Krishan Kumar1, Simon Wigfield2, Harriet E. Gee2,5, Cecilia M. Devlin1, Dean Singleton2, Ji-Liang Li2, Francesca Buffa2, Melanie Huffman1, Anthony L. Sinn3, Jayne Silver3, Helen Turley2, Russell Leek2, Adrian L. Harris2 & Mircea Ivan4
1 Dipartimento di Medicina, Indiana University, Indianapolis, IN 46202, USA
2 Dipartimento di Oncologia, Weatherall Institute of Molecular
Medicine, University of Oxford, John Radcliffe Hospital, Oxford OX3 9DS, UK
e-mail: [email protected]
3 In Vivo Therapeutics Core, Indiana University, Indianapolis, IN 46202, USA
4 Dipartimento di Medicina, Immunologia e Microbiologia, Indiana University, 980W. Walnut Street, Room C225, Indianapolis, IN 46202, USA
e-mail: [email protected]
5 Dipartimento di radio-oncologia, Sydney Cancer Centre, Royal Prince Alfred Hospital, Camperdown, New South Wales 2050, Australia
Ricevuto: 7 novembre 2012
Revisione: 20 dicembre 2012
Accettato: 2 gennaio 2013
Pubblicato online: 30 gennaio 2013
Abstract
L’inibizione del fattore di crescita endoteliale vascolare aumenta i tassi di risposta alla chemioterapia e la sopravvivenza libera da progressione nel glioblastoma. Tuttavia, si verifica invariabilmente una resistenza, che rende urgente l’identificazione di agenti sinergizzanti. Una possibile strategia consiste nel comprendere l’adattamento del tumore ai cambiamenti microambientali indotti dai farmaci antiangiogenici e testare agenti che sfruttino questo processo. Abbiamo utilizzato un modello di xenotrapianto derivato da glioblastoma in vivo di fuga del tumore in presenza di un trattamento continuo con bevacizumab. Le cellule U87-MG o U118-MG sono state impiantate sottocute in topi BALB/c SCID o atimici nudi. Bevacizumab è stato somministrato per iniezione intraperitoneale ogni 3 giorni (2,5 mg/kg/dose) e/o il dicloroacetato (DCA) è stato somministrato per via orale due volte al giorno (50 mg/kg/dose) quando il volume del tumore ha raggiunto 0,3 cm3 e ha continuato fino a quando i tumori hanno raggiunto circa 1,5-2,0 cm3. L’analisi microarray dei tumori U87 resistenti ha rivelato cambiamenti coordinati a livello di geni metabolici, in particolare un aumento del divario tra glicolisi e respirazione mitocondriale. È stata riscontrata una differenza altamente significativa tra i topi atimici nudi U87-MG impiantati 1 settimana dopo il trattamento farmacologico. Dopo 2 settimane di trattamento, bevacizumab e DCA insieme hanno bloccato drasticamente la crescita tumorale rispetto a uno dei due farmaci da solo. Risultati simili sono stati osservati in topi nudi atimici impiantati con cellule U118-MG. Dimostriamo per la prima volta che l’inversione dello spostamento del metabolismo indotto da bevacizumab utilizzando il DCA è dannosa per la crescita neoplastica in vivo. Poiché il DCA è considerato un agente promettente che agisce sul metabolismo dei tumori, i nostri dati dimostrano che la sua combinazione con la terapia antiangiogenica rappresenta una potente strategia antineoplastica.
Parole chiave: Dicloroacetato; Ipossia; Bevacizumab; Fosforilazione ossidativa; Glicolisi
© Springer-Verlag Berlin Heidelberg 2013
INTRODUZIONE
Le terapie molecolari che hanno come bersaglio la neo-angiogenesi e, in particolare, il fattore di crescita dell’endotelio vascolare (VEGF) hanno dimostrato attività antitumorale in diversi contesti clinici [1]. Il glioblastoma (GBM) è un tumore cerebrale primario altamente vascolarizzato e letale, con una sopravvivenza mediana di circa 12-14 mesi, e rappresenta quindi un importante bersaglio per i farmaci antiangiogenici [2]. Bevacizumab, un anticorpo umanizzato anti-VEGF, è attualmente approvato dalla Food and Drug Administration come trattamento di seconda linea del GBM e gli studi clinici in corso mirano a valutarne il potenziale come agente di prima linea [3]. Tuttavia, poiché il blocco del VEGF prolunga la sopravvivenza libera da progressione ma non la sopravvivenza globale, è indispensabile individuare strategie che ne aumentino l’impatto e ritardino l’insorgenza della resistenza [4]. Ad esempio, sono stati ottenuti progressi limitati in combinazione con l’irinotecan; tuttavia, non è stato possibile dimostrare alcun impatto sulla sopravvivenza globale [5]. Un limite importante nello sviluppo di combinazioni sinergiche incentrate su agenti anti-VEGF è la mancanza di dati clinici solidi, poiché i tumori che stanno diventando resistenti a questi agenti non sono disponibili di routine per ulteriori analisi. Inoltre, le conseguenze cellulari e molecolari del trattamento anti-VEGF non sono ancora sufficientemente comprese. Informazioni dettagliate a livello molecolare su come bevacizumab influisce sul GBM in un arco di tempo prolungato sono essenziali non solo per la comprensione delle risposte adattative del tumore e del conseguente fallimento del trattamento, ma anche per lo sviluppo di terapie di combinazione razionali.
Abbiamo quindi cercato di determinare la risposta tumorale a bevacizumab a livello fenotipico e molecolare in modelli di xenotrapianto derivati da linee cellulari di GBM. Estendendo i modelli fino all’eventuale fallimento terapeutico nonostante il trattamento continuo con bevacizumab, abbiamo cercato di cogliere i programmi di adattamento del tumore e il corrispondente ricablaggio delle vie molecolari utilizzando l’analisi microarray. Abbiamo ipotizzato che i meccanismi fondamentali della resistenza si riflettano nell’alterazione di queste vie e che le piccole molecole che interferiscono con questi processi rappresentino candidati realistici per aumentare l’efficacia di bevacizumab. L’analisi bioinformatica ha rivelato che i tumori resistenti presentano una forte firma del fattore inducibile dell’ipossia (HIF) e un passaggio dalla respirazione mitocondriale alla glicolisi. La riattivazione della respirazione mitocondriale con il farmaco orfano dicloroacetato (DCA) potenzia l’effetto transitorio del bevacizumab, a differenza dell’assenza di effetto additivo del 2-deossiglucosio (2-DG), che mira principalmente alla glicolisi. I nostri dati forniscono indicazioni sulla plasticità del metabolismo tumorale in risposta alle sfide terapeutiche e suggeriscono nuove opportunità per interventi sinergici.
Materiali e metodi
Tumorigenicità in vivo
Tutti i protocolli sono stati eseguiti in conformità al Comitato per la cura e l’uso degli animali dell’Indiana University e ai protocolli e alle normative approvate dal Ministero dell’Interno del Regno Unito.107 cellule U87-MG (acquistate dall’ATCC) sono state impiantate in topi femmina BALB/c SCID (Harlan Sprague Dawley, Inc.) di 6-8 settimane per via sottocutanea, sotto forma di sospensioni cellulari da 100 μL con un uguale volume di Matrigel (BD Bioscience). I tumori sono stati misurati due volte alla settimana con un calibro e i volumi sono stati calcolati utilizzando la formula lunghezza × larghezza × altezza × 0,52. Una volta che il volume del tumore ha raggiunto i 150 mm3, i topi sono stati randomizzati in due gruppi con coorti iniziali di cinque topi per gruppo e si è iniziato il trattamento con bevacizumab (Roche) iniettato per via intraperitoneale ogni 3 giorni alla dose di 10 mg/kg o con il controllo salino. Il trattamento è stato continuato fino a quando i tumori sono cresciuti fino a raggiungere un volume di circa 600-800 mm3; a quel punto i topi sono stati eutanasia e i tumori sono stati rapidamente escissi chirurgicamente. Per i modelli di xenotrapianto su topi nudi atimici, le cellule U87-MG sono state impiantate come sopra in topi femmina di 4-8 settimane (Harlan Laboratories, Indianapolis, IN, USA), oppure sono state innestate 7 ×106 cellule U118-MG (acquistate da ATCC). I frammenti tumorali sono stati trattati mediante fissazione in formalina prima dell’inclusione in paraffina per l’IHC o congelati per la successiva estrazione dell’RNA, come descritto in precedenza [6].
Protocolli di somministrazione dei farmaci per gli studi di combinazione
Il DCA è stato somministrato per via orale due volte al giorno a 50 mg/kg/dose in acqua sterile (il veicolo di controllo era acqua sterile). Questa dose si è basata su rapporti pubblicati e sulla scala allometrica. Pertanto, 100 mg/kg al giorno per topo si traducono in circa 13 mg/kg nell’uomo (http://home.fuse.net/clymer/minor/allometry.html), il che è coerente con le dosi utilizzate in ambito clinico. Bevacizumab è stato somministrato per via intraperitoneale a una concentrazione di 2,5 mg/kg/dose (U87-MG) o 2,0 mg/kg/dose (U118-MG). Il trattamento è stato continuato fino a quando i tumori sono cresciuti fino a circa 20 mm di diametro; a quel punto i topi sono stati eutanasia e i tumori sono stati rapidamente escissi. il 2-deossiglucosio (Sigma, 500 mg/kg) è stato somministrato giornalmente tramite iniezione intraperitoneale.
I dettagli della coltura cellulare, dell’analisi dell’array genico, dell’analisi istologica e dell’immunoistochimica, degli sferoidi trattati con DCA e dell’isolamento dell’RNA e dell’analisi quantitativa RT-PCR (QPCR) sono descritti nel materiale supplementare elettronico.
Analisi statistica
La significatività statistica delle differenze osservate tra i diversi gruppi sperimentali è stata calcolata con il test t a due code. I valori di P <0,05 sono stati considerati statisticamente significativi.
Risultati
Creazione del modello tumorale di resistenza al bevacizumab
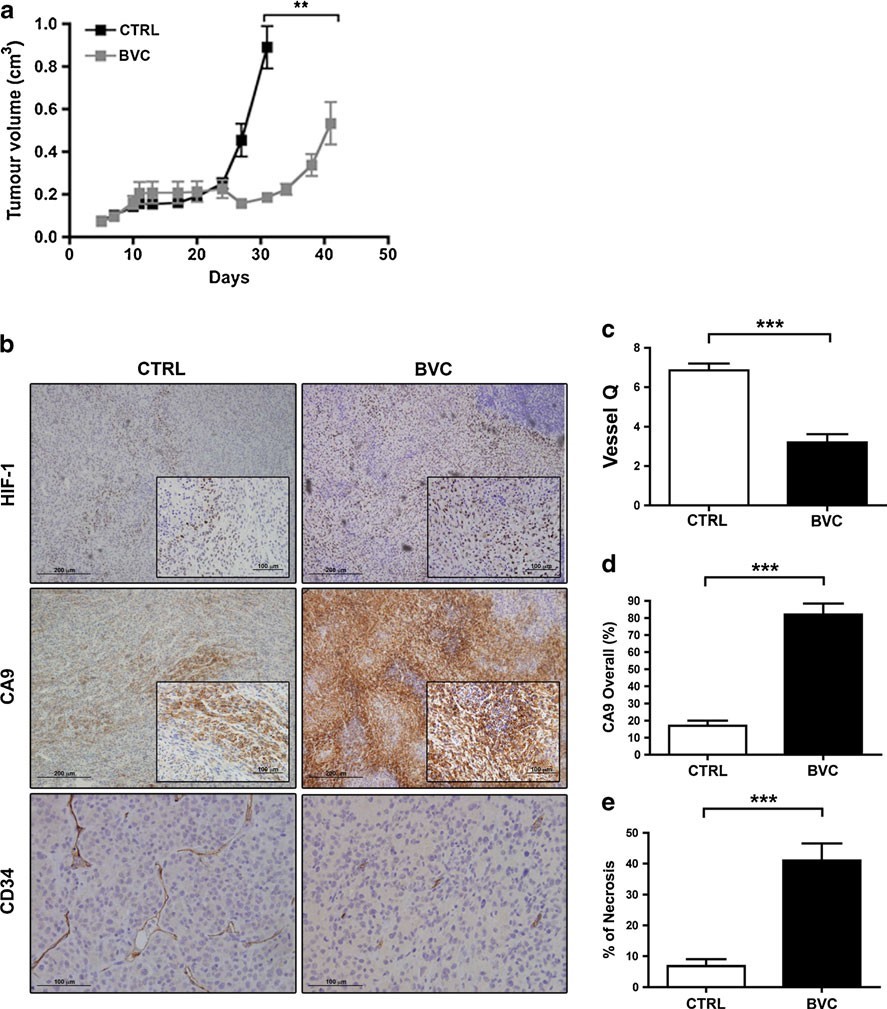
Le sospensioni di cellule U87-MG sono state iniettate per via sottocutanea nel fianco destro di topi SCID. Il trattamento con bevacizumab (iniezione intraperitoneale di 10 mg/kg ogni 3 giorni) è stato iniziato quando la media dei tumori era di 100-200 mm3. La divergenza tra le coorti trattate e non trattate è stata evidente una settimana dopo e, approssimativamente al 40° giorno, è stata raggiunta la completa resistenza al bevacizumab (Fig. 1). I tumori sono stati asportati separatamente per i controlli e le coorti trattate in momenti in cui i tassi di crescita e le dimensioni medie erano simili. Una risposta analoga (cioè una risposta iniziale seguita da resistenza) è stata osservata utilizzando una dose submassimale di bevacizumab (2,5 mg/kg; ogni 3 giorni per iniezione intraperitoneale) in topi nudi atimici. Questo aspetto sarà descritto in dettaglio nell’ambito degli studi di combinazione che seguono. Eseguendo l’immunoistochimica per i marcatori vascolari standard (CD31 e CD34) seguita dal conteggio della densità dei microvasi, abbiamo confermato che i tumori resistenti presentano una vascolarizzazione significativamente più scarsa (Fig. 1). Pertanto, la resistenza terapeutica non sembra essere principalmente legata a una nuova vascolarizzazione dovuta a un passaggio a fattori di crescita angiogenici alternativi, come dimostrato in altri modelli di resistenza al bevacizumab [7]. L’espressione di HIF-1α e dell’anidrasi carbonica IX (CA9), un robusto bersaglio di HIF e un marker consolidato di ipossia, è aumentata drasticamente nei tumori resistenti, indicando che la loro crescita è continuata in un ambiente significativamente più povero di ossigeno, in linea con i dati riportati da Rapisarda et al. [8].
Caratterizzazione molecolare del modello di resistenza a bevacizumab
Al fine di ottenere una comprensione completa dei processi molecolari associati e potenzialmente critici per il processo di resistenza, l’RNA totale di tumori trattati e non trattati è stato sottoposto ad analisi di espressione utilizzando gli array Affymetrix HGU133plus2. Il set completo di dati per l’analisi dell’espressione degli array Affymetrix è disponibile all’indirizzo http://www.ncbi.nlm.gog/geo/query/acc.cgi?acc=GSE37956. Un tema centrale nei tumori resistenti al bevacizumab è l’attivazione coordinata del programma di trascrizione guidato da HIF (Fig. 2). Un’ampia percentuale di target HIF ha mostrato un’upregulation coordinata, nella maggior parte dei casi con più di una sonda array. In particolare, i target glicolitici dell’HIF, tra cui le aldolasi A e C, l’isomerasi 1 del triosiofosfato e la 6-fosfrutto-2-chinasi/fruttosio-2,6-bifosfatasi 3, nonché i trasportatori inducibili del glucosio GLUT1/SLC2A1 e GLUT3/SLC2A3, sono stati robustamente indotti, indicando una maggiore dipendenza dall’utilizzo glicolitico del glucosio. Al contrario, nei tumori resistenti al bevacizumab è stata osservata una significativa repressione a livello dei geni della piruvato deidrogenasi (PDH) alfa 1 e beta, che regolano l’ingresso del piruvato nel ciclo dell’acido tricarbossilico (TCA) [9]. L’attività della PDH è inibita dalla fosforilazione da parte delle piruvato deidrogenasi chinasi (PDK). Le isoforme PDK1 e PDK3, che sono bersagli ben documentati dell’HIF [10-12], sono state fortemente upregolate nei tumori resistenti al bevacizumab, a ulteriore sostegno di uno spostamento biochimico dalla fosforilazione ossidativa (OXPHOS) (Fig. 2). Probabilmente, il cambiamento di espressione genica più evidente nei tumori resistenti al bevacizumab è stata la downregulation globale dei geni del metabolismo mitocondriale, in particolare dei membri di tutti e cinque i complessi OXPHOS mitocondriali (Fig. 2). Infine, diversi componenti del ciclo TCA, tra cui la fumarato idratasi (FH) e la succinato deidrogenasi (SDH), sono stati downregolati (Fig. 2), il che potrebbe contribuire alla crescita tumorale in quanto funzionano anche come soppressori tumorali [13].
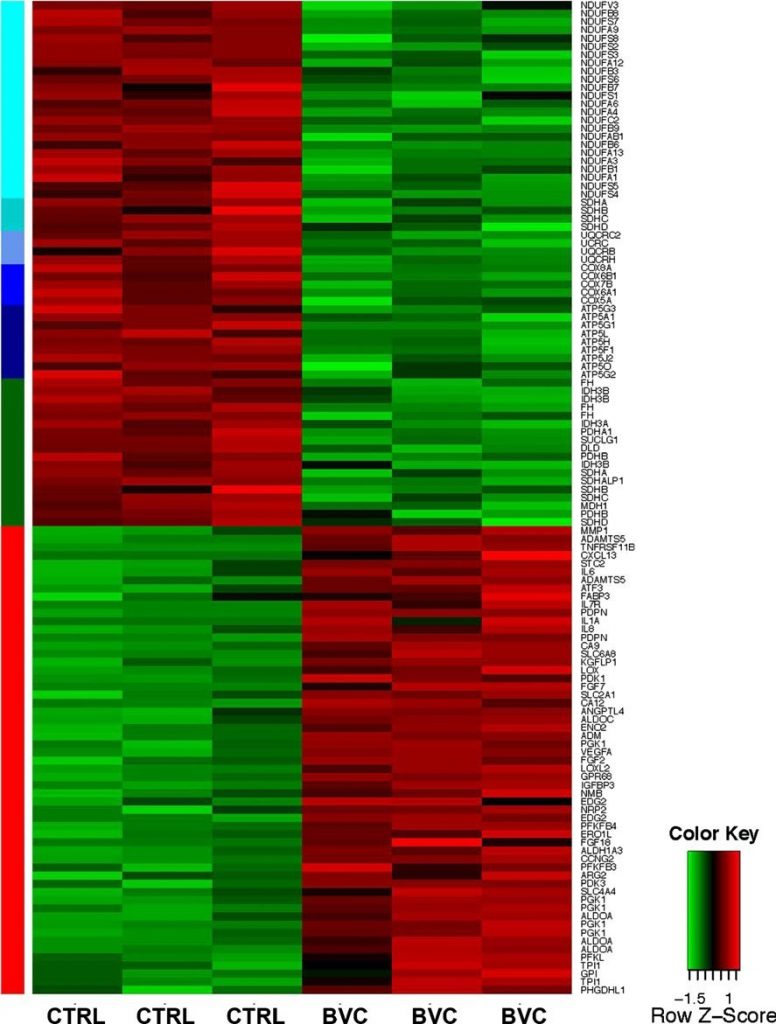
L’analisi dei percorsi KEGG ha confermato che il metabolismo energetico rappresenta uno dei cambiamenti dominanti associati ai tumori resistenti al bevacizumab. Pertanto, i geni glicolitici erano più abbondanti tra i geni upregolati, insieme ai geni appartenenti al metabolismo del fruttosio e del mannosio, alla via del pentoso fosfato, al metabolismo degli zuccheri amminici e degli zuccheri nucleotidici e al metabolismo del fosfato di inositolo (Tabella supplementare S1A). I valori di p ipergeometrici e ipergeometrici corretti erano inferiori a 0,01 per tutti questi percorsi. Al contrario, tra i geni downregolati, l’OXPHOS è stata la principale via repressa, con valori di p inferiori a 1,00E-28, con il metabolismo del piruvato e il ciclo TCA che hanno mostrato una repressione molto significativa (Tabella supplementare S1B). Un’ulteriore firma osservata nei tumori resistenti al bevacizumab è stata l’aumento dello stress del reticolo endoplasmatico e della risposta alle proteine dispiegate. In particolare, i mediatori chiave di queste risposte ATF4, 5, 6 e DDIT3/CHOP erano tutti fortemente sovraespressi (Tabella supplementare S2), coerentemente con le nostre precedenti osservazioni che l’ipossia più grave che si sviluppa durante il corso della terapia antiangiogenica attiva vie non HIF [14].
Effetti in vitro dell’ipossia cronica sull’espressione genica dell’OXPHOS mitocondriale
Per verificare se l’ipossia rappresenti la causa principale della downregulation dei geni della respirazione mitocondriale e del ciclo di Krebs, abbiamo intrapreso un’indagine completa in vitro degli effetti del basso ossigeno nelle cellule U87. È interessante notare che diversi geni OXPHOS e componenti del ciclo di Krebs che sono stati trovati downregolati negli array hanno risposto con una downregulation anche in ipossia, tra cui ATP5A1, ATP5G3, NDUFA9, FH e MDH1 (Fig. 3). Anche MRPL36 e MRPS11, due geni ritenuti critici nell’assemblaggio dei complessi OXPHOS [15, 16], hanno mostrato una downregulation in ipossia, simile all’effetto del bevacizumab (Fig. 3). Tuttavia, la maggior parte dei geni downregolati negli array non ha mostrato una downregulation durante l’esposizione all’ipossia, suggerendo che altri fattori possono essere i principali responsabili [17]. Il riesame degli array ha rivelato che diversi fattori di trascrizione che regolano l’espressione genica mitocondriale (compresa l’OXPHOS) sono stati downregolati: NRF1, TFAM e TFB2M [18-20] (Tabella supplementare S3).

Sfruttamento dell’aumento del gap glicolitico: sinergia in vivo tra bevacizumab e DCA
Sulla base della marcata firma di un aumento della segnalazione HIF e di una diminuzione dell’OXPHOS mitocondriale, abbiamo ipotizzato che i tumori resistenti al bevacizumab sarebbero stati particolarmente sensibili ai riattivatori mitocondriali. La principale molecola candidata di questa classe è il DCA, che inibisce l’attività della PDK, aumentando così il flusso di piruvato nei mitocondri e promuovendo l’ossidazione del glucosio rispetto alla glicolisi [21, 22]. Tenendo conto dei ben noti effetti collaterali della terapia antiangiogenica e del DCA, per la valutazione della combinazione di farmaci abbiamo scelto una dose submassimale dell’agente antiangiogenico (2,5 mg/kg; ogni 3 giorni per iniezione intraperitoneale), una strategia ampiamente utilizzata in vivo [23]. Questo approccio dovrebbe anche aumentare la nostra capacità di individuare il sinergismo tra i due farmaci, sia dal punto di vista della risposta tumorale che degli effetti sulla segnalazione di HIF. In effetti, il trattamento con bevacizumab e DCA ha bloccato drasticamente la crescita tumorale rispetto a ciascun farmaco da solo (Fig. 4A e Fig. S1 supplementare). Per valutare la generalità della risposta, abbiamo studiato un altro tipo di cellula GBM, la U118. Anche la risposta negli innesti basati su U118 è stata molto più robusta con la combinazione, rispetto ai singoli farmaci (Fig. 4B e Fig. S1 supplementare).
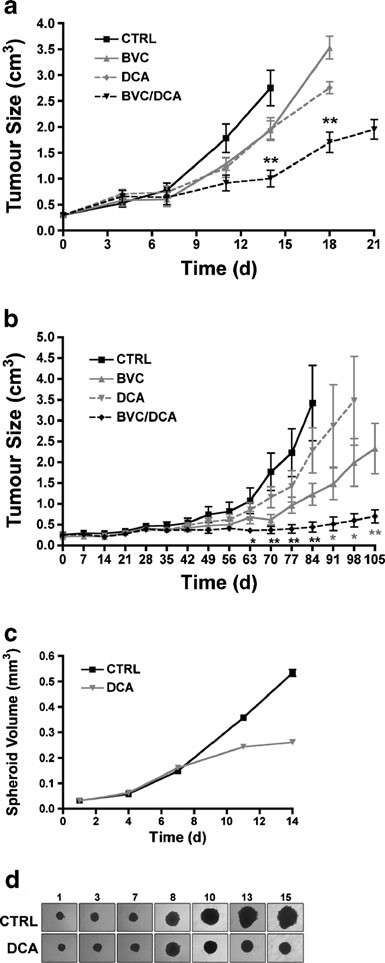
L’efficacia degli inibitori glicolitici nel superare la resistenza al bevacizumab è stata proposta in precedenza [24] senza conferme sperimentali. DCA e 2-DG sono stati discussi insieme come parte di questo concetto. Tuttavia, quando il 2-DG è stato testato in parallelo al DCA, non è stato notato alcun effetto additivo al bevacizumab. Questo nonostante l’effetto transitorio del 2-DG come agente singolo, somministrato alla concentrazione descritta in letteratura (Fig. S2 supplementare). Pertanto, la riattivazione mitocondriale e l’inibizione diretta della glicolisi hanno effetti diversi in combinazione con bevacizumab.
Effetti in vitro del DCA
I modelli sferoidi rappresentano un sistema di complessità intermedia tra i sistemi di coltura bidimensionali standard e i tumori in vivo, grazie ai gradienti di ossigeno e nutrienti. A differenza dei sistemi monostrato, gli sferoidi in espansione imitano la maggiore avascolarità delle strutture in vivo che crescono in presenza di bevacizumab. Gli sferoidi U87 sono stati generati, come descritto [25], e fatti crescere per 7 giorni fino a raggiungere 0,2 mm3. Sferoidi di queste dimensioni sono abbastanza grandi da consentire la diffusione di un farmaco attraverso lo sferoide, ma iniziano anche a formare una piccola area centrale di ipossia e a mostrare gradienti di nutrienti, pH e O2. Dopo 3 giorni di trattamento si è notata una forte divergenza della cinetica di crescita tra i gruppi trattati con DCA e quelli non trattati. Questo effetto era persistente e, a partire da 6 giorni, il DCA ha compromesso in modo significativo l’espansione degli sferoidi (Fig. 4C, D ).
Effetto della combinazione di bevacizumab e DCA sui target HIF e sui marcatori istologici della crescita tumorale
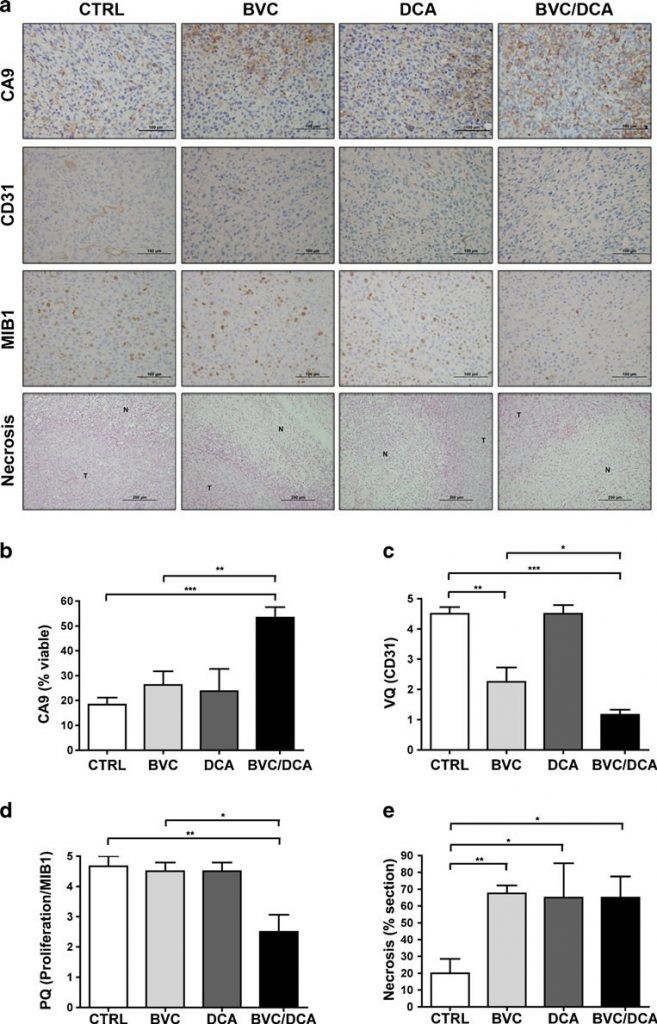
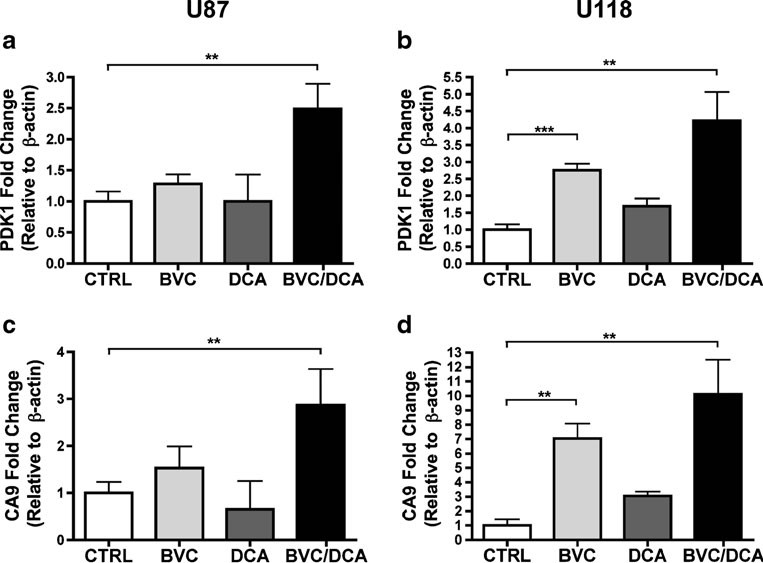
Per stabilire se l’effetto della combinazione sulla crescita tumorale fosse determinato principalmente da un aumento della morte cellulare o da una diminuzione della proliferazione, abbiamo eseguito analisi istologiche su questi tumori, utilizzando marcatori ben consolidati. La necrosi era maggiore nei tumori trattati rispetto a quelli non trattati, ma non è stata osservata alcuna differenza significativa tra i farmaci da soli o in combinazione (Fig. 5). Il tasso di proliferazione, valutato attraverso la quantificazione della colorazione Ki-67 (MIB-1), è risultato significativamente più basso nel gruppo di combinazione rispetto ai tumori non trattati e a quelli trattati con solo bevacizumab, suggerendo che l’effetto della combinazione era prevalentemente citostatico (Fig. 5). Abbiamo quindi valutato gli effetti della combinazione di farmaci sulla segnalazione di HIF, utilizzando una combinazione di immunoistochimica e RT-PCR quantitativa (Figg. 5 e 6). Sorprendentemente, pur essendo noto per aumentare il consumo di ossigeno in vari sistemi sperimentali, il DCA da solo non è stato sufficiente ad aumentare in modo misurabile l’espressione dei target HIF in vivo. Tuttavia, in combinazione con bevacizumab, l’espressione di CA9 è aumentata notevolmente nelle cellule tumorali U87 vitali. Nell’U118, invece, il bevacizumab submassimale da solo ha portato a un aumento drammatico di tutti i target HIF testati, senza un ulteriore aumento misurabile nei tumori trattati in combinazione (Fig. 6 e Fig. S3 supplementare). Tuttavia, l’avvertenza principale è che i tumori sopravvissuti in presenza della combinazione erano drasticamente più piccoli e praticamente stazionari, fattori che dovrebbero attenuare l’entità dell’ipossia.
Discussione
L’obiettivo principale del nostro studio è stato quello di comprendere più a fondo l’adattamento del tumore a bevacizumab, identificando le vie associate alla resistenza, con particolare attenzione alle risposte metaboliche. Ci siamo concentrati su un modello eterotopico piuttosto che ortotopico, principalmente per garantire la fattibilità del monitoraggio della crescita e della fuga neoplastica. Inoltre, poiché i tumori sottocutanei raggiungono volumi significativamente maggiori rispetto alle loro controparti ortotopiche, sono probabilmente più rilevanti per la modellazione di neoplasie avanzate e ipossiche.
In un modello ortotopico di GBM pubblicato di recente, è stato segnalato un aumento dell’ipossia dopo l’uso di Bevacizumab [24, 26]; tuttavia, lo studio delle differenze di crescita a lungo termine è meno fattibile in questi sistemi. Nonostante lo status di “classico” del metabolismo tumorale nella ricerca sul cancro e il suo recente rilancio, nessun farmaco che agisca principalmente a questo livello è stato approvato per l’uso clinico di routine [27]. Il DCA, una piccola molecola che attraversa la barriera ematoencefalica [22], ha mostrato risultati promettenti in uno studio clinico sul GBM in combinazione con la chirurgia, la temozolomide e le radiazioni [21], con ulteriori studi attualmente in corso http://clinicaltrials.gov/ct2/results?term=+Dicloroacetato). Tuttavia, l’effetto del DCA come agente singolo è al massimo transitorio e i nostri modelli tumorali riflettono certamente questa limitazione. Uno studio recente ha discusso la possibilità che il trattamento con bevacizumab sensibilizzi i tumori sia al 2-DG che al DCA [24], ma queste previsioni sono state confermate solo per il DCA nelle nostre mani. È improbabile che la mancanza di un effetto additivo o sinergico misurabile del 2-DG sia dovuta a un’inattività biologica o a un dosaggio inadeguato, dal momento che l’efficacia come agente singolo è stata transitoria. Rimangono dubbi sul meccanismo dell’inibizione tumorale mediata dal DCA in presenza di bevacizumab. Sebbene sia stato riportato che il DCA blocca l’angiogenesi da solo [21], la misurazione della densità vasale media non ha confermato questo dato nel nostro sistema.
È stato dimostrato che il DCA mostra maggiori effetti citotossici in condizioni di ipossia in diverse linee cellulari [28]. Il paradigma attuale è che il DCA acceleri il consumo di ossigeno attraverso la riattivazione mitocondriale e riduca ulteriormente la tensione di ossigeno locale [29], ponendo così un’ulteriore sfida alle cellule tumorali per sopravvivere e/o proliferare. Inoltre, è stato dimostrato che nelle cellule “cablate” per utilizzare selettivamente la glicolisi per la generazione di ATP a causa di mutazioni del DNA mitocondriale, l’OXPHOS forzata indotta dal DCA ha un effetto tossico. Il DCA mostra anche una citotossicità sinergica in vitro in combinazione con cisplatino e topotecan, due agenti antineoplastici noti per danneggiare il DNA mitocondriale [30]. Si potrebbe ipotizzare che la downregulation dei geni mitocondriali nei tumori resistenti al bevacizumab rappresenti una forma di disfunzione mitocondriale che sensibilizza agli effetti del DCA.
È interessante notare che l’effetto del DCA, come agente singolo, sui bersagli HIF negli xenotrapianti è stato al massimo impercettibile, nonostante il suo ben noto effetto positivo sul consumo di ossigeno. Al contrario, in combinazione con bevacizumab, il DCA ha portato a un aumento dell’espressione della maggior parte dei target HIF testati nei tumori sopravvissuti rispetto al solo bevacizumab. Una possibile spiegazione di questi risultati è che l’OXPHOS forzata indotta dal DCA in presenza di livelli molto bassi di ossigeno può portare a un aumento della produzione di specie reattive dell’ossigeno che, a sua volta, può contribuire a un’ulteriore induzione di HIF [31]. Inoltre, o in alternativa, il DCA in presenza di bevacizumab può portare a un’ulteriore diminuzione della concentrazione locale di ossigeno a livelli in cui l’induzione dei target di HIF diventa più evidente con il saggio quantitativo RT-PCR. Oltre all’aumento dell’espressione di CA9, è stata osservata una maggiore espressione dei target HIF PDK1, 3 e GLUT1 nei tumori sopravvissuti in presenza della combinazione di farmaci sia nelle cellule U87 che U118. Ciò potrebbe riflettere un cambiamento metabolico “di ultima istanza” più drammatico, critico per la sopravvivenza delle cellule tumorali. In particolare, l’aumento dell’espressione di PDK1/3 potrebbe essere parte di un “tentativo finale” delle cellule tumorali di contrastare in parte l’effetto del DCA e di inattivare l’OXPHOS mitocondriale. Questi cambiamenti metabolici nei tumori che sopravvivono in presenza di bevacizumab e DCA possono anche fornire importanti indizi su come aumentare ulteriormente l’efficacia di questa combinazione. Ad esempio, si potrebbe ipotizzare che i tumori resistenti possano mostrare almeno una certa sensibilità a un ulteriore aumento della concentrazione di DCA, sebbene la tossicità di questo composto possa diventare un fattore limitante. Anche l’aumento della CA9 specifica per il tumore può svolgere un ruolo importante nella sopravvivenza dei tumori trattati con la combinazione, poiché accelera l’eliminazione dellaCO2 in eccesso generata dalla riattivazione del ciclo di Krebs [25]. Pertanto, gli inibitori di CA9, che hanno recentemente mostrato promettenti effetti antitumorali [32, 33], possono essere considerati candidati realistici per un terzo componente di una strategia di combinazione.
In conclusione, la dissezione molecolare dell’adattamento del tumore agli agenti anti-VEGF può offrire indizi preziosi per costruire combinazioni più efficienti che includano il bersaglio del metabolismo del cancro.
Contributi dell’autore
Ideazione e progettazione: M.I. e A.L.H. Acquisizione dei dati: K.K., S.W., H.E.G., H.T., J.L., J.S., A.L.S., R.L., D.S., C.M.D. e M.H. Analisi e interpretazione dei dati: M.I. e A.L.H. Stesura, revisione e/o revisione del manoscritto: M.I. e A.L.H. Supporto amministrativo, tecnico o materiale: F.B. Supervisione dello studio: M.I. e A.L.H.
Fonte di finanziamento
Il lavoro è stato sostenuto da sovvenzioni del Cancer Research United Kingdom [S.W., A.L.H., H.T., R.L., J.L.], METOXIA p-Medicine European Union Framework 7 [D.S., F.B.], Rhodes Scholar [H.G.], fondi di avviamento dell’Indiana University Cancer Center e American Cancer Society [M.I., C.M.D., K.K.].
Conflitto di interessi
Nessun potenziale conflitto di interesse da dichiarare.
RIFERIMENTI
1 Cao Y, Arbiser J, D’Amato RJ, D’Amore PA, Ingber DE, Kerbel R, Klagsbrun M, Lim S, Moses MA, Zetter B et al (2011) Forty-year journey of angiogenesis translational research. Sci Transl Med 3:114rv3
2 Wick W, Wick A, Weiler M, Weller M (2011) Patterns of progression in malignant glioma following anti-VEGF therapy: perceptions and evidence. Curr Neurol Neurosci Rep 11:305-312
3 Raizer JJ, Grimm S, Chamberlain MC, Nicholas MK, Chandler JP, Muro K, Dubner S, Rademaker AW, Renfrow J, Bredel M (2010) A phase 2 trial of single-agent bevacizumab given in an every-3-week schedule for patients with recurrent high-grade gliomas. Cancro 116:5297-5305
4 Bergers G, Hanahan D (2008) Modalità di resistenza alla terapia anti-angiogenica. Nat Rev Cancer 8:592-603
5 Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R et al (2009) Bevacizumab da solo e in combinazione con irinotecan nel glioblastoma ricorrente. J Clin Oncol 27:4733-4740
6 Winter SC, Shah KA, Campo L, Turley H, Leek R, Corbridge RJ, Cox GJ, Harris AL (2005) Relation of erythropoietin and erythropoietin receptor expression to hypoxia and anemia in head and neck squamous cell carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res 11:7614-7620
7 Davies S, Dai D, Pickett G, Thiel KW, Korovkina VP, Leslie KK (2011) Effects of bevacizumab in mouse model of endometrial cancer: defining the molecular basis for resistance. Oncol Rep 25:855-862
8 Rapisarda A, Hollingshead M, Uranchimeg B, Bonomi CA, Borgel SD, Carter JP, Gehrs B, Raffeld M, Kinders RJ, Parchment R et al (2009) Incremento dell’attività antitumorale di bevacizumab in combinazione con l’inibizione dell’hypoxia inducible factor-1. Mol Cancer Ther 8:1867-1877
9 Harris RA, Bowker-Kinley MM, Huang B, Wu P (2002) Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul 42:249-259
10 Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL (2008) PDK-1 regola la produzione di lattato in ipossia ed è associato a una prognosi sfavorevole nel cancro squamoso della testa e del collo. Br J Cancer 98:1975-1984
11 Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177-185
12 Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ (2008) Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 283:28106-28114
13 Frezza C, Pollard PJ, Gottlieb E (2011) Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) 89:213-220
14 Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL (2010) Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 29:4424-4435
15 Prestele M, Vogel F, Reichert AS, Herrmann JM, Ott M (2009) Mrpl36 è importante per la generazione di proteine competenti per l’assemblaggio durante la traduzione mitocondriale. Mol Biol Cell 20:2615-2625
16 Emdadul Haque M, Grasso D, Miller C, Spremulli LL, Saada A (2008) L’effetto delle proteine ribosomiali mitocondriali mutate S16 e S22 sull’assemblaggio delle subunità ribosomiali piccole e grandi nei mitocondri umani. Mitocondrio 8:254-261
17 Tang X, Lucas JE, Chen JL, Lamonte G, Wu J, Wang MC, Koumenis C, Chi JT (2012) L’interazione funzionale tra le risposte all’acidosi lattica e all’ipossia regola gli output trascrizionali genomici. Cancer Res 72:491-502
18 Scarpulla RC (2002) Attivatori e coattivatori nucleari nella biogenesi mitocondriale dei mammiferi. Biochim Biophys Acta 1576:1-14
19 Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM (2002) Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet 31:289-294
20 Larsson NG, Barsh GS, Clayton DA (1997) Struttura e localizzazione cromosomica del gene del fattore di trascrizione mitocondriale A del topo (Tfam). Mamm Genome 8:139-140
21 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:31ra34
22 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99:989-994
23 Pechman KR, Donohoe DL, Bedekar DP, Kurpad SN, Hoffmann RG, Schmainda KM (2011) Characterization of bevacizumab dose response relationship in U87 brain tumors using magnetic resonance imaging measures of enhancing tumor volume and relative cerebral blood volume. J Neurooncol 105:233-239
24 Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R et al (2011) Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A 108:3749-3754
25 Swietach P, Patiar S, Supuran CT, Harris AL, Vaughan-Jones RD (2009) The role of carbonic anhydrase 9 in regulating extracellular and intracellular ph in three-dimensional tumor cell growths. J Biol Chem 284:20299-20310
26 de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA (2010) Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro Oncol 12:233-242
27 Kaelin WG Jr, Thompson CB (2010) Domande e risposte: cancro: indizi dal metabolismo cellulare. Nature 465:562-564
28 Anderson KM, Jajeh J, Guinan P, Rubenstein M (2009) Effetti in vitro del dicloroacetato e della CO2 sulle cellule HeLa ipossiche. Anticancer Res 29:4579-4588
29 Chen Y, Cairns R, Papandreou I, Koong A, Denko NC (2009) Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS One 4:e7033
30 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL (2010) Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 127:2510-2519
31 Park JH, Kim TY, Jong HS, Kim TY, Chun YS, Park JW, Lee CT, Jung HC, Kim NK, Bang YJ (2003) Gastric epithelial reactive oxygen species prevent normoxic degradation of hypoxia-inducible factor-1alpha in gastric cancer cells. Clin Cancer Res Off J Am Assoc Cancer Res 9:433-440
32 Dubois L, Peeters S, Lieuwes NG, Geusens N, Thiry A, Wigfield S, Carta F, McIntyre A, Scozzafava A, Dogne JM et al (2011) L’inibizione specifica dell’attività dell’anidrasi carbonica IX potenzia l’effetto terapeutico in vivo dell’irradiazione tumorale. Radiother Oncol 99:424-431
33 v Morris JC, Chiche J, Grellier C, Lopez M, Bornaghi LF, Maresca A, Supuran CT, Pouyssegur J, Poulsen SA (2011) Targeting hypoxic tumor cell viability with carbohydrate-based carbonic anhydrase IX and XII inhibitors. J Med Chem 54:6905-6918