 en HN9 (HN4R en HN9R) cellen werden ontwikkeld uit de ouderlijke cisplatine-gevoelige AMC-HN4 en HN9 cellen, respectievelijk door blootstelling aan oplopende concentraties cisplatine (cis-platina (II) diamine dichloride [CDDP]; Sigma-Aldrich, Louis, MO, USA) [16]. De cisplatineresistentie in de gevestigde cellijnen werd geëvalueerd door middel van levensvatbaarheidstests en vergeleken met die van de oudercellen.
Levensvatbaarheidstest
De levensvatbaarheid van de cellen werd bepaald door trypanblauwexclusie, MTT en klonogene tests. Trypan blue exclusie werd uitgevoerd bij HNC-cellen die met 1 x105 in 6-wellsplaten waren uitgezaaid. De cellen mochten 60-70% confluentie bereiken en werden gedurende 72 uur blootgesteld aan dichlooracetaat (DCA; Sigma-Aldrich). De cellen werden vervolgens getrypsiniseerd, gekleurd met 0,4% trypanblauw (Life TechnologiesTM ) en geteld met een hemacytometer. De MTT-test werd uitgevoerd bij HNC-cellen met 3-5 x103 cellen per well in 96-wellsplaten. De cellen werden ’s nachts geïncubeerd en vervolgens blootgesteld aan DCA en cisplatine, alleen of in combinatie, gedurende 72 uur. De cellen werden vervolgens blootgesteld aan de tetrazoliumverbinding 3-[4,5-dimethyl-2-thiazolyl]-2,5-difenyl-2H-tetrazoliumbromide (MTT; Sigma-Aldrich) gedurende 4 uur, waarna gedurende 2 uur oplosbuffer werd toegevoegd. De absorptie in elk putje werd gemeten bij 570 nm met een SpectraMax M2 microplaatlezer (Molecular Devices, Sunnyvale, CA, USA). Voor klonogene tests werden de cellen gedurende 72 uur blootgesteld aan DCA of medium en vervolgens gedurende 7-10 dagen geïncubeerd in een geneesmiddelvrij medium. De putjes werden gekleurd met 0,5% kristalvioletoplossing en het aantal kolonies werd geteld. Alle tests werden uitgevoerd in drievoud en driemaal herhaald.
Cisplatine cytotoxiciteit werd beoordeeld door MTT na 72 uur, en de halfmaximale remmende concentratie (IC50) van elke HNC-cel werd berekend. De interactie van twee geneesmiddelen werd als synergetisch beschouwd wanneer de groeiremming door de combinatie van geneesmiddelen groter was dan de som van de remmingen door een van beide geneesmiddelen alleen: combinatie-index (CI) = 1, additieve interactie; CI < 1, synergetische interactie; en CI > 1, antagonistische interactie [17].
Meting van bio-energetica en ROS-productie
HNC-cellen (1 x104 cellen per well) werden uitgezaaid in 24-wellsplaten, en de volgende dag werden ze gewassen en geïncubeerd in serumvrij DMEM aangevuld met of zonder 100 ng/mL oligomycine (een ATP-synthaseremmer; Sigma-Aldrich) gedurende 6 uur. Het kweekmedium (50 µL) werd van elke plaat verzameld, en de lactaatconcentratie van het kweekmedium werd gemeten met een lactaat assay kit (Biovision, Mountain View, CA, USA). Lac (o) en Lac (c) staan voor de lactaatconcentratie in het medium na 6 uur incubatie in aanwezigheid en afwezigheid van oligomycine. De bijdrage van glycolyse en oxidatieve fosforylering (OXPHOS) aan de cellulaire bio-energetica werd berekend met de volgende formule: glycolyse (%) = Lac (c)/Lac (o) x 100; OXPHOS (%) = 100 – glycolyse (%) [14].
De cellen werden gedurende 24 uur blootgesteld aan 15 of 30 mM DCA, en ROS-generatie werd gedetecteerd in cellen geïncubeerd met 10 µM 2′,7′-dichloorfluoresceïne-diacetaat (DCF-DA) (Enzo Life Sciences, Farmingdale, NY, USA). De cellen werden gedurende 30 minuten bij 37 °C geïncubeerd met 10 µM DCF-DA, tweemaal gewassen met PBS en geanalyseerd in een FACScalibur-flowcytometer. De cellen werden ook voorbehandeld met 3 mM N-acetyl-l-cysteïne (NAC; Sigma-Aldrich) gedurende 1 uur of 10 µM superoxide dismutase (SOD)-remmer diethyl-dithio-carbamaat (Sigma-Aldrich) vóór blootstelling aan 30 mM DCA. De ROS-niveaus werden gemeten door flowcytometrie met DCF-DA en worden weergegeven als de vouwverandering ten opzichte van de controle (basisniveau).
Celcyclus- en celdoodtests
Voor celcyclustests werden de cellen gedurende 72 uur blootgesteld aan DCA. De cellen werden vervolgens getrypsiniseerd, een nacht gefixeerd in ijskoude ethanol, en gedurende 30 minuten gekleurd met propidiumjodide (Sigma-Aldrich) bij 37 °C. Het cellulaire DNA-gehalte werd gemeten met een FACScalibur flowcytometer (BD Bioscience, San Jose, CA, USA). Voor celdoodtests werden de cellen gekweekt met cisplatine en DCA, alleen of in combinatie, of een gelijkwaardige hoeveelheid DMSO (voertuigcontrole). Na 72 uur werden de cellen geoogst, gewassen met ijskoud PBS en geresuspendeerd in bindingsbuffer. De cellen werden gekleurd met annexine V-FITC (fluoresceïne-isothiocyanaat) en propidiumjodide met behulp van een annexine V-FITC apoptosedetectiekit (BD Biosciences, Franklin Lakes, NJ, USA), en vervolgens geanalyseerd met flowcytometrie. Alle gegevens werden geanalyseerd met de Cell Quest software (BD Biosciences). De statistische significantie tussen verschillende behandelingsgroepen werd beoordeeld met een tweekoppige Mann-Whitney U-test of Student’s t-test.
Voor caspase-activiteitstests werden HN4R- en HN9R-cellen, gezaaid in een 96-wells plaat, blootgesteld aan 100 µL medium met cisplatine en DCA alleen of in combinatie, gedurende 72 uur, met of zonder 3 mM NAC of 50 µM Z-VAD-fmk (R&D Systems, Minneapolis, MN, USA) vóór blootstelling aan 30 mM DCA. De assays werden uitgevoerd in drievoudige wells met behulp van de fluorimetrische Homogene Caspase Assay (Roche Life Science, Basel, Zwitserland). De werkende substraatoplossing werd toegevoegd en de plaat werd in het donker geïncubeerd bij 37 °C gedurende 2-8 uur of bij kamertemperatuur ’s nachts. De absorptie in elk putje werd gemeten bij een excitatiegolflengte van 485 nm en een emissiegolflengte van 520 nm met een SpectraMax M2 microplaatlezer.
Voor het meten van de mitochondriale membraanpotentiaal (ΔΨm) werden HN4R- en HN9R-cellen in een 96-wells-plaat blootgesteld aan 100 µL medium met cisplatine en DCA alleen of in combinatie gedurende 36 uur. De cellen werden gedurende 20 minuten gekleurd met 200 nM tetramethylrhodamine ethylester (TMRE, Life Technologies TM ) en vervolgens geanalyseerd met flowcytometrie. De gemiddelde fluorescentie-intensiteit (MFI) van elke behandelingsgroep werd genormaliseerd ten opzichte van de controlegroep.
