Jong-Lyel Roh a, *, Jin Young Park a, Eun Hye Kim a, Hye Jin Jang a, Minsu Kwon b
a Abteilung für Hals-Nasen-Ohrenheilkunde, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republik Korea
b Abteilung für Hals-Nasen-Ohrenheilkunde, Gyeongsang National University Hospital School of Medicine, Jinju, Republik
KoreaKorrespondenzautor: Tel.: +82 2 3010 3965; fax: +82 2 489 2773. E-mail-Adresse: [email protected] (J.-L. Roh).
Received: 9. September 2015Revised
: revised form 13 November 2015Accepted
: 14 November 2015
Zusammenfassung
Dichloracetat (DCA), ein Arzneimittel für seltene Krankheiten, das eine Verlagerung von der Glykolyse zur oxidativen Phosphorylierung fördert, wurde für die Krebstherapie wiederverwendet. In der vorliegenden Studie wurde untersucht, ob DCA die Cisplatinresistenz bei Kopf- und Halskrebs (HNC) überwinden kann. Es wurden zwei cisplatinresistente HNC-Zelllinien (AMC-HN4R und -HN9R), ihre Elternlinien und andere menschliche HNC-Linien verwendet. Die Wirkung von DCA, allein und in Kombination mit Cisplatin, wurde durch Messung des Zellzyklus, der Lebensfähigkeit, des Todes, der Produktion reaktiver Sauerstoffspezies (ROS), des mitochondrialen Membranpotenzials (ΔΨm) und der Proteinexpression in präklinischen Mäusetumor-Xenograft-Modellen untersucht. Eine erhöhte Glykolyse korrelierte mit einer geringeren Empfindlichkeit gegenüber Cisplatin und wurde durch DCA reduziert. Cisplatin-resistente Zellen überexprimierten Pyruvat-Dehydrogenase-Kinase 2 (PDK2). DCA löste den Tod von HNC-Zellen aus, indem es ΔΨm verringerte und die mitochondriale ROS-Produktion förderte. Diese Wirkung wurde durch das Antioxidans N-Acetyl-L-Cystein oder durch die Hemmung der Caspase-vermittelten Apoptose verringert. Die Aktivierung der mitochondrialen Glukoseoxidation durch DCA aktivierte schließlich die nachgelagerten mitochondrialen apoptotischen Signalwege, die zum Tod der chemoresistenten Krebszellen führten. Daher sensibilisierte DCA resistente HNC-Zellen in vitro und in vivo erheblich für Cisplatin. Eine hohe Glykolyse und eine Überexpression von PDK2 stehen in engem Zusammenhang mit der Cisplatinresistenz von HNC-Zellen; letztere kann durch DCA überwunden werden.
Schlüsselwörter: Kopf-Hals-Krebs, Cisplatin-Resistenz, PDK2, Dichloracetat, MitochondrienumbauAbkürzungen
: HNC, Kopf- und Halskrebs; DCA, Dichloracetat; CDDP, Cisplatin; OXPHOS, oxidative Phosphorylierung; PDK2, Pyruvat-Dehydrogenase-Kinase 2; PDHE1α, Pyruvat-Dehydrogenase-Isoform E1α; ROS, reaktive Sauerstoffspezies; ΔΨm, mitochondriales Membranpotenzial; NAC, N-Acetyl-L-Cystein; DCF-DA, 2′,7′-Dichlorfluoresceindiacetat; PARP, Poly(ADP-Ribose)-Polymerase; siRNA, short interfering RNA; 18F-FDG, 18F-Fluordesoxyglucose; PET, Positronenemissionstomographie; SUV, standardisierter Aufnahmewert; MTV, metabolisches Tumorvolumen; TUNEL, terminale Desoxynukleotidyltransferase-vermittelte dUTP-Nick-End-Markierung.
© 2015 Elsevier Ireland Ltd. All rights reserved.
EINFÜHRUNG
Kopf- und Halskrebs (HNC) ist die achthäufigste Krebsart weltweit, und jedes Jahr werden mehr als eine halbe Million neuer Fälle diagnostiziert [1] . Die Tumore entstehen im oberen Aerodigestivtrakt, einschließlich der Mund- oder Nasenhöhle, des Pharynx und des Larynx, und metastasieren in regionale Lymphknoten und entfernte Stellen. Die derzeitige Standardtherapie bei HNC umfasst einen multimodalen Ansatz mit Operation, Chemotherapie und Strahlentherapie, insbesondere bei fortgeschrittenem HNC. Im Zuge des jüngsten Interesses an organerhaltenden Strategien werden zunehmend nicht-chirurgische Verfahren, wie z. B. Strahlentherapie in Kombination mit Chemotherapie, als Erstlinientherapie bei HNC eingesetzt [2]. Bei der Behandlung von Patienten mit fortgeschrittenem HNC ist die systemische Chemotherapie heute ein zentraler Bestandteil mehrerer kurativer Ansätze, einschließlich der Kombination mit definitiver Strahlentherapie oder Induktionstherapie, und geht über die reine Palliation hinaus [3]. Cisplatin, das Platinderivat Cis-Diamindichloroplatin (II) (CDDP), ist trotz der jüngsten Fortschritte bei der zielgerichteten Therapie nach wie vor das Chemotherapeutikum der ersten Wahl bei nicht-chirurgischen Behandlungsmethoden des HNC [4]. In den letzten drei Jahrzehnten hat sich jedoch die Gesamtüberlebensrate bei HNC trotz diagnostischer und therapeutischer Fortschritte nicht wesentlich verändert, was auf die hartnäckige Resistenz der Krebszellen gegenüber der Therapie, einschließlich Cisplatin und Bestrahlung, zurückzuführen ist [2].
Stoffwechselveränderungen sind ein gemeinsames Merkmal von Krebszellen: Die Verlagerung der zellulären Energieerzeugung von der oxidativen Phosphorylierung in den Mitochondrien zur aeroben Glykolyse verschafft den Krebszellen einen biosynthetischen Vorteil [5]. Die verstärkte Glykolyse in Krebszellen, der so genannte Warburg-Effekt, steht in der Regel in Zusammenhang mit phänotypischen Veränderungen, einschließlich der Anpassung an Hypoxie und Nährstoffmangel, der Resistenz gegenüber oxidativem Stress und apoptotischen Reizen sowie einer erhöhten Biomassesynthese [6]. Deregulierter Stoffwechsel wird mit Behandlungsresistenz in der Krebstherapie in Verbindung gebracht [7]. Im glykolytischen Stoffwechselweg wird die Hochregulierung des Glukosetransporters (GLUT), der Hexokinase (HK), der Pyruvatkinase M2 (PKM2), der Pyruvatdehydrogenase-Kinase (PDK), der Laktatdehydrogenase-A (LDHA), der Fettsäuresynthese (FASN) und der Glutaminase u. a. mit der Resistenz gegen Krebsmedikamente in Verbindung gebracht [7]. Es gibt Hinweise darauf, dass die Regulierung des Stoffwechsels von Krebszellen die Therapie verbessern und die Resistenz gegen Chemo- oder Strahlentherapie überwinden kann [8,9].
Dichloracetat (DCA), ein Orphan Drug gegen Laktatazidose, verlagert den Krebsstoffwechsel von der Glykolyse zur oxidativen Phosphorylierung [10]. DCA hemmt selektiv die PDK, die bei vielen Krebsarten aktiviert ist, was zu einer Aktivierung der Pyruvatdehydrogenase (PDH) führt, einem Komplex von Enzymen, die zytosolisches Pyruvat in mitochondriales Acetyl-CoA umwandeln. Die Hemmung von PDK mit small interfering RNA (siRNA) oder die Behandlung mit DCA verändert die Bioenergetik von Krebs und stellt die mitochondrienabhängige Apoptose in Krebszellen wieder her [11]. DCA ist wirksam bei der Behandlung von Krebs mit aggressiven Phänotypen in vitro und in vivo[12,13] und überwindet die Sorafenib-Resistenz bei hepatozellulärem Karzinom durch Aktivierung der oxidativen Phosphorylierung in den Mitochondrien [14]. Daher könnte DCA ein geeignetes Krebsmedikament sein, um die Wirksamkeit der Chemotherapie zu erhöhen und die Resistenz gegen Chemotherapeutika bei menschlichem Krebs zu überwinden; obwohl DCA umfassend bei Krebs untersucht wurde, wurde es nur selten bei HNC-Zellen getestet [15]getestet, insbesondere bei Resistenz gegen Chemotherapeutika. Weitere Untersuchungen der Mechanismen von DCA und der Synergie mit herkömmlichen Chemotherapeutika sind erforderlich. Hier zeigen wir, dass DCA die Bioenergetik von HNC-Zellen auf mitochondriale Glukoseoxidation verlagert und zu einer zellulären Anhäufung mitochondrialer reaktiver Sauerstoffspezies (mROS) führt, wodurch chemoresistente HNC-Zellen in vitro und in vivo gegenüber Cisplatin sensibilisiert werden.
Materialien und Methoden
Zellkultur und Etablierung von Cisplatin-resistenten HNC-Zellen
Die menschlichen Kopf- und Halskrebszellen (HNC) AMC-HN2, -HN3, -HN4, -HN5, -HN9 und -HN10 wurden in Eagle’s Minimum Essential Medium (Life TechnologiesTM , Carlsbad, CA, USA) gezüchtet; SNU-1041, -1066 und -1076 Zellen wurden in Roswell Park Memorial Institute Medium (Life TechnologiesTM ) gezüchtet; und UMSCC1 Zellen wurden in Dulbecco’s modified Eagle Medium (Life TechnologiesTM ) gezüchtet. Die Medien wurden mit 10 % fötalem Rinderserum angereichert. Alle Krebszelllinien wurden durch DNA-Profilierung (short-tandem-repeat, STR) authentifiziert, die von der koreanischen Zelllinienbank bereitgestellt wurde. Die Zellen wurden bei 37 °C in einer befeuchteten Atmosphäre mit 5 % CO2 inkubiert.
Cisplatin-resistente AMC-HN4- und HN9-Zellen (HN4R und HN9R) wurden aus den Cisplatin-empfindlichen AMC-HN4- bzw. HN9-Elternzellen entwickelt, indem sie steigenden Konzentrationen von Cisplatin (Cis-Platin(II)diamin-Dichlorid [CDDP]; Sigma-Aldrich, Louis, MO, USA) ausgesetzt wurden [16] . Die Cisplatin-Resistenz der etablierten Zelllinien wurde anhand von Zelllebensfähigkeitstests bewertet und mit derjenigen der Elternzellen verglichen.
Test der Zelllebensfähigkeit
Die Lebensfähigkeit der Zellen wurde mit Trypanblau-Ausschluss, MTT und klonogenen Tests bestimmt. Der Trypanblau-Ausschluss wurde an HNC-Zellen durchgeführt, die zu 1 x105 in 6-Well-Platten ausgesät worden waren. Die Zellen erreichten eine Konfluenz von 60-70 % und wurden 72 h lang Dichloracetat (DCA; Sigma-Aldrich) ausgesetzt. Anschließend wurden die Zellen trypsiniert, mit 0,4 % Trypanblau (Life TechnologiesTM ) angefärbt und mit einem Hämacytometer gezählt. Der MTT-Assay wurde mit HNC-Zellen durchgeführt, die mit 3-5 x103 Zellen pro Vertiefung in 96-Well-Platten ausgesät wurden. Die Zellen wurden über Nacht inkubiert und dann 72 Stunden lang DCA und Cisplatin, allein oder in Kombination, ausgesetzt. Anschließend wurden die Zellen 4 Stunden lang der Tetrazoliumverbindung 3-[4,5-Dimethyl-2-thiazolyl]-2,5-diphenyl-2H-tetrazoliumbromid (MTT; Sigma-Aldrich) ausgesetzt, wonach 2 Stunden lang Solubilisierungspuffer hinzugefügt wurde. Die Absorption in jeder Vertiefung wurde bei 570 nm mit einem SpectraMax M2 Mikroplatten-Lesegerät (Molecular Devices, Sunnyvale, CA, USA) gemessen. Für die klonogenen Tests wurden die Zellen 72 Stunden lang mit DCA oder Vehikel behandelt und dann 7-10 Tage lang in arzneimittelfreiem Medium inkubiert. Die Vertiefungen wurden mit 0,5%iger Kristallviolettlösung angefärbt, und die Anzahl der Kolonien wurde gezählt. Alle Tests wurden mit dreifachen Proben durchgeführt und dreimal wiederholt.
Die Zytotoxizität von Cisplatin wurde nach 72 Stunden mit MTT bestimmt, und die halbe maximale Hemmkonzentration (IC50) jeder HNC-Zelle wurde berechnet. Die Interaktion von zwei Arzneimitteln wurde als synergistisch betrachtet, wenn die durch die Arzneimittelkombination hervorgerufene Wachstumshemmung größer war als die Summe der Hemmungen, die durch jedes Arzneimittel allein hervorgerufen wurden: Kombinationsindex (CI) = 1, additive Interaktion; CI < 1, synergistische Interaktion; und CI > 1, antagonistische Interaktion [17] .
Messung der Bioenergetik und der ROS-Produktion
HNC-Zellen (1 x104 Zellen pro Vertiefung) wurden in 24-Well-Platten ausgesät und am nächsten Tag gewaschen und 6 Stunden lang in serumfreiem DMEM mit oder ohne 100 ng/mL Oligomycin (ein ATP-Synthase-Inhibitor; Sigma-Aldrich) inkubiert. Das Kulturmedium (50 µL) wurde aus jeder Platte entnommen, und die Laktatkonzentration des Kulturmediums wurde mit einem Laktatassay-Kit (Biovision, Mountain View, CA, USA) gemessen. Lac (o) und Lac (c) bezeichnen die Laktatkonzentration im Medium nach 6 Stunden Inkubation in Gegenwart bzw. Abwesenheit von Oligomycin. Der Beitrag von Glykolyse und oxidativer Phosphorylierung (OXPHOS) zur zellulären Bioenergetik wurde mit folgender Formel berechnet: Glykolyse (%) = Lac (c)/Lac (o) x 100; OXPHOS (%) = 100 – Glykolyse (%) [14] .
Die Zellen wurden 24 Stunden lang 15 oder 30 mM DCA ausgesetzt, und die ROS-Produktion wurde in den mit 10 µM 2′,7′-Dichlorfluoresceindiacetat (DCF-DA) (Enzo Life Sciences, Farmingdale, NY, USA) inkubierten Zellen nachgewiesen. Die Zellen wurden 30 Minuten lang mit 10 µM DCF-DA bei 37 °C inkubiert, zweimal mit PBS gewaschen und in einem FACScalibur-Durchflusszytometer analysiert. Die Zellen wurden auch mit 3 mM N-Acetyl-l-Cystein (NAC; Sigma-Aldrich) für 1 h oder 10 µM Superoxiddismutase (SOD)-Inhibitor Diethyl-Dithio-Carbamat (Sigma-Aldrich) vorbehandelt, bevor sie 30 mM DCA ausgesetzt wurden. Die ROS-Konzentrationen wurden mittels Durchflusszytometrie unter Verwendung von DCF-DA gemessen und sind in Form von Fold-Change im Vergleich zu den Kontrollkonzentrationen (basal) angegeben.
Zellzyklus- und Zelltod-Assays
Für die Zellzyklusuntersuchungen wurden die Zellen 72 Stunden lang DCA ausgesetzt. Anschließend wurden die Zellen trypsinisiert, über Nacht in eiskaltem Ethanol fixiert und 30 Minuten lang mit Propidiumiodid (Sigma-Aldrich) bei 37 °C gefärbt. Der zelluläre DNA-Gehalt wurde mit einem FACScalibur-Durchflusszytometer (BD Bioscience, San Jose, CA, USA) gemessen. Für die Zelltoduntersuchungen wurden die Zellen mit Cisplatin und DCA, allein oder in Kombination, oder einer entsprechenden Menge DMSO (Vehikelkontrolle) kultiviert. Nach 72 Stunden wurden die Zellen geerntet, mit eiskaltem PBS gewaschen und in Bindungspuffer resuspendiert. Die Zellen wurden mit Annexin V-FITC (Fluoresceinisothiocyanat) und Propidiumjodid unter Verwendung eines Annexin V-FITC-Apoptose-Detektionskits (BD Biosciences, Franklin Lakes, NJ, USA) angefärbt und anschließend durchflusszytometrisch analysiert. Alle Daten wurden mit der Software Cell Quest (BD Biosciences) ausgewertet. Die statistische Signifikanz zwischen den verschiedenen Behandlungsgruppen wurde mit einem zweiseitigen Mann-Whitney-U-Test oder einem Student’s t-Test ermittelt.
Für die Caspase-Aktivitäts-Assays wurden HN4R- und HN9R-Zellen, die in einer 96-Well-Platte ausgesät worden waren, 72 Stunden lang 100 µL Medium ausgesetzt, das Cisplatin und DCA allein oder in Kombination enthielt, mit oder ohne 3 mM NAC oder 50 µM Z-VAD-fmk (R&D Systems, Minneapolis, MN, USA) vor der Exposition mit 30 mM DCA. Die Tests wurden in dreifachen Vertiefungen mit dem fluorimetrischen Homogeneous Caspase Assay (Roche Life Science, Basel, Schweiz) durchgeführt. Die Arbeitssubstratlösung wurde zugegeben, und die Platte wurde im Dunkeln bei 37 °C für 2-8 Stunden oder über Nacht bei Raumtemperatur inkubiert. Die Absorption in jeder Vertiefung wurde bei einer Anregungswellenlänge von 485 nm und einer Emissionswellenlänge von 520 nm mit einem SpectraMax M2 Mikroplattenlesegerät gemessen.
Zur Messung des mitochondrialen Membranpotenzials (ΔΨm) wurden HN4R- und HN9R-Zellen, die in einer 96-Well-Platte ausgesät worden waren, 36 Stunden lang 100 µl Medium ausgesetzt, das Cisplatin und DCA allein oder in Kombination enthielt. Die Zellen wurden 20 Minuten lang mit 200 nM Tetramethylrhodaminethylester (TMRE, Life Technologies TM ) angefärbt und dann mittels Durchflusszytometrie analysiert. Die mittlere Fluoreszenzintensität (MFI) der einzelnen Behandlungsgruppen wurde auf die Kontrollgruppe normiert.
Quantitative reverse Transkriptions-PCR in Echtzeit
Die gesamte zelluläre RNA wurde mit dem QIAzol-Lysereagenz und dem RNeasy Mini Kit (Qiagen, Valencia, CA, USA) extrahiert. cDNA wurde aus der gereinigten RNA mit einem QuantiTect Reverse Transcription Kit (Qiagen) gemäß den Anweisungen des Herstellers erzeugt. Pyruvat-Dehydrogenase-Kinase 2 (PDK2) cDNA wurde durch PCR mit den folgenden Primern amplifiziert: 5′-ATGGCAGTCCTCCTCTCTGA-3′ (vorwärts) und 5′-CACCCACCCTCTTCCTAACA-3′ (rückwärts). Für β-Actin (eine endogene Kontrolle) wurden die folgenden Primer verwendet: 5′-ACCCCCCCACTGAAAAAGATGA-3′ (vorwärts) und 5′-ATCTTCAAACCTCCATGATG-3′ (rückwärts). Quantitative reverse Transkriptions-PCR (qRT-PCR) in Echtzeit wurde mit SYBR Green (Qiagen) auf einem 7900HT Fast Real-time PCR System (Applied Bioscience, Foster, CA, USA) durchgeführt. Die relativen Ziel-mRNA-Spiegel wurden auf die β-Actin-Expression normalisiert.
siRNA
Für den Knockdown von PDK2 und PDHE1α wurden AMC-HN4-cisR und HN9-cisR auf 60-mm-Platten in Medium ohne Antibiotika ausgesät und 18 Stunden später mit 100 nmol/L small interfering RNA (siRNA), die auf das menschliche PDK2- oder PDHE1α-Gen abzielt, oder einer verschlüsselten Kontroll-siRNA (Life Technologies TM ) transfiziert. Nach 72 Stunden wurden die Zellen für weitere 72 Stunden DCA ausgesetzt und anschließend auf die Proteinexpression untersucht. Der Knockdown wurde durch Western Blotting mit Anti-PDK2- oder PDHE1-Antikörpern bestätigt.
Immunoblotting
Die Zellen wurden bei 4 °C in Radioimmunopräzipitationspuffer (RIPA) (Life TechnologiesTM ) lysiert. Das Immunoblotting wurde nach den Standardverfahren durchgeführt. Kurz gesagt wurden insgesamt 50 µg Protein durch Natriumdodecylsulfat-Polyacrylamid-Gelelektrophorese (SDS-PAGE) auf 10-12%igen Gelen aufgelöst, auf Nitrocellulose- oder Polyvinylidendifluorid-Membranen übertragen und mit primären und sekundären Antikörpern sondiert. Es wurden die folgenden primären Antikörper verwendet: p53 (DO1) (Santa Cruz Biotechnology, Santa Cruz, CA, USA); Hexokinase 2 (HK2), p21 WAF1/CIP1, PUMA, gespaltene Poly(ADP-Ribose)-Polymerase (PARP), phospho-p53-Ser15, und gespaltene Caspase-3 (Cell Signaling Technology, Danvers, MA, USA); PDK2 (Abcam, Cambridge, MA, USA); und PDHE1α und phospho-PDHE1α (ser293) (Calbiochem, Billerica, MA, USA). β-Actin (Sigma-Aldrich) wurde als Ladekontrolle verwendet. Alle Antikörper wurden zwischen 1:250 und 1:5000 verdünnt.
Konfokale Mikroskopie
ΔΨm wurde in lebenden Krebszellen mit TMRM (Life TechnologiesTM ), einem mitochondrien- und spannungsempfindlichen Farbstoff auf Rhodaminbasis (rote Fluoreszenz), abgebildet. Das während der Zellatmung gebildete mitochondriale Superoxid (mROS, Life TechnologiesTM ) wurde mit MitoSOX (rote Fluoreszenz) gemessen. TMRM- oder MitoSOX-Fluoreszenzbilder wurden mit einem konfokalen Zwei-Photonen-Mikroskop LSM 510 von Zeiss (Carl Zeiss AG, Heidenheim, Deutschland) aufgenommen, und die mittlere Fluoreszenz wurde mit Hilfe der Zen Imaging Software (Carl Zeiss AG) in Krebszellen in Kultur quantifiziert (arbiträre Fluoreszenzeinheiten, AFU). Die Bildaufnahme und -analyse wurde von zwei Wissenschaftlern durchgeführt, die gegenüber der Quelle der Zellproben verblindet waren.
Positronen-Emissions-Tomographie (PET)-Bildgebung
Die Glukoseaufnahme wurde in vivo in Nacktmäusen nach Transplantation von HN4R- oder HN9R-Zellen unter Isofluran-Gasnarkose (20 % Sauerstoff) und 18 F-Fluordesoxyglukose(18 F-FDG) als Radiotracer abgebildet. Die PET-Bildgebung wurde mit dem microPET FOCUS 120 (Concorde Microsystem Inc., Knoxville, TN, USA) durchgeführt. Die Mäuse fasteten über Nacht und bekamen dann intravenös 0,15 mCi injiziert. Nach 1 Stunde wurde ein 18 F-FDG-PET-Scan des gesamten Körpers durchgeführt. Die Bilder der PET-Datenerfassung sind in einer Pseudofarbkarte dargestellt, wobei die rote Farbe eine hohe 18 F-FDG-Aufnahme anzeigt.
Der maximale und der mittlere standardisierte Aufnahmewert (SUVmax und SUVmean) wurden zur Bestimmung der 18 F-FDG-PET-Aktivität verwendet. Das SUV wurde anhand der Gleichung SUV = A/(ID/BW) analysiert, wobei A die zerfallskorrigierte Aktivität im Gewebe (in mCi/mL), ID die injizierte FDG-Dosis (in mCi) und BW das Körpergewicht der Maus (in Gramm) ist. Über die auf den PET-Bildern sichtbaren Läsionen wurden kugelförmige oder elliptische Interessengebiete (ROIs) gelegt. SUVmax und SUVmean wurden berechnet, indem automatisch eine ROI über den intensivsten Teil der auf den PET-Bildern sichtbaren Tumoren gezogen wurde. Die MTVs wurden aus den abschwächungskorrigierten PET-Daten mit Hilfe eines kommerziellen Softwarepakets (INFINITT PACS; INFINITT Healthcare Co., Ltd) berechnet. die 18 F-FDG-PET-Daten wurden im DICOM-Format in die Workstation eingespeist, und die Intensitätswerte wurden automatisch in SUVs umgerechnet. Für die MTV-Berechnungen wurden die Konturierungsränder des Tumors mit einem festen SUV von 2,0 definiert, und das Tumorvolumen wurde dann mit der SUV-2,0-Isokontur abgegrenzt. Die PET-Bilder wurden bei tumorimplantierten Mäusen am Tag 21 nach Behandlungsbeginn aufgenommen. Die Mittelwerte der Tumor-SUVmax- und MTV-Werte wurden zwischen den verschiedenen Behandlungsgruppen verglichen.
Präklinische Studien
Alle Tierversuche wurden gemäß den Protokollen durchgeführt, die vom Institutional Animal Care and Use Committee unserer Einrichtung genehmigt worden waren. Sechs Wochen alte athymische männliche BALB/c-Nacktmäuse (nu/nu) wurden von Central Lab Animal Inc. (Seoul, Republik Korea) erworben. AMC-HN4R- oder HN9R-Zellen (5 x 10 6 ) wurden subkutan in die Flanke injiziert. Tumorvolumen und Körpergewicht wurden alle 3 Tage gemessen. Die Tumore wurden mit einer Schieblehre gemessen, und das Volumen wurde als (Länge x Breite2 )/2 berechnet. Die Behandlung begann, wenn die Zellimplantate zu tastbaren Knötchen wurden (= Tag 0). Die Mäuse wurden randomisiert in vier Behandlungsgruppen eingeteilt: Vehikel, DCA, Cisplatin und DCA plus Cisplatin.
Die Mäuse wurden mit Trinkwasser, das mit 0,5 g/L DCA angereichert war, oder mit einer i.p.-Injektion von 5 mg/kg Cisplatin einmal pro Woche oder mit einer Kombination aus DCA und Cisplatin nach denselben Schemata behandelt. Die Mäuse wurden am 24. Tag getötet, und die Tumoren wurden isoliert und mittels Immunoblotting und In-situ-Terminal-Desoxynukleotidyl-Transferase-vermitteltem dUTP-Nick-End-Labeling (TUNEL)-Test (EMD Millipore, Billerica, MA, USA) analysiert. Die Anzahl der apoptotischen Körper wurde in zehn zufällig ausgewählten High-Power-Feldern blind gezählt. Die statistische Signifikanz zwischen den verschiedenen Behandlungsgruppen wurde mit einem zweiseitigen Mann-Whitney-U-Test oder einem Student’s t-Test ermittelt.
Ergebnisse
Erhöhte Glykolyse in HNC-Zellen ist mit Resistenz gegen Cisplatin verbunden und wird durch DCA aufgehoben
Alle in unserer Studie verwendeten Zelllinien waren menschliche HNC-Zellen. Die zytotoxische Wirkung von DCA wurde in HNC-Zellen durch Trypanblau-Ausschluss, Kristallviolett-Färbung und MTT-Assay untersucht. DCA, das 72 Stunden lang mit bis zu 30 mM getestet wurde, hemmte das Wachstum von HNC-Zellen deutlich (MTT-Assay, Abb. 1A). Die Bioenergetik der HNC-Zellen wurde durch DCA verändert: Die zelluläre Glykolyse nahm deutlich ab, während die oxidative Phosphorylierung zunahm (Abb. 1B ). Die Veränderung der Bioenergetik variierte zwischen den HNC-Zelllinien und stand in signifikantem Zusammenhang mit der Empfindlichkeit gegenüber Cisplatin: HNC-Zellen mit hoher Glykolyse zeigten Resistenz gegenüber Cisplatin (R2 = 0,859, P < 0,001; Abb. 1C ). Außerdem waren die Krebszellen mit der größten Veränderung in der Bioenergetik bei DCA-Exposition wahrscheinlich stärker apoptotisch (R2 = 0,769, P = 0,002; Abb. 1D ). Dies deutet darauf hin, dass DCA durch die Verringerung der Glykolyse in HNC-Zellen eine krebszellspezifische Apoptose auslöst.
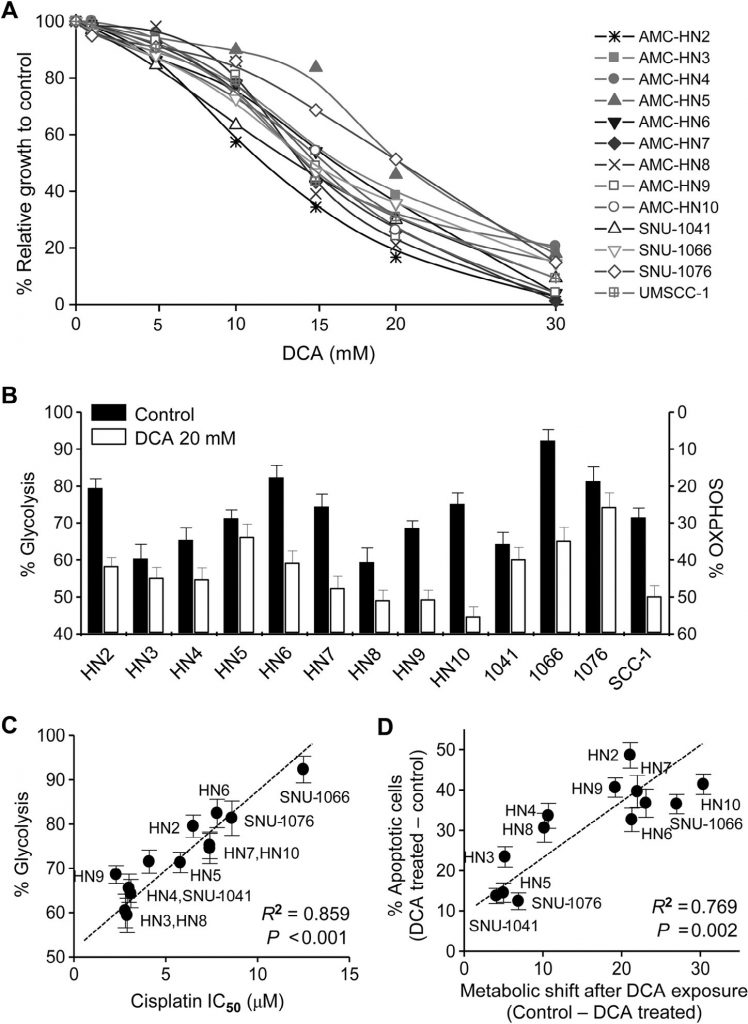
PDK2-Expression ist mit Cisplatin-Resistenz bei HNC verbunden
Die zytotoxischen Wirkungen von Cisplatin wurden in kultivierten HN4-cisR- und HN9-cisR-Zellen sowie in den elterlichen Krebszelllinien getestet (Abb. 2A). Cisplatin-resistente HN4-cisR- und HN9-cisR-Zellen zeigten eine 12- bzw. 18-fache Erhöhung der IC50 im Vergleich zu ihren jeweiligen Elternlinien. Die Western-Blot-Analyse zeigte, dass HK2 und PDK2 sowohl in HN4-cisR- als auch in HN9-cisR-Zellen im Vergleich zu den jeweiligen Elternzellen stark exprimiert wurden, während die Expression von PDHE1α in den cisplatinresistenten Zellen gering war (Abb. 2B). Auch die Expression von PDK2 mRNA war in den resistenten Zellen höher als in den empfindlichen Zellen (P < 0,01) (Abb. 2C). DCA hemmte das Wachstum von cisplatinresistenten HNC-Zellen ebenso stark wie das von cisplatinsensitiven HNC-Zellen (Abb. 2D). Western-Blot-Analysen zeigten, dass DCA die Spiegel von PDK2 und Phospho-PDHE1α (pPDHE1α) signifikant verringerte, aber die Spiegel von proapoptotischen Proteinen, einschließlich gespaltenem PARP (cPARP) und PUMA, sowie von p21 in HN4-cisR und HN9-cisR erhöhte (Abb. 2E). Dies deutet darauf hin, dass DCA sowohl das Wachstum von Cisplatin-resistenten HNC-Zellen als auch von Cisplatin-empfindlichen HNC-Zellen wirksam hemmen kann.
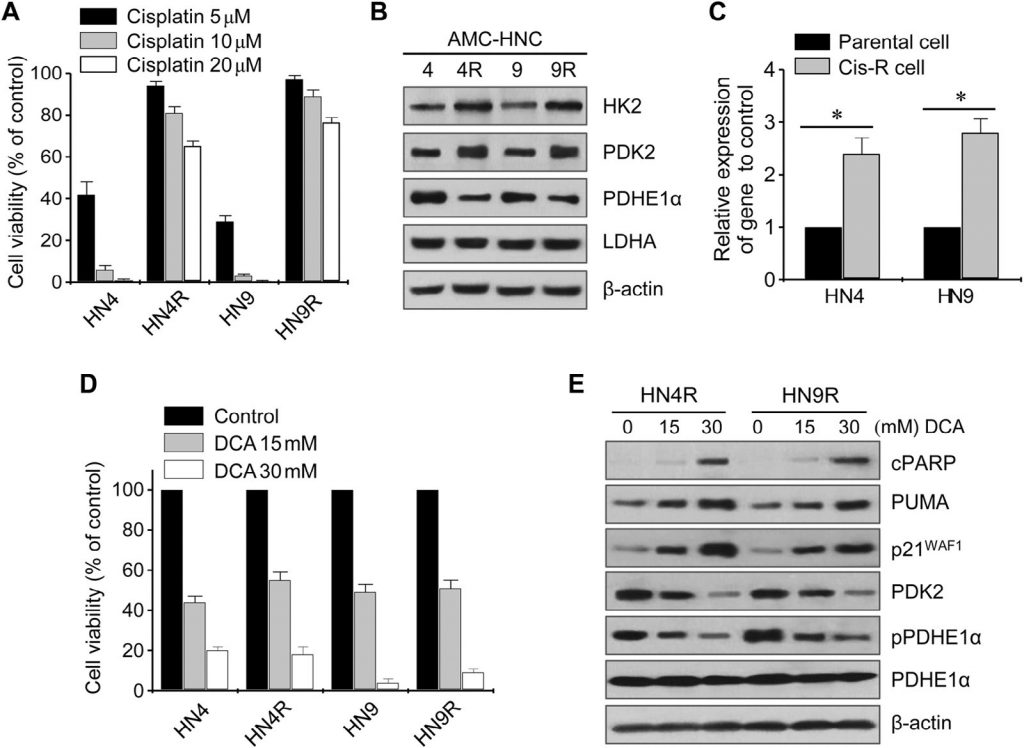
DCA induziert ROS-Akkumulation in HNC-Zellen
Die Veränderung der zellulären ROS durch DCA wurde mittels Durchflusszytometrie unter Verwendung der redoxsensitiven Fluoreszenzsonde DCF-DA bestimmt. Die Exposition mit DCA verursachte einen signifikanten Anstieg der ROS-Konzentration in HNC-Zellen (P < 0,01), der durch die gleichzeitige Exposition mit NAC oder dem SOD-Inhibitor Diethyl-Dithio-Carbamat blockiert wurde (Abb. 3A). Die Glykolyse nahm in den cisplatinresistenten HNC-Zellen im Vergleich zu den Elternzellen signifikant zu (P < 0,01), aber DCA bewirkte sowohl in den cisplatinempfindlichen als auch in den cisplatinresistenten HNC-Zellen einen signifikanten Rückgang der Glykolyse und einen Anstieg der oxidativen Phosphorylierung (P < 0,01) (Abb. 3B). Das mitochondriale Membranpotenzial (ΔΨm) wurde mit TMRM, einem mitochondrien- und spannungsempfindlichen Farbstoff auf Rhodaminbasis, und mitochondriales ROS (mROS, mitochondriales Superoxid) wurde mit MitoSOX red gemessen. DCA-behandelte HNC-Zellen zeigten einen Rückgang von ΔΨm und einen Anstieg von mROS (P < 0,01) (Abb. 3C). Die Veränderung von ΔΨm in Cisplatin-resistenten HNC-Zellen nach DCA-Exposition wurde durch die Vorbehandlung mit NAC blockiert (Abb. 3D). Dies deutet darauf hin, dass DCA die ROS-Akkumulation in HNC-Zellen durch Aktivierung der oxidativen Phosphorylierung induzieren kann.
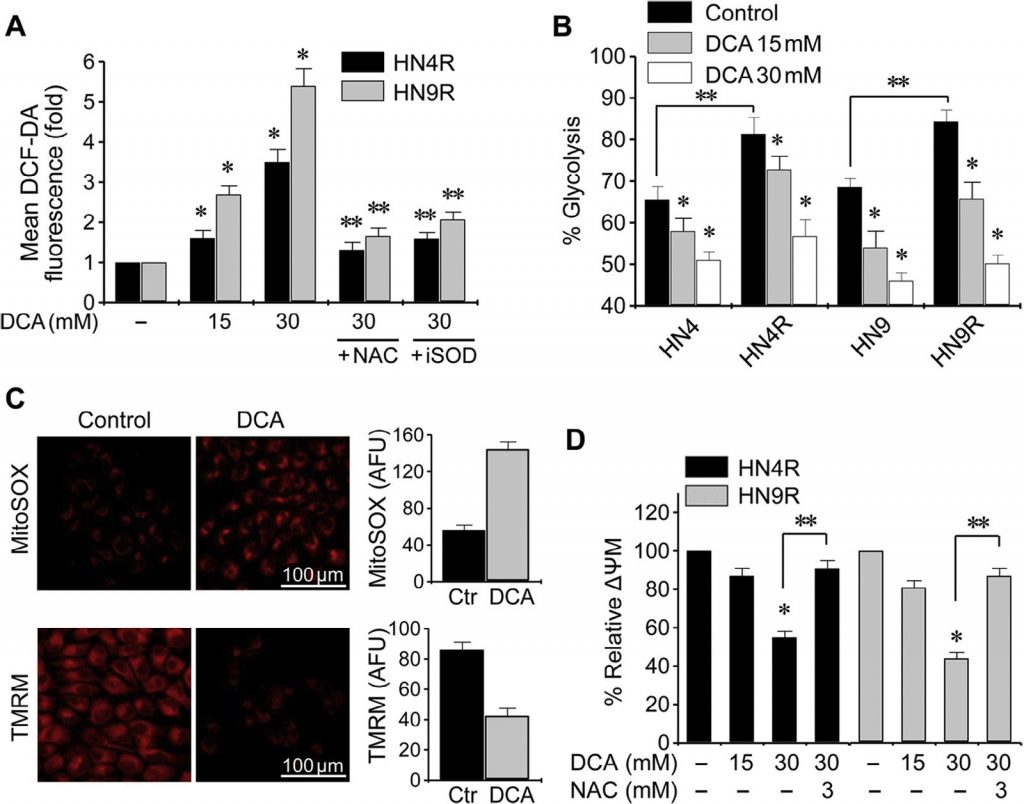
DCA fördert den Zellzyklus-Stillstand und die Apoptose in HNC-Zellen
Klonogene Assays ergaben eine deutliche Abnahme der Anzahl der HN4-cisR- und HN9-cisR-Kolonien nach DCA-Exposition (P < 0,01) (Abb. 4A ). Die Durchflusszytometrie mit Propidiumjodid-Färbung zeigte eine signifikante Veränderung des Zellzyklus in HNC-Zellen: Wogonin erhöhte die apoptotische Population sub-G1 (P < 0,05), und dieser Effekt wurde durch die gleichzeitige Exposition mit NAC blockiert (Abb. 4B ). Die Western-Blot-Analyse zeigte, dass DCA pPDHE1α verringerte, aber cPARP, p21, phospho-p53 und gespaltene Caspase-3 (cCasp3) zeitabhängig erhöhte (Abb. 4C ). Die PDHE1α-Aktivierung durch Knockdown von PDK2 in AMC-HN4-cisR-Zellen führte nicht zu einem signifikanten Anstieg der proapoptotischen Proteine cPARP und cCasp3(Abb. 4D ); der Knockdown von PDHE1α verringerte jedoch die Expression von p21 und die Phosphorylierung von p53. Die Durchflusszytometrie unter Verwendung von Propidiumjodid und Annexin-V-Färbung zeigte eine effektive Induktion von Apoptose und Zelltod in Cisplatin-resistenten HNC-Zellen durch DCA; der Effekt wurde durch die gleichzeitige Exposition mit DCA und dem Antioxidans NAC oder dem Pan-Caspase-Inhibitor Z-VAD-fmk verringert (Abb. 4E). Dies wurde durch die Messung der Caspase-Aktivität bestätigt, die durch DCA in einer konzentrationsabhängigen Weise signifikant erhöht wurde (Abb. 4F ).
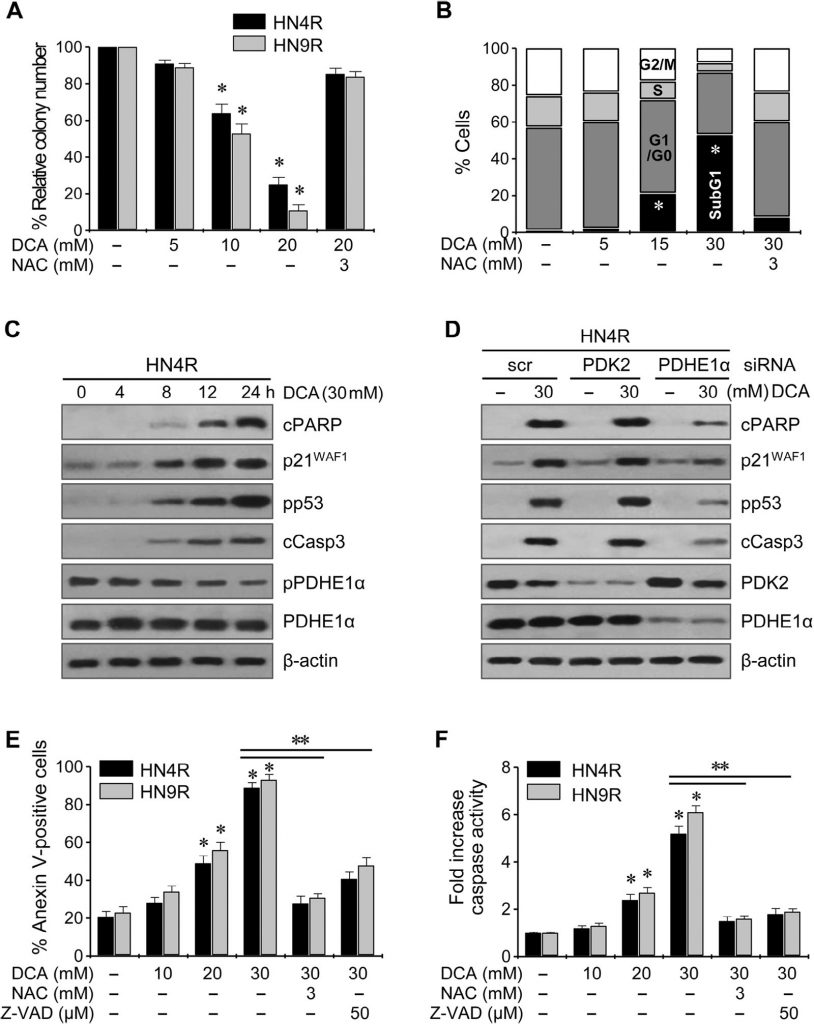
DCA sensibilisiert cisplatinresistente HNC-Zellen für Cisplatin in vitro und in vivo
Cisplatin (10 µM) induzierte keine signifikante Zytotoxizität oder apoptotische Proteinexpression in den cisplatinresistenten HNC-Linien HN4-cisR und HN9-cisR im Vergleich zu den cisplatinsensitiven Elternlinien HN4 und HN9 (Abb. 5A); DCA induzierte jedoch eine deutliche Abnahme der Überlebensrate in den cisplatinresistenten HNC-Zellen (P < 0,05), die durch eine Vorbehandlung mit NAC blockiert wurde. DCA induzierte die Expression apoptotischer Proteine und erhöhte die Cisplatin-induzierte Zytotoxizität und die Expression apoptotischer Proteine in HN4-cisR-Zellen (Abb. B). In Kombination steigerte DCA die Zytotoxizität von Cisplatin in HN4-cisR-Zellen durch Erhöhung der Caspase-Aktivität in einem Ausmaß, das größer war als die Summe der Wirkungen jedes einzelnen Wirkstoffs (CI < 1, P < 0,01), die durch PDHE1α-Gensilencing abgeschwächt wurden (Abb. 5C). Die ΔΨm war in den cisplatinresistenten HN4-cisR-Zellen höher als in den cisplatinsensitiven HN4-Elternzellen (ΔΨm, mittlere Fluoreszenzintensität [MFI]: 1 ± 0 vs. 0,54 ± 0,09, P < 0,001) und wurde durch DCA oder die Kombination von Cisplatin und DCA reduziert, was durch PDHE1α-Gensilencing aufgehoben wurde (Abb. 5D).
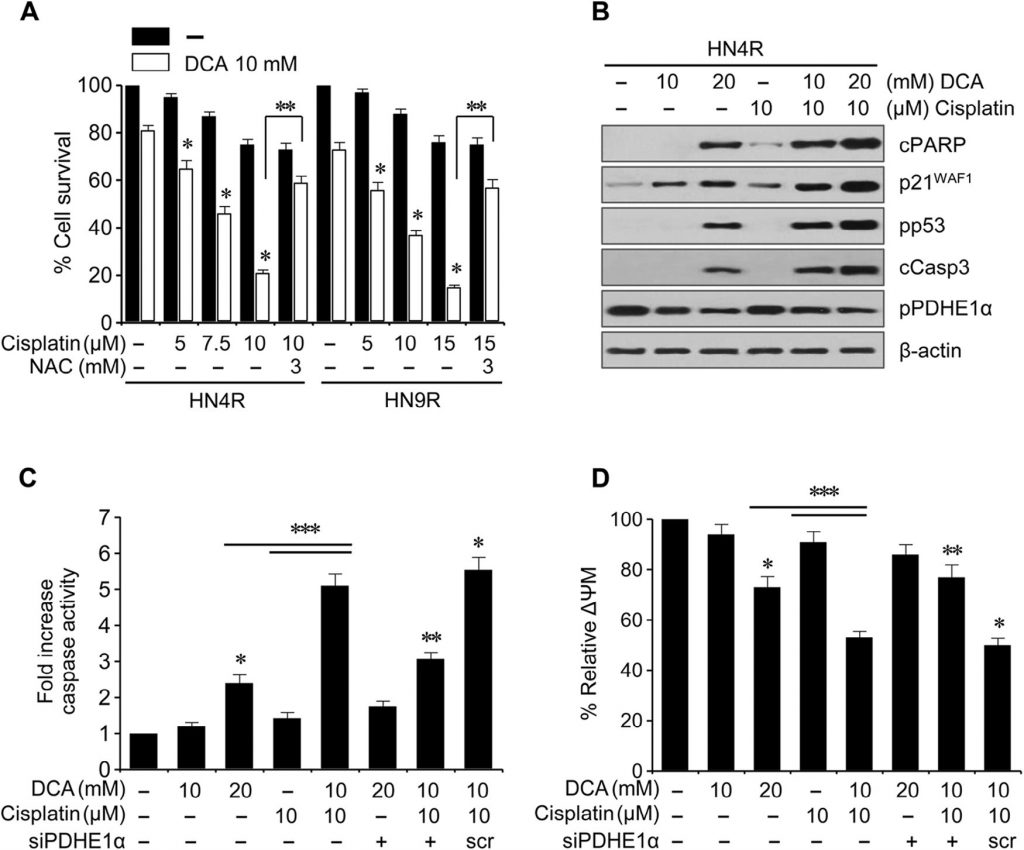
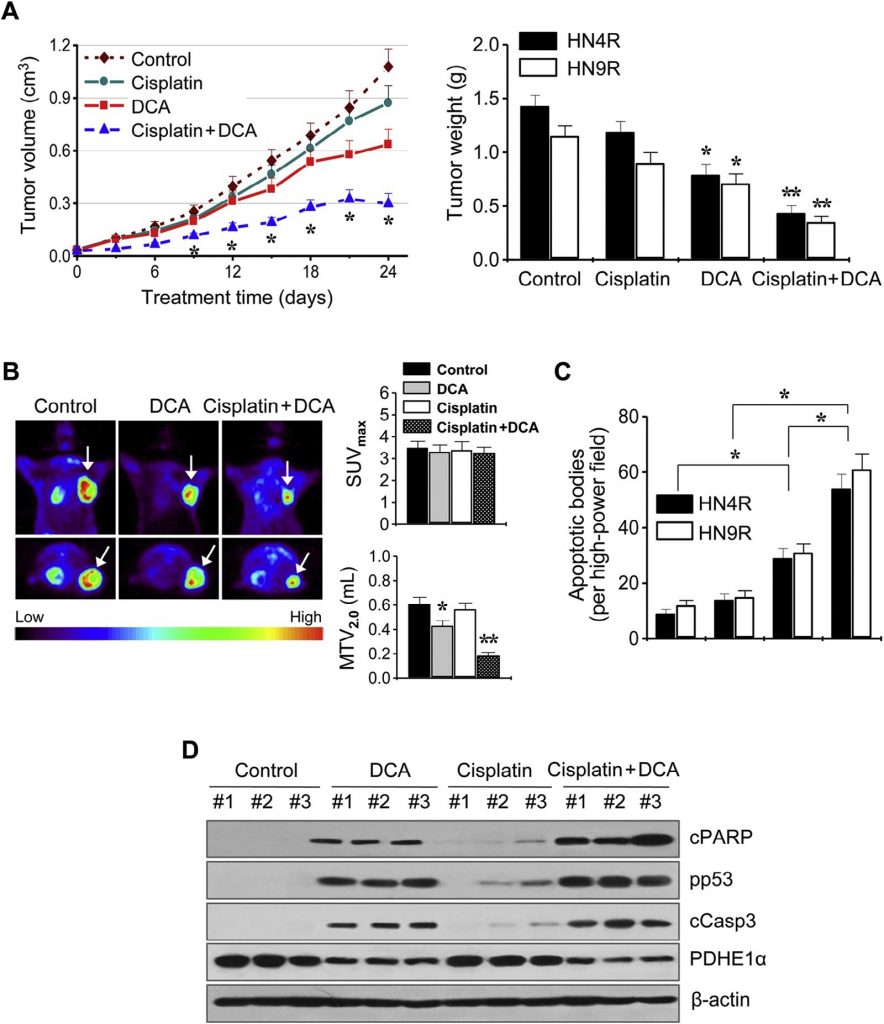
Diese Ergebnisse wurden in In-vivo-Tumor-Xenograft-Mausmodellen weiter untersucht. Athymische BALB/c-Nacktmäuse mit AMC-HN4-cisR- oder HN9-cisR-Tumoren wurden mit DCA, Cisplatin, DCA plus Cisplatin oder einem Vehikel behandelt. Die Kombination von Cisplatin und DCA unterdrückte synergistisch das Tumorwachstum (Abb. 6A). Die In-vivo-Darstellung des Tumorwachstums wurde am Tag 21 der Behandlung mittels 18 F-FDG-PET durchgeführt. Fokale 18 F-FDG-Aufnahmen wurden an den Stellen der Tumorimplantation beobachtet, wo die maximale standardisierte Aufnahme (SUVmax) und das metabolische Tumorvolumen bei SUV 2,0 (MTV2,0) gemessen wurden. Die SUVmax war zwischen den Behandlungsgruppen nicht unterschiedlich (P > 0,1), aber die MTV2,0 war in den mit DCA und der Kombination behandelten Gruppen signifikant niedriger als in den anderen Gruppen (P < 0,05) (Abb. 6B). In-situ-Apoptosetests zeigten, dass TUNEL-positive apoptotische Körper in Tumoren, die mit DCA und Cisplatin plus DCA behandelt wurden, häufiger zu sehen waren als in denen, die mit Vehikel behandelt wurden (P < 0,01) (Abb. 6C). Western-Blot-Analysen von Tumorgewebe zeigten, dass die apoptotischen Proteinkonzentrationen in HN4-cisR-Zellen, die mit der Kombination von Cisplatin und DCA behandelt wurden, stärker zunahmen als in Tumoren, die mit den einzelnen Wirkstoffen behandelt wurden (Abb. 6D).
Diskussion
Eine erhöhte Glykolyse, die häufig bei Krebserkrankungen auftritt, ist eng mit einer Beeinträchtigung der Mitochondrien oder einer defekten oxidativen Phosphorylierung verbunden und trägt zur Therapieresistenz bei [18,19]. Die aerobe Glykolyse wurde mit der Resistenz gegenüber Chemotherapie [20] oder Strahlentherapie [21]. In der vorliegenden Studie weisen cisplatinresistente Zelllinien eine erhöhte Glykolyse auf, was auf einen biochemischen Zusammenhang zwischen Glykolyse und Chemoresistenz hinweist. Medikamentenresistente Zellen haben einen höheren ATP-Bedarf als normale Zellen, um die zelluläre Homöostase aufrechtzuerhalten und die Überlebenswege zu aktivieren, die es ermöglichen, dem Zelltod unter genotoxischem Stress zu entgehen [20]. Resistente Krebszellen erhöhen die Glykolyse, um schnell ATP zu erzeugen und den intrazellulären Bedarf zu decken. Dies wurde in unseren cisplatinresistenten Zellen im Vergleich zu den empfindlichen Elternzellen eindeutig festgestellt, was darauf hindeutet, dass die Arzneimittelresistenz bei HNC direkt mit einer Zunahme der Glykolyse verbunden ist. Dies deutet darauf hin, dass die bioenergetische Veränderung der Krebszellen in Richtung einer erhöhten Glykolyse mit der Chemoresistenz zusammenhängt [14,22]. Daher könnte das Glykolyse-Niveau bei menschlichen Krebserkrankungen ein Biomarker für Chemoresistenz sein und bedarf der klinischen Validierung in menschlichem Krebsgewebe.
Die vorliegende Studie zeigt, dass DCA bei HNC die Energiegewinnung von der Glykolyse auf die mitochondriale Glukoseoxidation verlagert. Diese DCA-induzierte Umkehrung der Stoffwechselverschiebung in Richtung Glykolyse beseitigt den Proliferationsvorteil der Krebszellen und führt schließlich zum Zelltod [23]. DCA hemmt als strukturelles Analogon von Pyruvat die PDK und reaktiviert die PDH, ein Enzym, das Teil des Komplexes ist, der Pyruvat in Acetyl-CoA umwandelt, das primäre Substrat des Krebszyklus [10,23] . Da die meisten Krebsarten ein hypoxisches Umfeld schaffen, sind Krebszellen auf die anaerobe Glykolyse als primäre Energiequelle angewiesen. Die Aktivierung des Hypoxie-induzierbaren Faktors (HIF) induziert die mitochondriale PDK [24]. Die mitochondriale PDH-Aktivität wird bei Krebs durch PDK blockiert, was zu einer geringeren Verfügbarkeit von Acetyl-CoA für die mitochondriale Glukoseoxidation führt [24]. Eine erhöhte Expression von PDK wird mit Arzneimittelresistenz bei Krebs in Verbindung gebracht [25,26] . Eine Hochregulierung von PDK2 durch mitochondriale Mutationen und HIF1α-Stabilisierung wird auch in HNC-Zellen beobachtet [27] . In der vorliegenden Studie wurde der Zusammenhang zwischen der Überexpression von PDK2 und der Cisplatin-Resistenz in HNC-Zellen ebenfalls bestätigt. Diese Ergebnisse weisen auf die Bedeutung von PDK als neues molekulares Ziel in der Krebstherapie hin [10] . Frühere Daten haben gezeigt, dass eine genetische oder pharmakologische Hemmung von PDK die Bioenergetik von Krebszellen verändert und die mitochondrienabhängige Apoptose in Krebszellen wiederherstellt [11,13]. Daher ist DCA wirksam bei der Überwindung von Behandlungsresistenzen bei Krebserkrankungen mit aggressivem Phänotyp in vitro und in vivo[12-14] .
Die Aktivierung der PDH durch DCA induziert die Akkumulation von mROS in Krebszellen. Krebszellen haben einen verringerten mitochondrialen Glukosestoffwechsel, was zu einer verringerten Aktivität der Elektronentransportkette (ETC) und einer verringerten mROS [10], [28]. Der mitochondriale Umbau der ΔΨm-Hyperpolarisation und die verringerte mROS-Produktion bei Krebs führen zu einer Resistenz gegen Apoptose unter genotoxischem Stress. In resistenten Krebszellen mit einem eher glykolytischen Phänotyp führt HK2 zu seiner Verlagerung an die äußere Mitochondrienmembran und dem Anstieg von ΔΨm [29]. Die Hemmung der Glykolyse und der HK2-Translokation verringert das ΔΨm der Krebszellen und kehrt die Resistenz gegen Apoptose um [29]. In der vorliegenden Studie wurden eine erhöhte HK2- und PDK2-Expression und ein erhöhtes mitochondriales Membranpotenzial auch in Cisplatin-resistenten Krebszellen festgestellt. DCA erzwingt den Eintritt von Pyruvat in die Mitochondrien über die Aktivierung von PDH, dem Gatekeeping-Regulator der mitochondrialen Glukoseoxidation, und senkt ΔΨm und erhöht mROS [13]. In der vorliegenden Studie kehrte DCA den erhöhten mitochondrialen Umbau in Cisplatin-resistenten HNC-Zellen einfach um, was zum Tod der Krebszellen beitrug. Die Aktivierung der mitochondrialen Glukoseoxidation durch DCA induzierte mROS und die Aktivierung nachgeschalteter mitochondrialer Signalwege, was zur Aktivierung von p53 und den damit verbundenen proapoptotischen Wegen führte und den Tod der chemoresistenten Krebszellen bewirkte. In unserer Studie schwächte die pharmakologische Hemmung der mROS-Produktion oder der Caspase-vermittelten Apoptose die zytotoxische Wirkung von DCA in HNC-Zellen ab, was die bekannten Mechanismen des Medikaments bestätigt.
Präklinische und klinische Studien unterstützen den Einsatz von DCA bei Krebspatienten und bei Patienten mit Laktatazidose im Zusammenhang mit mitochondrialen Erkrankungen [10,23] . Eine Reihe von Studien belegen die zytotoxische Wirkung von DCA, allein oder in Kombination mit anderen Behandlungen, bei einer Vielzahl von Tumoren aus allen drei Keimschichten [23]. Eine frühere Studie hat gezeigt, dass das Glioblastom, eine der aggressivsten Krebsarten beim Menschen, in Krebsgeweben von 49 Patienten eine Überexpression von PDK2 aufweist und sich bei fünf Patienten nach einer Behandlung mit DCA zurückbildete, was den klinischen Nutzen von DCA bei resistenten Krebsarten beweist [13]. Bei HNC wurde in einer kürzlich durchgeführten Studie die Wirkung von DCA bei drei Zelllinien von oralen Plattenepithelkarzinomen (OSCC) verglichen [15] . Die OSCC-Zellen, die einen Mangel an mitochondrialer oxidativer Phosphorylierung (d. h. eine hohe Glykolyse) aufwiesen, waren empfindlicher gegenüber DCA als andere [15,30]. Die vorliegende Studie bestätigte, dass HNC-Zellen mit starken bioenergetischen Veränderungen empfindlicher auf DCA reagierten. Da chemoresistente Krebszellen in der Regel eine hohe Glykolyse aufweisen, könnten diese Zellen für Stoffwechselhemmer interessant sein [31]. DCA ist daher ein guter Kandidat für die Behandlung von Krebszellen mit aggressivem Phänotyp, einschließlich chemoresistenter HNC-Zellen. Da die potenziellen krebshemmenden Wirkungen von DCA jedoch noch umstritten sind, insbesondere bei Tumoren unter Hypoxie, sind weitere präklinische und klinische Studien zu DCA und Krebs erforderlich [32]. Eine kürzlich durchgeführte systematische Übersichtsarbeit zeigt, dass DCA mit vielen Standard-Krebsmitteln synergistisch wirkt, und klinische Studien der ersten Phase deuten darauf hin, dass chronisches DCA bei einer oralen Verabreichung von 12,5 mg/kg zweimal täglich sicher und gut verträglich ist [23].
Unsere Studie ergab, dass DCA mit Cisplatin synergiert und dadurch die Resistenz gegen Cisplatin in HNC-Zellen umgeht. Da Cisplatin ein Chemotherapeutikum der ersten Wahl bei HNC ist, könnte die Kombination von Cisplatin und DCA im klinischen Umfeld wirksam sein, um die Toxizität zu verringern und die Resistenz gegen Krebsmedikamente zu überwinden. Frühere Berichte haben gezeigt, dass DCA die zytotoxische Wirkung von Cisplatin verstärkt, indem es eine von den Mitochondrien abhängige Apoptose auslöst [33,34]. Die Verknüpfung von zwei DCA-Molekülen mit Cisplatin, Mitaplatin genannt, führt zur selektiven Abtötung von Krebszellen [35,36]. Diese Ergebnisse belegen, dass DCA in Kombination mit aktuellen Krebstherapien zusätzliche Vorteile haben kann. In unserer Studie wirkten sich DCA und seine Kombination mit Cisplatin nicht auf die SUVmax aus, verringerten jedoch MTV2.0 signifikant. Dies bedeutet, dass DCA das In-vivo-Tumorwachstum wirksam unterdrückt, obwohl es keine signifikante Veränderung der maximalen Glukoseaufnahme in den tumornahen ROIs gibt. In der vorliegenden Studie wurde erstmals gezeigt, dass DCA die zytotoxische Wirkung von Cisplatin in arzneimittelresistenten HNC-Zellen in vitro und in vivo wiederherstellt. DCA induzierte in cisplatinresistenten HNC-Zellen eine starke Zunahme der durch Cisplatin vermittelten Apoptose über die Aktivierung von p53, PARP und Caspase. DCA sensibilisierte arzneimittelresistente HNC-Zellen für Cisplatin, was zu einer erhöhten Zytotoxizität und einer wirksameren Therapie für aggressive HNC führte. Zusammengenommen könnten diese Ergebnisse von größter klinischer Bedeutung sein: Die Herbeiführung des Absterbens resistenter Zellen mit DCA könnte die in der klinischen Praxis erforderliche Cisplatin-Dosis verringern und damit die potenziellen Nebenwirkungen der Cisplatin-Chemotherapie minimieren.
Zusammenfassend deuten unsere Daten darauf hin, dass eine hohe Glykolyse und eine PDK-Überexpression eng mit der Cisplatin-Resistenz in HNC-Zellen verbunden sind. DCA schaltet die Bioenergetik von HNC-Zellen auf mitochondriale oxidative Phosphorylierung um. Dies führt zu einem Rückgang von ΔΨm und einem Anstieg von mROS, wodurch chemoresistente HNC-Zellen in vitro und in vivo für Cisplatin sensibilisiert werden. Diese Daten rechtfertigen weitere präklinische und klinische Untersuchungen von DCA, einem vielversprechenden Kandidaten für Krebsmedikamente, bei HNC mit aggressivem Phänotyp. Unsere Schlussfolgerung sollte jedoch in einem umgekehrten Szenario mit Vorsicht betrachtet werden. Die gestörte mitochondriale oxidative Phosphorylierung stand in Zusammenhang mit der Empfindlichkeit der Krebszellen gegenüber niedrigem Glukosegehalt, was als Biomarker für die Krebstherapie genutzt werden könnte [37]. Die Krebszelle ist auch so angepasst, dass sie ihre Vermehrung durch die Aufnahme und den Einbau von Nährstoffen in die Biomasse und nicht durch eine effiziente ATP-Produktion fördert [38]. Daher sollten die präklinischen und klinischen Auswirkungen von DCA auf HNC und andere Krebsarten weiter untersucht werden.
Danksagung
Diese Studie wurde durch ein Stipendium (2015R1A2A1A15054540) des Basic Science Research Program durch die National Research Foundation of Korea (NRF), Ministry of Science, ICT, and Future Planning, und ein Stipendium (HI14C23050000) des Korean Health Technology R&D Project, Ministry of Health & Welfare, Seoul, Republic of Korea (J.-L. Roh) unterstützt.
Interessenkonflikt
Die Autoren erklären, dass keine Interessenkonflikte bestehen.
REFERENZEN
1 A. Jemal, F. Bray, M.M. Center, J. Ferlay, E. Ward, D. Forman, Global cancer statistics, CA Cancer J. Clin, Vol. 61, 2011, 69-90
2 R.I. Haddad, D.M. Shin, Aktuelle Fortschritte bei Kopf- und Halskrebs, N. Engl. J. Med, Vol. 359, 2008, 1143-1154
3 E.B. Lamont, E.E. Vokes, Chemotherapy in the management of squamous-cell carcinoma of the head and neck, Lancet Oncol, Vol. 2, 2001, 261-269
4 F. Petrelli, A. Coinu, V. Riboldi, K. Borgonovo, M. Ghilardi, M. Cabiddu, Concomitant platinum-based chemotherapy or cetuximab with radiotherapy for locally advanced head and neck cancer: a systematic review and meta-analysis of published studies, Oral Oncol, Vol. 50, 2014, 1041-1048
5 D. Hanahan, R.A. Weinberg, Hallmarks of cancer: the next generation, Cell, Vol. 144, 2011, 646-674
6 J.R. Cantor, D.M. Sabatini, Cancer cell metabolism: one hallmark, many faces, Cancer Discov, Vol. 2, 2012, 881-898
7 Y. Zhao, E.B. Butler, M. Tan, Targeting cellular metabolism to improve cancer therapeutics, Cell Death Dis, Vol. 4, 2013, e532
8 R.A. Cairns, I.S. Harris, T.W. Mak, Regulation of cancer cell metabolism, Nat. Rev. Cancer, Vol. 11, 2011, 85-95
9 R.H. Xu, H. Pelicano, Y. Zhou, J.S. Carew, L. Feng, K.N. Bhalla, Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia, Cancer Res, Vol. 65, 2005, 613-621
10 G. Sutendra, E.D. Michelakis, Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology, Front. Oncol, Vol. 3, 2013, 38
11 G. Sutendra, P. Dromparis, A. Kinnaird, T.H. Stenson, A. Haromy, J.M. Parker, Mitochondriale Aktivierung durch Hemmung von PDKII unterdrückt HIF1a-Signalisierung und Angiogenese bei Krebs, Oncogene, Vol. 32, 2013, 1638-1650
12 R.C. Sun, M. Fadia, J.E. Dahlstrom, C.R. Parish, P.G. Board, A.C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo, Breast Cancer Res. Treat, Vol. 120, 2010, 253-260
13 E.D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, Metabolic modulation of glioblastoma with dichloroacetate, Sci. Transl. Med, Vol. 2, 2010, 31ra34
14 Y.C. Shen, D.L. Ou, C. Hsu, K.L. Lin, C.Y. Chang, C.Y. Lin, Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma, Br. J. Cancer, Vol. 108, 2013, 72-81
15 V. Ruggieri, F. Agriesti, R. Scrima, I. Laurenzana, D. Perrone, T. Tataranni, Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment, Oncotarget, Vol. 6, 2015, 1217-1230
16 M. Nakamura, K. Nakatani, K. Uzawa, K. Ono, H. Uesugi, K. Ogawara, Establishment and characterization of a cisplatin-resistant oral squamous cell carcinoma cell line, H-1R, Oncol. Rep, Vol. 14, 2005, 1281-1286
17 T.C. Chou, Drug combination studies and their synergy quantification using the Chou-Talalay method, Cancer Res, Vol. 70, 2010, 440-446
18 N. Guaragnella, S. Giannattasio, L. Moro, Mitochondrial dysfunction in cancer chemoresistance, Biochem. Pharmacol, Vol. 92, 2014, 62-72
19 S. Ganapathy-Kanniappan, J.F. Geschwind, Tumor Glycolysis as a target for cancer therapy: progress and prospects, Mol. Cancer, Vol. 12, 2013, 152
20 Y. Zhou, F. Tozzi, J. Chen, F. Fan, L. Xia, J. Wang, Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells, Cancer Res, Vol. 72, 2012, 304-314
21 S.P. Pitroda, B.T. Wakim, R.F. Sood, M.G. Beveridge, M.A. Beckett, D.M. MacDermed, STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect, BMC Med, Vol. 7, 2009, 68
22 B. Bhattacharya, S.H. Low, C. Soh, N. Kamal Mustapa, M. Beloueche-Babari, K.X. Koh, Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype, Br. J. Pharmacol, Vol. 171, 2014, 3255-3267
23 S. Kankotia, P.W. Stacpoole, Biochim. Biophys. Acta, Vol. 1846, 2014, 617-629
24 J.W. Kim, I. Tchernyshyov, G.L. Semenza, C.V. Dang, HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia, Cell Metab, Vol. 3, 2006, 177-185
25 Y. Sun, A. Daemen, G. Hatzivassiliou, D. Arnott, C. Wilson, G. Zhuang, Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells, Cancer Metab, Vol. 2, 2014, 20
26 C.W. Lu, S.C. Lin, C.W. Chien, S.C. Lin, C.T. Lee, B.W. Lin, Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer, Am. J. Pathol, Vol. 179, 2011, 1405-1414
27 W. Sun, S. Zhou, S.S. Chang, T. McFate, A. Verma, J.A. Califano, Mitochondrial mutations contribute to HIF1alpha accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma, Clin. Cancer Res, Vol. 15, 2009, 476-484
28 S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, Channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth, Cancer Cell, Vol. 11, 2007, 37-51
29 J.G. Pastorino, J.B. Hoek, N. Shulga, Activation of glycogen synthase kinase 3beta disruptes the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity, Cancer Res, Vol. 65, 2005, 10545-10554
30 L.H. Stockwin, S.X. Yu, S. Borgel, C. Hancock, T.L. Wolfe, L.R. Phillips, Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC, Int. J. Cancer, Vol. 127, 2010, 2510-2519
31 E.J. Sullivan, M. Kurtoglu, R. Brenneman, H. Liu, T.J. Lampidis, Targeting cisplatin-resistenter menschlicher Tumorzellen mit Stoffwechselhemmern, Cancer Chemother. Pharmacol, Vol. 73, 2014, 417-427
32 S. Shahrzad, K. Lacombe, U. Adamcic, K. Minhas, B.L. Coomber, Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia, Cancer Lett, Vol. 297, 2010, 75-83
33 D. Heshe, S. Hoogestraat, C. Brauckmann, U. Karst, J. Boos, C. Lanvers-Kaminsky, Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs, Cancer Chemother. Pharmacol, Vol. 67, 2011, 647-655
34 J. Xie, B.S. Wang, D.H. Yu, Q. Lu, J. Ma, H. Qi, Dichloracetate shifts the metabolism from glycolysis to glucose oxidation and exhibits synergistic growth inhibition with cisplatin in HeLa cells, Int. J. Oncol, Vol. 38, 2011, 409-417
35 S. Dhar, S.J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate, Proc. Natl. Acad. Sci. U.S.A., Vol. 106, 2009, 22199-22204
36 X. Xue, S. You, Q. Zhang, Y. Wu, G.Z. Zou, P.C. Wang, Mitaplatin increases sensitivity of tumor cells to cisplatin by inducing mitochondrial dysfunction, Mol. Pharm, Vol. 9, 2012, 634-644
37 K. Birsoy, R. Possemato, F.K. Lorbeer, E.C. Bayraktar, P. Thiru, B. Yucel, Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides, Nature, Vol. 508, 2014, 108-112
38 M.G. Vander Heiden, L.C. Cantley, C.B. Thompson, Understanding the Warburg effect: the metabolic requirements of cell
Verwandte Inhalte: