Jong-Lyel Roh a, *, Jin Young Park a, Eun Hye Kim a, Hye Jin Jang a, Minsu Kwon b
a Département d’oto-rhino-laryngologie, Asan Medical Center, University of Ulsan College of Medicine, Séoul, République de Corée
b Département d’oto-rhino-laryngologie, Gyeongsang National University Hospital School of Medicine, Jinju, République de Corée
Auteur de la correspondance : Tél : +82 2 3010 3965 ; fax : +82 2 489 2773. Adresse électronique : [email protected] (J.-L. Roh).
Reçu : 9 septembre 2015
Révisé : forme révisée 13 novembre 2015
Accepté : 14 novembre 2015
Résumé
Le dichloroacétate (DCA), un médicament orphelin qui favorise le passage de la glycolyse à la phosphorylation oxydative, a été réorienté vers le traitement du cancer. La présente étude vise à déterminer si le DCA peut surmonter la résistance au cisplatine dans le cancer de la tête et du cou (HNC). Deux lignées cellulaires HNC résistantes au cisplatine (AMC-HN4R et -HN9R), leurs lignées parentales et d’autres lignées HNC humaines ont été utilisées. L’effet du DCA, seul et en combinaison avec le cisplatine, a été évalué en mesurant le cycle cellulaire, la viabilité, la mort, la production d’espèces réactives de l’oxygène (ROS), le potentiel de la membrane mitochondriale (ΔΨm) et l’expression des protéines dans des modèles précliniques de xénogreffe de tumeurs chez la souris. L’augmentation de la glycolyse était corrélée à une diminution de la sensibilité au cisplatine et était réduite par le DCA. Les cellules résistantes au cisplatine surexpriment la pyruvate déshydrogénase kinase 2 (PDK2). Le DCA a induit la mort des cellules HNC en diminuant le ΔΨm et en favorisant la production de ROS mitochondriaux. Cet effet a été diminué par l’antioxydant N-acétyl-l-cystéine ou par l’inhibition de l’apoptose médiée par les caspases. L’activation de l’oxydation mitochondriale du glucose par le DCA a finalement activé la signalisation apoptotique mitochondriale en aval, entraînant la mort des cellules cancéreuses chimiorésistantes. Par conséquent, le DCA a considérablement sensibilisé les cellules HNC résistantes au cisplatine in vitro et in vivo. La glycolyse élevée et la surexpression de PDK2 sont étroitement liées à la résistance au cisplatine dans les cellules HNC ; cette dernière peut être surmontée par le DCA.
Mots clés : Cancer de la tête et du cou, résistance au cisplatine, PDK2, Dichloroacétate, remodelage mitochondrial
Abréviations : HNC, cancer de la tête et du cou ; DCA, dichloroacétate ; CDDP, cisplatine ; OXPHOS, phosphorylation oxydative ; PDK2, pyruvate déshydrogénase kinase 2 ; PDHE1α, pyruvate déshydrogénase isoforme E1α ; ROS, espèces réactives de l’oxygène ; ΔΨm, potentiel de la membrane mitochondriale ; NAC, N-acétyl-l-cystéine ; DCF-DA, 2′,7′- dichlorofluorescéine diacétate ; PARP, poly(ADP-ribose) polymérase ; siRNA, short interfering RNA ; 18F-FDG, 18F-fluorodésoxyglucose ; TEP, tomographie par émission de positons ; SUV, standardized uptake value ; MTV, metabolic tumor volume ; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling.
© 2015 Elsevier Ireland Ltd. Tous droits réservés.
INTRODUCTION
Le cancer de la tête et du cou (HNC) est le huitième cancer le plus fréquent dans le monde, avec plus d’un demi-million de nouveaux cas diagnostiqués chaque année [1]. Les tumeurs se développent dans les voies aérodigestives supérieures, y compris la cavité buccale ou nasale, le pharynx et le larynx, et se métastasent dans les ganglions lymphatiques régionaux et les sites distants. La norme actuelle de soins pour les HNC implique une approche multimodale comprenant la chirurgie, la chimiothérapie et la radiothérapie, en particulier pour les HNC avancés. Parallèlement à l’intérêt récent pour les stratégies de préservation des organes, les modalités non chirurgicales, telles que la radiothérapie associée à la chimiothérapie, ont été de plus en plus utilisées comme traitement de première intention dans les CNH [2]. Dans la prise en charge des patients atteints de CNH avancée, la chimiothérapie systémique est désormais un élément central de plusieurs approches curatives, y compris en association avec la radiothérapie définitive ou le traitement d’induction, dépassant la simple réserve palliative [3]. Le cisplatine, dérivé du platine, le cis-diamminedichloroplatine (II) (CDDP), reste un agent chimiothérapeutique de première ligne dans les modalités non chirurgicales contre le HNC, malgré les progrès récents de la thérapie ciblée [4]. Cependant, au cours des trois dernières décennies, malgré les progrès diagnostiques et thérapeutiques, le taux de survie global dans les HNC n’a pas changé de manière substantielle, résultat de la résistance tenace des cellules cancéreuses aux traitements, y compris le cisplatine et les radiations [2].
Les modifications métaboliques sont une caractéristique commune des cellules cancéreuses : le passage de la production d’énergie cellulaire de la phosphorylation oxydative mitochondriale à la glycolyse aérobie procure un avantage biosynthétique aux cellules cancéreuses [5]. L’augmentation de la glycolyse dans les cellules cancéreuses, appelée effet Warburg, est généralement liée à des changements phénotypiques, notamment l’adaptation à l’hypoxie et à une faible teneur en nutriments, la résistance au stress oxydatif et aux stimuli apoptotiques, et une synthèse élevée de la biomasse [6]. La dérégulation du métabolisme est liée à la résistance aux traitements anticancéreux [7]. Dans la voie glycolytique, la régulation à la hausse du transporteur de glucose (GLUT), de l’hexokinase (HK), de la pyruvate kinase M2 (PKM2), de la pyruvate déshydrogénase kinase (PDK), de la lactate déshydrogénase-A (LDHA), de la synthèse des acides gras (FASN) et de la glutaminase, entre autres, est associée à la résistance aux médicaments anticancéreux [7]. Il est prouvé que la régulation du métabolisme des cellules cancéreuses peut améliorer la thérapie et surmonter la résistance à la chimiothérapie ou à la radiothérapie [8,9].
Le dichloroacétate (DCA), un médicament orphelin contre l’acidose lactique, fait passer le métabolisme du cancer de la glycolyse à la phosphorylation oxydative [10]. Le DCA inhibe sélectivement la PDK, qui est activée dans de nombreux cancers, ce qui entraîne l’activation de la pyruvate déshydrogénase (PDH), un complexe d’enzymes qui transforme le pyruvate cytosolique en acétyl-CoA mitochondrial. L’inhibition de la PDK à l’aide d’un petit ARN interférent (siRNA) ou le traitement au DCA modifie la bioénergétique du cancer et rétablit l’apoptose dépendante des mitochondries dans les cellules cancéreuses [11]. Le DCA est efficace dans le traitement des cancers au phénotype agressif in vitro et in vivo[12,13], et surmonte la résistance au sorafénib dans le carcinome hépatocellulaire en activant la phosphorylation oxydative mitochondriale [14]. Par conséquent, le DCA peut être un médicament anticancéreux approprié pour augmenter l’efficacité de la chimiothérapie et surmonter la résistance aux agents chimiothérapeutiques dans le cancer humain ; cependant, bien que le DCA ait été largement étudié dans le cancer, il a rarement été testé dans les cellules HNC [15], en particulier dans le cadre d’une résistance aux agents chimiothérapeutiques. Il est nécessaire de poursuivre les recherches sur les mécanismes et la synergie du DCA avec les agents chimiothérapeutiques conventionnels. Nous montrons ici que le DCA fait basculer la bioénergétique des HNC vers l’oxydation mitochondriale du glucose et conduit à l’accumulation cellulaire d’espèces réactives de l’oxygène mitochondrial (mROS), sensibilisant ainsi les cellules HNC chimiorésistantes au cisplatine in vitro et in vivo.