Real-time kwantitatieve reverse transcription-PCR
Totaal cellulair RNA werd geëxtraheerd met behulp van het QIAzol lysis-reagens en de RNeasy Mini Kit (Qiagen, Valencia, CA, USA). cDNA werd gegenereerd uit gezuiverd RNA met behulp van een QuantiTect Reverse Transcription Kit (Qiagen) volgens de instructies van de fabrikant. Pyruvaat dehydrogenase kinase 2 (PDK2) cDNA werd geamplificeerd door PCR met de volgende primers: 5′-ATGGCAGTCCTCCTCTGA-3′ (voorwaarts) en 5′-CACCCACCCTCTCTAACA-3′ (achterwaarts). Voor β-actine (een endogene controle) werden de volgende primers gebruikt: 5′-ACCCCCACTGAAAGATGA-3′ (voorwaarts) en 5′-ATCTTCAAACCTCCATGATG-3′ (achterwaarts). Realtime kwantitatieve omgekeerde transcriptie-PCR (qRT-PCR) werd uitgevoerd met SYBR Green (Qiagen) op een 7900HT Fast Real-time PCR-systeem (Applied Bioscience, Foster, CA, USA). Relatieve doel-mRNA-niveaus werden genormaliseerd ten opzichte van β-actine-expressie.
siRNA
Voor knockdown van PDK2 en PDHE1α werden AMC-HN4-cisR en HN9-cisR uitgezaaid op 60 mm-platen in medium zonder antibiotica, en 18 uur later werden ze getransfecteerd met 100 nmol/L small interfering RNA (siRNA) gericht op het menselijke PDK2- of PDHE1α-gen of een gescrambeld controle-siRNA (Life Technologies TM ). Na 72 uur werden de cellen gedurende een extra periode van 72 uur blootgesteld aan DCA en vervolgens geanalyseerd op eiwitexpressie. De uitschakeling werd bevestigd door Western blotting met anti-PDK2- of PDHE1-antilichamen.
Immunoblotting
De cellen werden bij 4 °C gelyseerd in RIPA-buffer (Life TechnologiesTM ). Immunoblotting werd uitgevoerd volgens standaardprocedures. In het kort werd een totaal van 50 µg eiwit opgelost door natriumdodecylsulfaat-polyacrylamidegelelectroforese (SDS-PAGE) op 10-12%-gels, overgebracht op nitrocellulose- of polyvinylideendifluoridemembranen en geprobed met primaire en secundaire antilichamen. De volgende primaire antilichamen werden gebruikt: p53 (DO1) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); hexokinase 2 (HK2), p21 WAF1/CIP1, PUMA, gesplitst poly(ADP-ribose) polymerase (PARP), fosfo-p53-Ser15, en gesplitst caspase-3 (Cell Signaling Technology, Danvers, MA, USA); PDK2 (Abcam, Cambridge, MA, USA); en PDHE1α en fosfo-PDHE1α (ser293) (Calbiochem, Billerica, MA, USA). β-Actine (Sigma-Aldrich) werd gebruikt als laadcontrole. Alle antilichamen werden verdund tussen 1:250 en 1:5000.
Confocale microscopie
ΔΨm werd afgebeeld in levende kankercellen met TMRM (Life TechnologiesTM ), een mitochondriëngevoelige, spanningsgevoelige kleurstof op basis van rhodamine (rode fluorescentie). Mitochondriaal superoxide (mROS, Life TechnologiesTM ), geproduceerd tijdens de celademhaling, werd gemeten met MitoSOX (rode fluorescentie). TMRM- of mitoSOX-fluorescentiebeelden werden vastgelegd door een Zeiss LSM 510 confocale tweefotonmicroscoop (Carl Zeiss AG, Heidenheim, Duitsland), en de gemiddelde fluorescentie werd gekwantificeerd (arbitrary fluorescence units, AFU) in kankercellen in cultuur met behulp van de Zen imaging software (Carl Zeiss AG). De beeldvorming en de analyses werden uitgevoerd door twee wetenschappers die blind waren voor de bron van de celspecimens.
Positron emissie tomografie (PET) beeldvorming
Glucose opname werd in vivo in naakte muizen in beeld gebracht met transplantatie van HN4R of HN9R cellen met behulp van isofluraan gas anesthesie (20% zuurstof) en 18 F-fluorodeoxyglucose (18 F-FDG) als radiotracer. PET-beeldvorming werd uitgevoerd met microPET FOCUS 120 (Concorde Microsystem Inc., Knoxville, TN, USA). De muizen vastten een nacht en werden vervolgens intraveneus geïnjecteerd met 0,15 mCi. Na 1 uur werd 18 F-FDG PET scanning uitgevoerd in het hele lichaam. Beelden van de PET-gegevensverzameling worden getoond met een pseudokleurkaart, waarbij de rode kleur een hoge 18 F-FDG-opname aangeeft.
De maximale en gemiddelde gestandaardiseerde opnamewaarde (SUVmax en SUVmean) werden gebruikt om de 18 F-FDG-PET-activiteit te bepalen. De SUV werd geanalyseerd met behulp van de vergelijking SUV = A/(ID/BW), waarbij A de voor verval gecorrigeerde activiteit in weefsel is (in mCi/mL), ID de geïnjecteerde dosis FDG (in mCi), en BW het lichaamsgewicht van de muis (in gram). Sferische of elliptische interessegebieden (ROI’s) werden geplaatst over de op PET-beelden zichtbare laesies. De SUVmax en SUVmean werden berekend door automatisch een ROI te trekken over het meest intense deel van de tumoren die zichtbaar waren op PET-beelden. MTV’s werden berekend uit voor attenuatie gecorrigeerde PET-gegevens met behulp van een commercieel softwarepakket (INFINITT PACS; INFINITT Healthcare Co., Ltd). 18 F-FDG PET-gegevens werden in DICOM-formaat in het werkstation ingevoerd, en de intensiteitswaarden werden automatisch omgezet in SUV’s. Voor MTV-berekeningen werden de contourmarges van de tumor gedefinieerd met behulp van een vaste SUV van 2,0, en het tumorvolume werd vervolgens afgebakend met de SUV 2,0 isocontour. De PET-beelden werden verkregen bij muizen met een tumor op dag 21 na het begin van de behandeling. De gemiddelde waarden van de tumor SUVmax en MTV werden vergeleken tussen de verschillende behandelingsgroepen.
Preklinische studies
Alle onderzoeksprocedures bij dieren werden uitgevoerd volgens protocollen die waren goedgekeurd door het Institutional Animal Care and Use Committee van onze instelling. Zes weken oude athymische BALB/c mannelijke naaktmuizen (nu/nu) werden gekocht van Central Lab Animal Inc. (Seoul, Republiek Korea). AMC-HN4R- of HN9R-cellen (5 x 10 6 ) werden subcutaan in de flank geïnjecteerd. Tumorvolume en lichaamsgewicht werden om de 3 dagen gemeten. Tumoren werden gemeten met een schuifmaat, en het volume werd berekend als (lengte x breedte2 )/2. De behandeling begon wanneer de celimplantaten tastbare knobbels werden (=dag 0). De muizen werden gerandomiseerd in vier behandelingsgroepen: voertuig, DCA, cisplatine, en DCA plus cisplatine.
