BM Madhok*,1, S Yeluri1, SL Perry1, TA Hughes2 e DG Jayne1
1 Sezione di anestesia e chirurgia traslazionale, Università di Leeds, Level 7 Clinical Sciences Building, St. James’s University Hospital, Leeds, UK
2 Leeds Institute of Molecular Medicine, Università di Leeds, St. James’s University Hospital, Leeds, UK
Corrispondenza: Dr BM Madhok; E-mail: [email protected]
Revisionato: 23 marzo 2010
Accettato: 26 aprile 2010
Pubblicato: 18 maggio 2010
Abstract
Background
Le cellule tumorali sono altamente dipendenti dalla glicolisi. Il nostro obiettivo era quello di determinare se il passaggio del metabolismo dalla glicolisi alla respirazione mitocondriale riducesse la crescita in modo preferenziale nelle cellule tumorali del colon-retto rispetto alle cellule normali e di esaminare i meccanismi sottostanti.
Metodi
Linee cellulari rappresentative del cancro colorettale e non cancerose sono state trattate con dicloroacetato (DCA), un inibitore della piruvato deidrogenasi chinasi.
Risultati
Il dicloroacetato (20 mM) non ha ridotto la crescita delle cellule non cancerose, ma ha causato una riduzione significativa della proliferazione delle cellule cancerose (P=0,009), associata ad apoptosi e arresto del ciclo cellulare in fase G2. L’effetto apoptotico maggiore è stato evidente nelle cellule metastatiche LoVo, in cui il DCA ha indotto un aumento fino a dieci volte del numero di cellule apoptotiche dopo 48 ore. L’arresto G2 più evidente è stato riscontrato nelle cellule HT29 ben differenziate, in cui il DCA ha causato un aumento di otto volte delle cellule in fase G2 dopo 48 ore. Il dicloroacetato ha ridotto i livelli di lattato nei mezzi di crescita e ha indotto la de-fosforilazione della subunità E1α del complesso piruvato deidrogenasi in tutte le linee cellulari, ma il potenziale di membrana mitocondriale intrinseco è stato ridotto solo nelle cellule tumorali (P=0,04).
Conclusioni
L’inibizione della piruvato deidrogenasi chinasi attenua la glicolisi e facilita la fosforilazione ossidativa mitocondriale, determinando una riduzione della crescita delle cellule tumorali del colon-retto ma non di quelle non tumorali.
Parole chiave: dicloroacetato, cancro colorettale, piruvato deidrogenasi, piruvato deidrogenasi chinasi
British Journal of Cancer (2010) 102, 1746 – 1752, www.bjcancer.com
doi: 10.1038/sj.bjc.6605701
© 2010 Cancer Research UK
INTRODUZIONE
Il cancro del colon-retto è il terzo tumore più comune al mondo e la quarta causa di morte per cancro (Shike et al, 1990). Nel 2007 il cancro del colon-retto ha causato 17,1 decessi ogni 100.000 persone nel Regno Unito (UK Bowel Cancer Statistics, 2009). Nonostante i recenti progressi, la prognosi dei pazienti con tumore del colon-retto avanzato e metastatico rimane scarsa. Il bersaglio del metabolismo tumorale per la terapia del cancro è un settore in rapido sviluppo (Pan e Mak, 2007). Le prime osservazioni sulle differenze metaboliche tra cellule tumorali e normali sono state fatte da Otto Warburg, che ha dimostrato che le cellule tumorali sono intrinsecamente dipendenti dalla glicolisi per la produzione di energia chimica (Warburg, 1956). Oggi è sempre più evidente che l’aumento della glicolisi deriva dall’influenza di molteplici vie molecolari, tra cui le risposte adattative al microambiente tumorale ipossico, la segnalazione oncogena e la disfunzione mitocondriale (Gatenby e Gillies, 2004; Gillies e Gatenby, 2007; Wu et al, 2007). Il fenotipo glicolitico offre vantaggi di crescita alle cellule tumorali, resistendo all’apoptosi e facilitando la diffusione del tumore e le metastasi (Yeluri et al, 2009).
Un regolatore chiave del metabolismo cellulare è la piruvato deidrogenasi (PDH). La piruvato deidrogenasi converte il piruvato, prodotto dalla glicolisi, in acetil-CoA, che viene ossidato nel ciclo degli acidi tricarbossilici all’interno dei mitocondri. L’attività della piruvato deidrogenasi è strettamente regolata dalla fosforilazione inibitoria della piruvato deidrogenasi chinasi (PDK). La fosforilazione avviene sulla subunità E1α della PDH (PDHE1α) in tre siti: Ser232, Ser293 e Ser300 (Rardin et al, 2009). Il dicloroacetato (DCA) è un inibitore di tutti e quattro gli isoenzimi della PDK(1-4) (Stacpoole, 1989) e recentemente ha dimostrato di ridurre la crescita di linee cellulari di cancro al polmone, all’endometrio e al seno (Bonnet et al, 2007; Wong et al, 2008; Sun et al, 2009). È stato riportato che riduce la crescita di queste cellule tumorali principalmente riducendo la fosforilazione inibitoria della PDH, promuovendo così la fosforilazione ossidativa mitocondriale e inducendo l’apoptosi attraverso vie mitocondriali, NFAT-Kv 1.5 e p53 upregulated modulator of apoptosis (PUMA).
È stato riscontrato che le cellule del cancro del colon-retto subiscono un aumento della glicolisi (Bi et al, 2006) e che il microambiente tumorale è ipossico e acidotico, soprattutto a causa di un apporto sanguigno poco sviluppato (Dewhirst et al, 1989; Milosevic et al, 2004). Abbiamo precedentemente dimostrato che ciò è particolarmente vero per il fenotipo più aggressivo (Thorn et al, 2009) e che l’espressione di importanti marcatori di ipossia è aumentata nel tumore del colon-retto, soprattutto in corrispondenza del margine invasivo (Rajaganeshan et al, 2008, 2009). Lo scopo di questo studio è stato quello di analizzare gli effetti del DCA sulla crescita delle cellule di cancro del colon-retto, nel tentativo di esaminare l’inibizione della PDK come nuova strategia terapeutica contro il cancro del colon-retto.
Materiali e metodi
Colture cellulari
Tutte le linee cellulari sono state acquistate dall’American Type Culture Collection (Manassas, VA, USA) o dalla European Collection of Cell Cultures (Salisbury, Wiltshire, UK): HB2 (cellule epiteliali mammarie di origine non tumorale), 293 (cellule epiteliali da rene embrionale umano), HT29 (adenocarcinoma colorettale primario ben differenziato), SW480 (adenocarcinoma colorettale primario scarsamente differenziato) e LoVo (linfonodo sovraclaveare sinistro metastatico da adenocarcinoma colorettale). le cellule 293 e HB2 sono state mantenute in terreno DMEM, le HT29 e SW480 in terreno RPMI 1640 e le LoVo in terreno F12 (tutti prodotti da Invitrogen, Carlsbad, CA, USA), integrati con il 10% di siero fetale di vitello, in un incubatore umidificato a 37°C, 5%CO2. Per gli esperimenti in condizioni di ipossia, abbiamo incubato le cellule in un incubatore ipossico umidificato (1% O2, 5%CO2, 94% N2, 37°C). Il dicloroacetato di sodio (Specials Lab, Prudhoe, UK) è stato donato dal Dipartimento di Farmacia del St. James’s University Hospital, Leeds, UK.
Saggi MTT
Le cellule (1 ×104) per pozzetto sono state seminate in piastre di coltura tissutale a 96 pozzetti. Dopo un’incubazione di una notte, abbiamo sostituito i terreni con terreni freschi contenenti dosi crescenti di DCA (0, 10, 15, 20, 30, 50 e 100 mM). Dopo 24 e 48 ore di incubazione, abbiamo eseguito il saggio MTT sostituendo i terreni con 50μldi soluzione di MTT 1 mg ml-1 e le piastre sono state incubate al buio per 3 ore. La soluzione di MTT è stata quindi rimossa e i precipitati di formazan blu scuro sono stati disciolti in 100μldi propan-1-olo. La densità ottica è stata misurata con un lettore di micropiastre (Opsys MR; Dynex Technologies Ltd, Worthing, West Sussex, UK) a 570 nm.
Saggi di Annexina V e 7-AAD
Le cellule sono state seminate in fiasche di coltura tissutale da 25 cm2 e incubate per una notte in condizioni standard. Il terreno è stato sostituito con terreno fresco contenente una serie di dosi di DCA (0, 10, 20 e 50 mM). L’analisi citometrica a flusso è stata eseguita dopo 24 e 48 ore di incubazione. Le cellule sono state lavate due volte con PBS freddo e risospese in 1 × tampone di legame (BD Bioscience, Franklin Lakes, NJ, USA) a 5 ×106 cellule per ml. 100μldi soluzione (5 ×105 cellule) sono stati trasferiti in provette da 5 ml. Le cellule sono state colorate con 5μl diannexina V-FITC e 10μl di7-AAD (BD Bioscience), agitate delicatamente e incubate a temperatura ambiente per 15 minuti al buio. Successivamente, sono stati aggiunti 400μldi tampone di legame 1 × a ciascuna provetta e sono stati analizzati entro un’ora sul citometro a flusso LSR II (BD Bioscience).
Saggi di ioduro di propidio
Le cellule sono state propagate come indicato per il saggio di apoptosi. È stato utilizzato dicloroacetato (50 mM) e confrontato con il controllo veicolo. Dopo la raccolta, abbiamo risospeso le cellule in 350μldi PBS a una concentrazione di 0,5-1,0 ×106 cellule per ml. alla sospensione cellulare sono stati aggiunti 100μldi 0,25 mg ml-1 di ioduro di propidio (PI)/5% di Triton (Sigma, St Louis, MO, USA). sono stati quindi aggiunti 50μldi 1 mg ml-1 di ribonucleasi A (Sigma). Le provette di campione sono state accuratamente agitate e incubate per 10 minuti al buio a temperatura ambiente. La citometria a flusso è stata eseguita su citometro a flusso LSR II (BD Bioscience) e i dati sono stati analizzati con il software FlowJo (FlowJo, Ashland, OR, USA).
Misurazioni del lattato
Le misurazionidel lattatonei mezzi di crescita sono state eseguite dal dipartimento di patologia chimica del General Infirmary, Leeds Teaching Hospitals NHS Trust. Le cellule sono state incubate in fiasche da 25 cm2 per una notte in normossia. Il giorno successivo il terreno è stato sostituito con una serie di dosi di DCA (0, 10, 20 e 50 mM). Dopo 48 ore di incubazione, abbiamo raccolto 2 ml di terreno in provette al fluoro e trasferito immediatamente al laboratorio di patologia chimica. Le provette sono state mantenute in ghiaccio durante il trasferimento. I livelli di lattato sono stati misurati con un analizzatore automatico (Advia 1200 Chemistry system; Siemens Healthcare Diagnostics, Camberley, Surrey, UK).
Saggi TMRM
Le cellule sono state trattate con DCA come descritto per il saggio di apoptosi. Dopo 24 e 48 ore di incubazione, abbiamo lavato le cellule in PBS e sospeso 1 ×106 cellule per ml in soluzione salina tamponata di Hank con 50 nM di tetrametil rodamina metil estere (TMRM) (Invitrogen). 100μldella sospensione cellulare (1 ×105 cellule per pozzetto) sono stati trasferiti in piastre opache a 96 pozzetti, incubati per 30 minuti e la fluorescenza è stata misurata a 530/620 nm a 37°C utilizzando un lettore di piastre (Mithras LB 40; Berthold Technologies, Bad, Wildbad, Germania).
Western blotting
Le cellule sono state trattate con DCA come descritto sopra. Dopo 8 ore di trattamento, abbiamo estratto le proteine dalle cellule in tampone Laemmli (2% SDS, 10% glicerolo, 0,7% 2-mercaptoetanolo, 0,05% blu di bromofenolo e 0,5 M Tris-HCl). I lisati sono stati risolti mediante elettroforesi su gel NuPAGE Novex 12% Bis-Tris (Invitrogen) in tampone di corsa MOPS-SDS (Invitrogen). Le proteine sono state trasferite su una membrana di fluoruro di polivinile (GE Healthcare, Chalford St Giles, Bucks, UK). La membrana è stata bloccata per 1 ora a temperatura ambiente con latte scremato al 5% in TBS-T (Tris-buffered saline with 0.1% Tween). La membrana è stata quindi testata con anticorpi primari in latte scremato all’1% in TBS-T per 90 minuti, lavata in TBS-T e quindi testata con l’anticorpo secondario appropriato coniugato con perossidasi di rafano (HRP) per 60 minuti. Anticorpi primari policlonali di coniglio fosfodetect anti-PDH-E1α (pSer293), 1 :500 (AP1062; EMD Chemicals, Darmstadt, Germania) e anticorpi monoclonali di topo anti-PDHE1α, 1 :500 (459400; Invitrogen). Anticorpi secondari anti-rabbit o anti-mouse coniugati HRP, 1 :1000 (Dako, Glostrup, Danimarca). Le proteine sono state visualizzate con il substrato chemiluminescente Supersignal West Pico o Femto (Pierce Biotechnology, Rockford, IL, USA) e il sistema Chemidoc XRS (Bio-Rad, Hercules, CA, USA). la β-actina è stata utilizzata come controllo di carico.
Analisi statistiche
I dati della citometria a flusso sono stati acquisiti utilizzando software specifici, BD FACSDiva 6.0 e FlowJo. Le analisi statistiche sono state eseguite utilizzando SPSS per Windows (SPSS versione 15.0, Chicago, IL, USA). Le differenze tra i gruppi trattati con DCA e quelli di controllo con veicolo sono state valutate utilizzando il test U di Mann-Whitney e gli intervalli di confidenza al 95% della differenza tra le medie dei due gruppi. Un valore P inferiore a 0,05 è stato considerato statisticamente significativo. I dati sono rappresentati come media di almeno tre esperimenti indipendenti e le barre di errore rappresentano la deviazione standard della media.
Risultati
IlDCA riduce la proliferazione delle cellule tumorali e l’effetto è simile in normossia e ipossia
In primo luogo, abbiamo voluto determinare se il trattamento con DCA inibisse la proliferazione cellulare e se vi fosse una risposta differenziale nelle cellule tumorali e non tumorali in condizioni di normossia e ipossia. Per quanto riguarda l’ipossia, la nostra ipotesi era che l’influenza del DCA sarebbe stata particolarmente potente con livelli di ossigeno insufficienti a sostenere un’ulteriore fosforilazione ossidativa. Tutte le linee cellulari (HB2, 293, HT29, SW480 e LoVo) sono state trattate con una serie di dosi di DCA per 24-48 ore in condizioni di normossia e ipossia. Il numero relativo di cellule è stato valutato mediante saggi MTT.
Il trattamento con dosi crescenti di DCA ha ridotto la proliferazione cellulare in modo dose-dipendente (Figura 1A-D). Contrariamente alle nostre aspettative, i profili di riduzione della crescita cellulare erano simili in ipossia e normoxia. A 24 e 48 ore, fino a 20 mM di DCA non ha influenzato la crescita delle colture di cellule non cancerose, HB2 e 293. Tuttavia, 20 mM di DCA hanno ridotto significativamente la crescita delle colture di tutte e tre le linee cellulari di cancro colorettale (P⩽0,009). L’effetto del DCA è stato maggiore sulle cellule SW480 scarsamente differenziate e sulle cellule LoVo metastatiche rispetto alle cellule HT29 ben differenziate. La crescita delle colture di cellule LoVo trattate con 20 mM DCA è stata ridotta fino al 40% rispetto alle cellule trattate con il controllo veicolo. Poiché la differenza nella riduzione della crescita delle colture trattate con DCA in condizioni di ipossia e normossia era relativamente bassa, sono stati eseguiti ulteriori esperimenti solo in normossia.
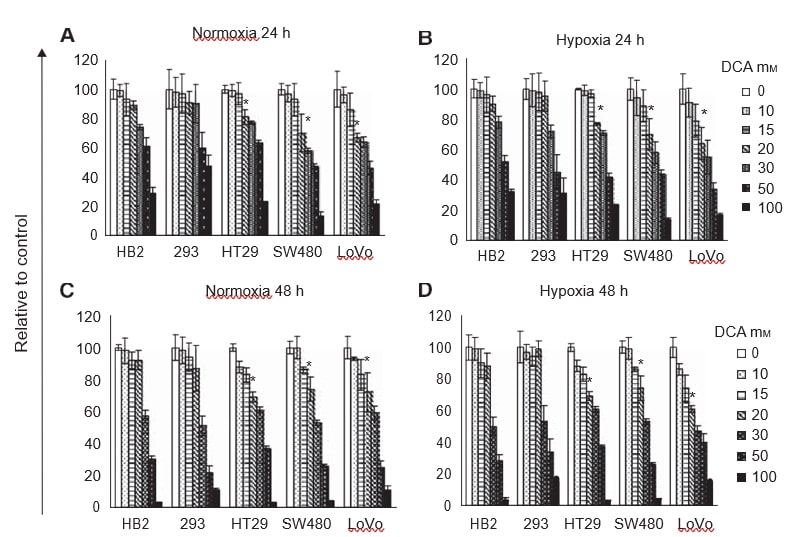
IlDCA promuove l’apoptosi nelle cellule cancerose risparmiando quelle non cancerose
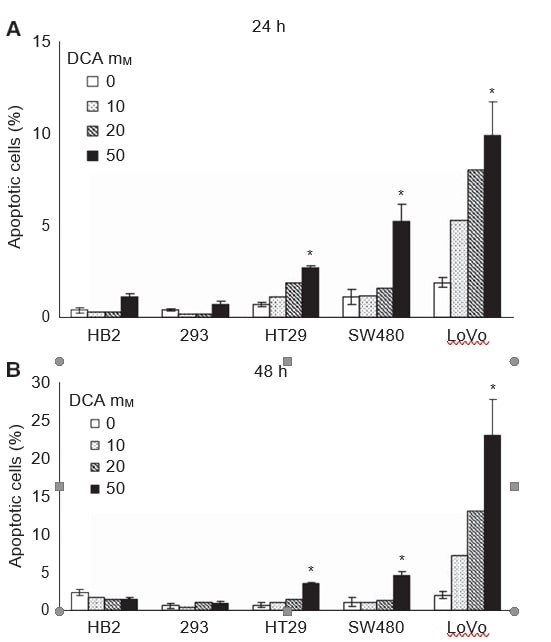
Successivamente, abbiamo voluto indagare se la ridotta crescita delle colture in seguito al trattamento con DCA fosse associata all’induzione dell’apoptosi. Le cellule sono state trattate con una serie di dosi di DCA (0, 10, 20 e 50 mM) per 24 e 48 ore e la percentuale di cellule sottoposte ad apoptosi è stata valutata rilevando la fosfatidilserina di membrana con l’annexina V-FITC. Le cellule sono state colorate con annexina V-FITC e colorante vitale 7-AAD e analizzate con la citometria a flusso. Si è osservata un’induzione dose-dipendente dell’apoptosi nelle linee cellulari tumorali dopo 24 e 48 ore di trattamento, mentre l’apoptosi indotta nelle cellule non cancerose è stata minima (Figura 2A e B). L’effetto maggiore è stato osservato nelle cellule metastatiche LoVo; il DCA 50 mM ha causato un aumento di dieci volte della percentuale di cellule apoptotiche dopo 48 ore, mentre nelle cellule HT29 e SW480 l’aumento è stato rispettivamente di sette e cinque volte. L’aumento della percentuale media di cellule apoptotiche totali con 50 mM DCA è stato: 2,8 (95% CI: 2-3) nelle cellule HT29, 3,5 (95% CI: 2-5) nelle cellule SW480 e 21 (95% CI: 8-34) nelle cellule LoVo. L’apoptosi indotta nelle cellule 293 è stata minima anche con 50 mM di DCA, 0,2 (95% CI: da -0,2 a 0,6). Nelle cellule HB2, la percentuale di cellule apoptotiche è diminuita in modo non significativo con il trattamento con 50 mM DCA, -0,9 (95% CI: da -2,2 a 0,4).
IlDCA induce l’arresto della fase G2 in cellule di cancro del colon-retto, ma non ha alcun effetto sul profilo del ciclo cellulare di cellule 293 non cancerose
Abbiamo inoltre voluto esaminare se la riduzione della crescita delle colture in seguito al trattamento con DCA fosse associata all’induzione dell’arresto della crescita. Le cellule sono state trattate con 50 mM di DCA per 24 o 48 ore e i profili del ciclo cellulare sono stati analizzati mediante valutazione citometrica a flusso del contenuto di DNA dopo colorazione con PI. Il trattamento con dicloroacetato ha causato cambiamenti nei profili del ciclo cellulare di tutte le cellule tumorali, ma non ha influenzato le cellule non tumorali. I cambiamenti nel profilo del ciclo cellulare erano rilevabili dopo 24 ore di trattamento e persistevano a 48 ore (Figura 3A e B).
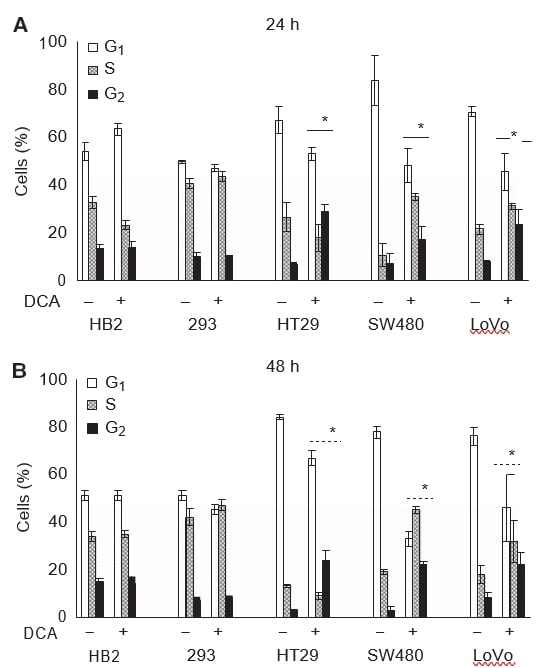
Dopo 48 ore di trattamento con 50 mM DCA, si è registrato un aumento di otto volte delle cellule in fase G2 nelle cellule HT29 e SW480 e di tre volte nelle cellule LoVo. L’aumento della percentuale media di tutte le cellule tumorali in fase G2 è stato: 21 (95% CI: 13-30) per le cellule HT29, 19 (95% CI: 13-24) per le cellule SW480 e 14 (95% CI: 10-21) per le cellule LoVo; mentre non vi è stata alcuna differenza nelle cellule 293, 1 (95% CI: da -4 a 7), e nelle cellule HB2, -0,3 (95% CI: da -9 a 9). In tutte le linee cellulari tumorali si è registrata una corrispondente diminuzione delle cellule in fase G0/G1. È interessante notare che nelle cellule HT29 si è verificata una piccola diminuzione, mentre nelle cellule SW480 e LoVo si è registrato un aumento significativo della proporzione di cellule considerate in fase S (vedere la sezione Discussione). Il profilo del ciclo cellulare delle cellule 293 e HB2 è cambiato in minima parte con il trattamento con DCA.
Il DCA riduce i livelli di lattato extracellulare nei terreni di crescita
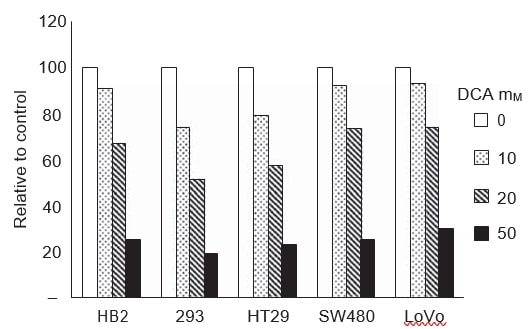
Per stabilire se i cambiamenti nella crescita e nell’apoptosi indotti dal DCA fossero correlati alla riduzione della glicolisi, abbiamo misurato i livelli di lattato nei mezzi di crescita. L’acido lattico è il prodotto finale della glicolisi. Se il DCA inducesse la fosforilazione ossidativa mitocondriale, il piruvato verrebbe decarbossilato ad acetil-CoA e non ridotto a lattato, per cui i livelli di lattato nei terreni di crescita diminuirebbero. I livelli di lattato nei mezzi di crescita di tutte le linee cellulari sono stati misurati dopo 48 ore di trattamento con una serie di dosi di DCA (Figura 4). I livelli di lattato sono stati determinati con un auto-analizzatore utilizzato di routine per la misurazione biochimica dei livelli di lattato; i saggi si basano su una reazione colorimetrica catalizzata dalla lattato ossidasi. Il trattamento con DCA ha ridotto i livelli di lattato extracellulare nei terreni di coltura in modo dose-dipendente in tutte le linee cellulari cancerose e non cancerose.
IlDCA depolarizza la membrana mitocondriale intrinseca nelle cellule di cancro del colon-retto ma non in quelle non cancerose
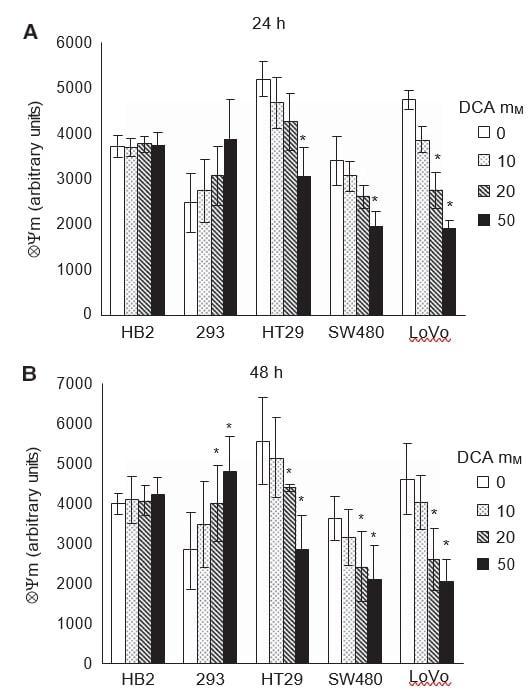
Per verificare se l’induzione dell’apoptosi nelle cellule cancerose in seguito al trattamento con DCA fosse associata alla promozione della fosforilazione ossidativa mitocondriale, abbiamo misurato il potenziale di membrana mitocondriale intrinseco (ΔΨm). L’aumento della respirazione mitocondriale riattiverebbe la catena di trasporto degli elettroni e ridurrebbe il ΔΨm iperpolarizzato nelle cellule tumorali. Le cellule sono state trattate con dosi di DCA per 24 e 48 ore e colorate con il colorante TMRM, che consente di misurare in fluorescenza il ΔΨm.
Come negli esperimenti precedenti, l’effetto del DCA era evidente dopo 24 ore di trattamento e persisteva a 48 ore (Figura 5A e B). Il trattamento con dicloroacetato ha ridotto il ΔΨm iperpolarizzato in tutte le cellule tumorali in modo dose-dipendente. Il dicloroacetato non ha avuto alcun effetto sul ΔΨm delle cellule HB2 non cancerose, mentre, sorprendentemente, il ΔΨm delle cellule 293 non cancerose è aumentato in modo dose-dipendente. A 24 ore di trattamento, 50 mM di DCA hanno ridotto significativamente il ΔΨm in tutte le cellule tumorali; tuttavia, nelle cellule LoVo si è registrata una riduzione significativa anche con 20 mM di DCA (Figura 5A, P=0,02). Nelle cellule 293 non cancerose, c’è stata una tendenza all’aumento di ΔΨm con il trattamento con DCA, anche se non statisticamente significativa (P=0,08). A 48 ore di trattamento, si è osservata una riduzione significativa di ΔΨm in tutte le cellule tumorali e un aumento nelle cellule 293, con DCA 20-50 mM (Figura 5B, P⩽0,04).
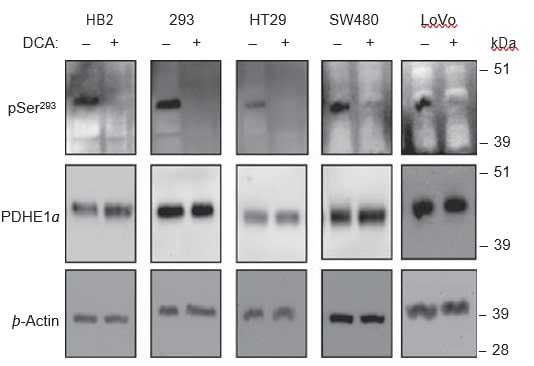
Iltrattamento con DCA porta alla de-fosforilazione della subunità PDHE1α
Si ritiene che ilDCAinibisca tutti e quattro gli isoenzimi della PDK e quindi riduca la fosforilazione della subunità PDHE1α, portando a sua volta all’attivazione del complesso PDH. Per verificare se la de-fosforilazione di PDHE1α avvenisse con il trattamento con DCA nelle linee cellulari utilizzate, abbiamo utilizzato analisi western blot su lisati di cellule trattate con DCA e non trattate. In tutte le linee cellulari, il trattamento con 20 mM di DCA per 8 ore ha causato una drastica riduzione del segnale di fosforilazione nel sito pSer293, ma non è stato rilevato alcun cambiamento nei livelli di PDHE1α totale (Figura 6). Gli anticorpi fosfo-specifici per gli altri due siti di fosforilazione, Ser232 e Ser300, non sono ancora disponibili in commercio.
Discussione
Abbiamo dimostrato che il DCA induce una riduzione dose-dipendente della crescita di colture in vitro di cellule tumorali del colon-retto e di cellule non tumorali. Tuttavia, le cellule cancerose sono risultate più sensibili al DCA, con una dose di 20 mM che ha provocato una significativa inibizione della crescita delle cellule cancerose, mentre ha avuto scarso effetto sulle cellule non cancerose. Abbiamo dimostrato che le componenti di questo effetto differenziale sono le seguenti: una potente induzione dell’apoptosi e dell’arresto del ciclo cellulare nelle cellule cancerose, ma non in quelle non cancerose.
Queste conclusioni supportano un semplice modello di sensibilità differenziale al DCA. Tuttavia, alcuni dati richiedono ulteriori discussioni. In primo luogo, 50 mM di DCA hanno ridotto la crescita delle colture di cellule non cancerose 293 e HB2, ma non è stato osservato un aumento delle cellule apoptotiche o un cambiamento nel profilo del ciclo cellulare di queste cellule. Una possibile spiegazione di questi risultati potrebbe essere che questa dose di DCA ha portato a un transito più lento di queste cellule non cancerose attraverso tutti gli stadi del ciclo cellulare, senza modificare le proporzioni relative all’interno di ciascuno stadio. In secondo luogo, i nostri risultati indicano che il DCA ha indotto l’arresto G2 nelle cellule del cancro colorettale. Ciò è in contrasto con studi precedenti, che hanno mostrato un arrestoG1 o nessun cambiamento nel profilo del ciclo cellulare con il trattamento con DCA (Cao et al, 2008; Wong et al, 2008). Wong et al (2008) hanno mostrato un aumento dell’espressione di PUMA in tutte le linee cellulari di cancro endometriale che hanno avuto una risposta apoptotica al DCA e hanno concluso che l’attivazione di p53 ha portato all’arrestoG1. Tuttavia, nel nostro studio le cellule di cancro del colon-retto si sono arrestate in fase G2 dopo il trattamento con DCA e non abbiamo riscontrato alcuna induzione di p53 da parte di DCA nelle nostre linee cellulari di cancro del colon-retto (dati non mostrati). È interessante notare che Cao et al. (2008) hanno scoperto che la combinazione di DCA e radioterapia arrestava le cellule di cancro alla prostata nella fase G2, mentre il DCA da solo non influiva sul profilo del ciclo cellulare. In terzo luogo, nelle cellule SW480 e LoVo, il trattamento con DCA ha determinato un aumento della percentuale di cellule considerate in fase S. Ciò suggerisce un aumento della proliferazione. Ciò suggerisce un aumento della proliferazione e l’induzione dell’apoptosi. Un risultato simile è stato riportato da Wong et al. (2008) in una delle diverse cellule di cancro endometriale analizzate. Una spiegazione alternativa è che una parte delle cellule osservate in “fase S” dopo il trattamento con DCA delle linee cellulari tumorali rappresenti in realtà cellule apoptotiche nella regione “sub-G2“, come è stato riportato in precedenza nelle cellule di linfoma (Klucar e Al-Rubeai, 1997).
Cambiamenti nel metabolismo cellulare con il trattamento con DCA
Il DCAsembra sopprimere la produzione di acido lattico dal piruvato sia nelle cellule cancerose che in quelle non cancerose. Inoltre, il trattamento con DCA ha portato alla de-fosforilazione di PDHE1α e quindi all’attivazione di PDH in tutte le linee cellulari analizzate. Pertanto, la base dell’effetto differenziato del DCA sulle cellule cancerose e non cancerose potrebbe risiedere nella sua influenza sulla funzione mitocondriale. Il trattamento con DCA ha ridotto l’elevato ΔΨm di tutte le cellule tumorali ma non di quelle non tumorali. Ciò suggerisce che il DCA, inibendo la PDK e quindi attivando la PDH, promuove la respirazione mitocondriale che porta alla depolarizzazione della membrana mitocondriale intrinseca e induce l’apoptosi attraverso la via mitocondriale prossimale, come descritto negli studi precedenti (Bonnet et al, 2007; Cao et al, 2008; Wong et al, 2008). L’induzione dell’apoptosi e i cambiamenti nella funzione mitocondriale sono stati più pronunciati nelle cellule LoVo altamente invasive e metastatiche rispetto alle cellule HT29 e SW480, meno invasive. Ciò potrebbe avere implicazioni cliniche per il trattamento del tumore del colon-retto metastatico, poiché di solito sono i tumori metastatici altamente invasivi ad essere più resistenti alla chemioterapia convenzionale e ad essere più sensibili all’inibizione della PDK. A sostegno di ciò, un recente studio ha riportato che i tumori del colon-retto resistenti al 5-fluorouracile hanno maggiori probabilità di avere una glicolisi upregolata, e quindi sono più adatti a una terapia mirata al metabolismo del cancro (Shin et al, 2009). A questo proposito, i nostri risultati contrastano con quelli di Wong et al (2008), che hanno riscontrato che le cellule del cancro endometriale altamente invasive sono più resistenti al trattamento con DCA.
Inibizione della PDK come terapia antitumorale contro il cancro del colon-retto
Abbiamo riscontrato che dosi di DCA di 20-50 mM hanno dato risposte diverse tra cellule cancerose e non cancerose. Pertanto, le potenziali dosi terapeutiche di DCA sarebbero comprese tra 20 e 50 mM. Inoltre, uno studio recente ha riportato che l’IC50 del DCA per le cellule del cancro al seno è compreso tra 20 e 30 mM (Ko e Allalunis-Turner, 2009). Ciò è in contrasto con studi precedenti che hanno riportato che il DCA riduce la proliferazione e induce l’apoptosi nelle cellule tumorali con dosi fino a 0,5-10 mM (Bonnet et al, 2007; Wong et al, 2008; Sun et al, 2009). Il dicloroacetato è risultato relativamente sicuro nell’uomo quando viene utilizzato per il trattamento dell’acidosi lattica (Stacpoole et al, 2003). I principali effetti collaterali di una dose di DCA fino a 100 mg kg-1 si manifestano a livello del sistema nervoso e del fegato, causando una lieve sedazione o sonnolenza, una neuropatia periferica reversibile e un lieve innalzamento asintomatico delle transaminasi sieriche che riflette un danno epatocellulare (Stacpoole et al, 1998). Inoltre, studi recenti hanno riportato che il DCA riduce efficacemente la crescita tumorale a dosi clinicamente raggiungibili sia in vitro che in vivo (Bonnet et al, 2007; Sun et al, 2009). È stato suggerito che il DCA potrebbe essere rapidamente tradotto in studi clinici sul cancro in fase iniziale (Michelakis et al, 2008). Tuttavia, è improbabile che la dose di DCA necessaria per inibire la crescita delle cellule di cancro del colon-retto nel nostro studio possa essere raggiunta clinicamente senza causare effetti collaterali significativi. La dose di DCA necessaria per ottenere concentrazioni plasmatiche equivalenti in vivo sarebbe da cinque a dieci volte superiore a quella utilizzata negli studi clinici contro l’acidosi lattica. Sembra che le cellule di cancro del colon-retto utilizzate nel nostro studio siano più resistenti al DCA rispetto alle cellule di cancro del polmone, dell’endometrio e della mammella. È interessante notare che Sun et al. (2009), nel loro studio sulle cellule del cancro al seno, hanno scoperto che il DCA inibisce la proliferazione delle cellule tumorali, ma non induce apoptosi o morte cellulare. Questi risultati sono nettamente diversi dagli effetti del DCA osservati su cellule di cancro del polmone (Bonnet et al, 2007), dell’endometrio (Wong et al, 2008) e del colon-retto nel nostro studio. Pertanto, sebbene il DCA inibisca la crescita di una varietà di cellule tumorali, l’effetto e i meccanismi sottostanti sembrano dipendere dal tipo di cellula. Una probabile spiegazione di questi effetti differenziati potrebbe essere la diversa espressione degli isoenzimi PDK nelle cellule tumorali esaminate. Il dicloroacetato è un inibitore non specifico della PDK (Whitehouse e Randle, 1973) e ha unKi diverso per ciascuno dei quattro isoenzimi PDK (Bowker-Kinley et al, 1998). Inoltre, è noto che i quattro isoenzimi PDK sono espressi in modo diverso nei vari tessuti. Pertanto, è necessario sviluppare inibitori dei singoli isoenzimi PDK che consentano una manipolazione metabolica specifica per ogni tipo di cellula tumorale.
RIFERIMENTI
1 Bi X, Lin Q, Foo TW, Joshi S, You T, Shen HM, Ong CN, Cheah PY, Eu KW, Hew CL (2006) Proteomic analysis of colorectal cancer reveals alterations in metabolic pathways: mechanism of tumorigenesis. Mol Cell Proteomics 5: 1119-1130
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11: 37-51
3 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Evidenza dell’esistenza di una regolazione tessuto-specifica del complesso della piruvato deidrogenasi nei mammiferi. Biochem J 329 (Part 1): 191-196
4 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Il dicloroacetato (DCA) sensibilizza alle radiazioni sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2 in vitro. Prostata 68: 1223-1231
4 Dewhirst MW, Tso CY, Oliver R, Gustafson CS, Secomb TW, Gross JF (1989) Morphologic and hemodynamic comparison of tumor and healing normal tissue microvasculature. Int J Radiat Oncol Biol Phys 17: 91-99
5 Gatenby RA, Gillies RJ (2004) Perché i tumori hanno un’elevata glicolisi aerobica? Nat Rev Cancer 4: 891-899
6 Gillies RJ, Gatenby RA (2007) Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? J Bioenerg Biomembr 39: 251-257
7 Klucar J, Al-Rubeai M (1997) L’arresto del ciclo cellulare G2 e l’apoptosi sono indotti nelle cellule di linfoma di Burkitt dall’agente antitumorale oracina. FEBS Lett 400: 127-130
8 Ko L, Allalunis-Turner J (2009) Investigation on the mechanism of dichloroacetate (DCA) induced apoptosis in breast cancer. J Clin Oncol 27 (Suppl 15): e14637
9 Michelakis ED, Webster L, Mackey JR (2008) Il dicloroacetato (DCA) come potenziale terapia a bersaglio metabolico per il cancro. Br J Cancer 99: 989-994
10 Milosevic M, Fyles A, Hedley D, Hill R (2004) Il microambiente tumorale umano: misurazione invasiva (con ago) dell’ossigeno e della pressione del fluido interstiziale. Semin Radiat Oncol 14: 249-258
11 Pan JG, Mak TW (2007) Il targeting metabolico come strategia antitumorale: l’alba di una nuova era? Sci STKE 2007: e14
12 Rajaganeshan R, Prasad R, Guillou PJ, Poston G, Scott N, Jayne DG (2008) Il ruolo dell’ipossia nella recidiva dopo resezione del tumore colorettale B di Dukes. Int J Colorectal Dis 23: 1049-1055
13 Rajaganeshan R, Prasad R, Guillou PJ, Scott N, Poston G, Jayne DG (2009) Pattern di espressione dei marcatori ipossici al margine invasivo dei tumori colorettali e delle metastasi epatiche. Eur J Surg Oncol 35 (12): 1286-1294
14 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE (2009) Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 389: 157-164
15 Shike M, Winawer SJ, Greenwald PH, Bloch A, Hill MJ, Swaroop SV (1990) Prevenzione primaria del cancro colorettale. Il Centro di collaborazione dell’OMS per la prevenzione del cancro colorettale. Bollettino dell’Organizzazione Mondiale della Sanità 68: 377-385
16 Shin YK, Yoo BC, Hong YS, Chang HJ, Jung KH, Jeong SY, Park JG (2009) Upregulation of glycolytic enzymes in proteins secreted from human colon cancer cells with 5-fluorouracil resistance. Elettroforesi 30: 2182-2192
17 Stacpoole PW (1989) La farmacologia del dicloroacetato. Metabolismo 38: 1124-1144
18 Stacpoole PW, Henderson GN, Yan Z, James MO (1998) Farmacologia clinica e tossicologia del dicloroacetato. Environ Health Perspect 106 (Suppl 4): 989-994
19 Stacpoole PW, Nagaraja NV, Hutson AD (2003) Efficacia del dicloroacetato come farmaco per la riduzione del lattato. J Clin Pharmacol 43: 683-691
20SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC (2009) L’inversione del fenotipo glicolitico da parte del dicloroacetato inibisce la crescita delle cellule metastatiche del cancro al seno in vitro e in vivo. Breast Cancer Res Treat 120 (1): 253-260
21 Thorn CC, Freeman TC, Scott N, Guillou PJ, Jayne DG (2009) Laser microdissection expression profiling of marginal edges of colorectal tumours reveals evidence of increased lactate metabolism in the aggressive phenotype. Gut 58: 404-412
22 Statistiche del Regno Unito sul cancro all’intestino (2009) http://info.cancerresearchuk.org/cancerstats/types/bowel/
23 Warburg O (1956) Sull’origine delle cellule cancerose. Science 123: 309-314
24 Whitehouse S, Randle PJ (1973) Attivazione della piruvato deidrogenasi nel cuore di ratto perfuso da parte del dicloroacetato. Biochem J 134: 651-653
25 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Il dicloroacetato induce l’apoptosi nelle cellule del cancro endometriale. Gynecol Oncol 109: 394-402
26 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA (2007) L’analisi metabolica multiparametrica rivela uno stretto legame tra l’attenuazione della funzione bioenergetica mitocondriale e la maggiore dipendenza dalla glicolisi nelle cellule tumorali umane. Am J Physiol Cell Physiol 292: C125-C136
27 Yeluri S, Madhok B, Prasad KR, Quirke P, Jayne DG (2009) Cancer’s craving for sugar: an opportunity for clinical exploitation. J Cancer Res Clin Oncol 135: 867-877