Matériel et méthodes
Culture cellulaire et établissement de cellules HNC résistantes au cisplatine
Les cellules humaines de cancer de la tête et du cou (HNC) AMC-HN2, -HN3, -HN4, -HN5, -HN9, et -HN10 ont été cultivées dans le milieu minimum essentiel d’Eagle (Life TechnologiesTM, Carlsbad, CA, USA) ; Les cellules SNU-1041, -1066 et -1076 ont été cultivées dans le milieu Roswell Park Memorial Institute (Life TechnologiesTM ) ; et les cellules UMSCC1 ont été cultivées dans le milieu Eagle modifié de Dulbecco (Life TechnologiesTM ). Les milieux ont été complétés par du sérum bovin fœtal à 10 %. Toutes les lignées de cellules cancéreuses ont été authentifiées par un profilage de l’ADN (STR, short-tandem-repeat) fourni par la Korean Cell Line Bank. Les cellules ont été incubées à 37 °C dans une atmosphère humidifiée contenant 5 % de CO2.
Les cellules AMC-HN4 et HN9 (HN4R et HN9R) résistantes au cisplatine ont été développées à partir des cellules parentales AMC-HN4 et HN9 sensibles au cisplatine, respectivement, par exposition à des concentrations croissantes de cisplatine (dichlorure de cis-platine (II) diamine [CDDP] ; Sigma-Aldrich, Louis, MO, USA) [16]. La résistance au cisplatine des lignées cellulaires établies a été évaluée par des tests de viabilité cellulaire et comparée à celle des cellules parentales.
Test de viabilité cellulaire
La viabilité cellulaire a été déterminée par exclusion au bleu trypan, MTT et tests clonogéniques. L’exclusion au bleu trypan a été réalisée sur des cellules HNC ensemencées à 1 x105 dans des plaques à 6 puits. Les cellules ont atteint une confluence de 60 à 70 % et ont été exposées au dichloroacétate (DCA ; Sigma-Aldrich) pendant 72 heures. Les cellules ont ensuite été trypsinées, colorées avec du bleu trypan à 0,4 % (Life TechnologiesTM ) et comptées à l’aide d’un hématimètre. Le test MTT a été réalisé sur des cellules HNC ensemencées à 3-5 x103 cellules par puits dans des plaques à 96 puits. Les cellules ont été incubées pendant une nuit puis exposées au DCA et au cisplatine, seuls ou en combinaison, pendant 72 h. Les cellules ont ensuite été exposées au composé de tétrazolium, le bromure de 3-[4,5-diméthyl-2-thiazolyl]-2,5-diphényl-2H-tétrazolium (MTT ; Sigma-Aldrich) pendant 4 h, après quoi un tampon de solubilisation a été ajouté pendant 2 h. L’absorbance dans chaque puits a été mesurée à 570 nm en utilisant un lecteur de microplaques SpectraMax M2 (Molecular Devices, Sunnyvale, CA, USA). Pour les tests clonogéniques, les cellules ont été exposées au DCA ou au véhicule pendant 72 h, puis incubées dans un milieu sans médicament pendant 7 à 10 jours. Les puits ont été colorés avec une solution de cristal violet à 0,5 %, et le nombre de colonies a été compté. Tous les tests ont été réalisés avec des échantillons triples et répétés trois fois.
La cytotoxicité du cisplatine a été évaluée par MTT après 72 heures, et la concentration inhibitrice semi-maximale (IC50) de chaque cellule HNC a été calculée. L’interaction de deux médicaments a été considérée comme synergique lorsque l’inhibition de la croissance induite par l’association de médicaments était supérieure à la somme des inhibitions induites par l’un ou l’autre des médicaments seuls : indice de combinaison (IC) = 1, interaction additive ; IC < 1, interaction synergique ; et IC > 1, interaction antagoniste [17].
Mesure de la bioénergétique et de la production de ROS
Les cellules HNC (1 x104 cellules par puits) ont été ensemencées dans des plaques à 24 puits, et le lendemain, elles ont été lavées et incubées dans du DMEM sans sérum complété par ou sans 100 ng/mL d’oligomycine (un inhibiteur de l’ATP synthase ; Sigma-Aldrich) pendant 6 h.
Le milieu de culture (50 µL) a été prélevé sur chaque plaque, et la concentration en lactate du milieu de culture a été mesurée avec un kit de dosage du lactate (Biovision, Mountain View, CA, USA). Lac (o) et Lac (c) indiquent la concentration de lactate dans le milieu après 6 h d’incubation en présence et en l’absence d’oligomycine, respectivement. La contribution de la glycolyse et de la phosphorylation oxydative (OXPHOS) à la bioénergétique cellulaire a été calculée selon la formule suivante : glycolyse (%) = Lac (c)/Lac (o) x 100 ; OXPHOS (%) = 100 – glycolyse (%) [14].
Les cellules ont été exposées à 15 ou 30 mM de DCA pendant 24 h, et la génération de ROS a été détectée dans les cellules incubées avec 10 µM de 2′,7′-dichlorofluorescéine diacétate (DCF-DA) (Enzo Life Sciences, Farmingdale, NY, USA). Les cellules ont été incubées avec 10 µM de DCF-DA pendant 30 minutes à 37 °C, lavées deux fois avec du PBS, et analysées dans un cytomètre en flux FACScalibur. Les cellules ont également été prétraitées avec 3 mM de N-acétyl-l-cystéine (NAC ; Sigma-Aldrich) pendant 1 h ou 10 µM de diéthyl-dithio-carbamate (Sigma-Aldrich), un inhibiteur de la superoxyde dismutase (SOD), avant d’être exposées à 30 mM de DCA. Les niveaux de ROS ont été mesurés par cytométrie de flux en utilisant le DCF-DA et sont indiqués en tant que changement de pli par rapport aux niveaux de contrôle (basal).
Analyses du cycle cellulaire et de la mort cellulaire
Pour les analyses du cycle cellulaire, les cellules ont été exposées au DCA pendant 72 h. Les cellules ont ensuite été trypsinisées, fixées pendant une nuit dans de l’éthanol glacé et colorées pendant 30 min avec de l’iodure de propidium (Sigma-Aldrich) à 37 °C. Le contenu en ADN cellulaire a été mesuré à l’aide d’un cytomètre en flux FACScalibur (BD Bioscience, San Jose, CA, USA). Pour les tests de mort cellulaire, les cellules ont été cultivées avec du cisplatine et du DCA, seuls ou en combinaison, ou avec une quantité équivalente de DMSO (contrôle du véhicule). Après 72 heures, les cellules ont été récoltées, lavées avec du PBS glacé et remises en suspension dans un tampon de liaison. Les cellules ont été colorées avec de l’annexine V-FITC (isothiocyanate de fluorescéine) et de l’iodure de propidium à l’aide d’un kit de détection de l’apoptose à l’annexine V-FITC (BD Biosciences, Franklin Lakes, NJ, USA), puis analysées par cytométrie en flux. Toutes les données ont été analysées à l’aide du logiciel Cell Quest (BD Biosciences). La signification statistique entre les différents groupes de traitement a été évaluée par un test U de Mann-Whitney à deux intervalles ou un test t de Student.
Pour les tests d’activité caspasique, les cellules HN4R et HN9R ensemencées dans une plaque à 96 puits ont été exposées à 100 µL de milieu contenant du cisplatine et du DCA, seuls ou en combinaison, pendant 72 h, avec ou sans 3 mM de NAC ou 50 µM de Z-VAD-fmk (R&D Systems, Minneapolis, MN, USA) avant l’exposition à 30 mM de DCA. Les tests ont été effectués dans des puits triples en utilisant le test fluorimétrique Homogeneous Caspase Assay (Roche Life Science, Basel, Suisse). La solution de substrat de travail a été ajoutée, et la plaque a été incubée dans l’obscurité à 37 °C pendant 2-8 h ou à température ambiante pendant la nuit. L’absorbance dans chaque puits a été mesurée à une longueur d’onde d’excitation de 485 nm et une longueur d’onde d’émission de 520 nm en utilisant un lecteur de microplaques SpectraMax M2.
Pour mesurer le potentiel de la membrane mitochondriale (ΔΨm), les cellules HN4R et HN9R ensemencées dans une plaque à 96 puits ont été exposées à 100 µL de milieu contenant du cisplatine et du DCA, seuls ou en combinaison, pendant 36 h. Les cellules ont été colorées avec 200 nM d’ester éthylique de tétraméthylrhodamine (TMRE, Life Technologies TM ) pendant 20 min, puis analysées par cytométrie en flux. L’intensité de fluorescence moyenne (IFM) de chaque groupe de traitement a été normalisée par rapport au groupe témoin.
Transcription inverse quantitative en temps réel-PCR
L’ARN cellulaire total a été extrait à l’aide du réactif de lyse QIAzol et du mini kit RNeasy (Qiagen, Valencia, CA, USA). L’ADNc a été généré à partir de l’ARN purifié à l’aide du kit de transcription inverse QuantiTect (Qiagen) conformément aux instructions du fabricant. L’ADNc de la pyruvate déshydrogénase kinase 2 (PDK2) a été amplifié par PCR en utilisant les amorces suivantes : 5′-ATGGCAGTCCTCCTCTCTGA-3′ (sens direct) et 5′-CACCCACCCTCTTCCTAACA-3′ (sens inverse). Pour la β-actine (un contrôle endogène), les amorces suivantes ont été utilisées : 5′-ACCCCCACTGAAAAAGATGA-3′ (sens direct) et 5′-ATCTTCAAACCTCCATGATG-3′ (sens inverse). La transcription inverse quantitative en temps réel (qRT-PCR) a été réalisée en utilisant SYBR Green (Qiagen) sur un système 7900HT Fast Real-time PCR (Applied Bioscience, Foster, CA, USA). Les niveaux relatifs des ARNm cibles ont été normalisés par rapport à l’expression de la β-actine.
siRNA
Pour l’élimination de PDK2 et PDHE1α, les cellules AMC-HN4-cisR et HN9-cisR ont été ensemencées sur des plaques de 60 mm dans un milieu sans antibiotique, et 18 heures plus tard, elles ont été transfectées avec 100 nmol/L de petit ARN interférent (siRNA) ciblant le gène humain PDK2 ou PDHE1α ou un siRNA de contrôle brouillé (Life Technologies TM ). Après 72 h, les cellules ont été exposées au DCA pendant une période supplémentaire de 72 h, puis analysées pour l’expression des protéines. Le knockdown a été confirmé par Western blotting en utilisant des anticorps anti-PDK2 ou PDHE1.
Immunoblotting
Les cellules ont été lysées à 4 °C dans un tampon d’analyse de radio-immunoprécipitation (RIPA) (Life TechnologiesTM ). L’immunoblotting a été réalisé selon les procédures standard. En bref, un total de 50 µg de protéines a été résolu par électrophorèse sur gel de dodécylsulfate de sodium-polyacrylamide (SDS-PAGE) sur des gels à 10-12 %, transféré sur des membranes de nitrocellulose ou de difluorure de polyvinylidène, et sondé avec des anticorps primaires et secondaires. Les anticorps primaires suivants ont été utilisés : p53 (DO1) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) ; hexokinase 2 (HK2), p21 WAF1/CIP1, PUMA, poly(ADP-ribose) polymérase (PARP) clivée, phospho-p53-Ser15, et caspase-3 clivée (Cell Signaling Technology, Danvers, MA, USA) ; PDK2 (Abcam, Cambridge, MA, USA) ; et PDHE1α et phospho-PDHE1α (ser293) (Calbiochem, Billerica, MA, USA). la β-Actine (Sigma-Aldrich) a été utilisée comme contrôle de charge. Tous les anticorps ont été dilués entre 1:250 et 1:5000.
Microscopie confocale
ΔΨm a été imagé dans des cellules cancéreuses vivantes à l’aide du TMRM (Life TechnologiesTM ), un colorant à base de rhodamine sensible aux mitochondries et au voltage (fluorescence rouge). Le superoxyde mitochondrial (mROS, Life TechnologiesTM ) produit pendant la respiration cellulaire a été mesuré par MitoSOX (fluorescence rouge). Les images de fluorescence TMRM ou mitoSOX ont été capturées par un microscope confocal à deux photons Zeiss LSM 510 (Carl Zeiss AG, Heidenheim, Allemagne), et la fluorescence moyenne a été quantifiée (unités de fluorescence arbitraires, AFU) dans les cellules cancéreuses en culture à l’aide du logiciel d’imagerie Zen (Carl Zeiss AG). L’acquisition et l’analyse des images ont été effectuées par deux scientifiques qui ne connaissaient pas la source des échantillons de cellules.
Imagerie par tomographie par émission de positons (TEP)
L’absorption de glucose a été imagée in vivo chez des souris nudes avec transplantation de cellules HN4R ou HN9R sous anesthésie par gaz isoflurane (20 % d’oxygène) et avec
18 F-fluorodéoxyglucose (18 F-FDG) comme radiotraceur. L’imagerie TEP a été réalisée à l’aide du microPET FOCUS 120 (Concorde Microsystem Inc., Knoxville, TN, USA). Les souris ont été mises à jeun pendant la nuit et ont ensuite reçu une injection intraveineuse de 0,15 mCi. Après 1 h, une TEP au 18 F-FDG a été réalisée dans le corps entier. Les images d’acquisition des données TEP sont représentées à l’aide d’une carte pseudo-colorée, la couleur rouge indiquant une forte captation de 18 F-FDG.
La valeur maximale et la valeur moyenne de la captation normalisée (SUVmax et SUVmoy) ont été utilisées pour déterminer l’activité de la TEP au 18 F-FDG. Le SUV a été analysé à l’aide de l’équation SUV = A/(ID/BW), où A est l’activité corrigée de la décroissance dans le tissu (en mCi/mL), ID est la dose injectée de FDG (en mCi) et BW est le poids corporel de la souris (en grammes). Des régions d’intérêt (ROI) sphériques ou elliptiques ont été placées sur les lésions visibles sur les images TEP. Le SUVmax et le SUVmoyen ont été calculés en dessinant automatiquement une ROI sur la tranche la plus intense des tumeurs visibles sur les images TEP. Les MTV ont été calculés à partir des données TEP corrigées de l’atténuation à l’aide d’un logiciel commercial (INFINITT PACS ; INFINITT Healthcare Co., Ltd). les données TEP au 18 F-FDG ont été introduites dans la station de travail au format DICOM, et les valeurs d’intensité ont été automatiquement converties en SUV. Pour les calculs MTV, les marges de contour de la tumeur ont été définies en utilisant un SUV fixe de 2,0, et le volume tumoral a ensuite été délimité avec l’isocontour SUV 2,0. Les images TEP ont été obtenues chez des souris porteuses de tumeurs au jour 21 après le début du traitement. Les valeurs moyennes des valeurs SUVmax et MTV de la tumeur ont été comparées entre les différents groupes de traitement.
Études précliniques
Toutes les procédures d’étude sur les animaux ont été réalisées conformément aux protocoles approuvés par le Comité institutionnel de soins et d’utilisation des animaux de notre institution. Des souris nude mâles BALB/c athymiques âgées de six semaines (nu/nu) ont été achetées auprès de Central Lab Animal Inc. (Séoul, République de Corée). Les cellules AMC-HN4R ou HN9R (5 x 10 6 ) ont été injectées par voie sous-cutanée dans le flanc. Le volume des tumeurs et le poids corporel ont été mesurés tous les 3 jours. Les tumeurs ont été mesurées à l’aide d’un pied à coulisse, et le volume a été calculé comme suit : (longueur x largeur2 )/2. Le traitement a commencé lorsque les implants cellulaires sont devenus des nodules palpables (=jour 0). Les souris ont été réparties au hasard en quatre groupes de traitement : véhicule, DCA, cisplatine et DCA plus cisplatine.
Les souris ont été traitées avec de l’eau potable additionnée de 0,5 g/L de DCA, ou par injection i.p. de 5 mg/kg de cisplatine une fois par semaine, ou avec une combinaison de DCA et de cisplatine selon les mêmes schémas. Les souris ont été sacrifiées le 24e jour, et les tumeurs ont été isolées et analysées par immunoblotting et par un test in situ de marquage in situ par la désoxynucléotidyltransférase terminale médiée par le dUTP nick-end (TUNEL) (EMD Millipore, Billerica, MA, USA). Le nombre de corps apoptotiques a été compté à l’aveugle dans dix champs de haute puissance sélectionnés au hasard. La signification statistique entre les différents groupes de traitement a été évaluée par un test U de Mann-Whitney à deux intervalles ou un test t de Student.
Résultats
L‘augmentation de la glycolyse dans les cellules HNC est associée à la résistance au cisplatine et inversée par le DCA
Toutes les lignées cellulaires utilisées dans notre étude étaient des HNC humaines. L’effet cytotoxique du DCA a été évalué dans les cellules HNC par exclusion au bleu trypan, coloration au cristal violet et test MTT. Le DCA, testé jusqu’à 30 mM pendant 72 h, a inhibé de façon marquée la croissance des cellules HNC (test MTT, Fig. 1A). La bioénergétique des cellules HNC a été modifiée par le DCA : la glycolyse cellulaire a diminué de manière significative, tandis que la phosphorylation oxydative a augmenté (Fig. 1B). La modification de la bioénergétique variait entre les lignées cellulaires HNC et était associée de manière significative à la sensibilité au cisplatine : les cellules HNC présentant une glycolyse élevée ont montré une résistance au cisplatine (R2 = 0,859, P < 0,001 ; Fig. 1C). En outre, lors de l’exposition au DCA, les cellules cancéreuses présentant la plus grande variation bioénergétique étaient susceptibles de devenir plus apoptotiques (R2 = 0,769, P = 0,002 ; Fig. 1D). Cela suggère que le DCA induit une apoptose spécifique aux cellules cancéreuses en diminuant la glycolyse dans les cellules HNC.
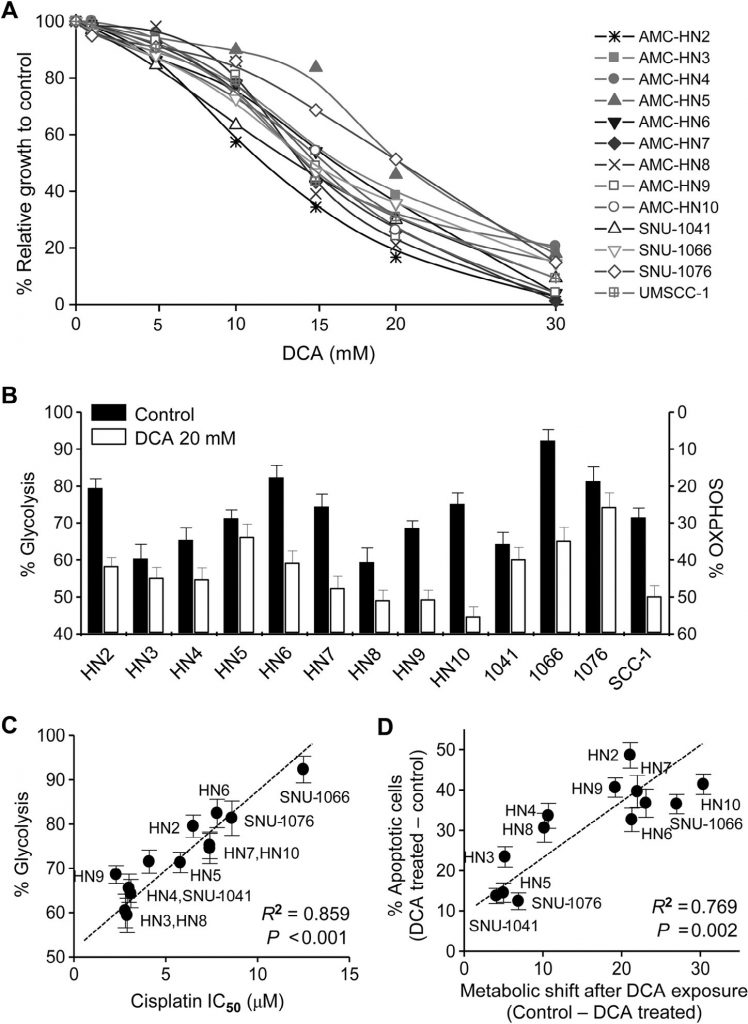
L‘expression de PDK2 est associée à la résistance au cisplatine dans les HNC
Les effets cytotoxiques du cisplatine ont été testés dans des lignées cellulaires cancéreuses HN4-cisR et HN9-cisR cultivées et parentales (Fig. 2A). Les cellules HN4-cisR et HN9-cisR résistantes au cisplatine ont montré une augmentation de 12 et 18 fois de la CI50, respectivement, par rapport à leurs lignées parentales respectives. Une analyse Western blot a montré que HK2 et PDK2 étaient fortement exprimés dans les cellules HN4-cisR et HN9-cisR par rapport aux cellules parentales respectives, tandis que l’expression de PDHE1α était faible dans les cellules résistantes au cisplatine ( Fig. 2B ). Le niveau d’expression de l’ARNm de PDK2 était également plus élevé dans les cellules résistantes que dans les cellules sensibles (P < 0,01) (Fig. 2C). Le DCA a inhibé la croissance des cellules HNC résistantes au cisplatine autant que celle des cellules HNC sensibles au cisplatine (Fig. 2D). Une analyse Western blot a montré que le DCA diminuait significativement les niveaux de PDK2 et de phospho-PDHE1α (pPDHE1α) mais augmentait le niveau des protéines proapoptotiques, y compris la PARP clivée (cPARP) et la PUMA, ainsi que celui de p21 dans les cellules HN4-cisR et HN9-cisR (Fig. 2E). Cela suggère que le DCA peut inhiber efficacement la croissance des cellules HNC résistantes au cisplatine ainsi que celle des cellules HNC sensibles au cisplatine.
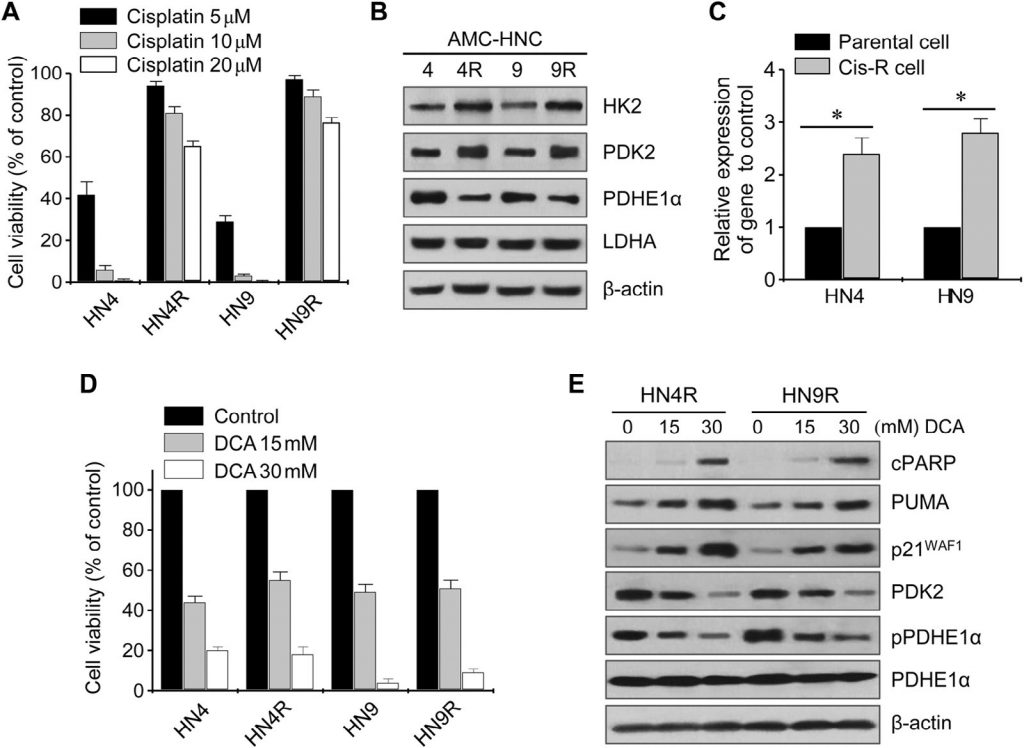
Le DCA induit une accumulation de ROS dans les cellules HNC
La modification des ROS cellulaires par le DCA a été évaluée par cytométrie en flux à l’aide de la sonde fluorescente redox-sensible DCF-DA. L’exposition au DCA a provoqué une augmentation significative des niveaux de ROS dans les cellules HNC (P < 0,01), qui a été bloquée par une co-exposition à la NAC ou à l’inhibiteur de la SOD, le diéthyl-dithio-carbamate (Fig. 3A). La glycolyse a significativement augmenté dans les cellules HNC résistantes au cisplatine par rapport aux cellules parentales (P < 0,01), mais le DCA a induit une diminution significative de la glycolyse et une augmentation de la phosphorylation oxydative dans les cellules HNC sensibles et résistantes au cisplatine (P < 0,01) (Fig. 3B). Le potentiel de la membrane mitochondriale (ΔΨm) a été mesuré à l’aide du TMRM, un colorant à base de rhodamine sensible aux mitochondries et sensible au voltage, et les ROS mitochondriaux (mROS, superoxyde mitochondrial) ont été mesurés à l’aide du rouge MitoSOX. Les cellules HNC traitées au DCA ont montré une diminution de ΔΨm et une augmentation de mROS (P < 0,01) (Fig. 3C). La modification du ΔΨm dans les cellules HNC résistantes au cisplatine après exposition au DCA a été bloquée par un prétraitement au NAC (Fig. 3D). Cela suggère que le DCA peut induire une accumulation de ROS dans les cellules HNC en activant la phosphorylation oxydative.
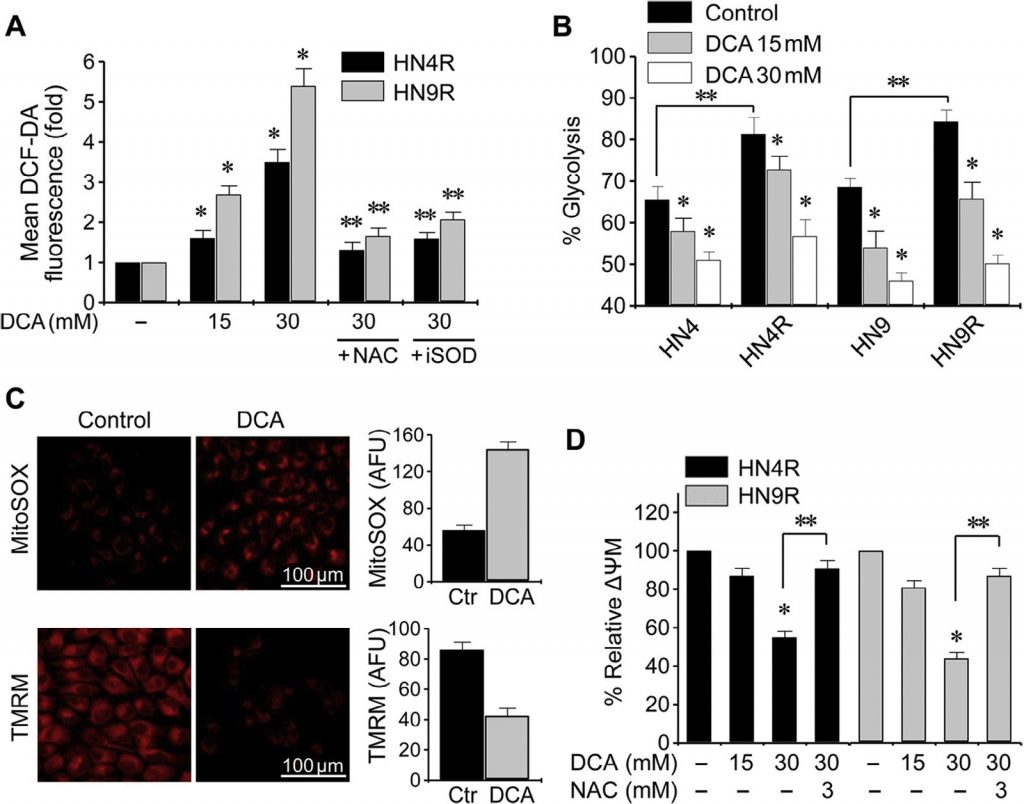
Le DCA favorise l’arrêt du cycle cellulaire et l’apoptose dans les cellules HNC
Les essais clonogéniques ont entraîné une diminution marquée du nombre de colonies HN4-cisR et HN9-cisR après exposition au DCA (P < 0,01) (Fig. 4A ). La cytométrie en flux utilisant la coloration à l’iodure de propidium a montré un changement significatif du cycle cellulaire dans les cellules HNC : la wogonine a augmenté la population apoptotique sub-G1 (P < 0,05), et cet effet a été bloqué par la co-exposition à la NAC (Fig. 4B). Une analyse Western blot a montré que le DCA diminuait pPDHE1α mais augmentait cPARP, p21, phospho-p53 et caspase-3 clivée (cCasp3) de manière dépendante du temps (Fig. 4C ). L’activation de PDHE1α par le knockdown de PDK2 dans les cellules AMC-HN4-cisR n’a pas augmenté de manière significative les protéines proapoptotiques cPARP et cCasp3 (Fig. 4D ) ; cependant, le knockdown de PDHE1α a diminué l’expression de p21 et la phosphorylation de p53. La cytométrie en flux utilisant l’iodure de propidium et la coloration de l’annexine-V a montré une induction efficace de l’apoptose et de la mort cellulaire dans les cellules HNC résistantes au cisplatine par le DCA ; l’effet était réduit lors de la co-exposition au DCA et à l’antioxydant NAC ou à l’inhibiteur de la pan-caspase Z-VAD-fmk (Fig. 4E). Ces résultats ont été confirmés par la mesure de l’activité des caspases, qui a été augmentée de manière significative par le DCA en fonction de la concentration (fig. 4F).
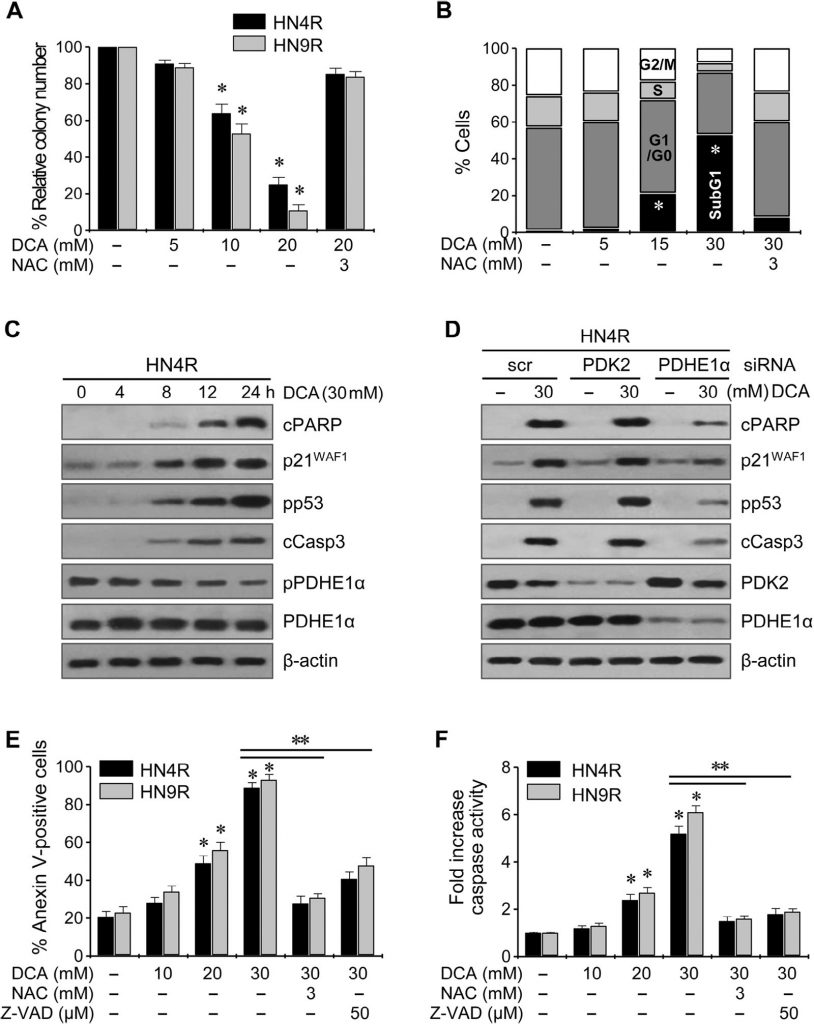
Le DCA sensibilise les cellules HNC résistantes au cisplatine au cisplatine in vitro et in vivo
Le cisplatine (10 µM) n’a pas induit de cytotoxicité significative ou d’expression de protéines apoptotiques dans les lignées HNC HN4-cisR et HN9-cisR résistantes au cisplatine par rapport aux lignées parentales HN4 et HN9 sensibles au cisplatine (Fig. 5A) ; toutefois, le DCA a entraîné une diminution marquée de la survie des cellules HNC résistantes au cisplatine (P < 0,05), qui a été bloquée par un prétraitement au NAC. Le DCA a induit l’expression de protéines apoptotiques et a augmenté la cytotoxicité induite par la cisplatine et l’expression des protéines apoptotiques dans les cellules HN4-cisR (Fig. B). En association, le DCA a augmenté la cytotoxicité du cisplatine dans les cellules HN4-cisR en augmentant l’activité des caspases dans une mesure supérieure à la somme des effets de chaque agent seul (CI < 1, P < 0,01), qui ont été affaiblis par l’extinction du gène PDHE1α (Fig. 5C). Le ΔΨm était plus élevé dans les cellules HN4-cisR résistantes à la cisplatine que dans les cellules HN4 parentales sensibles à la cisplatine (ΔΨm, intensité de fluorescence moyenne [IFM] : 1 ± 0 contre 0,54 ± 0,09, P < 0,001) et réduit par le DCA ou la combinaison de cisplatine et de DCA, ce qui a été abrogé par l’extinction du gène PDHE1α (Fig. 5D).
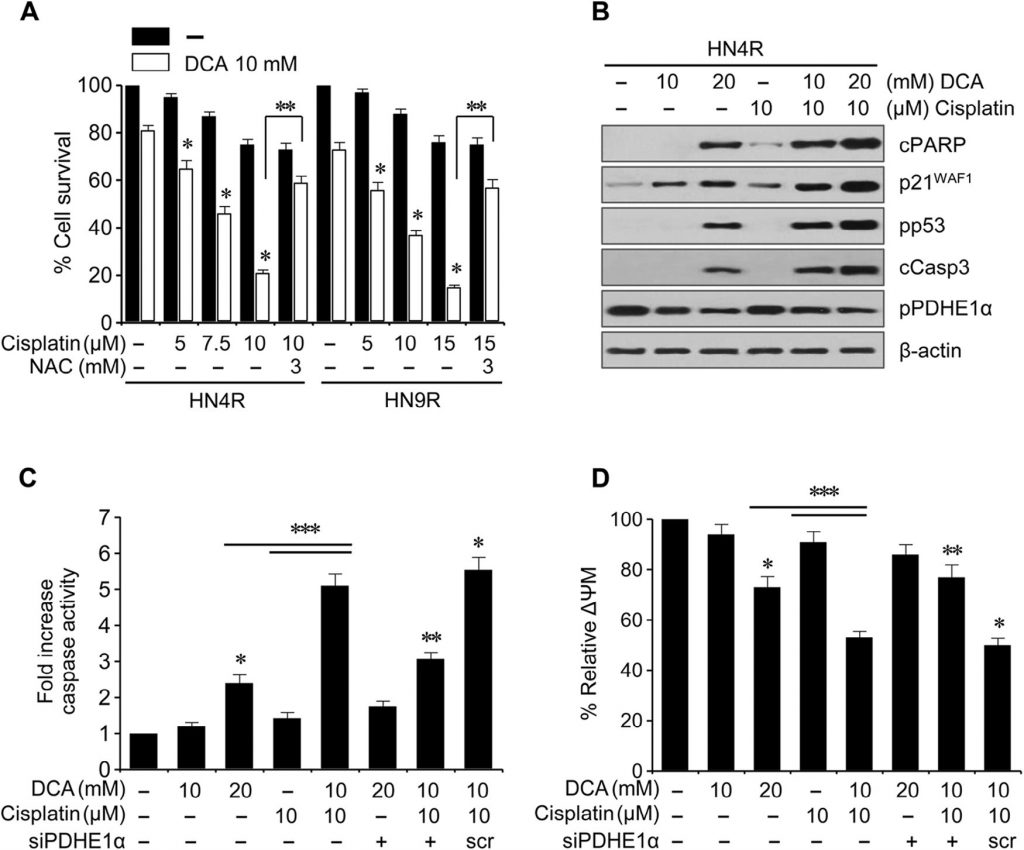
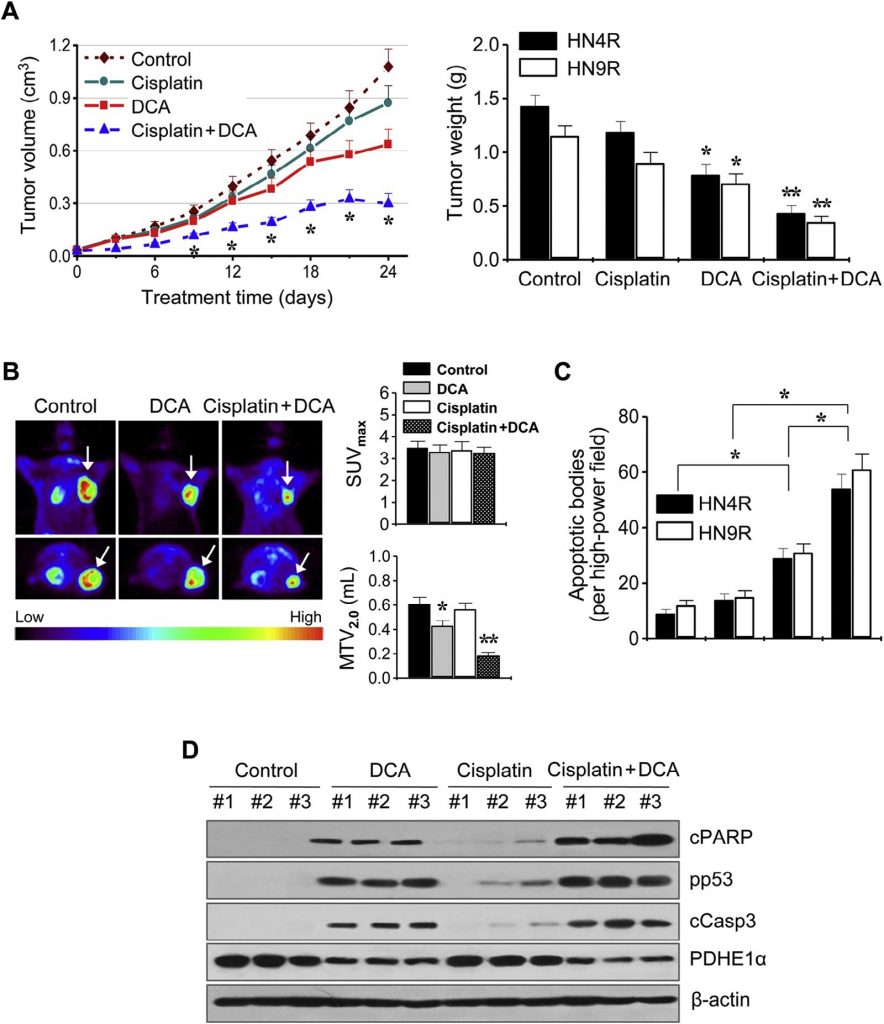
Ces résultats ont été examinés plus en détail dans des modèles de xénogreffes de tumeurs in vivo. Des souris nude BALB/c athymiques portant des tumeurs AMC-HN4-cisR ou HN9-cisR ont été traitées avec du DCA, du cisplatine, du DCA plus du cisplatine, ou un véhicule. L’association du cisplatine et du DCA a supprimé de manière synergique la croissance tumorale (Fig. 6A). L’imagerie in vivo de la croissance tumorale a été réalisée par TEP au 18 F-FDG au jour 21 du traitement. Des absorptions focales de 18 F-FDG ont été observées aux sites d’implantation des tumeurs, où l’absorption maximale normalisée (SUVmax) et le volume tumoral métabolique à SUV 2,0 (MTV2,0) ont été mesurés. Le SUVmax n’était pas différent entre les groupes de traitement (P > 0,1), mais le MTV2.0 était significativement plus faible dans les groupes traités par le DCA et l’association que dans les autres groupes (P < 0,05) (Fig. 6B). Les tests d’apoptose in situ ont montré que des corps apoptotiques TUNEL-positifs étaient plus fréquemment observés dans les tumeurs traitées par le DCA et le cisplatine plus DCA que dans celles traitées par le véhicule (P < 0,01) (Fig. 6C). Les analyses Western blot des tissus tumoraux ont montré que les taux de protéines apoptotiques augmentaient davantage dans les cellules HN4-cisR traitées par l’association de cisplatine et de DCA que dans les tumeurs traitées par un seul agent (fig. 6D).
Discussion
L’augmentation de la glycolyse, fréquemment retrouvée dans le cancer, est étroitement liée à une atteinte mitochondriale ou à une phosphorylation oxydative défectueuse et contribue à la résistance thérapeutique [18,19]. La glycolyse aérobie a été impliquée dans la résistance à la chimiothérapie [20] ou à la radiothérapie [21]. Dans la présente étude, les lignées cellulaires résistantes au cisplatine présentent une glycolyse élevée, indiquant un lien biochimique entre la glycolyse et la chimiorésistance. Les cellules résistantes aux médicaments ont un plus grand besoin d’ATP que les cellules normales pour maintenir l’homéostasie cellulaire et activer les voies de survie qui permettent d’échapper à la mort cellulaire en cas de stress génotoxique [20]. Les cellules cancéreuses résistantes augmentent la glycolyse pour générer rapidement de l’ATP afin de répondre à la demande intracellulaire. Ce phénomène a été clairement constaté dans nos cellules résistantes au cisplatine par rapport aux cellules sensibles parentales, ce qui indique que la résistance aux médicaments dans les HNC est directement associée à une augmentation de la glycolyse. Cela implique que le changement bioénergétique des cellules cancéreuses vers une augmentation de la glycolyse est lié à la chimiorésistance [14,22]. Par conséquent, le niveau de glycolyse dans les cancers humains peut être un biomarqueur de chimiorésistance et nécessite une validation clinique dans les tissus cancéreux humains.
La présente étude montre que le DCA fait passer la production d’énergie de la glycolyse à l’oxydation mitochondriale du glucose dans les HNC. Cette inversion du changement métabolique vers la glycolyse induite par le DCA élimine l’avantage prolifératif des cellules cancéreuses et conduit finalement à la mort cellulaire [23]. Le DCA, en tant qu’analogue structurel du pyruvate, inhibe la PDK et réactive la PDH, une enzyme faisant partie du complexe qui convertit le pyruvate en acétyl-CoA, le principal substrat du cycle de Krebs [10,23]. Comme la plupart des types de cancer produisent un environnement hypoxique, les cellules cancéreuses dépendent de la glycolyse anaérobie comme principale source d’énergie. L’activation du facteur inductible de l’hypoxie (HIF) induit la PDK mitochondriale [24]. Dans le cancer, l’activité de la PDH mitochondriale est bloquée par la PDK, ce qui entraîne une moindre disponibilité de l’acétyl-CoA pour l’oxydation mitochondriale du glucose [24]. L’expression accrue de la PDK est associée à la résistance aux médicaments dans le cancer [25,26]. La régulation à la hausse de PDK2 par des mutations mitochondriales et la stabilisation de HIF1α est également observée dans les cellules HNC [27]. Dans la présente étude, l’association entre la surexpression de PDK2 et la résistance au cisplatine dans les cellules HNC a également été confirmée. Ces résultats indiquent l’importance de PDK comme nouvelle cible moléculaire dans la thérapie du cancer [10]. Des données antérieures ont montré que l’inhibition génétique ou pharmacologique de la PDK modifie la bioénergétique du cancer et rétablit l’apoptose dépendante des mitochondries dans les cellules cancéreuses [11,13]. Par conséquent, la PDK est efficace pour surmonter la résistance au traitement dans les cancers à phénotype agressif in vitro et in vivo[12-14].
L’activation de la PDH par le DCA induit l’accumulation de mROS dans les cellules cancéreuses. Les cellules cancéreuses présentent une diminution du métabolisme mitochondrial du glucose, ce qui entraîne une diminution de l’activité de la chaîne de transport des électrons (ETC) et une diminution du mROS [10], [28]. Le remodelage mitochondrial de l’hyperpolarisation ΔΨm et la diminution de la production de mROS dans le cancer conduisent à une résistance à l’apoptose sous stress génotoxique. Dans les cellules cancéreuses résistantes ayant un phénotype plus glycolytique, la HK2 entraîne sa translocation vers la membrane mitochondriale externe et l’augmentation du ΔΨm [29]. L’inhibition de la glycolyse et de la translocation de HK2 diminue le ΔΨm du cancer et inverse la résistance à l’apoptose [29]. Dans la présente étude, l’augmentation de l’expression de HK2 et PDK2 et du potentiel de la membrane mitochondriale a également été constatée dans les cellules cancéreuses résistantes au cisplatine. Le DCA force l’entrée du pyruvate dans la mitochondrie via l’activation de la PDH, le régulateur de maintien de la porte de l’oxydation mitochondriale du glucose, et diminue le ΔΨm et augmente le mROS [13]. Dans la présente étude, le DCA a simplement inversé le remodelage mitochondrial élevé dans les cellules HNC résistantes au cisplatine, ce qui a contribué à la mort des cellules cancéreuses. L’activation de l’oxydation mitochondriale du glucose par le DCA a induit le mROS et l’activation de la signalisation mitochondriale en aval, ce qui a entraîné l’activation de p53 et de ses voies proapoptotiques connexes et a conduit à la mort des cellules cancéreuses chimiorésistantes. Dans notre étude, l’inhibition pharmacologique de la production de mROS ou de l’apoptose médiée par les caspases a atténué l’effet cytotoxique du DCA dans les cellules HNC, ce qui confirme les mécanismes connus du médicament.
Des études précliniques et cliniques soutiennent l’utilisation du DCA chez les patients cancéreux et chez les patients souffrant d’acidose lactique associée à des maladies mitochondriales [10,23]. Un certain nombre d’études démontrent les effets cytotoxiques du DCA, seul ou en combinaison avec d’autres traitements, dans une variété de tumeurs dérivées des trois couches germinales [23]. Une étude antérieure a montré que le glioblastome, l’un des cancers humains les plus agressifs, surexprime PDK2 dans les tissus cancéreux prélevés chez 49 patients et qu’il a régressé chez cinq patients après un traitement au DCA, ce qui prouve le bénéfice clinique du DCA dans les cancers résistants [13]. Dans le cas de l’HNC, une étude récente a comparé l’effet du DCA sur trois lignées cellulaires de carcinome épidermoïde oral (OSCC) [15]. Les cellules OSCC présentant une phosphorylation oxydative mitochondriale déficiente (c’est-à-dire une glycolyse élevée) étaient plus sensibles au DCA que les autres [15,30]. La présente étude a confirmé que les cellules de HNC présentant des changements bioénergétiques élevés étaient plus sensibles au DCA. En fait, comme les cellules cancéreuses chimiorésistantes ont tendance à avoir une glycolyse élevée, elles pourraient être ciblées par des inhibiteurs métaboliques [31]. Le DCA est donc un bon candidat pour traiter les cellules cancéreuses au phénotype agressif, y compris les cellules HNC chimiorésistantes. Cependant, comme les effets anticancéreux potentiels du DCA sont encore controversés, en particulier dans les tumeurs hypoxiques, des études précliniques et cliniques supplémentaires sur le DCA et le cancer sont nécessaires [32]. Une étude systématique récente montre que le DCA agit en synergie avec de nombreux agents anticancéreux standard, et des essais cliniques de phase initiale suggèrent que le DCA chronique est sûr et bien toléré lorsqu’il est administré par voie orale à raison de 12,5 mg/kg deux fois par jour [23].
Notre étude a révélé que le DCA est synergique avec le cisplatine et contourne ainsi la résistance au cisplatine dans les cellules HNC. Étant donné que le cisplatine est un agent chimiothérapeutique de première ligne dans les HNC, l’association du cisplatine et du DCA peut être efficace dans le cadre clinique pour réduire la toxicité et surmonter la résistance aux médicaments anticancéreux. Des rapports antérieurs ont montré que le DCA potentialise l’effet cytotoxique du cisplatine en induisant une apoptose dépendante des mitochondries [33,34]. La liaison de deux molécules de DCA au cisplatine, appelée mitaplatine, induit la destruction sélective des cellules cancéreuses [35,36]. Ces résultats prouvent que le DCA peut présenter des avantages supplémentaires en association avec les thérapies anticancéreuses actuelles. Dans notre étude, le DCA et son association avec le cisplatine n’ont pas affecté le SUVmax mais ont significativement diminué le MTV2.0. Cela signifie que le DCA supprime efficacement la croissance tumorale in vivo malgré le fait qu’il n’y ait pas de changement significatif de l’absorption maximale de glucose dans les ROIs localisés dans la tumeur. La présente étude est la première à montrer que le DCA restaure l’effet cytotoxique du cisplatine dans les cellules HNC résistantes aux médicaments in vitro et in vivo. Le DCA a induit une forte augmentation de l’apoptose médiée par le cisplatine via l’activation de p53, PARP et caspase dans les cellules HNC résistantes au cisplatine. Le DCA a sensibilisé les cellules HNC résistantes aux médicaments au cisplatine, ce qui a conduit à une cytotoxicité accrue et à un traitement plus efficace des HNC agressifs. Dans l’ensemble, ces résultats peuvent avoir une importance clinique primordiale : induire la mort des cellules résistantes avec le DCA pourrait réduire la dose de cisplatine nécessaire dans le cadre clinique et ainsi minimiser les effets indésirables potentiels de la chimiothérapie au cisplatine.
En conclusion, nos données suggèrent que la glycolyse élevée et la surexpression de PDK sont étroitement liées à la résistance au cisplatine dans les cellules HNC. Le DCA fait basculer la bioénergétique des cellules HNC vers la phosphorylation oxydative mitochondriale. Cela entraîne une diminution de ΔΨm et une augmentation de mROS, sensibilisant ainsi les cellules HNC chimiorésistantes au cisplatine in vitro et in vivo. Ces données justifient la poursuite des études précliniques et cliniques du DCA, un candidat médicament anticancéreux prometteur, dans les HNC à phénotypes agressifs. Cependant, notre conclusion doit être prise en considération avec prudence, dans un scénario inverse. La phosphorylation oxydative mitochondriale défectueuse était liée à la sensibilité des cellules cancéreuses aux conditions de glucose faible, ce qui pourrait être utilisé comme biomarqueur pour la thérapie du cancer [37]. Les cellules cancéreuses sont également adaptées pour faciliter la prolifération par l’absorption et l’incorporation de nutriments dans la biomasse plutôt que pour une production efficace d’ATP [38]. Par conséquent, les effets précliniques et cliniques du DCA sur le HNC et d’autres types de cancer doivent être examinés plus en détail.
Remerciements
Cette étude a été financée par une subvention (2015R1A2A1A15054540) du Basic Science Research Program de la National Research Foundation of Korea (NRF), Ministry of Science, ICT, and Future Planning, et une subvention (HI14C23050000) du Korean Health Technology R&D Project, Ministry of Health & Welfare, Séoul, République de Corée (J.-L. Roh).
Conflit d’intérêts
Les auteurs ne déclarent aucun conflit d’intérêt.
RÉFÉRENCES
1 A. Jemal, F. Bray, M.M. Center, J. Ferlay, E. Ward, D. Forman, Global cancer statistics, CA Cancer J. Clin, Vol. 61, 2011, 69-90
2 R.I. Haddad, D.M. Shin, Recent advances in head and neck cancer, N. Engl. J. Med, Vol. 359, 2008, 1143-1154
3 E.B. Lamont, E.E. Vokes, Chemotherapy in the management of squamous-cell carcinoma of the head and neck, Lancet Oncol, Vol. 2, 2001, 261-269
4 F. Petrelli, A. Coinu, V. Riboldi, K. Borgonovo, M. Ghilardi, M. Cabiddu, Concomitant platinum-based chemotherapy or cetuximab with radiotherapy for locally advanced head and neck cancer : a systematic review and meta-analysis of published studies, Oral Oncol, Vol. 50, 2014, 1041-1048
5 D. Hanahan, R.A. Weinberg, Hallmarks of cancer : the next generation, Cell, Vol. 144, 2011, 646-674
6 J.R. Cantor, D.M. Sabatini, Cancer cell metabolism : one hallmark, many faces, Cancer Discov, Vol. 2, 2012, 881-898
7 Y. Zhao, E.B. Butler, M. Tan, Targeting cellular metabolism to improve cancer therapeutics, Cell Death Dis, Vol. 4, 2013, e532
8 R.A. Cairns, I.S. Harris, T.W. Mak, Regulation of cancer cell metabolism, Nat. Rev. Cancer, Vol. 11, 2011, 85-95
9 R.H. Xu, H. Pelicano, Y. Zhou, J.S. Carew, L. Feng, K.N. Bhalla, Inhibition of glycolysis in cancer cells : a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia, Cancer Res, Vol. 65, 2005, 613-621
10 G. Sutendra, E.D. Michelakis, Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology, Front. Oncol, Vol. 3, 2013, 38
11 G. Sutendra, P. Dromparis, A. Kinnaird, T.H. Stenson, A. Haromy, J.M. Parker, Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer, Oncogene, Vol. 32, 2013, 1638-1650
12 R.C. Sun, M. Fadia, J.E. Dahlstrom, C.R. Parish, P.G. Board, A.C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo, Breast Cancer Res. Treat, Vol. 120, 2010, 253-260
13 E.D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, Metabolic modulation of glioblastoma with dichloroacetate, Sci. Transl. Med, Vol. 2, 2010, 31ra34
14 Y.C. Shen, D.L. Ou, C. Hsu, K.L. Lin, C.Y. Chang, C.Y. Lin, Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma, Br. J. Cancer, Vol. 108, 2013, 72-81
15 V. Ruggieri, F. Agriesti, R. Scrima, I. Laurenzana, D. Perrone, T. Tataranni, Dichloroacetate, un médicament sélectif ciblant les mitochondries pour le carcinome spinocellulaire oral : une perspective métabolique du traitement, Oncotarget, Vol. 6, 2015, 1217-1230
16 M. Nakamura, K. Nakatani, K. Uzawa, K. Ono, H. Uesugi, K. Ogawara, Establishment and characterization of a cisplatin-resistant oral squamous cell carcinoma cell line, H-1R, Oncol. Rep, Vol. 14, 2005, 1281-1286
17 T.C. Chou, Drug combination studies and their synergy quantification using the Chou-Talalay method, Cancer Res, Vol. 70, 2010, 440-446
18 N. Guaragnella, S. Giannattasio, L. Moro, Mitochondrial dysfunction in cancer chemoresistance, Biochem. Pharmacol, Vol. 92, 2014, 62-72
19 S. Ganapathy-Kanniappan, J.F. Geschwind, La glycolyse tumorale comme cible pour la thérapie du cancer : progrès et perspectives, Mol. Cancer, Vol. 12, 2013, 152
20 Y. Zhou, F. Tozzi, J. Chen, F. Fan, L. Xia, J. Wang, Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells, Cancer Res, Vol. 72, 2012, 304-314
21 S.P. Pitroda, B.T. Wakim, R.F. Sood, M.G. Beveridge, M.A. Beckett, D.M. MacDermed, STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect, BMC Med, Vol. 7, 2009, 68
22 B. Bhattacharya, S.H. Low, C. Soh, N. Kamal Mustapa, M. Beloueche-Babari, K.X. Koh, Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype, Br. J. Pharmacol, Vol. 171, 2014, 3255-3267
23 S. Kankotia, P.W. Stacpoole, Biochim. Biophys. Acta, Vol. 1846, 2014, 617-629
24 J.W. Kim, I. Tchernyshyov, G.L. Semenza, C.V. Dang, HIF-1-mediated expression of pyruvate dehydrogenase kinase : a metabolic switch required for cellular adaptation to hypoxia, Cell Metab, Vol. 3, 2006, 177-185
25 Y. Sun, A. Daemen, G. Hatzivassiliou, D. Arnott, C. Wilson, G. Zhuang, Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells, Cancer Metab, Vol. 2, 2014, 20
26 C.W. Lu, S.C. Lin, C.W. Chien, S.C. Lin, C.T. Lee, B.W. Lin, Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer, Am. J. Pathol, Vol. 179, 2011, 1405-1414
27 W. Sun, S. Zhou, S.S. Chang, T. McFate, A. Verma, J.A. Califano, Mitochondrial mutations contribute to HIF1alpha accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma, Clin. Cancer Res, Vol. 15, 2009, 476-484
28 S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth, Cancer Cell, Vol. 11, 2007, 37-51
29 J.G. Pastorino, J.B. Hoek, N. Shulga, Activation of glycogen synthase kinase 3beta disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity, Cancer Res, Vol. 65, 2005, 10545-10554
30 L.H. Stockwin, S.X. Yu, S. Borgel, C. Hancock, T.L. Wolfe, L.R. Phillips, Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC, Int. J. Cancer, Vol. 127, 2010, 2510-2519
31 E.J. Sullivan, M. Kurtoglu, R. Brenneman, H. Liu, T.J. Lampidis, Targeting cisplatin-resistant human tumor cells with metabolic inhibitors, Cancer Chemother. Pharmacol, vol. 73, 2014, 417-427
32 S. Shahrzad, K. Lacombe, U. Adamcic, K. Minhas, B.L. Coomber, Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia, Cancer Lett, Vol. 297, 2010, 75-83
33 D. Heshe, S. Hoogestraat, C. Brauckmann, U. Karst, J. Boos, C. Lanvers-Kaminsky, Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs, Cancer Chemother. Pharmacol, Vol. 67, 2011, 647-655
34 J. Xie, B.S. Wang, D.H. Yu, Q. Lu, J. Ma, H. Qi, Dichloroacetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells, Int. J. Oncol, Vol. 38, 2011, 409-417
35 S. Dhar, S.J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate, Proc. Natl. Acad. Sci. U.S.A., Vol. 106, 2009, 22199-22204
36 X. Xue, S. You, Q. Zhang, Y. Wu, G.Z. Zou, P.C. Wang, Mitaplatin increases sensitivity of tumor cells to cisplatin by inducing mitochondrial dysfunction, Mol. Pharm, Vol. 9, 2012, 634-644
37 K. Birsoy, R. Possemato, F.K. Lorbeer, E.C. Bayraktar, P. Thiru, B. Yucel, Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides, Nature, Vol. 508, 2014, 108-112
38 M.G. Vander Heiden, L.C. Cantley, C.B. Thompson, Understanding the Warburg effect : the metabolic requirements of cell, Nature, Vol. 508, 2014, 108-112
Contenu connexe :