De muizen werden behandeld met drinkwater aangevuld met 0,5 g/L DCA, of door i.p. injectie van 5 mg/kg cisplatine eenmaal per week, of met een combinatie van DCA en cisplatine volgens hetzelfde schema. De muizen werden op dag 24 gedood, en de tumoren werden geïsoleerd en geanalyseerd door middel van immunoblotting en in situ terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay (EMD Millipore, Billerica, MA, USA). Het aantal apoptotische lichamen werd blind geteld in tien willekeurig gekozen velden met hoog vermogen. De statistische significantie tussen verschillende behandelingsgroepen werd beoordeeld met een tweekoppige Mann-Whitney U-test of Student’s t-test.
Resultaten
Verhoogde glycolyse in HNC-cellen is geassocieerd met resistentie tegen cisplatine en wordt omgekeerd door DCA
. Alle in ons onderzoek gebruikte cellijnen waren menselijke HNC-cellen. Het cytotoxische effect van DCA werd in HNC-cellen beoordeeld door trypan blue exclusie, kristalvioletkleuring en MTT-test. DCA, getest tot 30 mM gedurende 72 uur, remde de groei van HNC-cellen duidelijk af (MTT-test, fig. 1A ). De bio-energetica van HNC-cellen werd door DCA veranderd: de cellulaire glycolyse nam aanzienlijk af, terwijl de oxidatieve fosforylering toenam (fig. 1B ). De verandering in bio-energetica varieerde tussen de HNC-cellijnen en was significant geassocieerd met gevoeligheid voor cisplatine: HNC-cellen met een hoge glycolyse vertoonden weerstand tegen cisplatine (R2 = 0,859, P < 0,001;Fig. 1C ). Bij blootstelling aan DCA bleken de kankercellen met de grootste verandering in de bio-energetica bovendien meer apoptotisch te worden (R2 = 0,769, P = 0,002; Fig. 1D ). Dit suggereert dat DCA kankercelspecifieke apoptose induceert door verlaging van de glycolyse in HNC-cellen.
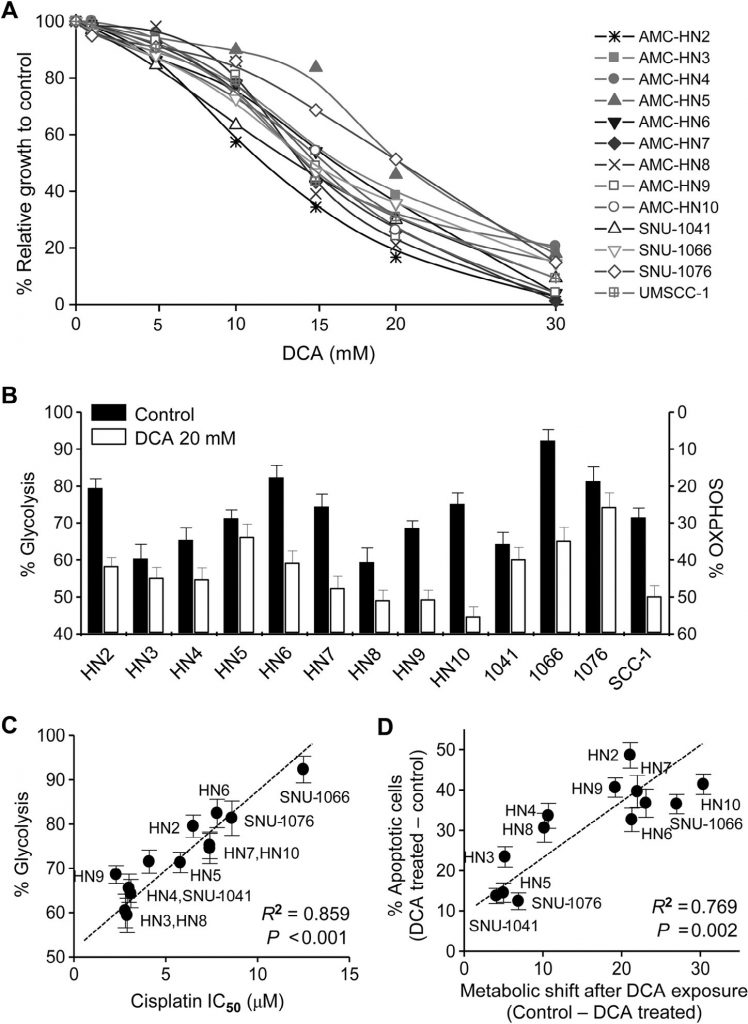
PDK2-expressie is geassocieerd met cisplatineresistentie in HNC
De cytotoxische effecten van cisplatine werden getest in gekweekte HN4-cisR en HN9-cisR en ouderlijke kankercellijnen (fig. 2A). Cisplatineresistente HN4-cisR- en HN9-cisR-cellen vertoonden een 12-voudige en 18-voudige verhoging van de IC50, respectievelijk in vergelijking met hun respectieve ouderlijnen. Western blot analyse toonde aan dat HK2 en PDK2 sterk tot expressie kwamen in zowel HN4-cisR als HN9-cisR cellen in vergelijking met de respectieve oudercellen, terwijl de expressie van PDHE1α laag was in de cisplatine-resistente cellen ( Fig. 2B ). Het expressieniveau van PDK2 mRNA was ook hoger in de resistente cellen dan in de gevoelige cellen (P < 0,01) (Fig. 2C). DCA remde de groei van cisplatine-resistente HNC-cellen evenzeer als die van cisplatine-gevoelige HNC-cellen (Fig. 2D). Western blot analyse toonde aan dat DCA de niveaus van PDK2 en fosfo-PDHE1α (pPDHE1α) aanzienlijk verminderde, maar het niveau van pro-apoptotische eiwitten, waaronder gekloofd PARP (cPARP) en PUMA, en dat van p21 in HN4-cisR en HN9-cisR verhoogde (Fig. 2E). Dit suggereert dat DCA de groei van cisplatine-resistente HNC-cellen effectief kan remmen, evenals die van cisplatine-gevoelige HNC-cellen.
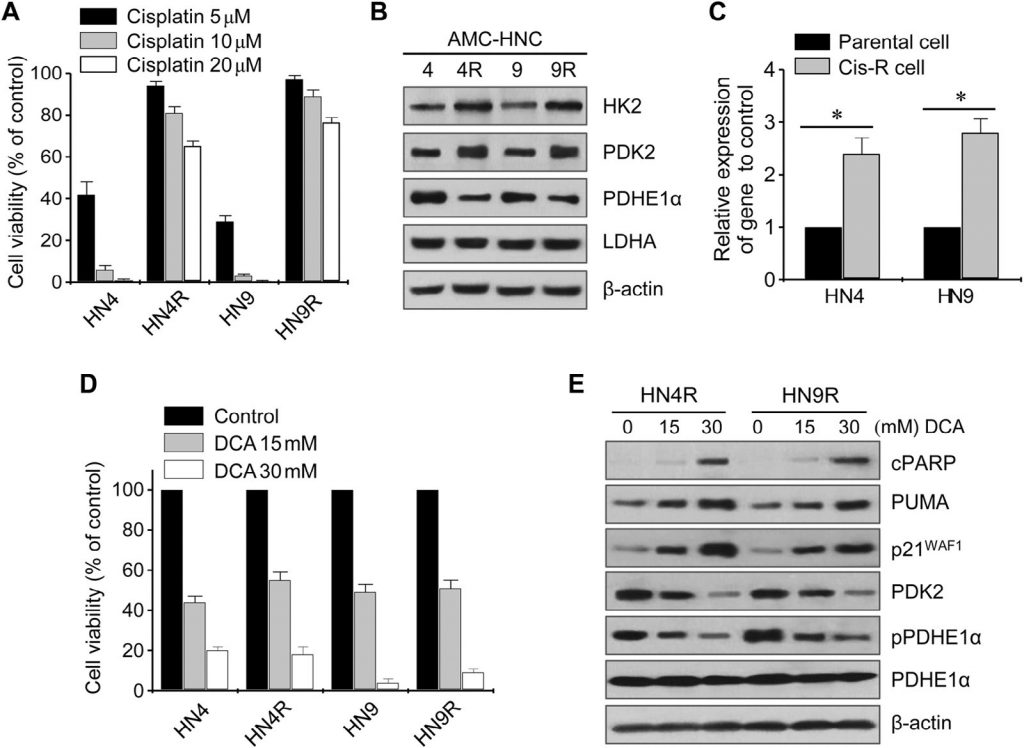
DCA induceert ROS-accumulatie in HNC-cellen
De verandering in cellulaire ROS door DCA werd beoordeeld door middel van flowcytometrie met de redoxgevoelige fluorescentieprobe DCF-DA. Blootstelling aan DCA veroorzaakte een significante stijging van de ROS-niveaus in HNC-cellen (P < 0,01), die werd geblokkeerd door gelijktijdige blootstelling aan NAC of de SOD-remmer diethyl-dithio-carbamaat (fig. 3A). Glycolyse nam significant toe in cisplatine-resistente HNC-cellen vergeleken met hun oudercellen (P < 0,01), maar DCA induceerde een significante afname van glycolyse en toename van oxidatieve fosforylering in zowel cisplatine-gevoelige als cisplatine-resistente HNC-cellen (P < 0,01) ( Fig. 3B ). Mitochondriaal membraanpotentiaal (ΔΨm) werd gemeten met TMRM, een mitochondriale, spanningsgevoelige kleurstof op basis van rhodamine, en mitochondriale ROS (mROS, mitochondriaal superoxide) werd gemeten met MitoSOX rood. Met DCA behandelde HNC-cellen vertoonden een verlaagde ΔΨm en een verhoogde mROS (P < 0,01) (fig. 3C). De verandering in ΔΨm in cisplatine-resistente HNC-cellen bij blootstelling aan DCA werd geblokkeerd door voorbehandeling met NAC (Fig. 3D). Dit suggereert dat DCA ROS accumulatie kan induceren in HNC-cellen door het activeren van oxidatieve fosforylering.
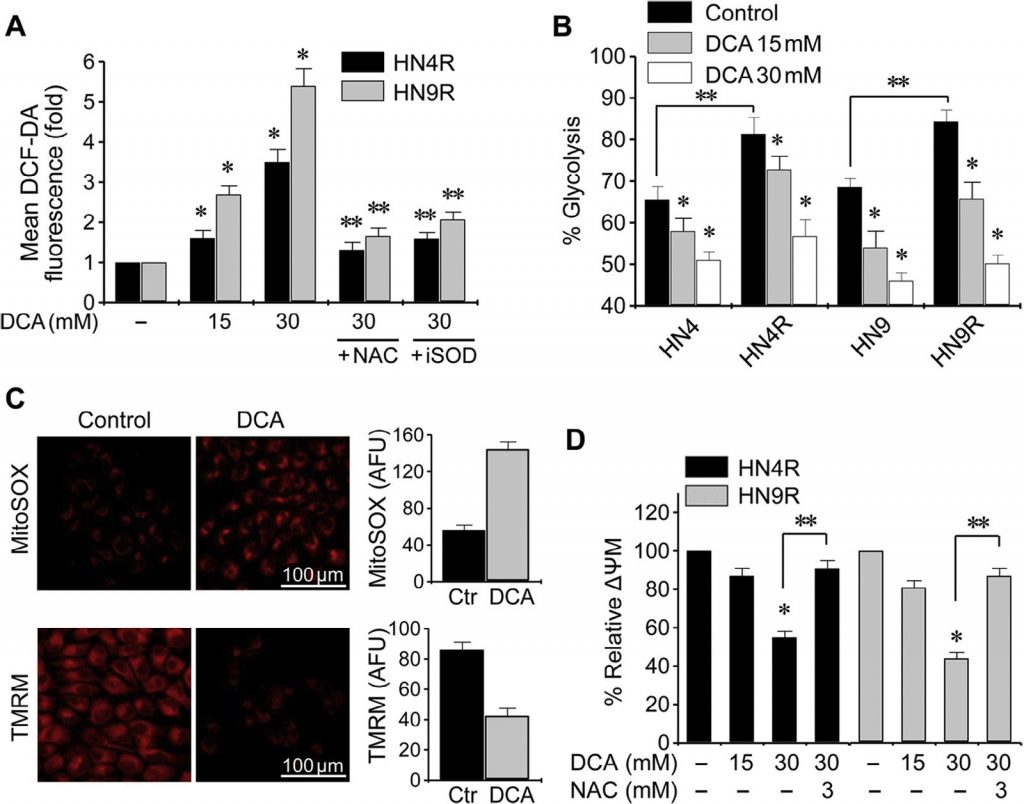
DCA bevordert celcyclusstilstand en apoptose in HNC-cellen
Klonogene tests resulteerden in een duidelijke afname van het aantal HN4-cisR- en HN9-cisR-kolonies bij blootstelling aan DCA (P <0,01) (fig. 4A ). Flowcytometrie met propidiumjodidekleuring toonde een significante celcyclusverandering in HNC-cellen: wogonine verhoogde de sub-G1 apoptotische populatie (P < 0,05), en dit effect werd geblokkeerd door gelijktijdige blootstelling aan NAC (fig. 4B ). Western blot analyse toonde aan dat DCA pPDHE1α verminderde, maar cPARP, p21, fosfo-p53 en geklaarde caspase-3 (cCasp3) verhoogde op een tijdsafhankelijke manier (Fig. 4C ). Activering van PDHE1α door uitschakeling van PDK2 in AMC-HN4-cisR-cellen leidde niet tot een significante toename van de proapoptotische eiwitten cPARP en cCasp3 (fig. 4D ); uitschakeling van PDHE1α verminderde echter de expressie van p21 en de fosforylering van p53. Flowcytometrie met propidiumjodide en annexine-V kleuring toonde effectieve inductie van apoptose en celdood in cisplatine-resistente HNC-cellen door DCA; het effect werd verminderd bij gelijktijdige blootstelling aan DCA en het antioxidant NAC of de pan-caspase-remmer Z-VAD-fmk ( Fig. 4E ). Dit werd bevestigd door de meting van de caspase-activiteit, die door DCA op een concentratie-afhankelijke wijze aanzienlijk werd verhoogd (fig. 4F ).
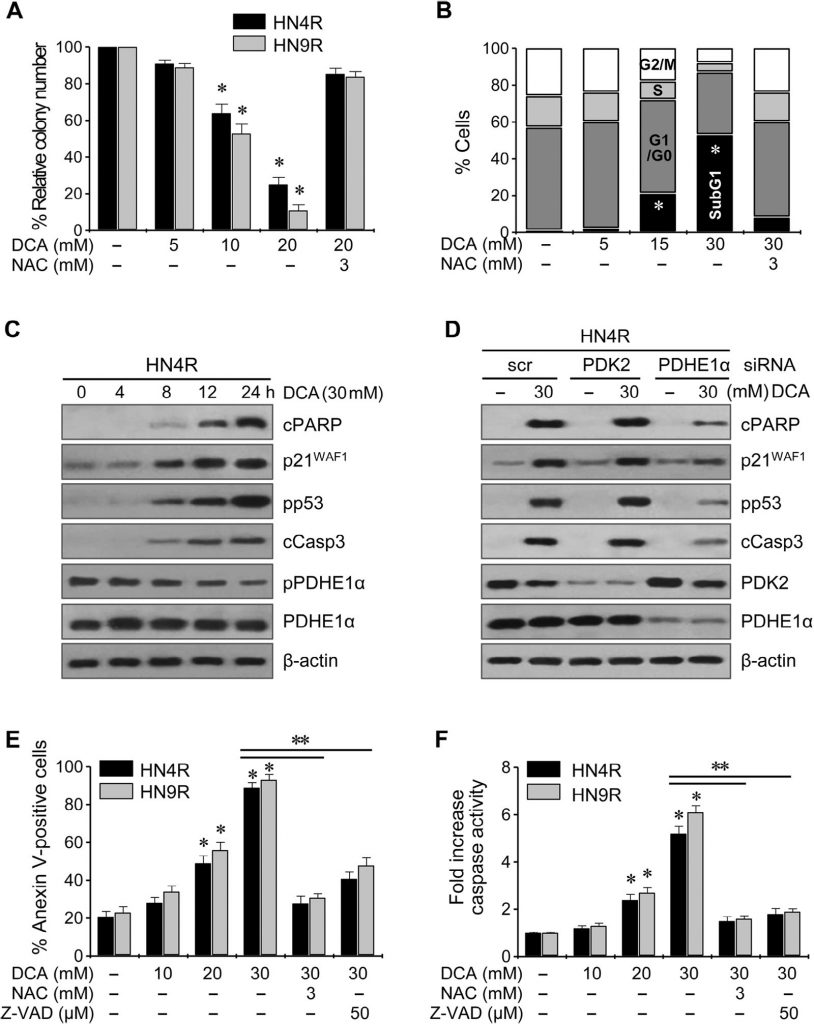
DCA sensibiliseert cisplatineresistente HNC-cellen voor cisplatine in vitro en in vivo
Cisplatine (10 µM) induceerde geen significante cytotoxiciteit of apoptotische eiwitexpressie in de cisplatineresistente HNC-lijnen HN4-cisR en HN9-cisR in vergelijking met de ouderlijke cisplatine-gevoelige lijnen HN4 en HN9 (Fig. 5A); DCA induceerde echter een duidelijke afname van de overleving in de cisplatineresistente HNC-cellen (P <0,05), die werd geblokkeerd door voorbehandeling met NAC. DCA induceerde de expressie van apoptotische eiwitten en verhoogde de cisplatine-geïnduceerde cytotoxiciteit en de expressie van apoptotische eiwitten in HN4-cisR-cellen (fig. B). In combinatie verhoogde DCA de cytotoxiciteit van cisplatine in HN4-cisR-cellen door verhoging van de caspase-activiteit in een mate die groter was dan de som van de effecten van beide agentia alleen (CI < 1, P < 0,01), die werden verzwakt door PDHE1α-gen silencing (Fig. 5C). De ΔΨm was hoger in de cisplatine-resistente HN4-cisR-cellen dan in de cisplatine-gevoelige ouderlijke HN4-cellen (ΔΨm, gemiddelde fluorescentie-intensiteit [MFI]: 1 ± 0 vs. 0,54 ± 0,09, P < 0,001) en werd verminderd door DCA of de combinatie van cisplatine en DCA, hetgeen werd tegengegaan door PDHE1α-gen silencing (Fig. 5D).
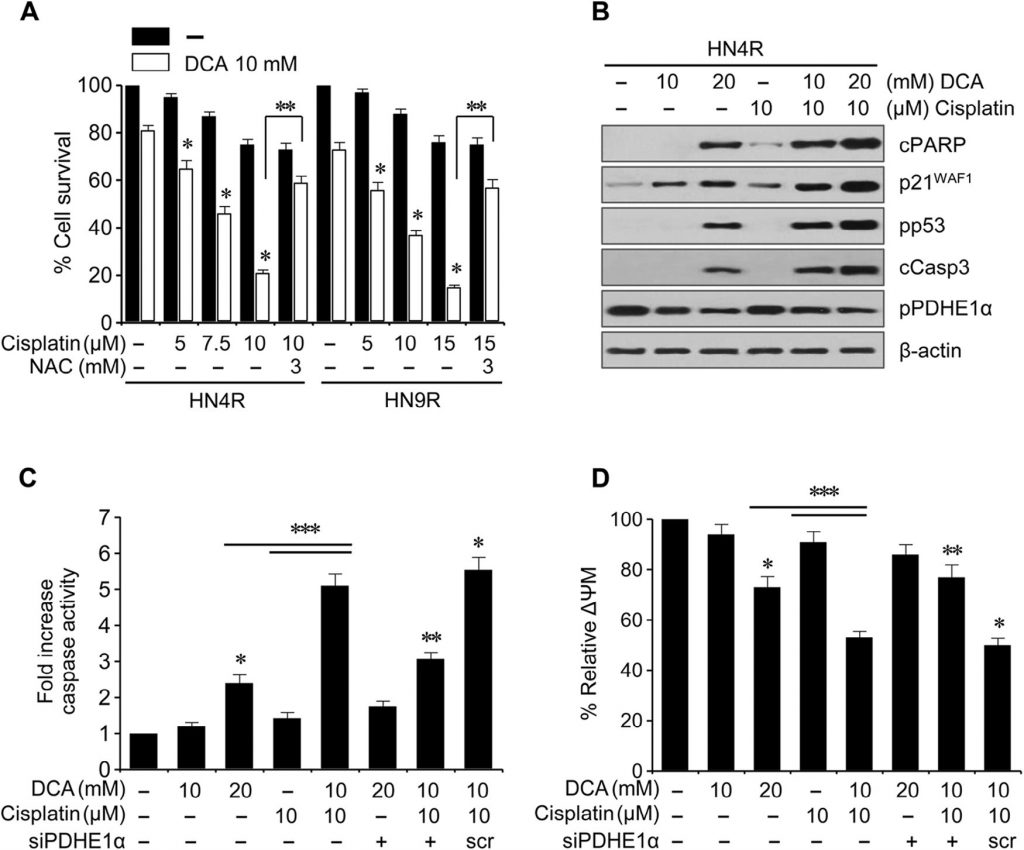
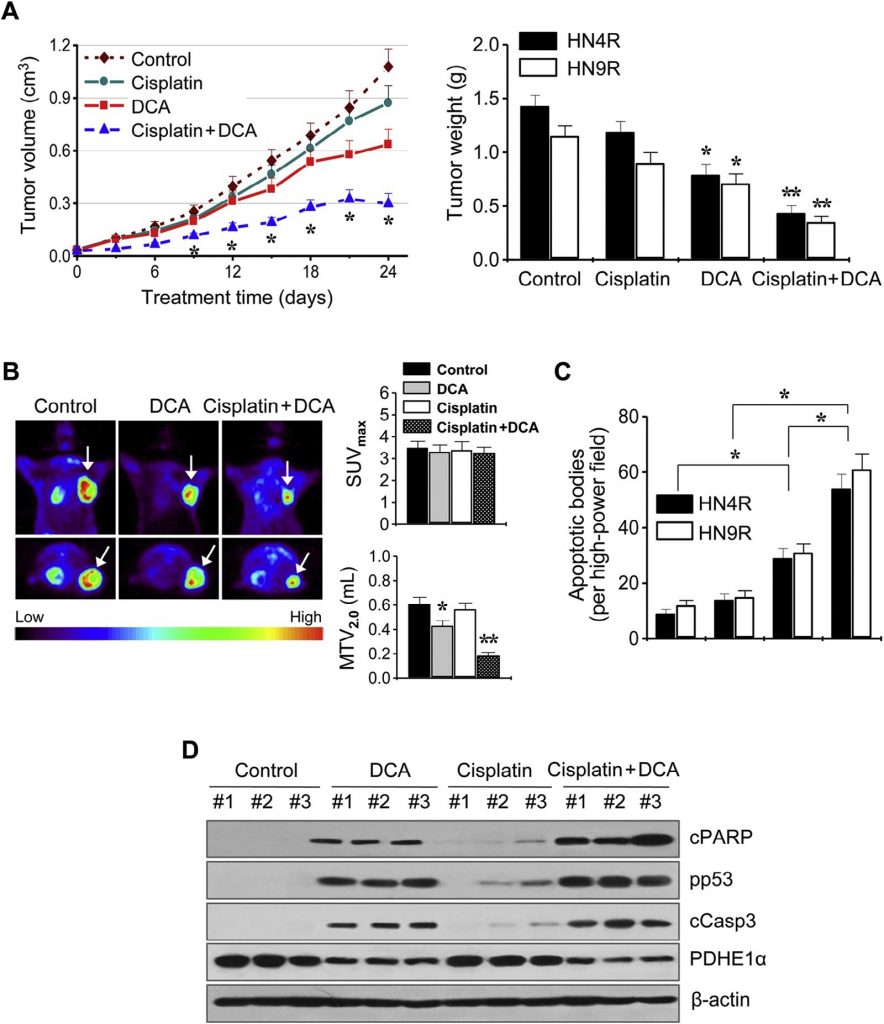
Deze bevindingen werden verder onderzocht in in vivo tumor xenograft muismodellen. BALB/c athymische naaktmuizen met AMC-HN4-cisR- of HN9-cisR-tumoren werden behandeld met DCA, cisplatine, DCA plus cisplatine, of medium. De combinatie van cisplatine en DCA onderdrukte synergetisch de tumorgroei (Fig. 6A). In vivo beeldvorming van de tumorgroei werd uitgevoerd met 18 F-FDG PET op dag 21 van de behandeling. Er werden focale 18 F-FDG-opnames waargenomen op de plaatsen van de tumorimplantatie, waar de maximale gestandaardiseerde opname (SUVmax) en het metabolisch tumorvolume bij SUV 2,0 (MTV2,0) werden gemeten. De SUVmax was niet verschillend tussen de behandelingsgroepen (P > 0,1), maar de MTV2,0 was significant lager in de met DCA en de combinatie behandelde groepen dan in andere groepen (P < 0,05) (fig. 6B). In situ apoptosetests toonden aan dat TUNEL-positieve apoptotische lichamen vaker werden gezien in tumoren behandeld met DCA en cisplatine plus DCA dan in die behandeld met medium (P < 0,01) (fig. 6C). Western blot analyses van tumorweefsels toonden aan dat de apoptotische eiwitniveaus sterker toenamen in HN4-cisR-cellen behandeld met de combinatie van cisplatine en DCA dan in tumoren behandeld met enkelvoudige middelen (fig. 6D).
Bespreking
Verhoogde glycolyse, die vaak voorkomt bij kanker, is nauw verbonden met mitochondriale aantasting of defecte oxidatieve fosforylering en draagt bij tot therapeutische resistentie [18,19]. Aërobe glycolyse is in verband gebracht met resistentie tegen chemotherapie [20] of radiotherapie [21]. In de huidige studie vertonen cisplatineresistente cellijnen een verhoogde glycolyse, wat wijst op een biochemisch verband tussen glycolyse en chemoresistentie. Geneesmiddelresistente cellen vertonen een grotere behoefte aan ATP dan normale cellen om de cellulaire homeostase te handhaven en de overlevingswegen te activeren die ontsnappen aan celdood onder genotoxische stress mogelijk maken [20]. Resistente kankercellen verhogen de glycolyse om snel ATP te genereren om aan de intracellulaire vraag te voldoen. Dit werd duidelijk gevonden in onze cisplatine-resistente cellen in vergelijking met de ouderlijke gevoelige cellen, wat aangeeft dat geneesmiddelenresistentie in HNC direct geassocieerd is met een toename van de glycolyse. Dit impliceert dat de bio-energetische verandering van kankercellen in de richting van verhoogde glycolyse verband houdt met chemoresistentie [14,22]. Daarom kan het niveau van glycolyse in menselijke kankers een biomarker van chemoresistentie zijn en is klinische validatie in menselijk kankerweefsel vereist.
De huidige studie toont aan dat DCA in HNC de energieproductie verschuift van glycolyse naar mitochondriale glucose-oxidatie. Deze door DCA geïnduceerde omkering van de metabole verschuiving naar glycolyse elimineert het proliferatievoordeel van kankercellen en leidt uiteindelijk tot celdood [23]. DCA, als structureel analoog van pyruvaat, remt PDK en reactiveert PDH, een enzym dat deel uitmaakt van het complex dat pyruvaat omzet in acetyl-CoA, het primaire substraat van de Krebscyclus [10,23]. Aangezien de meeste kankertypes een hypoxische omgeving produceren, vertrouwen kankercellen op anaërobe glycolyse als primaire energiebron. De activering van hypoxie-induceerbare factor (HIF) induceert mitochondriale PDK [24]. Mitochondriale PDH activiteit in kanker wordt geblokkeerd door PDK, wat resulteert in een lagere beschikbaarheid van acetyl-CoA voor mitochondriale glucose-oxidatie [24]. Een verhoogde expressie van PDK wordt in verband gebracht met resistentie tegen geneesmiddelen bij kanker [25,26]. Upregulatie van PDK2 door mitochondriale mutaties en stabilisatie van HIF1α wordt ook waargenomen in HNC-cellen [27]. In de huidige studie werd de associatie tussen overexpressie van PDK2 en cisplatineresistentie in HNC-cellen ook bevestigd. Deze bevindingen wijzen op het belang van PDK als een nieuw moleculair doelwit in kankertherapie [10]. Uit eerdere gegevens bleek dat genetische of farmacologische remming van PDK de bio-energetica van kanker verandert en de mitochondria-afhankelijke apoptose in kankercellen herstelt [11,13]. Daarom is DCA effectief in het overwinnen van behandelingsresistentie in kankers met agressieve fenotypes in vitro en in vivo[12-14].
Activering van PDH door DCA induceert de accumulatie van mROS in kankercellen. Kankercellen hebben een verminderd mitochondriaal glucosemetabolisme, wat resulteert in een verminderde activiteit van de elektronentransportketen (ETC) en verminderde mROS [10], [28]. De mitochondriale remodellering van ΔΨm hyperpolarisatie en verminderde mROS productie in kanker leidt tot weerstand tegen apoptose onder genotoxische stress. In resistente kankercel met een meer glycolytisch fenotype resulteert HK2 in zijn translocatie naar de buitenste mitochondriale membraan en de toename van ΔΨm [29]. Remming van de glycolyse en HK2 translocatie vermindert het ΔΨm van de kanker en keert de weerstand tegen apoptose om [29]. In de huidige studie werden de verhoogde HK2 en PDK2 expressie en mitochondriaal membraanpotentieel ook gevonden in cisplatine-resistente kankercellen. DCA dwingt pyruvaattoegang tot de mitochondriën af via activering van PDH, de poortwachter van de mitochondriale glucose-oxidatie, en vermindert ΔΨm en verhoogt mROS [13]. In de huidige studie keerde DCA eenvoudigweg de verhoogde mitochondriale remodellering in cisplatine-resistente HNC-cellen om, wat bijdroeg tot de kankerceldood. Activering van mitochondriale glucose-oxidatie door DCA induceerde mROS en activering van downstream mitochondriale signalering, hetgeen resulteerde in activering van p53 en de daarmee samenhangende pro-apoptotische routes en leidde tot de dood van chemoresistente kankercellen. In onze studie verzwakte farmacologische remming van de mROS-productie of de caspase-gemedieerde apoptose het cytotoxische effect van DCA in HNC-cellen, hetgeen de bekende mechanismen van het geneesmiddel bevestigt.
Preklinische en klinische studies ondersteunen het gebruik van DCA bij kankerpatiënten en bij patiënten met melkzuursyndroom geassocieerd met mitochondriale ziekten [10,23]. Een aantal studies toont de cytotoxische effecten aan van DCA, alleen of in combinatie met andere behandelingen, bij diverse tumoren afkomstig van de drie kiemlagen [23]. Een eerdere studie toonde aan dat glioblastoma, een van de meest agressieve kankersoorten bij de mens, PDK2 overexpresseert in kankerweefsel van 49 patiënten en bij vijf patiënten regressie vertoonde na behandeling met DCA, wat het klinische voordeel van DCA bij resistente kankers bewijst [13]. Bij HNC werd in een recente studie het effect van DCA vergeleken bij drie orale plaveiselcelcarcinomen (OSCC) cellijnen [15]. De OSCC-cellen met deficiënte mitochondriale oxidatieve fosforylering (d.w.z. hoge glycolyse) waren gevoeliger voor DCA dan andere [15,30]. De huidige studie bevestigde dat HNC-cellen met hoge bio-energetische veranderingen gevoeliger waren voor DCA. Aangezien chemoresistente kankercellen een hoge glycolyse hebben, zouden deze cellen wel eens doelwit kunnen zijn van metabole remmers [31]. Daarom is DCA een goede kandidaat om kankercellen met een agressief fenotype, waaronder chemoresistente HNC-cellen, te behandelen. Echter, omdat de potentiële anti-kanker effecten van DCA nog steeds controversieel zijn, vooral in tumoren onder hypoxie, zijn verdere preklinische en klinische studies naar DCA en kanker vereist [32]. Een recent systematisch overzicht toont aan dat DCA synergetisch werkt met vele standaard anti-kanker middelen, en klinische proeven in de vroege fase suggereren dat chronische DCA veilig is en goed wordt verdragen bij orale toediening van 12,5 mg/kg tweemaal daags [23].
Uit ons onderzoek bleek dat DCA synergetisch werkt met cisplatine en daarmee de resistentie tegen cisplatine in HNC-cellen omzeilt. Aangezien cisplatine een eerstelijns chemotherapeutisch middel is bij HNC, kan de combinatie van cisplatine en DCA effectief zijn in de klinische setting om de toxiciteit te verminderen en resistentie tegen kankermedicijnen te overwinnen. Uit eerdere rapporten bleek dat DCA het cytotoxische effect van cisplatine versterkt door mitochondria-afhankelijke apoptose te induceren [33,34]. De koppeling van twee DCA-moleculen aan cisplatine, mitaplatine genaamd, induceert de selectieve doding van kankercellen [35,36]. Deze resultaten leveren het bewijs dat DCA extra voordelen kan hebben in combinatie met de huidige kankertherapieën. In onze studie hadden DCA en de combinatie met cisplatine geen invloed op de SUVmax, maar verminderde MTV2.0 aanzienlijk. Dit betekent dat DCA effectief de in vivo tumorgroei onderdrukt ondanks het feit dat er geen significante verandering is van de maximale glucoseopnames in de tumor gelokaliseerde ROI’s. Deze studie is de eerste die aantoont dat DCA het cytotoxische effect van cisplatine in geneesmiddelenresistente HNC-cellen in vitro en in vivo herstelt. DCA induceerde een robuuste toename van cisplatine-gemedieerde apoptose via activering van p53, PARP en caspase in cisplatine-resistente HNC-cellen. DCA sensibiliseerde geneesmiddelenresistente HNC-cellen voor cisplatine, wat leidde tot verhoogde cytotoxiciteit en een effectievere therapie voor agressieve HNC. Al met al kunnen deze bevindingen van groot klinisch belang zijn: het induceren van de dood van resistente cellen met DCA zou de dosis cisplatine die nodig is in de klinische setting kunnen verminderen en daarmee de potentiële nadelige effecten van cisplatine chemotherapie kunnen minimaliseren.
Concluderend suggereren onze gegevens dat hoge glycolyse en PDK-overexpressie nauw verbonden zijn met cisplatineresistentie in HNC-cellen. DCA schakelt de bio-energetica van HNC-cellen in de richting van mitochondriale oxidatieve fosforylering. Dit leidt tot een afname van ΔΨm en een toename van mROS, waardoor chemoresistente HNC-cellen in vitro en in vivo gevoelig worden voor cisplatine. Deze gegevens rechtvaardigen verder preklinisch en klinisch onderzoek van DCA, een veelbelovend kandidaat-geneesmiddel, in HNC met agressief fenotype. Onze conclusie moet echter met voorzichtigheid in aanmerking worden genomen bij een omgekeerd scenario. De defecte mitochondriale oxidatieve fosforylering werd in verband gebracht met de gevoeligheid van kankercellen voor lage glucosecondities, wat gebruikt zou kunnen worden als biomarker voor kankertherapie [37]. Kankercellen zijn ook aangepast om proliferatie te vergemakkelijken door opname en incorporatie van voedingsstoffen in de biomassa in plaats van voor efficiënte ATP-productie [38]. Daarom moeten de preklinische en klinische effecten van DCA op HNC en andere kankertypes verder worden onderzocht.
Dankbetuigingen
Deze studie werd ondersteund door een subsidie (2015R1A2A1A15054540) van Basic Science Research Program via de National Research Foundation of Korea (NRF), Ministerie van Wetenschap, ICT en Toekomstplanning, en een subsidie (HI14C23050000) van het Korean Health Technology R&D Project, Ministerie van Gezondheid & Welzijn, Seoul, Republiek Korea (J.-L. Roh).
Belangenverstrengeling
De auteurs verklaren geen belangenconflicten te hebben.
VERWIJZINGEN
1 A. Jemal, F. Bray, M.M. Center, J. Ferlay, E. Ward, D. Forman, Global cancer statistics, CA Cancer J. Clin, Vol. 61, 2011, 69-90
2 R.I. Haddad, D.M. Shin, Recent advances in head and neck cancer, N. Engl. J. Med, Vol. 359, 2008, 1143-1154
3 E.B. Lamont, E.E. Vokes, Chemotherapy in the management of squamous-cell carcinoma of the head and neck, Lancet Oncol, Vol. 2, 2001, 261-269
4 F. Petrelli, A. Coinu, V. Riboldi, K. Borgonovo, M. Ghilardi, M. Cabiddu, Concomitant platinum-based chemotherapy or cetuximab with radiotherapy for locally advanced head and neck cancer: a systematic review and meta-analysis of published studies, Oral Oncol, Vol. 50, 2014, 1041-1048
5 D. Hanahan, R.A. Weinberg, Hallmarks of cancer: the next generation, Cell, Vol. 144, 2011, 646-674
6 J.R. Cantor, D.M. Sabatini, Cancer cell metabolism: one hallmark, many faces, Cancer Discov, Vol. 2, 2012, 881-898
7 Y. Zhao, E.B. Butler, M. Tan, Targeting cellular metabolism to improve cancer therapeutics, Cell Death Dis, Vol. 4, 2013, e532
8 R.A. Cairns, I.S. Harris, T.W. Mak, Regulation of cancer cell metabolism, Nat. Rev. Cancer, Vol. 11, 2011, 85-95
9 R.H. Xu, H. Pelicano, Y. Zhou, J.S. Carew, L. Feng, K.N. Bhalla, Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia, Cancer Res, Vol. 65, 2005, 613-621
10 G. Sutendra, E.D. Michelakis, Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology, Front. Oncol, Vol. 3, 2013, 38
11 G. Sutendra, P. Dromparis, A. Kinnaird, T.H. Stenson, A. Haromy, J.M. Parker, Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer, Oncogene, Vol. 32, 2013, 1638-1650
12 R.C. Sun, M. Fadia, J.E. Dahlstrom, C.R. Parish, P.G. Board, A.C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo, Breast Cancer Res. Treat, Vol. 120, 2010, 253-260
13 E.D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, Metabolic modulation of glioblastoma with dichloroacetate, Sci. Transl. Med, Vol. 2, 2010, 31ra34
14 Y.C. Shen, D.L. Ou, C. Hsu, K.L. Lin, C.Y. Chang, C.Y. Lin, Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma, Br. J. Cancer, Vol. 108, 2013, 72-81
15 V. Ruggieri, F. Agriesti, R. Scrima, I. Laurenzana, D. Perrone, T. Tataranni, Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment, Oncotarget, Vol. 6, 2015, 1217-1230
16 M. Nakamura, K. Nakatani, K. Uzawa, K. Ono, H. Uesugi, K. Ogawara, Establishment and characterization of a cisplatin-resistant oral squamous cell carcinoma cell line, H-1R, Oncol. Rep, Vol. 14, 2005, 1281-1286
17 T.C. Chou, Drug combination studies and their synergy quantification using the Chou-Talalay method, Cancer Res, Vol. 70, 2010, 440-446
18 N. Guaragnella, S. Giannattasio, L. Moro, Mitochondrial dysfunction in cancer chemoresistance, Biochem. Pharmacol, Vol. 92, 2014, 62-72
19 S. Ganapathy-Kanniappan, J.F. Geschwind, Tumor glycolysis as a target for cancer therapy: progress and prospects, Mol. Cancer, Vol. 12, 2013, 152
20 Y. Zhou, F. Tozzi, J. Chen, F. Fan, L. Xia, J. Wang, Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells, Cancer Res, Vol. 72, 2012, 304-314
21 S.P. Pitroda, B.T. Wakim, R.F. Sood, M.G. Beveridge, M.A. Beckett, D.M. MacDermed, STAT1-dependent expression of energy metabolic pathways links tumor growth and radioresistance to the Warburg effect, BMC Med, Vol. 7, 2009, 68
22 B. Bhattacharya, S.H. Low, C. Soh, N. Kamal Mustapa, M. Beloueche-Babari, K.X. Koh, Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype, Br. J. Pharmacol, Vol. 171, 2014, 3255-3267
23 S. Kankotia, P.W. Stacpoole, Biochim. Biophys. Acta, Vol. 1846, 2014, 617-629
24 J.W. Kim, I. Tchernyshyov, G.L. Semenza, C.V. Dang, HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia, Cell Metab, Vol. 3, 2006, 177-185
25 Y. Sun, A. Daemen, G. Hatzivassiliou, D. Arnott, C. Wilson, G. Zhuang, Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells, Cancer Metab, Vol. 2, 2014, 20
26 C.W. Lu, S.C. Lin, C.W. Chien, S.C. Lin, C.T. Lee, B.W. Lin, Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer, Am. J. Pathol, Vol. 179, 2011, 1405-1414
27 W. Sun, S. Zhou, S.S. Chang, T. McFate, A. Verma, J.A. Califano, Mitochondrial mutations contribute to HIF1alpha accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma, Clin. Cancer Res, Vol. 15, 2009, 476-484
28 S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, Channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth, Cancer Cell, Vol. 11, 2007, 37-51
29 J.G. Pastorino, J.B. Hoek, N. Shulga, Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity, Cancer Res, Vol. 65, 2005, 10545-10554
30 L.H. Stockwin, S.X. Yu, S. Borgel, C. Hancock, T.L. Wolfe, L.R. Phillips, Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC, Int. J. Cancer, Vol. 127, 2010, 2510-2519
31 E.J. Sullivan, M. Kurtoglu, R. Brenneman, H. Liu, T.J. Lampidis, Targeting cisplatin-resistant human tumor cells with metabolic inhibitors, Cancer Chemother. Pharmacol, Vol. 73, 2014, 417-427
32 S. Shahrzad, K. Lacombe, U. Adamcic, K. Minhas, B.L. Coomber, Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia, Cancer Lett, Vol. 297, 2010, 75-83
33 D. Heshe, S. Hoogestraat, C. Brauckmann, U. Karst, J. Boos, C. Lanvers-Kaminsky, Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs, Cancer Chemother. Pharmacol, Vol. 67, 2011, 647-655
34 J. Xie, B.S. Wang, D.H. Yu, Q. Lu, J. Ma, H. Qi, Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells, Int. J. Oncol, Vol. 38, 2011, 409-417
35 S. Dhar, S.J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate, Proc. Natl. Acad. Sci. U.S.A., Vol. 106, 2009, 22199-22204
36 X. Xue, S. You, Q. Zhang, Y. Wu, G.Z. Zou, P.C. Wang, Mitaplatin increases sensitivity of tumor cells to cisplatin by inducing mitochondrial dysfunction, Mol. Pharm, Vol. 9, 2012, 634-644
37 K. Birsoy, R. Possemato, F.K. Lorbeer, E.C. Bayraktar, P. Thiru, B. Yucel, Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides, Nature, Vol. 508, 2014, 108-112
38 M.G. Vander Heiden, L.C. Cantley, C.B. Thompson, Understanding the Warburg effect: the metabolic requirements of cell
Gerelateerde inhoud: