Krishan Kumar1, Simon Wigfield2, Harriet E. Gee2,5, Cecilia M. Devlin1, Dean Singleton2, Ji-Liang Li2, Francesca Buffa2, Melanie Huffman1, Anthony L. Sinn3, Jayne Silver3, Helen Turley2, Russell Leek2, Adrian L. Harris2 & Mircea Ivan4
1 Département de médecine, Université d’Indiana, Indianapolis, IN 46202, USA
2 Département d’oncologie, Weatherall Institute of Molecular
Medicine, Université d’Oxford, John Radcliffe Hospital, Oxford OX3 9DS, UK
e-mail : [email protected]
3 In Vivo Therapeutics Core, Université d’Indiana, Indianapolis, IN 46202, USA
4 Département de médecine, immunologie et microbiologie, Université d’Indiana, 980W. Walnut Street, Room C225, Indianapolis, IN 46202, USA
e-mail : [email protected]
5 Département de radio-oncologie, Sydney Cancer Centre, Royal Prince Alfred Hospital, Camperdown, New South Wales 2050, Australie
Reçu : 7 novembre 2012
Révisé : 20 décembre 2012
Accepté : 2 janvier 2013
Publié en ligne : 30 janvier 2013
Résumé
L’inhibition du facteur de croissance endothélial vasculaire augmente les taux de réponse à la chimiothérapie et la survie sans progression dans le glioblastome. Cependant, une résistance apparaît invariablement, d’où le besoin urgent d’identifier des agents synergiques. Une stratégie possible consiste à comprendre l’adaptation des tumeurs aux changements micro-environnementaux induits par les médicaments antiangiogéniques et à tester des agents qui exploitent ce processus. Nous avons utilisé un modèle de xénogreffe in vivo dérivé du glioblastome, où la tumeur s’échappe en présence d’un traitement continu au bevacizumab. Des cellules U87-MG ou U118-MG ont été implantées par voie sous-cutanée dans des souris BALB/c SCID ou des souris nude athymiques. Le bevacizumab a été administré par injection intrapéritonéale tous les 3 jours (2,5 mg/kg/dose) et/ou le dichloroacétate (DCA) a été administré par gavage oral deux fois par jour (50 mg/kg/dose) lorsque le volume des tumeurs a atteint 0,3 cm3 et a continué jusqu’à ce que les tumeurs atteignent environ 1,5-2,0 cm3. L’analyse des microréseaux des tumeurs U87 résistantes a révélé des changements coordonnés au niveau des gènes métaboliques, en particulier, un écart croissant entre la glycolyse et la respiration mitochondriale. Une semaine après le traitement médicamenteux, une différence hautement significative a été observée entre les souris nude athymiques implantées de U87-MG. Après deux semaines de traitement, l’association du bevacizumab et du DCA a bloqué de façon spectaculaire la croissance tumorale par rapport à l’un ou l’autre médicament seul. Des résultats similaires ont été observés chez des souris nude athymiques implantées avec des cellules U118-MG. Nous démontrons pour la première fois que l’inversion de la modification du métabolisme induite par le bevacizumab à l’aide du DCA nuit à la croissance néoplasique in vivo. Le DCA étant considéré comme un agent prometteur ciblant le métabolisme des tumeurs, nos données établissent la preuve de concept opportune que sa combinaison avec un traitement antiangiogénique représente une stratégie antinéoplasique puissante.
Mots clés : Dichloroacétate ; Hypoxie ; Bevacizumab ; Phosphorylation oxydative ; Glycolyse
© Springer-Verlag Berlin Heidelberg 2013
INTRODUCTION
Les thérapies moléculaires ciblant la néo-angiogenèse et, en particulier, le facteur de croissance endothélial vasculaire (VEGF) ont montré une activité antitumorale dans divers contextes cliniques [1]. Le glioblastome (GBM) est une tumeur cérébrale primaire hautement vascularisée et létale, avec une survie médiane d’environ 12-14 mois, et représente donc une cible importante pour les médicaments antiangiogéniques [2]. Le bévacizumab, un anticorps anti-VEGF humanisé, est actuellement approuvé par la Food and Drug Administration comme traitement de deuxième intention du GBM, et les essais cliniques en cours visent à évaluer son potentiel comme agent de première intention [3]. Cependant, comme le blocage du VEGF prolonge la survie sans progression mais pas la survie globale, il est impératif d’identifier des stratégies qui augmentent son impact et retardent l’apparition de la résistance [4]. Par exemple, des progrès limités ont été réalisés en association avec l’irinotécan ; cependant, aucun impact sur la survie globale n’a pu être démontré [5]. L’une des principales limites au développement de combinaisons synergiques centrées sur les agents anti-VEGF est le manque de données cliniques solides, car les tumeurs qui deviennent résistantes à ces agents ne sont pas systématiquement disponibles pour des analyses plus poussées. De plus, les conséquences cellulaires et moléculaires du traitement anti-VEGF sont encore insuffisamment comprises. Des informations détaillées au niveau moléculaire sur la façon dont le bevacizumab affecte les GBM pendant une période prolongée sont non seulement essentielles à notre compréhension des réponses adaptatives des tumeurs et de l’échec ultérieur du traitement, mais aussi au développement de thérapies combinées rationnelles.
Nous avons donc cherché à déterminer la réponse tumorale au bevacizumab au niveau phénotypique et moléculaire dans des modèles de xénogreffe dérivés de lignées cellulaires de GBM. En prolongeant les modèles jusqu’à un éventuel échec thérapeutique malgré un traitement continu au bevacizumab, nous avons cherché à saisir les programmes d’adaptation de la tumeur et le recâblage correspondant des voies moléculaires à l’aide d’une analyse de microréseau. Nous avons émis l’hypothèse que les principaux mécanismes de résistance se refléteraient dans l’altération de ces voies, et que les petites molécules qui interfèrent avec ces processus représentent des candidats réalistes pour augmenter l’efficacité du bevacizumab. Une analyse bioinformatique a révélé que les tumeurs résistantes présentent une forte signature du facteur inductible de l’hypoxie (HIF) et un passage de la respiration mitochondriale à la glycolyse. La réactivation de la respiration mitochondriale par le dichloroacétate (DCA), un médicament orphelin, renforce l’effet transitoire du bevacizumab, contrairement à l’absence d’effet additif du 2-désoxyglucose (2-DG), qui cible principalement la glycolyse. Nos données donnent un aperçu de la plasticité du métabolisme tumoral en réponse aux défis thérapeutiques et suggèrent de nouvelles possibilités d’interventions synergiques.
Matériel et méthodes
Tumorigénicité in vivo
Tous les protocoles ont été réalisés dans le cadre du comité institutionnel de soins et d’utilisation des animaux de l’Université de l’Indiana, et des protocoles et réglementations approuvés par le ministère de l’Intérieur du Royaume-Uni.107 cellules U87-MG (achetées à l’ATCC) ont été implantées chez des souris SCID femelles BALB/c âgées de 6 à 8 semaines (Harlan Sprague Dawley, Inc.) par voie sous-cutanée, sous forme de suspensions cellulaires de 100-μL avec un volume égal de Matrigel (BD Bioscience). Les tumeurs ont été mesurées deux fois par semaine avec un pied à coulisse, et les volumes ont été calculés à l’aide de la formule longueur × largeur × hauteur × 0,52. Une fois que le volume de la tumeur a atteint 150 mm3, les souris ont été réparties de manière aléatoire en deux groupes avec des cohortes de départ de cinq souris par groupe et le traitement au bevacizumab (Roche) injecté par voie intrapéritonéale tous les 3 jours à une dose de 10 mg/kg ou un contrôle salin a commencé. Le traitement a été poursuivi jusqu’à ce que les tumeurs atteignent un volume d’environ 600-800 mm3, après quoi les souris ont été euthanasiées et les tumeurs ont été excisées rapidement par voie chirurgicale. Pour les modèles de xénogreffes de souris nude athymiques, des cellules U87-MG ont été implantées comme ci-dessus dans des souris femelles âgées de 4 à 8 semaines (Harlan Laboratories, Indianapolis, IN, USA), ou 7 ×106 cellules U118-MG (achetées à l’ATCC) ont été greffées. Les fragments de tumeur ont été soit traités par fixation au formol avant inclusion en paraffine pour l’IHC, soit congelés pour une extraction ultérieure de l’ARN, comme décrit précédemment [6].
Protocoles d’administration du médicament pour les études de combinaison
Le DCA a été administré par gavage oral deux fois par jour à raison de 50 mg/kg/dose dans de l’eau stérile (le véhicule témoin était de l’eau stérile). Cette dose était basée sur des rapports publiés et une mise à l’échelle allométrique. Ainsi, 100 mg/kg par souris et par jour se traduisent par environ 13 mg/kg chez l’homme (http://home.fuse.net/clymer/minor/allometry.html), ce qui correspond aux doses utilisées en milieu clinique. Le bevacizumab a été administré par voie intrapéritonéale à une concentration de 2,5 mg/kg/dose (U87-MG) ou de 2,0 mg/kg/dose (U118-MG). Le traitement a été poursuivi jusqu’à ce que les tumeurs atteignent un diamètre d’environ 20 mm, après quoi les souris ont été euthanasiées et les tumeurs rapidement excisées. le 2-désoxyglucose (Sigma, 500 mg/kg) a été administré quotidiennement par injection intrapéritonéale.
Les détails de la culture cellulaire, de l’analyse des réseaux de gènes, de l’analyse histologique et de l’immunohistochimie, des sphéroïdes traités au DCA, de l’isolement de l’ARN et de l’analyse quantitative par RT-PCR (QPCR) sont décrits dans le matériel supplémentaire électronique.
Analyse statistique
La signification statistique des différences observées entre les différents groupes expérimentaux a été calculée à l’aide d’un test t bilatéral. Les valeurs P <0,05 ont été considérées comme statistiquement significatives.
