BM Madhok*,1, S Yeluri1 , SL Perry1 , TA Hughes2 und DG Jayne1
1 Abteilung für translationale Anästhesie und Chirurgie, Universität Leeds, Level 7 Clinical Sciences Building, St. James’s University Hospital, Leeds, UK2
Leeds Institute of Molecular Medicine, Universität Leeds, St. James’s University Hospital, Leeds, UKKorrespondenz
: Dr. BM Madhok; E-Mail:
[email protected]: 23. März 2010Akzeptiert
: 26. April 2010Veröffentlicht
: 18. Mai 2010
Zusammenfassung
Hintergrund
Krebszellen sind in hohem Maße von der Glykolyse abhängig. Unser Ziel war es, festzustellen, ob eine Umstellung des Stoffwechsels von der Glykolyse auf die mitochondriale Atmung das Wachstum von Darmkrebszellen gegenüber normalen Zellen bevorzugt reduzieren würde, und die zugrunde liegenden Mechanismen zu untersuchen.
Methoden
Repräsentative kolorektale und nicht-kolorektale Zelllinien wurden mit Dichloracetat (DCA), einem Inhibitor der Pyruvat-Dehydrogenase-Kinase, behandelt.
Ergebnisse
Dichloracetat (20 mM) verringerte das Wachstum der nicht-kanzerösen Zellen nicht, verursachte jedoch eine signifikante Verringerung der Proliferation der Krebszellen(P=0,009), die mit Apoptose und einem Stillstand des Zellzyklus in der G2-Phase einherging. Die stärkste apoptotische Wirkung zeigte sich bei metastasierenden LoVo-Zellen, bei denen DCA nach 48 Stunden einen bis zu zehnfachen Anstieg der apoptotischen Zellzahlen bewirkte. Der auffälligste G2-Arrest zeigte sich bei gut differenzierten HT29-Zellen, bei denen DCA nach 48 Stunden einen achtfachen Anstieg der Zellen in der G2-Phase bewirkte. Dichloracetat verringerte den Laktatgehalt im Wachstumsmedium und induzierte die Dephosphorylierung der E1α-Untereinheit des Pyruvatdehydrogenase-Komplexes in allen Zelllinien, aber das intrinsische mitochondriale Membranpotenzial war nur in Krebszellen reduziert(P=0,04).
Schlussfolgerungen
Die Hemmung der Pyruvat-Dehydrogenase-Kinase dämpft die Glykolyse und erleichtert die oxidative Phosphorylierung in den Mitochondrien, was zu einer Verringerung des Wachstums von kolorektalen Krebszellen, aber nicht von nicht-kanzerösen Zellen führt.
Schlüsselwörter: Dichloracetat, Dickdarmkrebs, Pyruvatdehydrogenase, Pyruvatdehydrogenase-KinaseBritish
Journal of Cancer (2010) 102, 1746 – 1752,
www.bjcancer.comdoi: 10.1038/sj.bjc.6605701
© 2010 Cancer Research UK
EINLEITUNG
Darmkrebs ist weltweit die dritthäufigste Krebsart und die vierthäufigste krebsbedingte Todesursache (Shike et al., 1990). Im Jahr 2007 waren im Vereinigten Königreich 17,1 Todesfälle pro 100 000 Personen auf Darmkrebs zurückzuführen (UK Bowel Cancer Statistics, 2009). Trotz der jüngsten Fortschritte ist die Prognose für Patienten mit fortgeschrittenem und metastasiertem Darmkrebs nach wie vor schlecht. Der gezielte Einsatz des Tumorstoffwechsels in der Krebstherapie ist ein sich rasch entwickelnder Bereich (Pan und Mak, 2007). Die ersten Beobachtungen zu den Unterschieden im Stoffwechsel von Krebs- und normalen Zellen wurden von Otto Warburg gemacht, der zeigte, dass Krebszellen von Natur aus auf die Glykolyse zur Erzeugung chemischer Energie angewiesen sind (Warburg, 1956). Inzwischen gibt es immer mehr Belege dafür, dass diese verstärkte Glykolyse auf den Einfluss mehrerer molekularer Pfade zurückzuführen ist, darunter adaptive Reaktionen auf die hypoxische Mikroumgebung des Tumors, onkogene Signalübertragung und mitochondriale Dysfunktion (Gatenby und Gillies, 2004; Gillies und Gatenby, 2007; Wu et al, 2007). Der glykolytische Phänotyp bietet den Krebszellen Wachstumsvorteile, da er der Apoptose widersteht und die Ausbreitung und Metastasierung von Tumoren erleichtert (Yeluri et al., 2009).
Ein wichtiger Regulator des zellulären Stoffwechsels ist die Pyruvatdehydrogenase (PDH). Pyruvat-Dehydrogenase wandelt Pyruvat, das bei der Glykolyse entsteht, in Acetyl-CoA um, das im Tricarbonsäurezyklus in den Mitochondrien oxidiert wird. Die Aktivität der Pyruvat-Dehydrogenase wird durch hemmende Phosphorylierung durch die Pyruvat-Dehydrogenase-Kinase (PDK) streng reguliert. Die Phosphorylierung erfolgt an der E1α-Untereinheit der PDH (PDHE1α) an drei Stellen: Ser232, Ser293 und Ser300 (Rardin et al., 2009). Dichloracetat (DCA) ist ein Inhibitor aller vier Isoenzyme der PDK(1-4) (Stacpoole, 1989) und hat kürzlich gezeigt, dass es das Wachstum von Lungen-, Endometrium- und Brustkrebszelllinien reduziert (Bonnet et al., 2007; Wong et al., 2008; Sun et al., 2009). Es wurde berichtet, dass es das Wachstum dieser Krebszellen vor allem durch die Verringerung der hemmenden Phosphorylierung von PDH reduziert, wodurch die oxidative Phosphorylierung in den Mitochondrien gefördert und die Apoptose über mitochondriale, NFAT-Kv 1.5 und p53 upregulated modulator of apoptosis (PUMA)-vermittelte Wege eingeleitet wird.
Es wurde festgestellt, dass Darmkrebszellen eine erhöhte Glykolyse aufweisen (Bi et al., 2006), und die Mikroumgebung des Tumors ist hypoxisch und azidotisch, was hauptsächlich auf eine schlecht entwickelte Blutversorgung zurückzuführen ist (Dewhirst et al., 1989; Milosevic et al., 2004). Wir haben bereits gezeigt, dass dies insbesondere für den aggressiveren Phänotyp gilt (Thorn et al., 2009) und dass die Expression wichtiger Marker für Hypoxie bei Darmkrebs erhöht ist, insbesondere am invasiven Rand (Rajaganeshan et al., 2008, 2009). Ziel dieser Studie war es, die Auswirkungen von DCA auf das Wachstum von Darmkrebszellen zu untersuchen, um die PDK-Hemmung als neue therapeutische Strategie gegen Darmkrebs zu prüfen.
Materialien und Methoden
Zellkulturen
Alle Zelllinien wurden von der American Type Culture Collection (Manassas, VA, USA) oder der European Collection of Cell Cultures (Salisbury, Wiltshire, UK) erworben: HB2 (Brustepithelzellen, die nicht aus Krebs stammen), 293 (Epithelzellen aus der menschlichen Embryonalniere), HT29 (gut differenziertes primäres kolorektales Adenokarzinom), SW480 (schlecht differenziertes primäres kolorektales Adenokarzinom) und LoVo (metastasierter linker supraklavikulärer Lymphknoten aus kolorektalem Adenokarzinom). 293- und HB2-Zellen wurden in DMEM-Medium, HT29- und SW480-Zellen in RPMI 1640-Medium und LoVo-Zellen in F12-Medium (alle von Invitrogen, Carlsbad, CA, USA), ergänzt mit 10 % fetalem Kälberserum, in einem befeuchteten Inkubator bei 37 °C und 5 %CO2 gehalten. Für Experimente unter hypoxischen Bedingungen inkubierten wir die Zellen in einem befeuchteten, hypoxischen Inkubator (1%O2, 5%CO2, 94% N2, 37°C). Natriumdichloracetat (Specials Lab, Prudhoe, UK) wurde von der Apothekenabteilung des St. James’s University Hospital, Leeds, UK, gespendet.
MTT-Tests
Die Zellen (1 ×104) pro Vertiefung wurden in 96-Well-Gewebekulturplatten ausgesät. Nach der Inkubation über Nacht ersetzten wir die Medien durch frische Medien mit steigenden DCA-Dosen (0, 10, 15, 20, 30, 50 und 100 mM). Nach 24 und 48 Stunden Inkubation führten wir einen MTT-Test durch, indem wir die Medien durch 50μleiner 1 mg ml-1 MTT-Lösung ersetzten und die Platten 3 Stunden lang im Dunkeln inkubierten. Die MTT-Lösung wurde dann entfernt und die dunkelblauen Formazan-Ausfällungen wurden in 100μlPropan-1-ol gelöst. Die optische Dichte wurde mit einem Mikroplatten-Lesegerät (Opsys MR; Dynex Technologies Ltd, Worthing, West Sussex, UK) bei 570 nm gemessen.
Annexin V- und 7-AAD-Tests
Die Zellen wurden in 25 cm2-Gewebekulturflaschen ausgesät und über Nacht unter Standardbedingungen inkubiert. Das Medium wurde durch frisches Medium ersetzt, das eine Reihe von DCA-Dosen (0, 10, 20 und 50 mM) enthielt. Die durchflusszytometrische Analyse wurde nach 24 und 48 Stunden Inkubation durchgeführt. Die Zellen wurden zweimal mit kaltem PBS gewaschen und in 1 × Bindungspuffer (BD Bioscience, Franklin Lakes, NJ, USA) mit 5 ×106 Zellen pro ml resuspendiert. 100μlder Lösung (5 ×105 Zellen) wurden in 5-ml-Kulturröhrchen übertragen. Diese Zellen wurden mit 5μlAnnexin V-FITC und 10μl7-AAD (BD Bioscience) angefärbt, vorsichtig geschüttelt und bei Raumtemperatur 15 Minuten lang im Dunkeln inkubiert. Anschließend wurden 400μl1 × Bindungspuffer in jedes Röhrchen gegeben und innerhalb einer Stunde auf dem LSR II-Durchflusszytometer (BD Bioscience) analysiert.
Propidiumjodid-Assays
Die Zellen wurden wie für den Apoptose-Assay beschrieben vermehrt. Es wurde Dichloracetat (50 mM) verwendet und mit der Vehikelkontrolle verglichen. Nach der Ernte wurden die Zellen in 350μlPBS in einer Konzentration von 0,5-1,0 ×106 Zellen pro ml resuspendiert. 100μl0,25 mg ml-1 Propidiumiodid (PI)/5% Triton (Sigma, St Louis, MO, USA) wurden der Zellsuspension zugesetzt. anschließend wurden 50μlvon 1 mg ml-1 Ribonuklease A (Sigma) hinzugefügt. Die Probenröhrchen wurden gründlich geschüttelt und 10 Minuten lang im Dunkeln bei Raumtemperatur bebrütet. Die Durchflusszytometrie wurde mit einem LSR II-Durchflusszytometer (BD Bioscience) durchgeführt, und die Daten wurden mit der FlowJo-Software (FlowJo, Ashland, OR, USA) analysiert.
Laktatmessungen
Laktatmessungenin Wachstumsmedien wurden von der Abteilung für chemische Pathologie am General Infirmary, Leeds Teaching Hospitals NHS Trust, durchgeführt. Die Zellen wurden in 25 cm2-Flaschen über Nacht in Normoxie inkubiert. Das Medium wurde am nächsten Tag durch eine Reihe von DCA-Dosen (0, 10, 20 und 50 mM) ersetzt. Nach 48 Stunden Inkubation wurden 2 ml des Mediums in Fluoridröhrchen aufgefangen und sofort in das Labor für chemische Pathologie gebracht. Die Röhrchen wurden während des Transfers auf Eis gehalten. Der Laktatgehalt wurde mit einem automatischen Analysegerät (Advia 1200 Chemistry System; Siemens Healthcare Diagnostics, Camberley, Surrey, UK) gemessen.
TMRM-Assays
Die Zellen wurden mit DCA behandelt, wie für den Apoptosetest beschrieben. Nach 24 und 48 Stunden Inkubation wurden die Zellen in PBS gewaschen und 1 ×106 Zellen pro ml in Hank’s gepufferter Salzlösung mit 50 nM Tetramethylrhodaminmethylester (TMRM) (Invitrogen) suspendiert. 100μlder Zellsuspension (1 ×105 Zellen pro Vertiefung) wurden in undurchsichtige 96-Well-Platten übertragen, 30 Minuten lang inkubiert und die Fluoreszenz bei 530/620 nm bei 37 °C mit einem Plattenlesegerät (Mithras LB 40; Berthold Technologies, Bad, Wildbad, Deutschland) gemessen.
Western Blotting
Die Zellen wurden wie oben beschrieben mit DCA behandelt. Nach 8 Stunden Behandlung extrahierten wir die Proteine aus den Zellen in Laemmli-Puffer (2% SDS, 10% Glycerin, 0,7% 2-Mercaptoethanol, 0,05% Bromphenolblau und 0,5 M Tris-HCl). Die Lysate wurden durch Elektrophorese auf NuPAGE Novex 12% Bis-Tris-Gelen (Invitrogen) in MOPS-SDS-Laufpuffer (Invitrogen) aufgelöst. Die Proteine wurden auf eine Polyvinylidenfluorid-Membran (GE Healthcare, Chalford St Giles, Bucks, UK) übertragen. Die Membran wurde 1 Stunde lang bei Raumtemperatur in 5 % Magermilch in TBS-T (Tris-gepufferte Kochsalzlösung mit 0,1 % Tween) blockiert. Die Membran wurde dann 90 Minuten lang mit primären Antikörpern in 1 % Magermilch in TBS-T sondiert, in TBS-T gewaschen und anschließend 60 Minuten lang mit dem entsprechenden Meerrettichperoxidase (HRP)-konjugierten sekundären Antikörper sondiert. Primäre Antikörper: Kaninchen-Polyklonale phosphodetect anti-PDH-E1α (pSer293), 1 :500 (AP1062; EMD Chemicals, Darmstadt, Deutschland), und mausmonoklonaler Anti-PDHE1α, 1 :500 (459400; Invitrogen). Sekundäre Antikörper Anti-Kaninchen- oder Anti-Maus-HRP-Konjugate, 1 :1000 (Dako, Glostrup, Dänemark). Die Proteine wurden mit Supersignal West Pico oder Femto Chemilumineszenzsubstrat (Pierce Biotechnology, Rockford, IL, USA) und dem Chemidoc XRS System (Bio-Rad, Hercules, CA, USA) sichtbar gemacht. β-Actin wurde als Ladekontrolle verwendet.
Statistische Analysen
Durchflusszytometrische Daten wurden mit spezieller Software, BD FACSDiva 6.0 und FlowJo Software, erfasst. Statistische Analysen wurden mit SPSS für Windows (SPSS Version 15.0, Chicago, IL, USA) durchgeführt. Die Unterschiede zwischen den mit DCA behandelten Gruppen und den Kontrollgruppen wurden mit dem Mann-Whitney-U-Test und den 95%-Konfidenzintervallen der Mittelwertdifferenz zwischen den beiden Gruppen bewertet. Ein P-Wert von weniger als 0,05 wurde als statistisch signifikant angesehen. Die Daten sind als Mittelwerte von mindestens drei unabhängigen Experimenten dargestellt, und die Fehlerbalken stellen die Standardabweichung des Mittelwerts dar.
Ergebnisse
DCA reduziert die Proliferation von Krebszellen und die Wirkung ist bei Normoxie und Hypoxie ähnlich
Zunächst wollten wir feststellen, ob die Behandlung mit DCA die Zellproliferation hemmt und ob es eine unterschiedliche Reaktion bei Krebs- und Nicht-Krebszellen unter normoxischen und hypoxischen Bedingungen gibt. In Bezug auf Hypoxie war unsere Hypothese, dass der Einfluss von DCA besonders stark sein würde, wenn der Sauerstoffgehalt nicht ausreicht, um eine zusätzliche oxidative Phosphorylierung zu unterstützen. Alle Zelllinien (HB2, 293, HT29, SW480 und LoVo) wurden 24-48 Stunden lang unter normoxischen und hypoxischen Bedingungen mit einer Reihe von DCA-Dosen behandelt. Die relativen Zellzahlen wurden mit MTT-Tests bestimmt.
Die Behandlung mit steigenden DCA-Dosen verringerte die Zellproliferation dosisabhängig (Abbildung 1A-D). Entgegen unserer Erwartung waren die Profile des verringerten Zellwachstums in Hypoxie und Normoxie ähnlich. Nach 24 und 48 Stunden hatten bis zu 20 mM DCA keinen Einfluss auf das Wachstum der Kulturen der nicht krebsartigen Zellen HB2 und 293. 20 mM DCA verringerte jedoch das Wachstum der Kulturen aller drei Darmkrebs-Zelllinien erheblich(P⩽0,009). Die Wirkung von DCA war bei den schlecht differenzierten SW480-Zellen und den metastatischen LoVo-Zellen stärker als bei den gut differenzierten HT29-Zellen. Das Wachstum von Kulturen von LoVo-Zellen, die mit 20 mM DCA behandelt wurden, war im Vergleich zu Zellen, die mit der Vehikelkontrolle behandelt wurden, um bis zu 40 % reduziert. Da die Wachstumsverringerung der mit DCA behandelten Kulturen unter hypoxischen und normoxischen Bedingungen relativ gering war, wurden weitere Experimente nur unter normoxischen Bedingungen durchgeführt.
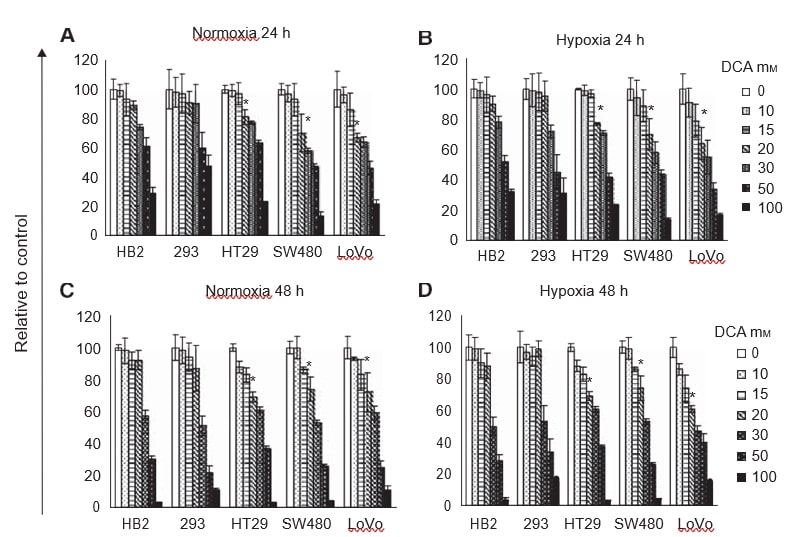
DCA fördert die Apoptose in Krebszellen und schont dabei nicht-krebsartige Zellen
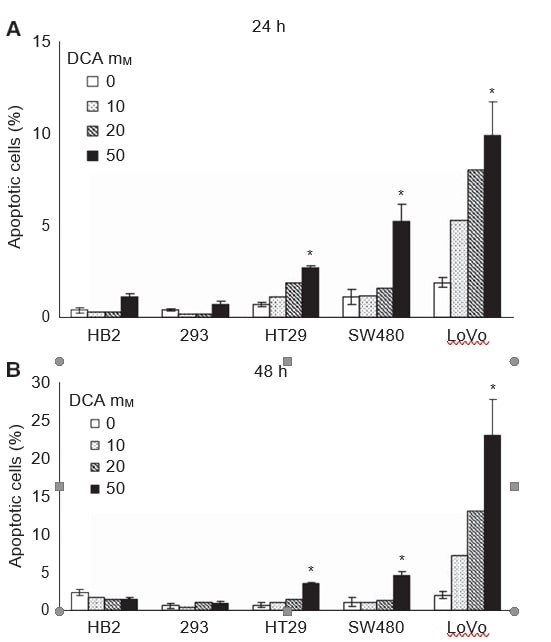
Als nächstes wollten wir untersuchen, ob das verringerte Wachstum der Kulturen nach der Behandlung mit DCA mit der Induktion von Apoptose verbunden ist. Die Zellen wurden 24 und 48 Stunden lang mit verschiedenen DCA-Dosen (0, 10, 20 und 50 mM) behandelt, und der Anteil der Zellen, die Apoptose durchlaufen, wurde durch den Nachweis von Membranphosphatidylserin mit Annexin V-FITC ermittelt. Die Zellen wurden mit Annexin V-FITC und dem Vitalfarbstoff 7-AAD angefärbt und mittels Durchflusszytometrie analysiert. In den Krebszelllinien wurde nach 24- und 48-stündiger Behandlung dosisabhängig Apoptose ausgelöst, während in den nicht krebsartigen Zellen, wenn überhaupt, nur wenig Apoptose ausgelöst wurde (Abbildung 2A und B). Die größte Wirkung wurde bei den metastatischen LoVo-Zellen beobachtet; 50 mM DCA bewirkte nach 48 Stunden einen zehnfachen Anstieg des Anteils apoptotischer Zellen, während bei HT29- und SW480-Zellen ein sieben- bzw. fünffacher Anstieg zu verzeichnen war. Die Zunahme des mittleren Prozentsatzes der gesamten apoptotischen Zellen mit 50 mM DCA betrug: 2,8 (95% CI: 2-3) in HT29-Zellen, 3,5 (95% CI: 2-5) in SW480-Zellen und 21 (95% CI: 8-34) in LoVo-Zellen. Bei den 293-Zellen wurde selbst mit 50 mM DCA nur eine minimale Apoptose ausgelöst, nämlich 0,2 (95% CI: -0,2 bis 0,6). Bei HB2-Zellen war ein nicht signifikanter Rückgang des Prozentsatzes apoptotischer Zellen bei der Behandlung mit 50 mM DCA zu verzeichnen, -0,9 (95% CI: -2,2 bis 0,4).
DCA induziert einen G2-Phasenstillstand in Darmkrebszellen, hat aber keine Auswirkungen auf das Zellzyklusprofil von nicht-kanzerösen 293-Zellen
Wir wollten auch untersuchen, ob die Wachstumsverringerung der Kulturen nach der Behandlung mit DCA mit der Induktion eines Wachstumsstillstands verbunden ist. Die Zellen wurden 24 oder 48 Stunden lang mit 50 mM DCA behandelt, und die Zellzyklusprofile wurden mittels durchflusszytometrischer Bewertung des DNA-Gehalts nach PI-Färbung analysiert. Die Behandlung mit Dichloracetat führte zu Veränderungen in den Zellzyklusprofilen aller Krebszellen, hatte aber keine Auswirkungen auf die nicht krebsartigen Zellen. Die Veränderungen im Zellzyklusprofil waren nach 24 Stunden Behandlung nachweisbar und blieben auch nach 48 Stunden bestehen (Abbildung 3A und B).
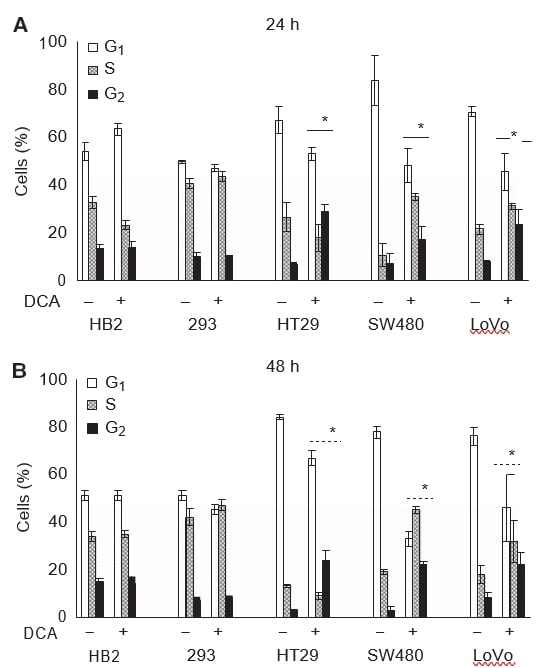
Nach 48 Stunden Behandlung mit 50 mM DCA stieg der Anteil der Zellen in der G2-Phase in HT29- und SW480-Zellen um das Achtfache und in LoVo-Zellen um das Dreifache an. Der durchschnittliche Prozentsatz aller Krebszellen in der G2-Phase stieg bei HT29 um 21 (95% CI: 13-30), bei SW480-Zellen um 19 (95% CI: 13-24) und bei LoVo-Zellen um 14 (95% CI: 10-21), während es bei 293-Zellen keinen Unterschied gab (1 (95% CI: -4 bis 7) und bei HB2-Zellen (-0,3 (95% CI: -9 bis 9)). Bei allen Krebszelllinien war ein entsprechender Rückgang der Zellen in der G0/G1-Phase festzustellen. Interessanterweise war bei HT29-Zellen ein geringer Rückgang zu verzeichnen, während bei SW480- und LoVo-Zellen der Anteil der Zellen, die sich in der S-Phase befanden, deutlich anstieg (siehe Abschnitt Diskussion). Das Zellzyklusprofil von 293- und HB2-Zellen änderte sich durch die Behandlung mit DCA nur geringfügig.
DCA reduziert die extrazellulären Laktatwerte in den Wachstumsmedien
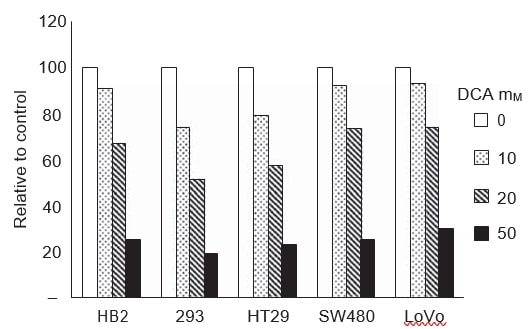
Um festzustellen, ob die durch DCA induzierten Veränderungen des Wachstums und der Apoptose mit einer verringerten Glykolyse korrelieren, haben wir den Laktatgehalt im Wachstumsmedium gemessen. Milchsäure ist das Endprodukt der Glykolyse. Würde DCA die oxidative Phosphorylierung in den Mitochondrien induzieren, würde Pyruvat zu Acetyl-CoA decarboxyliert und nicht zu Laktat reduziert werden, so dass der Laktatgehalt im Wachstumsmedium sinken würde. Die Laktatkonzentration in den Wachstumsmedien aller Zelllinien wurde nach 48 Stunden Behandlung mit verschiedenen DCA-Dosen gemessen (Abbildung 4). Der Laktatgehalt wurde mit einem Autoanalysator bestimmt, der routinemäßig für die biochemische Messung des Laktatgehalts verwendet wird; die Assays basieren auf einer kolorimetrischen Reaktion, die durch Laktatoxidase katalysiert wird. Die Behandlung mit DCA verringerte die extrazellulären Laktatwerte in den Wachstumsmedien in allen Krebs- und Nicht-Krebs-Zelllinien in dosisabhängiger Weise.
DCA depolarisiert die intrinsische mitochondriale Membran in kolorektalen Krebszellen, aber nicht in nicht-kanzerösen Zellen
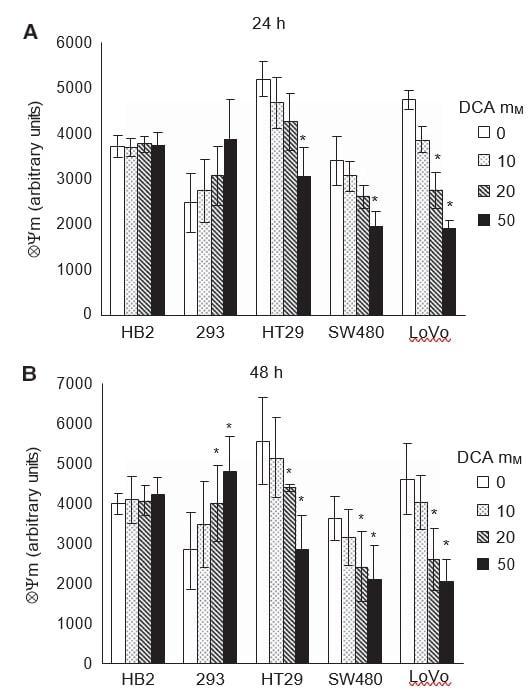
Um zu überprüfen, ob die Induktion der Apoptose in Krebszellen bei Behandlung mit DCA mit der Förderung der oxidativen Phosphorylierung in den Mitochondrien zusammenhängt, haben wir das intrinsische mitochondriale Membranpotenzial (ΔΨm) gemessen. Eine Eskalation der mitochondrialen Atmung würde die Elektronentransportkette reaktivieren und das hyperpolarisierte ΔΨm in Krebszellen verringern. Die Zellen wurden 24 und 48 Stunden lang mit DCA-Dosen behandelt und mit dem Farbstoff TMRM gefärbt, der eine Fluoreszenzmessung von ΔΨm ermöglicht.
Wie bei früheren Experimenten zeigte sich die Wirkung von DCA nach 24 Stunden Behandlung und hielt auch nach 48 Stunden an (Abbildung 5A und B). Die Behandlung mit Dichloracetat reduzierte das hyperpolarisierte ΔΨm in allen Krebszellen in einer dosisabhängigen Weise. Dichloracetat hatte keine Auswirkungen auf ΔΨm der nicht krebsartigen HB2-Zellen, während ΔΨm der nicht krebsartigen 293-Zellen überraschenderweise dosisabhängig anstieg. Nach 24 Stunden Behandlung verringerte sich ΔΨm mit 50 mM DCA in allen Krebszellen signifikant; in den LoVo-Zellen gab es jedoch auch mit 20 mM DCA eine signifikante Verringerung (Abbildung 5A, P=0,02). Bei den nicht krebsartigen 293-Zellen gab es einen Trend zu einem Anstieg von ΔΨm bei DCA-Behandlung, obwohl dies statistisch nicht signifikant war(P=0,08). Nach 48 Stunden Behandlung kam es zu einer signifikanten Verringerung von ΔΨm in allen Krebszellen und zu einem Anstieg in den 293-Zellen mit 20-50 mM DCA (Abbildung 5B, P⩽0,04).
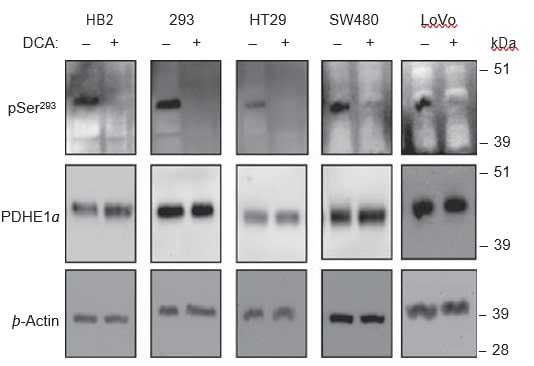
DCA-Behandlung führt zur Dephosphorylierung der PDHE1α-Untereinheit
Es wird angenommen, dass DCA alle vier Isoenzyme der PDK hemmt und somit die Phosphorylierung der PDHE1α-Untereinheit verringert, was wiederum zur Aktivierung des PDH-Komplexes führt. Um zu überprüfen, ob die Dephosphorylierung von PDHE1α bei der DCA-Behandlung in den verwendeten Zelllinien stattfand, führten wir Western-Blot-Analysen an Lysaten von DCA-behandelten und unbehandelten Zellen durch. Bei allen Zelllinien führte eine 8-stündige Behandlung mit 20 mM DCA zu einer drastischen Verringerung des Signals für die Phosphorylierung an der pSer293-Stelle, aber es wurde keine Veränderung der Gesamt-PDHE1α-Konzentration festgestellt (Abbildung 6). Phosphospezifische Antikörper für die beiden anderen Phosphorylierungsstellen, Ser232 und Ser300, sind noch nicht im Handel erhältlich.
Diskussion
Wir haben gezeigt, dass DCA eine dosisabhängige Verringerung des Wachstums von In-vitro-Kulturen kolorektaler Krebszellen und nicht-kanzeröser Zellen bewirkt. Die Krebszellen reagierten jedoch empfindlicher auf DCA, wobei eine Dosis von 20 mM eine signifikante Hemmung des Krebszellwachstums bewirkte, während sie auf die nicht krebsartigen Zellen kaum Auswirkungen hatte. Wir haben gezeigt, dass die Komponenten dieser unterschiedlichen Wirkung die folgenden sind: eine starke Induktion von Apoptose und Zellzyklus-Stillstand in den Krebszellen, aber nicht in den nicht-kanzerösen Zellen.
Diese Schlussfolgerungen unterstützen ein einfaches Modell der unterschiedlichen Empfindlichkeit gegenüber DCA. Einige Daten bedürfen jedoch weiterer Erörterung. Erstens verringerte 50 mM DCA das Wachstum der Kulturen der nicht kanzerösen 293- und HB2-Zellen, doch wurde keine Zunahme der apoptotischen Zellen oder eine Veränderung des Zellzyklusprofils dieser Zellen beobachtet. Eine mögliche Erklärung für diese Ergebnisse könnte sein, dass diese DCA-Dosis zu einem langsameren Durchgang dieser nicht kanzerösen Zellen durch alle Stadien des Zellzyklus führte, ohne die relativen Anteile innerhalb der einzelnen Stadien zu verändern. Zweitens deuten unsere Ergebnisse darauf hin, dass DCA bei Darmkrebszellen einen G2-Stillstand auslöst. Dies steht im Gegensatz zu früheren Studien, die einen G1-Arrest oder keine Veränderung des Zellzyklusprofils bei DCA-Behandlung gezeigt haben (Cao et al., 2008; Wong et al., 2008). Wong et al. (2008) zeigten eine erhöhte Expression von PUMA in allen Endometriumkrebs-Zelllinien, die eine apoptotische Reaktion auf DCA zeigten, und schlossen daraus, dass diese p53-Aktivierung zu einem G1-Arrest führte. Die Darmkrebszellen in unserer Studie verweilten jedoch nach der Behandlung mit DCA in der G2-Phase, und wir konnten in unseren Darmkrebszelllinien keine Induktion von p53 durch DCA feststellen (Daten nicht gezeigt). Interessanterweise stellten Cao et al. (2008) fest, dass die Kombination von DCA und Strahlentherapie Prostatakrebszellen in der G2-Phase zum Stillstand brachte, obwohl DCA allein keinen Einfluss auf das Zellzyklusprofil hatte. Drittens führte die DCA-Behandlung bei SW480- und LoVo-Zellen zu einem Anstieg des Anteils der Zellen, die sich in der S-Phase befanden. Dies deutet auf einen Anstieg der Proliferation und eine Induktion der Apoptose hin. Ein ähnlicher Befund wurde von Wong et al. (2008) bei einer von mehreren getesteten Endometriumkrebszellen gemeldet. Eine alternative Erklärung ist, dass ein Teil der Zellen, die sich nach der DCA-Behandlung der Krebszelllinien in der „S-Phase“ befinden, apoptotische Zellen in der „Sub-G2“-Region sind, wie bereits bei Lymphomzellen berichtet wurde (Klucar und Al-Rubeai, 1997).
Veränderungen des zellulären Stoffwechsels bei DCA-Behandlung
DCA scheint die Milchsäureproduktion aus Pyruvat sowohl in Krebs- als auch in Nicht-Krebszellen zu unterdrücken. Darüber hinaus führte die Behandlung mit DCA in allen untersuchten Zelllinien zu einer Dephosphorylierung von PDHE1α und damit zu einer Aktivierung von PDH. Der Grund für die unterschiedliche Wirkung von DCA auf Krebs- und Nicht-Krebszellen könnte also in seinem Einfluss auf die Mitochondrienfunktion liegen. Die Behandlung mit DCA reduzierte den hohen ΔΨm-Wert aller Krebszellen, nicht aber den der nicht krebsartigen Zellen. Dies deutet darauf hin, dass DCA durch Hemmung der PDK und damit Aktivierung der PDH die mitochondriale Atmung fördert, was zu einer Depolarisierung der intrinsischen Mitochondrienmembran führt und die Apoptose über den proximalen mitochondrialen Weg auslöst, wie in früheren Studien beschrieben (Bonnet et al, 2007; Cao et al, 2008; Wong et al, 2008). Die Induktion der Apoptose und die Veränderungen der Mitochondrienfunktion waren bei den stark invasiven und metastatischen LoVo-Zellen stärker ausgeprägt als bei den weniger invasiven HT29- und SW480-Zellen. Dies könnte klinische Auswirkungen auf die Behandlung von metastasierendem Darmkrebs haben, da in der Regel die hochinvasiven metastasierenden Krebsarten am resistentesten gegen herkömmliche Chemotherapien sind und möglicherweise am empfindlichsten auf eine PDK-Hemmung reagieren. Dies wird durch eine kürzlich durchgeführte Studie gestützt, in der festgestellt wurde, dass Darmtumore, die gegen 5-Fluorouracil resistent sind, mit größerer Wahrscheinlichkeit eine hochregulierte Glykolyse aufweisen und daher für eine Therapie, die auf den Krebsstoffwechsel abzielt, besser geeignet sind (Shin et al., 2009). In dieser Hinsicht stehen unsere Ergebnisse im Gegensatz zu den Erkenntnissen von Wong et al. (2008), die feststellten, dass hochinvasive Endometriumkrebszellen am resistentesten gegen eine DCA-Behandlung sind.
PDK-Hemmung als Krebstherapie gegen Darmkrebs
Wir fanden heraus, dass Dosen von 20-50 mM DCA zu unterschiedlichen Reaktionen zwischen Krebs- und Nicht-Krebszellen führten. Potenzielle therapeutische DCA-Dosen lägen daher zwischen 20 und 50 mM. Darüber hinaus wurde in einer aktuellen Studie berichtet, dass die IC50 von DCA für Brustkrebszellen zwischen 20 und 30 mM liegt (Ko und Allalunis-Turner, 2009). Dies steht im Gegensatz zu früheren Studien, in denen berichtet wurde, dass DCA die Proliferation reduziert und Apoptose in Krebszellen mit Dosen von nur 0,5-10 mM auslöst (Bonnet et al, 2007; Wong et al, 2008; Sun et al, 2009). Dichloracetat hat sich beim Menschen als relativ sicher erwiesen, wenn es zur Behandlung von Laktatazidose eingesetzt wird (Stacpoole et al., 2003). Die wichtigsten Nebenwirkungen von bis zu 100 mg/kg-1 DCA betreffen das Nervensystem und die Leber und verursachen eine leichte Sedierung oder Schläfrigkeit, eine reversible periphere Neuropathie und eine leichte asymptomatische Erhöhung der Serumtransaminasen, die eine hepatozelluläre Schädigung widerspiegelt (Stacpoole et al., 1998). Darüber hinaus wurde in jüngsten Studien berichtet, dass DCA das Tumorwachstum in klinisch erreichbaren Dosen sowohl in vitro als auch in vivo wirksam reduziert (Bonnet et al., 2007; Sun et al., 2009). Es wurde angedeutet, dass DCA rasch in klinische Studien für Krebserkrankungen in der Frühphase überführt werden könnte (Michelakis et al., 2008). Es ist jedoch unwahrscheinlich, dass die DCA-Dosis, die für die Hemmung des Wachstums von Darmkrebszellen in unserer Studie erforderlich ist, klinisch erreicht werden kann, ohne erhebliche Nebenwirkungen zu verursachen. Die DCA-Dosis, die erforderlich ist, um in vivo äquivalente Plasmakonzentrationen zu erreichen, wäre etwa fünf- bis zehnmal so hoch wie die in klinischen Studien gegen Laktatazidose verwendete Dosis. Es scheint, dass die in unserer Studie verwendeten Darmkrebszellen resistenter gegen DCA sind als Lungen-, Endometrium- und Brustkrebszellen. Interessanterweise stellten Sun et al. (2009) in ihrer Studie mit Brustkrebszellen fest, dass DCA die Proliferation von Krebszellen hemmt, aber keine Apoptose oder Zelltod auslöst. Diese Ergebnisse unterschieden sich deutlich von den Wirkungen von DCA, die in unserer Studie bei Lungen- (Bonnet et al., 2007), Endometrium- (Wong et al., 2008) und Darmkrebszellen beobachtet wurden. Obwohl DCA also das Wachstum einer Vielzahl von Krebszellen hemmt, scheinen die Wirkung und die zugrunde liegenden Mechanismen vom Zelltyp abhängig zu sein. Eine wahrscheinliche Erklärung für diese unterschiedlichen Wirkungen könnte die unterschiedliche Expression der PDK-Isoenzyme in den untersuchten Krebszellen sein. Dichloracetat ist ein unspezifischer Inhibitor der PDK (Whitehouse und Randle, 1973) und hat für jedes der vier PDK-Isoenzyme einen anderenKi-Wert (Bowker-Kinley et al., 1998). Darüber hinaus ist bekannt, dass die vier PDK-Isoenzyme in verschiedenen Geweben unterschiedlich exprimiert werden. Daher besteht die Notwendigkeit, Hemmstoffe für die einzelnen PDK-Isoenzyme zu entwickeln, die eine krebszelltypspezifische Stoffwechselbeeinflussung ermöglichen sollen.
REFERENZEN
1 Bi X, Lin Q, Foo TW, Joshi S, You T, Shen HM, Ong CN, Cheah PY, Eu KW, Hew CL (2006) Proteomic analysis of colorectal cancer reveals alterations in metabolic pathways: mechanism of tumorigenesis. Mol Cell Proteomics 5: 1119-1130
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle 11: 37-51
3 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Beweise für die Existenz einer gewebespezifischen Regulierung des Pyruvatdehydrogenase-Komplexes bei Säugetieren. Biochem J 329 (Part 1): 191-196
4 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Dichloracetat (DCA) sensibilisiert sowohl Wildtyp- als auch überexprimierende Bcl-2-Prostatakrebszellen in vitro für Strahlung. Prostate 68: 1223-1231
4 Dewhirst MW, Tso CY, Oliver R, Gustafson CS, Secomb TW, Gross JF (1989) Morphologischer und hämodynamischer Vergleich der Mikrovaskulatur von Tumor- und heilendem Normalgewebe. Int J Radiat Oncol Biol Phys 17: 91-99
5 Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4: 891-899
6 Gillies RJ, Gatenby RA (2007) Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? J Bioenerg Biomembr 39: 251-257
7 Klucar J, Al-Rubeai M (1997) G2-Zellzyklus-Arrest und Apoptose werden in Burkitt-Lymphom-Zellen durch das Krebsmittel Oracin induziert. FEBS Lett 400: 127-130
8 Ko L, Allalunis-Turner J (2009) Untersuchung des Mechanismus der durch Dichloracetat (DCA) induzierten Apoptose bei Brustkrebs. J Clin Oncol 27 (Suppl 15): e14637
9 Michelakis ED, Webster L, Mackey JR (2008) Dichloracetat (DCA) als potenzielles metabolisches Targeting für Krebs. Br J Cancer 99: 989-994
10 Milosevic M, Fyles A, Hedley D, Hill R (2004) The human tumor microenvironment: invasive (needle) measurement of oxygen and interstitial fluid pressure. Semin Radiat Oncol 14: 249-258
11 Pan JG, Mak TW (2007) Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE 2007: e14
12 Rajaganeshan R, Prasad R, Guillou PJ, Poston G, Scott N, Jayne DG (2008) The role of hypoxia in recurrence following resection of Dukes‘ B colorectal cancer. Int J Colorectal Dis 23: 1049-1055
13 Rajaganeshan R, Prasad R, Guillou PJ, Scott N, Poston G, Jayne DG (2009) Expressionsmuster von hypoxischen Markern am invasiven Rand von kolorektalen Karzinomen und Lebermetastasen. Eur J Surg Oncol 35 (12): 1286-1294
14 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE (2009) Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 389: 157-164
15 Shike M, Winawer SJ, Greenwald PH, Bloch A, Hill MJ, Swaroop SV (1990) Primary prevention of colorectal cancer. Das WHO-Kollaborationszentrum für die Prävention von Dickdarmkrebs. Bull World Health Organ 68: 377-385
16 Shin YK, Yoo BC, Hong YS, Chang HJ, Jung KH, Jeong SY, Park JG (2009) Upregulation of glycolytic enzymes in proteins secreted from human colon cancer cells with 5-fluorouracil resistance. Elektrophorese 30: 2182-2192
17 Stacpoole PW (1989) The pharmacology of dichloroacetate. Stoffwechsel 38: 1124-1144
18 Stacpoole PW, Henderson GN, Yan Z, James MO (1998) Clinical pharmacology and toxicology of dichloroacetate. Environ Health Perspect 106 (Suppl 4): 989-994
19 Stacpoole PW, Nagaraja NV, Hutson AD (2003) Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol 43: 683-691
20SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC (2009) Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 120 (1): 253-260
21 Thorn CC, Freeman TC, Scott N, Guillou PJ, Jayne DG (2009) Laser-Mikrodissektions-Expressionsprofilierung von marginalen Rändern kolorektaler Tumore zeigt Hinweise auf einen erhöhten Laktatstoffwechsel im aggressiven Phänotyp. Gut 58: 404-412
22 UK Bowel Cancer Statistics (2009) http://info.cancerresearchuk.org/cancerstats/types/bowel/
23 Warburg O (1956) On the origin of cancer cells. Wissenschaft 123: 309-314
24 Whitehouse S, Randle PJ (1973) Aktivierung der Pyruvat-Dehydrogenase im perfundierten Rattenherz durch Dichloracetat. Biochem J 134: 651-653
25 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichloracetat induziert Apoptose in Endometriumkrebszellen. Gynecol Oncol 109: 394-402
26 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA (2007) Multiparameter-Stoffwechselanalyse zeigt eine enge Verbindung zwischen abgeschwächter mitochondrialer bioenergetischer Funktion und verstärkter Glykolyse-Abhängigkeit in menschlichen Tumorzellen. Am J Physiol Cell Physiol 292: C125-C136
27 Yeluri S, Madhok B, Prasad KR, Quirke P, Jayne DG (2009) Cancer’s craving for sugar: an opportunity for clinical exploitation. J Cancer Res Clin Oncol 135: 867-877