Résultats
Établissement du modèle tumoral de résistance au bevacizumab
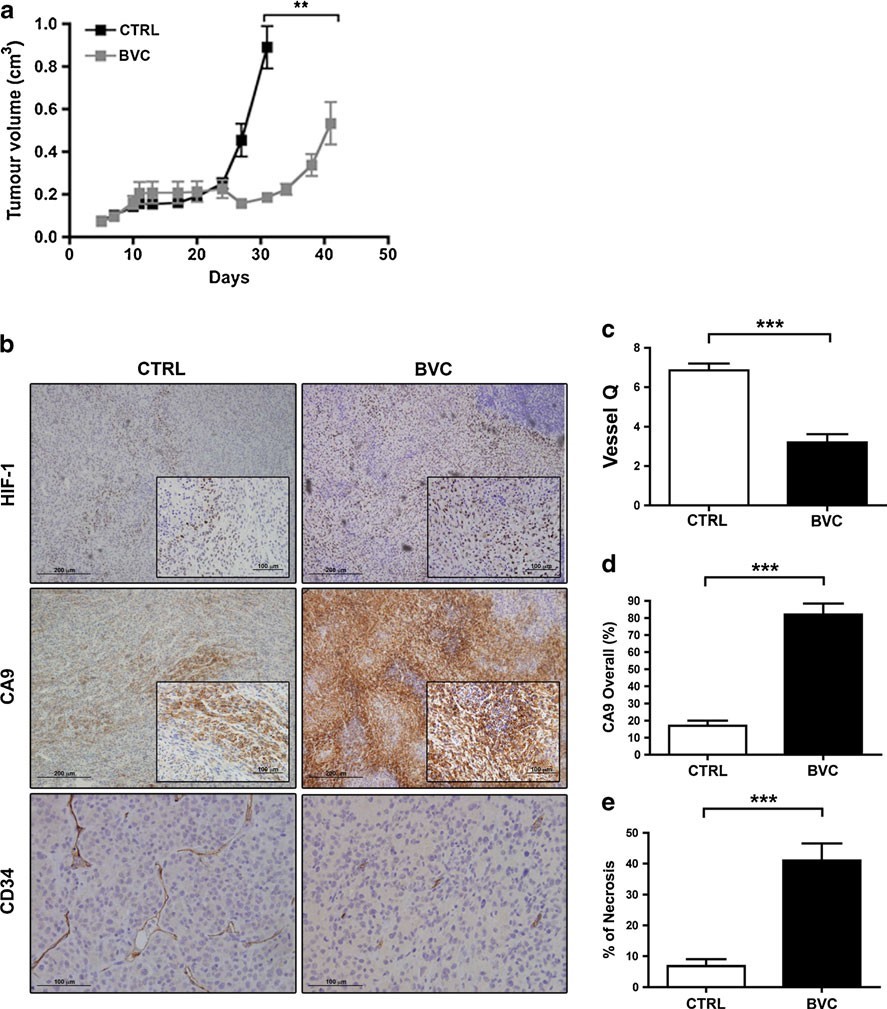
Des suspensions de cellules U87-MG ont été injectées par voie sous-cutanée dans le flanc droit de souris SCID. Le traitement au bevacizumab (injection intrapéritonéale de 10 mg/kg tous les 3 jours) a été initié lorsque les tumeurs atteignaient en moyenne 100-200 mm3. La divergence entre les cohortes traitées et non traitées était évidente une semaine plus tard, et à environ 40 jours, une résistance complète au bevacizumab était atteinte (Fig. 1). Les tumeurs ont été excisées séparément pour les témoins et les cohortes traitées à des moments où leurs taux de croissance moyens et leurs tailles étaient similaires. Une réponse analogue (c’est-à-dire une réponse initiale suivie d’une résistance) a été observée en utilisant une dose submaximale de bevacizumab (2,5 mg/kg ; tous les 3 jours par injection intrapéritonéale) chez des souris nude athymiques. Ceci sera décrit en détail dans le cadre des études de combinaison ci-dessous. En réalisant une immunohistochimie pour les marqueurs vasculaires standard (CD31 et CD34) suivie d’un comptage de la densité des microvaisseaux, nous avons confirmé que les tumeurs résistantes présentent une vascularisation nettement plus clairsemée (Fig. 1). Par conséquent, la résistance thérapeutique ne semble pas être principalement liée à un renouvellement de la vascularisation dû à un passage à des facteurs de croissance angiogéniques alternatifs, comme l’ont montré d’autres modèles de résistance au bevacizumab [7]. L’expression de HIF-1α, ainsi que de l’anhydrase carbonique IX (CA9), une cible robuste de HIF et un marqueur bien établi de l’hypoxie, était considérablement accrue dans les tumeurs résistantes, ce qui indique que leur croissance s’est poursuivie dans un environnement nettement plus appauvri en oxygène, conformément aux données rapportées par Rapisarda et al [8].
Caractérisation moléculaire du modèle de résistance au bevacizumab
Afin d’obtenir une compréhension complète des processus moléculaires associés au processus de résistance et potentiellement critiques pour celui-ci, l’ARN total des tumeurs traitées et non traitées a été soumis à une analyse d’expression à l’aide de matrices Affymetrix HGU133plus2. L’ensemble des données de l’analyse d’expression des matrices Affymetrix est disponible sur http://www.ncbi.nlm.gog/geo/query/acc.cgi?acc=GSE37956. Un thème central dans les tumeurs résistantes au bevacizumab était l’activation coordonnée du programme de transcription piloté par HIF (Fig. 2). Une grande proportion de cibles HIF a présenté une régulation coordonnée à la hausse, dans la plupart des cas avec plus d’une sonde. Notamment, les cibles glycolytiques de HIF, y compris les aldolases A et C, la triosephosphate isomérase 1 et la 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3, ainsi que les transporteurs de glucose inductibles GLUT1/SLC2A1 et GLUT3/SLC2A3, ont été fortement induits, ce qui indique une dépendance accrue de l’utilisation glycolytique du glucose. En revanche, une répression significative dans les tumeurs résistantes au bevacizumab a été observée au niveau des gènes alpha 1 et bêta de la pyruvate déshydrogénase (PDH) qui régissent l’entrée du pyruvate dans le cycle de l’acide tricarboxylique (TCA) [9]. L’activité de la PDH est inhibée par la phosphorylation par les pyruvate déshydrogénase kinases (PDK). Les isoformes PDK1 et PDK3, qui sont des cibles HIF bien documentées [10-12], ont été fortement régulées à la hausse dans les tumeurs résistantes au bevacizumab, ce qui confirme le changement biochimique de la phosphorylation oxydative (OXPHOS) (Fig. 2). Le changement d’expression génétique le plus frappant dans les tumeurs résistantes au bevacizumab est sans doute la régulation négative globale des gènes du métabolisme mitochondrial, en particulier des membres des cinq complexes OXPHOS mitochondriaux (Fig. 2). Enfin, plusieurs composants du cycle TCA, y compris la fumarate hydratase (FH) et la succinate déshydrogénase (SDH), étaient régulés à la baisse (Fig. 2), ce qui pourrait contribuer à la croissance tumorale, car ils fonctionnent également comme suppresseurs de tumeurs [13].
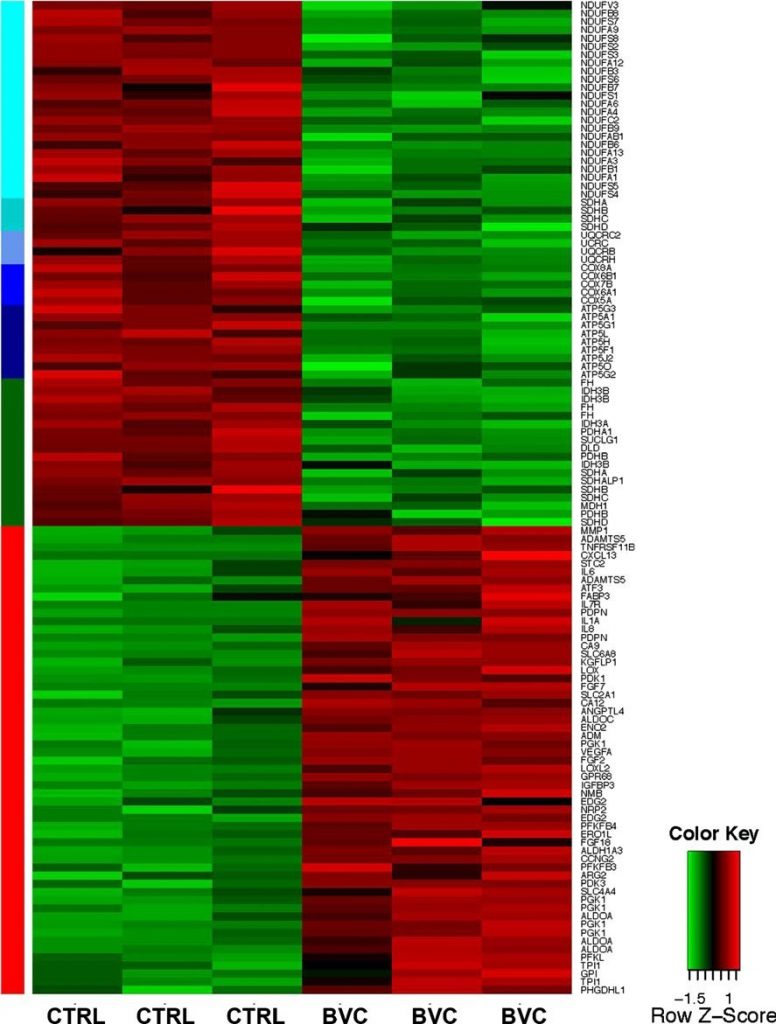
L’analyse des voies KEGG a confirmé que le métabolisme énergétique représentait l’un des changements dominants associés aux tumeurs résistantes au bevacizumab. Ainsi, les gènes glycolytiques étaient les plus abondants parmi les gènes régulés à la hausse, avec les gènes appartenant au métabolisme du fructose et du mannose, à la voie du pentose phosphate, au métabolisme des sucres aminés et des sucres nucléotidiques, et au métabolisme du phosphate d’inositol (tableau supplémentaire S1A). Les valeurs p hypergéométriques et hypergéométriques corrigées étaient inférieures à 0,01 pour toutes ces voies. À l’inverse, parmi les gènes régulés à la baisse, l’OXPHOS était la voie la plus réprimée, avec des valeurs p inférieures à 1,00E-28, le métabolisme du pyruvate et le cycle TCA présentant également une répression très significative (tableau supplémentaire S1B). Une autre signature observée dans les tumeurs résistantes au bevacizumab était une augmentation du stress du réticulum endoplasmique et de la réponse aux protéines non pliées. En particulier, les médiateurs clés de ces réponses, ATF4, 5, 6 et DDIT3/CHOP, étaient tous fortement surexprimés (tableau supplémentaire S2), ce qui est cohérent avec nos observations précédentes selon lesquelles une hypoxie plus sévère se développant au cours de la thérapie antiangiogénique active des voies non HIF [14].
Effets in vitro de l’hypoxie chronique sur l’expression des gènes de l’OXPHOS mitochondriale
Pour déterminer si l’hypoxie représente la cause principale de la régulation négative des gènes de la respiration mitochondriale et du cycle de Krebs, nous avons entrepris une étude in vitro complète des effets de l’oxygène faible sur les cellules U87. Il est intéressant de noter que plusieurs gènes OXPHOS et composants du cycle de Krebs qui ont été trouvés régulés à la baisse dans les matrices ont également réagi par une régulation à la baisse en hypoxie, notamment ATP5A1, ATP5G3, NDUFA9, FH et MDH1 (Fig. 3). MRPL36 et MRPS11, deux gènes considérés comme critiques dans l’assemblage des complexes OXPHOS [15, 16], ont également montré une régulation négative dans l’hypoxie, similaire à l’effet du bevacizumab (Fig. 3). Cependant, la majorité des gènes régulés à la baisse dans les réseaux n’ont pas montré de régulation à la baisse pendant l’exposition hypoxique, ce qui suggère que d’autres facteurs peuvent être principalement responsables [17]. Un réexamen des réseaux a révélé que plusieurs facteurs de transcription qui régulent l’expression des gènes mitochondriaux (y compris l’OXPHOS) étaient régulés à la baisse : NRF1, TFAM et TFB2M [18-20] (tableau supplémentaire S3).

Exploitation de l’écart glycolytique accru : synergie in vivo entre le bevacizumab et le DCA
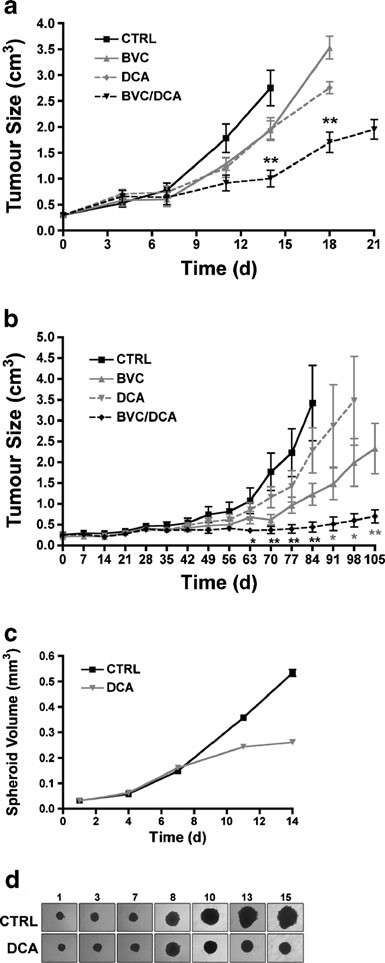
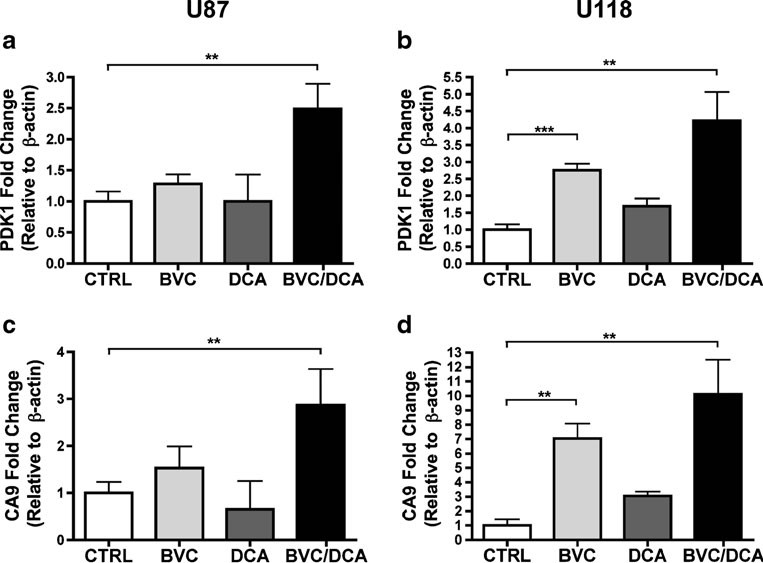
Sur la base de la signature marquée de l’augmentation de la signalisation HIF et de la diminution de l’OXPHOS mitochondriale, nous avons émis l’hypothèse que les tumeurs résistantes au bevacizumab seraient particulièrement sensibles aux réactivateurs mitochondriaux. La principale petite molécule candidate de cette classe est le DCA, qui inhibe l’activité de la PDK, augmentant ainsi le flux de pyruvate dans les mitochondries et favorisant l’oxydation du glucose par rapport à la glycolyse [21, 22]. Compte tenu des effets secondaires bien connus de la thérapie antiangiogénique et du DCA, nous avons choisi, pour l’évaluation de la combinaison de médicaments, une dose submaximale de l’agent antiangiogénique (2,5 mg/kg ; tous les 3 jours par injection intrapéritonéale), une stratégie largement utilisée in vivo [23]. Cette approche devrait également accroître notre capacité à détecter la synergie entre les deux médicaments, tant du point de vue de la réponse tumorale que des effets sur la signalisation HIF. En effet, le traitement par le bevacizumab et le DCA a considérablement bloqué la croissance tumorale par rapport à chaque médicament seul (Fig. 4A et Fig. supplémentaire S1). Afin d’évaluer la généralité de la réponse, nous avons étudié un autre type de cellule de GBM, U118. La réponse dans les greffons à base de U118 était également beaucoup plus robuste avec la combinaison, par rapport aux médicaments individuels (Fig. 4B et Supplementary Fig. S1).
L’efficacité des inhibiteurs glycolytiques pour surmonter la résistance au bevacizumab a été proposée précédemment [24] sans confirmation expérimentale. Le DCA et le 2-DG ont été examinés ensemble dans le cadre de ce concept. Cependant, lorsque le 2-DG a été testé en parallèle avec le DCA, aucun effet additif au bevacizumab n’a été remarqué. Et ce, malgré l’effet transitoire du 2-DG en tant qu’agent unique, administré à la concentration décrite dans la littérature (figure supplémentaire S2). Ainsi, la réactivation mitochondriale et l’inhibition directe de la glycolyse ont des effets différents en association avec le bevacizumab.
Effets in vitro du DCA
Les modèles sphéroïdes constituent un système d’une complexité intermédiaire entre les systèmes de culture bidimensionnels standard et les tumeurs in vivo en raison des gradients d’oxygène et de nutriments. Contrairement aux systèmes monocouches, les sphéroïdes en expansion imitent l’avascularisation accrue des structures in vivo qui se développent en présence de bevacizumab. Les sphéroïdes U87 ont été générés, comme décrit [25], et cultivés pendant 7 jours jusqu’à ce qu’ils atteignent 0,2 mm3. Les sphéroïdes de cette taille sont suffisamment grands pour permettre la diffusion d’un médicament à travers le sphéroïde mais commencent également à former une petite zone centrale d’hypoxie et à afficher des gradients de nutriments, de pH et d’O2. Une forte divergence de la cinétique de croissance entre les groupes traités au DCA et non traités a été constatée après 3 jours de traitement. Cet effet a été persistant et, à partir de 6 jours, le DCA a considérablement compromis l’expansion des sphéroïdes (Fig. 4C, D ).
Effet de l’association bevacizumab et DCA sur les cibles HIF et les marqueurs histologiques de la croissance tumorale
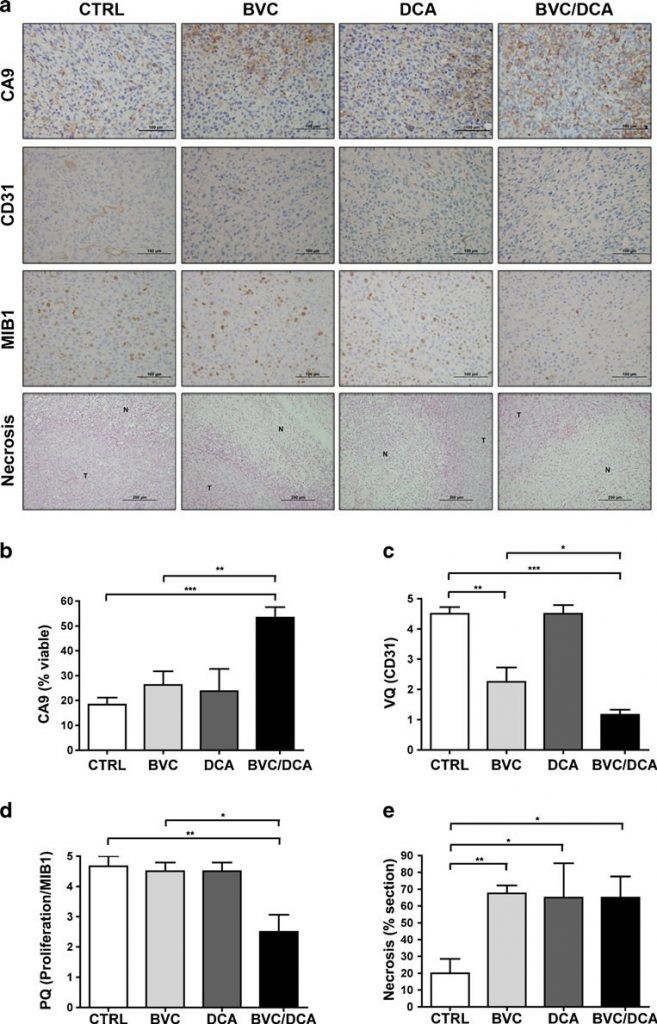
Afin de déterminer si l’effet de l’association sur la croissance tumorale était principalement déterminé par une augmentation de la mort cellulaire ou une diminution de la prolifération, nous avons effectué des analyses histologiques sur ces tumeurs, en utilisant des marqueurs bien établis. La nécrose était plus importante dans les tumeurs traitées que dans les tumeurs non traitées, mais aucune différence significative n’a été observée entre les médicaments seuls ou en association (Fig. 5). Le taux de prolifération, évalué par la quantification de la coloration Ki-67 (MIB-1), était significativement plus faible dans le groupe traité par rapport aux tumeurs non traitées et à celles traitées par le bevacizumab seul, ce qui suggère que l’effet de l’association est principalement cytostatique (Fig. 5). Nous avons ensuite évalué les effets de l’association médicamenteuse sur la signalisation HIF, en utilisant une combinaison d’immunohistochimie et de RT-PCR quantitative (Figs. 5, 6). De manière surprenante, bien que connu pour augmenter la consommation d’oxygène dans divers systèmes expérimentaux, le DCA seul n’était pas suffisant pour augmenter de manière mesurable l’expression des cibles HIF in vivo. Cependant, en association avec le bevacizumab, l’expression de CA9 a augmenté de façon spectaculaire dans les cellules tumorales U87 viables. Dans la tumeur U118, par contre, le bevacizumab submaximal seul a entraîné une augmentation spectaculaire de toutes les cibles HIF testées, sans autre augmentation mesurable dans les tumeurs traitées en association (figure 6 et figure supplémentaire S3). Toutefois, la principale mise en garde est que les tumeurs qui ont survécu en présence de la combinaison étaient nettement plus petites et pratiquement stationnaires, des facteurs qui devraient atténuer l’ampleur de l’hypoxie.
Discussion
L’objectif principal de notre étude était de mieux comprendre l’adaptation des tumeurs au bevacizumab, en identifiant les voies associées à la résistance, avec un accent particulier sur les réponses métaboliques. Nous nous sommes concentrés sur un modèle hétérotopique plutôt qu’orthotopique, principalement pour garantir la faisabilité du suivi de la croissance et de l’échappement néoplasique. De plus, comme les tumeurs sous-cutanées atteignent des volumes beaucoup plus importants que leurs homologues orthotopiques, elles sont sans doute plus pertinentes pour modéliser les malignités avancées et hypoxiques.
Dans un modèle orthotopique de GBM récemment publié, une hypoxie accrue après l’administration de bévacizumab a été signalée [24, 26]; cependant, l’étude des différences de croissance à long terme est moins réalisable dans ces systèmes. Malgré le statut « classique » du métabolisme tumoral dans la recherche sur le cancer et son récent renouveau, aucun médicament agissant principalement à ce niveau n’a été approuvé pour une utilisation clinique de routine [27]. Le DCA, une petite molécule qui traverse la barrière hémato-encéphalique [22], a donné des résultats prometteurs lors d’un essai clinique sur le GBM en association avec la chirurgie, le témozolomide et la radiothérapie [21], et plusieurs autres essais sont en cours http://clinicaltrials.gov/ct2/results?term=+Dichloroacetate). Cependant, l’effet du DCA en tant qu’agent unique est au mieux transitoire, et nos modèles tumoraux reflètent certainement cette limitation. Une étude récente a évoqué la possibilité que le traitement au bevacizumab sensibilise les tumeurs à la fois au 2-DG et au DCA [24], mais ces prédictions n’ont été confirmées que pour le DCA dans nos mains. L’absence d’un effet additif ou synergique mesurable du 2-DG n’était probablement pas due à une inactivité biologique ou à un dosage inadéquat, car il était transitoirement efficace en tant qu’agent unique. Des questions demeurent quant au mécanisme d’inhibition tumorale médiée par le DCA en présence de bevacizumab. Bien que le DCA ait été signalé comme bloquant l’angiogenèse par lui-même [21], la mesure de la densité moyenne des vaisseaux n’a pas confirmé cette hypothèse dans notre système.
Il a été démontré que le DCA présente des effets cytotoxiques accrus sous hypoxie dans diverses lignées cellulaires [28]. Le paradigme actuel est que le DCA accélère la consommation d’oxygène par la réactivation des mitochondries et diminue encore la tension locale en oxygène [29], ce qui pose un défi supplémentaire aux cellules tumorales pour survivre et/ou proliférer. En outre, il a été démontré que dans les cellules « câblées » pour utiliser sélectivement la glycolyse pour la production d’ATP en raison de mutations de l’ADN mitochondrial, l’OXPHOS forcée induite par le DCA avait un effet toxique. Le DCA présente également une cytotoxicité synergique in vitro en combinaison avec le cisplatine et le topotécan, deux agents antinéoplasiques connus pour endommager l’ADN mitochondrial [30]. On pourrait supposer que la régulation négative des gènes mitochondriaux dans les tumeurs résistantes au bevacizumab représente une forme de dysfonctionnement mitochondrial qui sensibilise aux effets du DCA.
Il est intéressant de noter que l’effet du DCA, en tant qu’agent unique, sur les cibles HIF dans les xénogreffes était au mieux subtil, malgré son effet positif bien connu sur la consommation d’oxygène. En revanche, associé au bevacizumab, le DCA a entraîné une augmentation de l’expression de la plupart des cibles HIF testées dans les tumeurs survivantes par rapport au bevacizumab seul. Une explication possible de ces résultats est que l’OXPHOS forcée induite par le DCA en présence de très faibles niveaux d’oxygène peut conduire à une production accrue d’espèces réactives de l’oxygène, qui, à son tour, peut contribuer à une induction supplémentaire de HIF [31]. En outre, ou alternativement, le DCA en présence de bevacizumab peut entraîner une nouvelle diminution de la concentration locale d’oxygène à des niveaux où l’induction des cibles HIF devient plus évidente par le test RT-PCR quantitatif. Outre l’augmentation de l’expression de CA9, une expression accrue des cibles HIF PDK1, 3 et GLUT1 a été observée dans les tumeurs survivant en présence de la combinaison de médicaments dans les cellules U87 ou U118. Cela peut refléter un changement métabolique de « dernier recours » plus spectaculaire, essentiel à la survie des cellules tumorales. En particulier, l’expression accrue de PDK1/3 peut faire partie d’une « tentative finale » des cellules tumorales pour contrecarrer en partie l’effet du DCA et désactiver l’OXPHOS mitochondriale. De tels changements métaboliques dans les tumeurs qui survivent en présence de bevacizumab et de DCA peuvent également fournir des indices importants sur la façon d’augmenter l’efficacité de cette combinaison. Par exemple, on peut supposer que les tumeurs résistantes peuvent présenter au moins une certaine sensibilité à une nouvelle augmentation de la concentration de DCA, bien que la toxicité de ce composé puisse devenir un facteur limitant. La régulation à la hausse du CA9 spécifique à la tumeur peut également jouer un rôle important dans la survie des tumeurs traitées par la combinaison, car elle accélère l’élimination de l’excès deCO2 généré par la réactivation du cycle de Krebs [25]. Par conséquent, les inhibiteurs de CA9 qui ont récemment montré des effets anticancéreux prometteurs [32, 33] peuvent être considérés comme des candidats réalistes pour un troisième composant d’une stratégie de combinaison.
En conclusion, la dissection moléculaire de l’adaptation des tumeurs aux agents anti-VEGF peut offrir des indices précieux pour construire des combinaisons plus efficaces qui incluent le ciblage du métabolisme du cancer.
Contributions de l’auteur
Conception et design : M.I. et A.L.H. Acquisition des données : K.K., S.W., H.E.G., H.T., J.L., J.S., A.L.S., R.L., D.S., C.M.D. et M.H. Analyse et interprétation des données : M.I. et A.L.H. Rédaction, examen et/ou révision du manuscrit : M.I. et A.L.H. Soutien administratif, technique ou matériel : F.B. Supervision de l’étude : M.I. et A.L.H.
Source de financement
Ce travail a été soutenu par des subventions de Cancer Research United Kingdom [S.W., A.L.H., H.T., R.L., J.L.], METOXIA p-Medicine European Union Framework 7 [D.S., F.B.], Rhodes Scholar [H.G.], fonds de démarrage de l’Indiana University Cancer Center, et American Cancer Society [M.I., C.M.D., K.K.].
Conflit d’intérêt
Aucun conflit d’intérêt potentiel à déclarer.
RÉFÉRENCES
1 Cao Y, Arbiser J, D’Amato RJ, D’Amore PA, Ingber DE, Kerbel R, Klagsbrun M, Lim S, Moses MA, Zetter B et al (2011) Forty-year journey of angiogenesis translational research. Sci Transl Med 3:114rv3
2 Wick W, Wick A, Weiler M, Weller M (2011) Patterns of progression in malignant glioma following anti-VEGF therapy : perceptions and evidence. Curr Neurol Neurosci Rep 11:305-312
3 Raizer JJ, Grimm S, Chamberlain MC, Nicholas MK, Chandler JP, Muro K, Dubner S, Rademaker AW, Renfrow J, Bredel M (2010) A phase 2 trial of single-agent bevacizumab given in an every-3-week schedule for patients with recurrent high-grade gliomas. Cancer 116:5297-5305
4 Bergers G, Hanahan D (2008) Modes de résistance à la thérapie anti-angiogénique. Nat Rev Cancer 8:592-603
5 Friedman HS, Prados MD, Wen PY, Mikkelsen T, Schiff D, Abrey LE, Yung WK, Paleologos N, Nicholas MK, Jensen R et al (2009) Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol 27:4733-4740
6 Winter SC, Shah KA, Campo L, Turley H, Leek R, Corbridge RJ, Cox GJ, Harris AL (2005) Relation entre l’expression de l’érythropoïétine et du récepteur de l’érythropoïétine, l’hypoxie et l’anémie dans le carcinome épidermoïde de la tête et du cou. Clin Cancer Res Off J Am Assoc Cancer Res 11:7614-7620
7 Davies S, Dai D, Pickett G, Thiel KW, Korovkina VP, Leslie KK (2011) Effects of bevacizumab in mouse model of endometrial cancer : defining the molecular basis for resistance. Oncol Rep 25:855-862
8 Rapisarda A, Hollingshead M, Uranchimeg B, Bonomi CA, Borgel SD, Carter JP, Gehrs B, Raffeld M, Kinders RJ, Parchment R et al (2009) Increased antitumor activity of bevacizumab in combination with hypoxia inducible factor-1 inhibition. Mol Cancer Ther 8:1867-1877
9 Harris RA, Bowker-Kinley MM, Huang B, Wu P (2002) Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul 42:249-259
10 Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL (2008) PDK-1 régule la production de lactate en hypoxie et est associé à un mauvais pronostic dans le cancer squameux de la tête et du cou. Br J Cancer 98:1975-1984
11 Kim JW, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase : a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177-185
12 Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ (2008) Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem 283:28106-28114
13 Frezza C, Pollard PJ, Gottlieb E (2011) Inborn and acquired metabolic defects in cancer. J Mol Med (Berl) 89:213-220
14 Rzymski T, Milani M, Pike L, Buffa F, Mellor HR, Winchester L, Pires I, Hammond E, Ragoussis I, Harris AL (2010) Regulation of autophagy by ATF4 in response to severe hypoxia. Oncogene 29:4424-4435
15 Prestele M, Vogel F, Reichert AS, Herrmann JM, Ott M (2009) Mrpl36 est important pour la génération de protéines compétentes pour l’assemblage pendant la traduction mitochondriale. Mol Biol Cell 20:2615-2625
16 Emdadul Haque M, Grasso D, Miller C, Spremulli LL, Saada A (2008) The effect of mutated mitochondrial ribosomal proteins S16 and S22 on the assembly of the small and large ribosomal subunits in human mitochondria. Mitochondrion 8:254-261
17 Tang X, Lucas JE, Chen JL, Lamonte G, Wu J, Wang MC, Koumenis C, Chi JT (2012) Functional interaction between responses to lactic acidosis and hypoxia regulates genomic transcriptional outputs. Cancer Res 72:491-502
18 Scarpulla RC (2002) Activateurs et coactivateurs nucléaires dans la biogenèse mitochondriale des mammifères. Biochim Biophys Acta 1576:1-14
19 Falkenberg M, Gaspari M, Rantanen A, Trifunovic A, Larsson NG, Gustafsson CM (2002) Mitochondrial transcription factors B1 and B2 activate transcription of human mtDNA. Nat Genet 31:289-294
20 Larsson NG, Barsh GS, Clayton DA (1997) Structure and chromosomal localization of the mouse mitochondrial transcription factor A gene (Tfam). Mamm Genome 8:139-140
21 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:31ra34
22 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99:989-994
23 Pechman KR, Donohoe DL, Bedekar DP, Kurpad SN, Hoffmann RG, Schmainda KM (2011) Characterization of bevacizumab dose response relationship in U87 brain tumors using magnetic resonance imaging measures of enhancing tumor volume and relative cerebral blood volume. J Neurooncol 105:233-239
24 Keunen O, Johansson M, Oudin A, Sanzey M, Rahim SA, Fack F, Thorsen F, Taxt T, Bartos M, Jirik R et al (2011) Anti-VEGF treatment reduces blood supply and increases tumor cell invasion in glioblastoma. Proc Natl Acad Sci U S A 108:3749-3754
25 Swietach P, Patiar S, Supuran CT, Harris AL, Vaughan-Jones RD (2009) The role of carbonic anhydrase 9 in regulating extracellular and intracellular ph in three-dimensional tumor cell growths. J Biol Chem 284:20299-20310
26 de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, Conrad CA (2010) Tumor invasion after treatment of glioblastoma with bevacizumab : radiographic and pathologic correlation in humans and mice. Neuro Oncol 12:233-242
27 Kaelin WG Jr, Thompson CB (2010) Q&A : cancer : clues from cell metabolism. Nature 465:562-564
28 Anderson KM, Jajeh J, Guinan P, Rubenstein M (2009) In vitro effects of dichloroacetate and CO2 on hypoxic HeLa cells. Anticancer Res 29:4579-4588
29 Chen Y, Cairns R, Papandreou I, Koong A, Denko NC (2009) Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS One 4:e7033
30 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL (2010) Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 127:2510-2519
31 Park JH, Kim TY, Jong HS, Kim TY, Chun YS, Park JW, Lee CT, Jung HC, Kim NK, Bang YJ (2003) Gastric epithelial reactive oxygen species prevent normoxic degradation of hypoxia-inducible factor-1alpha in gastric cancer cells. Clin Cancer Res Off J Am Assoc Cancer Res 9:433-440
32 Dubois L, Peeters S, Lieuwes NG, Geusens N, Thiry A, Wigfield S, Carta F, McIntyre A, Scozzafava A, Dogne JM et al (2011) Specific inhibition of carbonic anhydrase IX activity enhance the in vivo therapeutic effect of tumor irradiation. Radiother Oncol 99:424-431
33 v Morris JC, Chiche J, Grellier C, Lopez M, Bornaghi LF, Maresca A, Supuran CT, Pouyssegur J, Poulsen SA (2011) Targeting hypoxic tumor cell viability with carbohydrate-based carbonic anhydrase IX and XII inhibitors. J Med Chem 54:6905-6918