BM Madhok*,1, S Yeluri1, SL Perry1, TA Hughes2 et DG Jayne1
1 Section of Translational Anaesthesia & Surgery, University of Leeds, Level 7 Clinical Sciences Building, St. James’s University Hospital, Leeds, UK
2 Leeds Institute of Molecular Medicine, University of Leeds, St. James’s University Hospital, Leeds, UK
Correspondance : Dr BM Madhok ; E-mail : [email protected]
Révisé : 23 mars 2010
Accepté : 26 avril 2010
Publié : 18 mai 2010
Résumé
Contexte
Les cellules cancéreuses sont très dépendantes de la glycolyse. Notre objectif était de déterminer si le passage du métabolisme de la glycolyse à la respiration mitochondriale réduirait la croissance des cellules cancéreuses colorectales de manière préférentielle par rapport aux cellules normales, et d’examiner les mécanismes sous-jacents.
Méthodes
Des lignées cellulaires représentatives de cancer colorectal et de cellules non cancéreuses ont été traitées avec du dichloroacétate (DCA), un inhibiteur de la pyruvate déshydrogénase kinase.
Résultats
Le dichloroacétate (20 mM) n’a pas réduit la croissance des cellules non cancéreuses mais a provoqué une diminution significative de la prolifération des cellules cancéreuses (P=0,009), qui était associée à l’apoptose et à l’arrêt du cycle cellulaire en phase G2. L’effet apoptotique le plus important était évident dans les cellules métastatiques LoVo, dans lesquelles le DCA a induit une augmentation jusqu’à dix fois du nombre de cellules apoptotiques après 48 h. L’arrêt G2 le plus frappant était évident dans les cellules HT29 bien différenciées, dans lesquelles le DCA a provoqué une augmentation de huit fois du nombre de cellules en phase G2 après 48 h. Le dichloroacétate a réduit les niveaux de lactate dans les milieux de croissance et a induit la déphosphorylation de la sous-unité E1α du complexe pyruvate déshydrogénase dans toutes les lignées cellulaires, mais le potentiel intrinsèque de la membrane mitochondriale a été réduit uniquement dans les cellules cancéreuses (P=0,04).
Conclusions
L’inhibition de la pyruvate déshydrogénase kinase atténue la glycolyse et facilite la phosphorylation oxydative mitochondriale, entraînant une réduction de la croissance des cellules cancéreuses colorectales mais pas des cellules non cancéreuses.
Mots-clés : dichloroacétate, cancer colorectal, pyruvate déshydrogénase, pyruvate déshydrogénase kinase
British Journal of Cancer (2010) 102, 1746 – 1752, www.bjcancer.com
doi : 10.1038/sj.bjc.6605701
2010 Cancer Research UK
INTRODUCTION
Le cancer colorectal est le troisième cancer le plus fréquent dans le monde et la quatrième cause de décès liés au cancer (Shike et al, 1990). En 2007, le cancer colorectal était responsable de 17,1 décès pour 100 000 personnes au Royaume-Uni (UK Bowel Cancer Statistics, 2009). Malgré les progrès récents, le pronostic des patients atteints de cancer colorectal avancé et métastatique reste mauvais. Le ciblage du métabolisme tumoral pour le traitement du cancer est un domaine en plein essor (Pan et Mak, 2007). Les premières observations concernant les différences métaboliques entre les cellules cancéreuses et normales ont été faites par Otto Warburg, qui a montré que les cellules cancéreuses sont intrinsèquement dépendantes de la glycolyse pour la production d’énergie chimique (Warburg, 1956). Il est désormais de plus en plus évident que cette glycolyse accrue résulte de l’influence de multiples voies moléculaires, notamment des réponses adaptatives au microenvironnement tumoral hypoxique, de la signalisation oncogène et du dysfonctionnement mitochondrial (Gatenby et Gillies, 2004 ; Gillies et Gatenby, 2007 ; Wu et al, 2007). Le phénotype glycolytique offre des avantages de croissance aux cellules cancéreuses en résistant à l’apoptose et en facilitant la propagation de la tumeur et les métastases (Yeluri et al, 2009).
Un régulateur clé du métabolisme cellulaire est la pyruvate déshydrogénase (PDH). La pyruvate déshydrogénase convertit le pyruvate, produit par la glycolyse, en acétyl-CoA, qui est oxydé dans le cycle de l’acide tricarboxylique au sein des mitochondries. L’activité de la pyruvate déshydrogénase est étroitement régulée par la phosphorylation inhibitrice de la pyruvate déshydrogénase kinase (PDK). La phosphorylation se produit sur la sous-unité E1α de la PDH (PDHE1α) sur trois sites : Ser232, Ser293 et Ser300 (Rardin et al, 2009). Le dichloroacétate (DCA) est un inhibiteur des quatre isoenzymes de la PDK(1-4) (Stacpoole, 1989), et il a récemment été démontré qu’il réduisait la croissance de lignées cellulaires de cancer du poumon, de l’endomètre et du sein (Bonnet et al, 2007 ; Wong et al, 2008 ; Sun et al, 2009). Il a été rapporté qu’il réduit la croissance de ces cellules cancéreuses principalement en réduisant la phosphorylation inhibitrice de la PDH, favorisant ainsi la phosphorylation oxydative mitochondriale et induisant l’apoptose par des voies médiées par les mitochondries, NFAT-Kv 1.5 et le modulateur d’apoptose régulé par p53 (PUMA).
On a constaté que les cellules cancéreuses colorectales subissent une glycolyse accrue (Bi et al, 2006), et que le microenvironnement tumoral est hypoxique et acidotique, principalement en raison d’une irrigation sanguine peu développée (Dewhirst et al, 1989 ; Milosevic et al, 2004). Nous avons précédemment montré que cela est particulièrement vrai pour le phénotype le plus agressif (Thorn et al, 2009), et l’expression des marqueurs importants de l’hypoxie est augmentée dans le cancer colorectal, en particulier au niveau de la marge invasive (Rajaganeshan et al, 2008, 2009). L’objectif de cette étude était d’étudier les effets du DCA sur la croissance des cellules cancéreuses colorectales afin d’examiner l’inhibition de la PDK comme nouvelle stratégie thérapeutique contre le cancer colorectal.
Matériel et méthodes
Cultures cellulaires
Toutes les lignées cellulaires ont été achetées auprès de l’American Type Culture Collection (Manassas, VA, USA) ou de l’European Collection of Cell Cultures (Salisbury, Wiltshire, UK) : HB2 (cellules épithéliales mammaires d’origine non cancéreuse), 293 (cellules épithéliales de rein d’embryon humain), HT29 (adénocarcinome colorectal primaire bien différencié), SW480 (adénocarcinome colorectal primaire peu différencié) et LoVo (ganglion lymphatique supraclaviculaire gauche métastatique d’un adénocarcinome colorectal). les cellules 293 et HB2 ont été maintenues dans un milieu DMEM, les HT29 et SW480 dans un milieu RPMI 1640, et les LoVo dans un milieu F12 (tous provenant d’Invitrogen, Carlsbad, CA, USA), complété par 10% de sérum de veau fœtal, dans un incubateur humidifié à 37°C et 5% deCO2. Pour les expériences en conditions hypoxiques, nous avons incubé les cellules dans un incubateur hypoxique humidifié (1% O2, 5%CO2, 94% N2, 37°C). Le dichloroacétate de sodium (Specials Lab, Prudhoe, UK) a été donné par le département de pharmacie de l’hôpital universitaire St. James, Leeds, UK.
Tests MTT
Des cellules (1 ×104) par puits ont été ensemencées dans des plaques de culture tissulaire à 96 puits. Après une incubation d’une nuit, nous avons remplacé les milieux par des milieux frais contenant des doses croissantes de DCA (0, 10, 15, 20, 30, 50 et 100 mM). Après 24 et 48 h d’incubation, nous avons réalisé le test MTT en remplaçant les milieux par 50 μlde solution de MTT à 1 mg ml-1 et les plaques ont été incubées dans l’obscurité pendant 3 h. La solution de MTT a ensuite été retirée et les précipités de formazan bleu foncé ont été dissous dans 100 μlde propan-1-ol. La densité optique a été mesurée à l’aide d’un lecteur de microplaques (Opsys MR ; Dynex Technologies Ltd, Worthing, West Sussex, UK) à 570 nm.
Dosages de l’Annexin V et du 7-AAD
Les cellules ont été ensemencées dans des flacons de culture tissulaire de 25 cm2 et incubées pendant la nuit dans des conditions standard. Le milieu a été remplacé par un milieu frais contenant une gamme de doses de DCA (0, 10, 20 et 50 mM). Une analyse cytométrique en flux a été effectuée après 24 et 48 h d’incubation. Les cellules ont été lavées deux fois avec du PBS froid et remises en suspension dans 1 × tampon de liaison (BD Bioscience, Franklin Lakes, NJ, USA) à 5 ×106 cellules par ml. 100 μlde solution (5 ×105 cellules) ont été transférés dans des tubes de culture de 5 ml. Ces cellules ont été colorées avec 5 μld’annexine V-FITC et 10 μlde 7-AAD (BD Bioscience), doucement vortexées, et incubées à température ambiante pendant 15 min dans l’obscurité. Ensuite, 400 μlde tampon de liaison 1 × ont été ajoutés à chaque tube et analysés dans l’heure qui suit sur le cytomètre en flux LSR II (BD Bioscience).
Tests à l’iodure de propidium
Les cellules ont été propagées comme indiqué pour le test d’apoptose. Le dichloroacétate (50 mM) a été utilisé et comparé au contrôle véhicule. Après la récolte, nous avons remis en suspension les cellules dans 350 μlde PBS à une concentration de 0,5-1,0 ×106 cellules par ml. 100 μlde 0,25 mg ml-1 d’iodure de propidium (PI)/5% de Triton (Sigma, St Louis, MO, USA) ont été ajoutés à la suspension cellulaire. 50 μlde 1 mg ml-1 de ribonucléase A (Sigma) ont ensuite été ajoutés. Les tubes d’échantillons ont été soigneusement vortexés et incubés pendant 10 min dans l’obscurité à température ambiante. La cytométrie en flux a été réalisée sur un cytomètre en flux LSR II (BD Bioscience) et les données ont été analysées à l’aide du logiciel FlowJo (FlowJo, Ashland, OR, USA).
Mesures du lactate
Les mesures du lactate dans les milieux de croissance ont été effectuées par le service de pathologie chimique du General Infirmary, Leeds Teaching Hospitals NHS Trust. Les cellules ont été incubées dans des flacons de 25 cm2 pendant la nuit en normoxie. Le lendemain, le milieu a été remplacé par une gamme de doses de DCA (0, 10, 20 et 50 mM). Après 48 h d’incubation, nous avons recueilli 2 ml de milieu dans des tubes fluorés et transférés immédiatement au laboratoire de pathologie chimique. Les tubes ont été maintenus sur la glace pendant le transfert. Les niveaux de lactate ont été mesurés à l’aide d’un analyseur automatisé (Advia 1200 Chemistry system ; Siemens Healthcare Diagnostics, Camberley, Surrey, UK).
Essais TMRM
Les cellules ont été traitées avec du DCA comme décrit pour l’essai d’apoptose. Après 24 et 48 h d’incubation, nous avons lavé les cellules dans du PBS, et suspendu 1 ×106 cellules par ml dans une solution saline tamponnée de Hank avec 50 nM d’ester méthylique de tétraméthylrhodamine (TMRM) (Invitrogen). 100 μlde la suspension cellulaire (1 ×105 cellules par puits) ont été transférés dans des plaques opaques à 96 puits, incubés pendant 30 min, et la fluorescence a été mesurée à 530/620 nm à 37°C à l’aide d’un lecteur de plaques (Mithras LB 40 ; Berthold Technologies, Bad, Wildbad, Allemagne).
Western blotting
Les cellules ont été traitées au DCA comme décrit ci-dessus. Après 8 h de traitement, nous avons extrait les protéines des cellules dans le tampon Laemmli (2 % de SDS, 10 % de glycérol, 0,7 % de 2-mercaptoéthanol, 0,05 % de bleu de bromophénol et 0,5 M de Tris-HCl). Les lysats ont été résolus par électrophorèse sur des gels NuPAGE Novex 12 % Bis-Tris (Invitrogen) dans un tampon MOPS-SDS (Invitrogen). Les protéines ont été transférées sur une membrane en fluorure de polyvinylidène (GE Healthcare, Chalford St Giles, Bucks, UK). La membrane a été bloquée pendant 1 h à température ambiante dans du lait écrémé à 5 % dans du TBS-T (Tris-buffered saline avec 0,1 % de Tween). La membrane a ensuite été sondée avec des anticorps primaires dans du lait écrémé à 1 % dans du TBS-T pendant 90 minutes, puis lavée dans du TBS-T, et enfin sondée avec l’anticorps secondaire approprié conjugué à la peroxydase de raifort (HRP) pendant 60 minutes. Anticorps primaires polyclonaux de lapin phosphodétectés anti-PDH-E1α (pSer293), 1 :500 (AP1062 ; EMD Chemicals, Darmstadt, Allemagne), et anticorps monoclonal de souris anti-PDHE1α, 1 :500 (459400 ; Invitrogen). Les anticorps secondaires anti-lapin ou anti-souris conjugués HRP, 1 :1000 (Dako, Glostrup, Danemark). Les protéines ont été visualisées avec le substrat chimiluminescent Supersignal West Pico ou Femto (Pierce Biotechnology, Rockford, IL, USA) et le système Chemidoc XRS (Bio-Rad, Hercules, CA, USA). la β-Actine a été utilisée comme contrôle de charge.
Analyses statistiques
Les données de cytométrie en flux ont été acquises à l’aide de logiciels spécifiques, BD FACSDiva 6.0 et FlowJo. Les analyses statistiques ont été réalisées à l’aide de SPSS pour Windows (SPSS version 15.0, Chicago, IL, USA). Les différences entre les groupes traités au DCA et les groupes témoins avec véhicule ont été évaluées à l’aide du test U de Mann-Whitney et des intervalles de confiance à 95 % de la différence des moyennes entre les deux groupes. Une valeur P inférieure à 0,05 a été considérée comme statistiquement significative. Les données sont représentées par la moyenne d’au moins trois expériences indépendantes et les barres d’erreur représentent l’écart type de la moyenne.
Résultats
LeDCA réduit la prolifération des cellules cancéreuses et l’effet est similaire en normoxie et en hypoxie
Nous avons d’abord voulu déterminer si le traitement au DCA inhibait la prolifération cellulaire et s’il y aurait une réponse différentielle dans les cellules cancéreuses et non cancéreuses dans des conditions normoxiques et hypoxiques. En ce qui concerne l’hypoxie, notre hypothèse était que l’influence du DCA serait particulièrement puissante avec des niveaux d’oxygène qui sont insuffisants pour supporter une phosphorylation oxydative supplémentaire. Toutes les lignées cellulaires (HB2, 293, HT29, SW480 et LoVo) ont été traitées avec une gamme de doses de DCA pendant 24-48 h dans des conditions normoxiques et hypoxiques. Le nombre relatif de cellules a été évalué à l’aide de tests MTT.
Le traitement avec des doses croissantes de DCA a réduit la prolifération cellulaire de manière dose-dépendante (figure 1A-D). Contrairement à ce que nous avions prévu, les profils de réduction de la croissance cellulaire étaient similaires en hypoxie et en normoxie. À 24 et 48 heures, jusqu’à 20 mM de DCA n’ont pas affecté la croissance des cultures de cellules non cancéreuses, HB2 et 293. Cependant, 20 mM de DCA ont réduit de manière significative la croissance des cultures des trois lignées cellulaires de cancer colorectal (P⩽0,009). L’effet du DCA était plus important sur les cellules SW480 peu différenciées et les cellules LoVo métastatiques que sur les cellules HT29 bien différenciées. La croissance des cultures de cellules LoVo traitées avec 20 mM de DCA a été réduite jusqu’à 40 % par rapport aux cellules traitées avec le véhicule témoin. Comme il y avait relativement peu de différence dans la réduction de la croissance des cultures traitées au DCA dans des conditions hypoxiques et normoxiques, d’autres expériences ont été réalisées uniquement en normoxie.
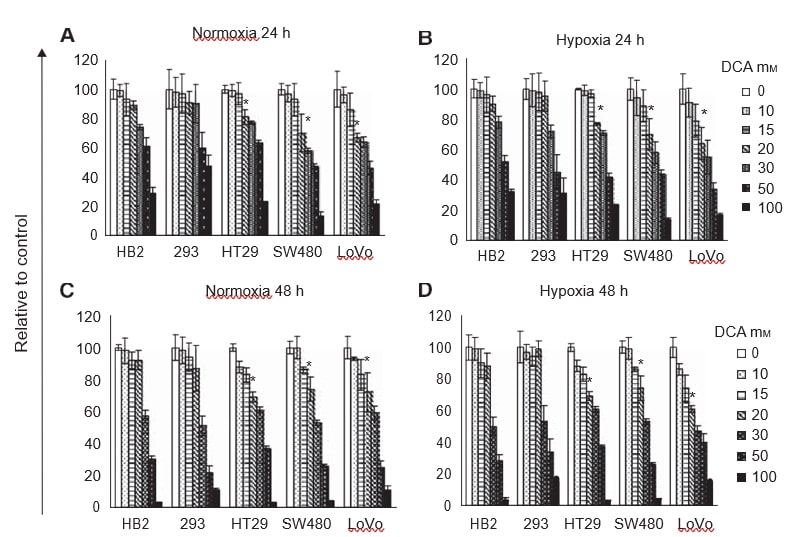
LeDCA favorise l’apoptose dans les cellules cancéreuses en épargnant les cellules non cancéreuses
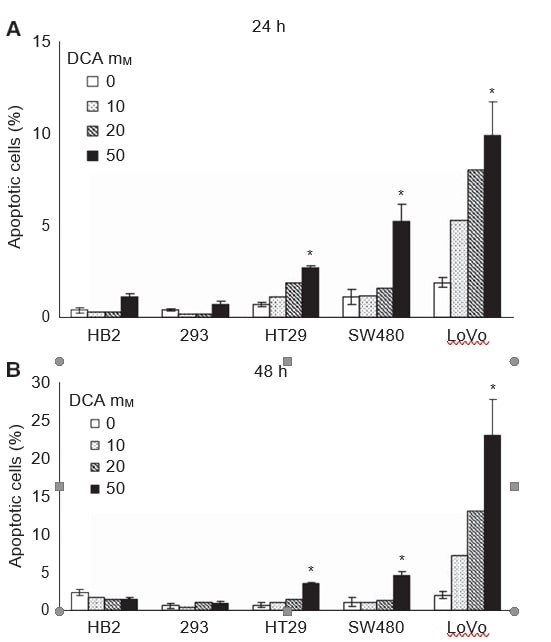
Ensuite, nous avons souhaité étudier si la croissance réduite des cultures lors du traitement au DCA était associée à l’induction de l’apoptose. Les cellules ont été traitées avec différentes doses de DCA (0, 10, 20 et 50 mM) pendant 24 et 48 h, et la proportion de cellules subissant une apoptose a été évaluée en détectant la phosphatidylsérine membranaire avec l’annexine V-FITC. Les cellules ont été colorées avec l’annexine V-FITC et le colorant vital 7-AAD, et analysées par cytométrie en flux. Une induction dose-dépendante de l’apoptose a été observée dans les lignées cellulaires cancéreuses après 24 et 48 heures de traitement, les cellules non cancéreuses n’induisant que peu ou pas d’apoptose (Figure 2A et B). L’effet le plus important a été observé dans les cellules métastatiques LoVo ; 50 mM de DCA ont provoqué une multiplication par dix de la proportion de cellules apoptotiques après 48 heures, tandis qu’une multiplication par sept et cinq a été observée dans les cellules HT29 et SW480, respectivement. L’augmentation du pourcentage moyen de cellules apoptotiques totales avec 50 mM de DCA était de : 2,8 (IC à 95 % : 2-3) dans les cellules HT29, 3,5 (IC à 95 % : 2-5) dans les cellules SW480 et 21 (IC à 95 % : 8-34) dans les cellules LoVo. L’apoptose induite dans les cellules 293 était minime, même avec 50 mM de DCA, soit 0,2 (IC 95 % : -0,2 à 0,6). Dans les cellules HB2, on a observé une diminution non significative du pourcentage de cellules apoptotiques lors du traitement par 50 mM de DCA, soit -0,9 (IC à 95 % : -2,2 à 0,4).
Le DCA induit l’arrêt de la phase G2 dans les cellules cancéreuses colorectales mais n’a aucun effet sur le profil du cycle cellulaire des cellules 293 non cancéreuses
Nous avons également souhaité examiner si la réduction de la croissance des cultures après traitement au DCA était associée à l’induction d’un arrêt de la croissance. Les cellules ont été traitées avec 50 mM de DCA pendant 24 ou 48 h, et les profils de cycle cellulaire ont été analysés en utilisant l’évaluation cytométrique en flux du contenu en ADN après coloration PI. Le traitement au dichloroacétate a provoqué des changements dans les profils de cycle cellulaire de toutes les cellules cancéreuses mais n’a pas affecté les cellules non cancéreuses. Les modifications du profil du cycle cellulaire étaient détectables après 24 h de traitement et persistaient après 48 h (figure 3A et B).
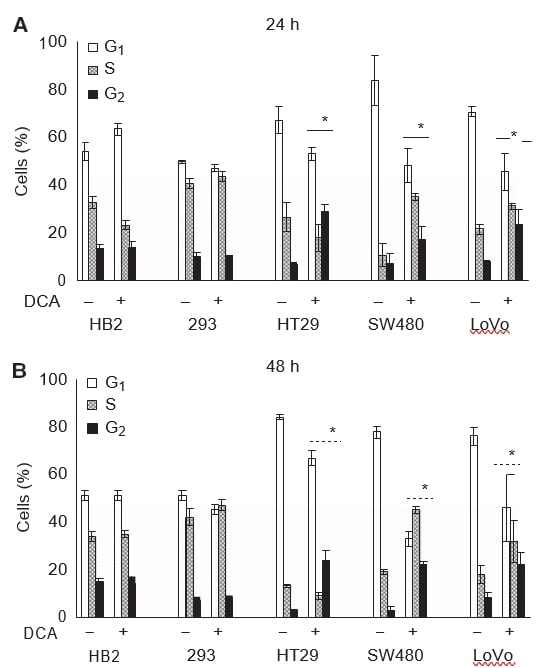
Après 48 heures de traitement par 50 mM de DCA, on a observé une multiplication par huit des cellules en phase G2 dans les cellules HT29 et SW480, et par trois dans les cellules LoVo. L’augmentation du pourcentage moyen de toutes les cellules cancéreuses en phase G2 était de 21 (IC à 95 % : 13-30) pour les cellules HT29, 19 (IC à 95 % : 13-24) pour les cellules SW480 et 14 (IC à 95 % : 10-21) pour les cellules LoVo, alors qu’il n’y avait pas de différence pour les cellules 293, 1 (IC à 95 % : -4 à 7), et les cellules HB2, -0,3 (IC à 95 % : -9 à 9). Une diminution correspondante des cellules en phase G0/G1 a été observée dans toutes les lignées cellulaires cancéreuses. Il est intriguant de constater qu’il y a eu une légère diminution dans les cellules HT29, mais une augmentation significative de la proportion de cellules considérées comme étant en phase S dans les cellules SW480 et LoVo (voir la section Discussion). Le profil du cycle cellulaire des cellules 293 et HB2 a peu changé lors du traitement au DCA.
Le DCA réduit les niveaux de lactate extracellulaire dans les milieux de croissance
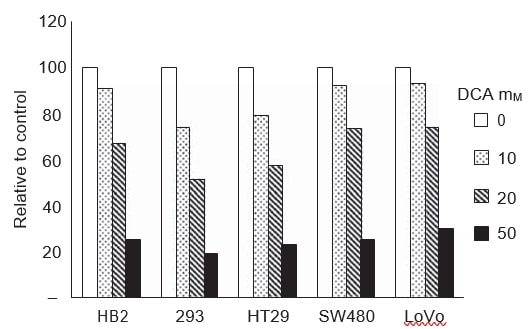
Pour établir si les changements dans la croissance et l’apoptose induits par le DCA sont corrélés à une réduction de la glycolyse, nous avons mesuré les niveaux de lactate dans les milieux de croissance. L’acide lactique est le produit final de la glycolyse. Si le DCA induisait une phosphorylation oxydative mitochondriale, le pyruvate serait décarboxylé en acétyl-CoA et non réduit en lactate, d’où une diminution des niveaux de lactate dans les milieux de croissance. Les niveaux de lactate dans les milieux de croissance de toutes les lignées cellulaires ont été mesurés après 48 heures de traitement avec une gamme de doses de DCA (figure 4). Les niveaux de lactate ont été déterminés à l’aide d’un auto-analyseur qui est utilisé en routine pour la mesure biochimique des niveaux de lactate ; les essais sont basés sur une réaction colorimétrique catalysée par la lactate oxydase. Le traitement au DCA a réduit les niveaux de lactate extracellulaire dans les milieux de croissance de manière dose-dépendante dans toutes les lignées cellulaires cancéreuses et non cancéreuses.
LeDCA dépolarise la membrane mitochondriale intrinsèque dans les cellules cancéreuses colorectales mais pas dans les cellules non cancéreuses
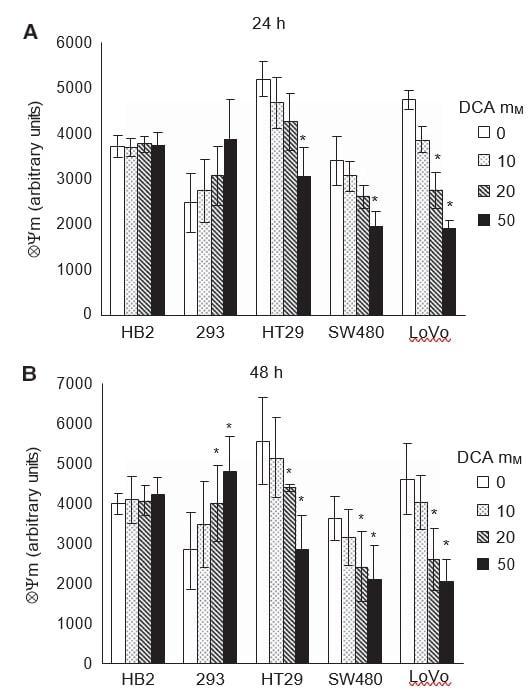
Pour vérifier si l’induction de l’apoptose dans les cellules cancéreuses lors du traitement avec le DCA était associée à la promotion de la phosphorylation oxydative mitochondriale, nous avons mesuré le potentiel de la membrane mitochondriale intrinsèque (ΔΨm). L’escalade de la respiration mitochondriale réactiverait la chaîne de transport d’électrons et réduirait le ΔΨm hyperpolarisé des cellules cancéreuses. Les cellules ont été traitées avec des doses de DCA pendant 24 et 48 h et colorées avec le colorant TMRM, qui permet de mesurer par fluorescence le ΔΨm.
Comme pour les expériences précédentes, l’effet du DCA était apparent après 24 h de traitement et persistait à 48 h (Figure 5A et B). Le traitement au dichloroacétate a réduit le ΔΨm hyperpolarisé dans toutes les cellules cancéreuses de manière dose-dépendante. Le dichloroacétate n’a eu aucun effet sur le ΔΨm des cellules HB2 non cancéreuses, alors que, de manière surprenante, le ΔΨm des cellules 293 non cancéreuses a augmenté de manière dose-dépendante. Après 24 heures de traitement, 50 mM de DCA ont réduit de manière significative le ΔΨm dans toutes les cellules cancéreuses ; cependant, dans les cellules LoVo, une réduction significative a été observée même avec 20 mM de DCA (Figure 5A, P=0,02). Dans les cellules 293 non cancéreuses, il y avait une tendance à l’augmentation du ΔΨm lors du traitement au DCA, bien que cela ne soit pas statistiquement significatif (P=0,08). À 48 h de traitement, on a observé une réduction significative de ΔΨm dans toutes les cellules cancéreuses et une augmentation dans les cellules 293, avec 20-50 mM de DCA (Figure 5B, P⩽0,04).
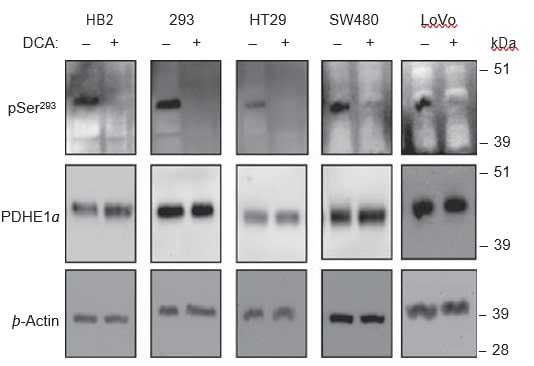
Letraitement au DCA entraîne la déphosphorylation de la sous-unité PDHE1α
Le DCAest censé inhiber les quatre isoenzymes de la PDK, et donc réduire la phosphorylation de la sous-unité PDHE1α, entraînant à son tour l’activation du complexe PDH. Pour vérifier si la déphosphorylation de PDHE1α se produisait avec le traitement au DCA dans les lignées cellulaires utilisées, nous avons utilisé des analyses par western blot sur des lysats de cellules traitées au DCA et non traitées. Dans toutes les lignées cellulaires, le traitement avec 20 mM de DCA pendant 8 h a provoqué une réduction spectaculaire du signal de phosphorylation au site pSer293, mais aucun changement n’a été détecté dans les niveaux de PDHE1α total (Figure 6). Les anticorps phospho-spécifiques pour les deux autres sites de phosphorylation, Ser232 et Ser300, ne sont pas encore disponibles dans le commerce.
Discussion
Nous avons montré que le DCA induit une réduction dose-dépendante de la croissance de cultures in vitro de cellules cancéreuses colorectales et de cellules non cancéreuses. Cependant, les cellules cancéreuses étaient plus sensibles au DCA, une dose de 20 mM provoquant une inhibition significative de la croissance des cellules cancéreuses, mais ayant peu d’effet sur les cellules non cancéreuses. Nous avons montré que les composantes de cet effet différentiel sont les suivantes : une induction puissante de l’apoptose et de l’arrêt du cycle cellulaire dans les cellules cancéreuses, mais pas dans les cellules non cancéreuses.
Ces conclusions soutiennent un modèle simple de sensibilité différentielle au DCA. Cependant, certaines données nécessitent une discussion plus approfondie. Tout d’abord, 50 mM de DCA ont réduit la croissance des cultures des cellules 293 et HB2 non cancéreuses, mais aucune augmentation des cellules apoptotiques ou modification du profil du cycle cellulaire de ces cellules n’a été observée. Une explication possible de ces résultats pourrait être que cette dose de DCA a entraîné un passage plus lent de ces cellules non cancéreuses à travers toutes les étapes du cycle cellulaire, sans modifier les proportions relatives à l’intérieur de chaque étape. Deuxièmement, nos résultats indiquent que le DCA a induit un arrêt G2 dans les cellules cancéreuses colorectales. Cela contraste avec les études précédentes, qui ont montré un arrêt G1 ou aucun changement du profil du cycle cellulaire avec le traitement au DCA (Cao et al, 2008 ; Wong et al, 2008). Wong et al (2008) ont montré une augmentation de l’expression de PUMA dans toutes les lignées cellulaires de cancer de l’endomètre qui ont eu une réponse apoptotique au DCA, et ont conclu que cette activation de p53 a conduit à un arrêt de G1. Cependant, les cellules de cancer colorectal de notre étude se sont arrêtées en phase G2 lors du traitement au DCA, et nous n’avons pas trouvé d’induction de p53 par le DCA dans nos lignées cellulaires de cancer colorectal (données non présentées). De manière intrigante, Cao et al (2008) ont découvert que la combinaison du DCA et de la radiothérapie arrêtait les cellules cancéreuses de la prostate en phase G2, bien que le DCA seul n’ait pas affecté le profil du cycle cellulaire. Troisièmement, dans les cellules SW480 et LoVo, le traitement au DCA a entraîné une augmentation de la proportion de cellules considérées comme étant en phase S. Cela suggère une augmentation de la prolifération. Cela suggère une augmentation de la prolifération ainsi qu’une induction de l’apoptose. Un résultat similaire a été rapporté par Wong et al (2008) dans l’une des cellules cancéreuses de l’endomètre testées. Une autre explication est qu’une partie des cellules observées comme étant en » phase S » après le traitement au DCA des lignées de cellules cancéreuses représentent en fait des cellules apoptotiques dans la région » sub-G2« , comme cela a été signalé précédemment dans les cellules de lymphome (Klucar et Al-Rubeai, 1997).
Modifications du métabolisme cellulaire avec le traitement au DCA
LeDCAsemble supprimer la production d’acide lactique à partir du pyruvate dans les cellules cancéreuses et non cancéreuses. En outre, le traitement au DCA a entraîné la déphosphorylation de PDHE1α, et donc l’activation de PDH dans toutes les lignées cellulaires étudiées. L’effet différentiel du DCA sur les cellules cancéreuses et non cancéreuses pourrait donc résider dans son influence sur la fonction mitochondriale. Le traitement au DCA a réduit le ΔΨm élevé de toutes les cellules cancéreuses mais pas celui des cellules non cancéreuses. Cela suggère que le DCA, en inhibant la PDK et donc en activant la PDH, favorise la respiration mitochondriale qui conduit à la dépolarisation de la membrane mitochondriale intrinsèque, et induit l’apoptose par la voie mitochondriale proximale comme décrit dans les études précédentes (Bonnet et al, 2007 ; Cao et al, 2008 ; Wong et al, 2008). L’induction de l’apoptose et les changements dans la fonction mitochondriale étaient plus prononcés dans les cellules LoVo hautement invasives et métastatiques que dans les cellules HT29 et SW480 moins invasives. Ces résultats pourraient avoir des implications cliniques pour le traitement du cancer colorectal métastatique, car ce sont généralement les cancers métastatiques hautement invasifs qui sont les plus résistants à la chimiothérapie conventionnelle, et qui pourraient être les plus sensibles à l’inhibition de la PDK. À l’appui de cette affirmation, une étude récente a signalé que les tumeurs colorectales résistantes au 5-fluorouracile sont plus susceptibles d’avoir une glycolyse régulée à la hausse, et donc de se prêter davantage à une thérapie ciblant le métabolisme du cancer (Shin et al, 2009). À cet égard, nos résultats contrastent avec ceux de Wong et al (2008), qui ont constaté que les cellules cancéreuses de l’endomètre hautement invasives étaient les plus résistantes au traitement par DCA.
L’inhibition de la PDK comme thérapie contre le cancer colorectal
Nous avons constaté que des doses de 20 à 50 mM de DCA donnaient des réponses différentielles entre les cellules cancéreuses et non cancéreuses. Ainsi, les doses thérapeutiques potentielles de DCA seraient comprises entre 20 et 50 mM. En outre, une étude récente a indiqué que la CI50 du DCA pour les cellules cancéreuses du sein se situe entre 20 et 30 mM (Ko et Allalunis-Turner, 2009). Cela contraste avec les études précédentes qui ont rapporté que le DCA réduisait la prolifération et induisait l’apoptose dans les cellules cancéreuses avec des doses aussi faibles que 0,5-10 mM (Bonnet et al, 2007 ; Wong et al, 2008 ; Sun et al, 2009). Le dichloroacétate s’est révélé relativement sûr chez l’homme lorsqu’il est utilisé pour le traitement de l’acidose lactique (Stacpoole et al, 2003). Les principaux effets secondaires d’une dose de DCA allant jusqu’à 100 mg kg-1 concernent le système nerveux et le foie, entraînant une légère sédation ou somnolence, une neuropathie périphérique réversible et une légère élévation asymptomatique des transaminases sériques reflétant des lésions hépatocellulaires (Stacpoole et al, 1998). En outre, des études récentes ont indiqué que le DCA réduisait efficacement la croissance tumorale à des doses cliniquement réalisables, tant in vitro qu’in vivo (Bonnet et al, 2007 ; Sun et al, 2009). Il a été suggéré que le DCA pourrait rapidement se traduire par des essais cliniques sur le cancer en phase précoce (Michelakis et al, 2008). Cependant, la dose de DCA nécessaire pour inhiber la croissance des cellules cancéreuses colorectales dans notre étude est peu susceptible d’être atteinte en clinique sans provoquer d’importants effets secondaires. La dose de DCA nécessaire pour atteindre les concentrations plasmatiques équivalentes in vivo serait environ cinq à dix fois supérieure à celle utilisée dans les essais cliniques contre l’acidose lactique. Il semble que les cellules cancéreuses colorectales utilisées dans notre étude soient plus résistantes au DCA que les cellules cancéreuses du poumon, de l’endomètre et du sein. De façon intrigante, Sun et al (2009), dans leur étude sur les cellules du cancer du sein, ont constaté que le DCA inhibait la prolifération des cellules cancéreuses, mais n’induisait pas l’apoptose ou la mort cellulaire. Ces résultats étaient très différents des effets du DCA observés sur les cellules cancéreuses du poumon (Bonnet et al, 2007), de l’endomètre (Wong et al, 2008) et du cancer colorectal dans notre étude. Ainsi, bien que le DCA inhibe la croissance d’une variété de cellules cancéreuses, l’effet et les mécanismes sous-jacents semblent dépendre du type de cellule. Une explication probable de ces effets différentiels pourrait être la différence d’expression des isoenzymes PDK dans les cellules cancéreuses examinées. Le dichloroacétate est un inhibiteur non spécifique de la PDK (Whitehouse et Randle, 1973), et sonKi est différent pour chacune des quatre isoenzymes de la PDK (Bowker-Kinley et al, 1998). En outre, on sait que les quatre isoenzymes de la PDK sont exprimées de manière différente dans divers tissus. Il est donc nécessaire de mettre au point des inhibiteurs des différentes isoenzymes de la PDK qui permettraient une manipulation métabolique spécifique du type de cellule cancéreuse.
RÉFÉRENCES
1 Bi X, Lin Q, Foo TW, Joshi S, You T, Shen HM, Ong CN, Cheah PY, Eu KW, Hew CL (2006) Proteomic analysis of colorectal cancer reveals alterations in metabolic pathways : mechanism of tumorigenesis. Mol Cell Proteomics 5 : 1119-1130
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cellule cancéreuse 11 : 37-51
3 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 329 (Part 1) : 191-196
4 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Le dichloroacétate (DCA) sensibilise in vitro les cellules cancéreuses de la prostate de type sauvage et sur-exprimant Bcl-2 aux radiations. Prostate 68 : 1223-1231
4 Dewhirst MW, Tso CY, Oliver R, Gustafson CS, Secomb TW, Gross JF (1989) Morphologic and hemodynamic comparison of tumor and healing normal tissue microvasculature. Int J Radiat Oncol Biol Phys 17 : 91-99
5 Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis ? Nat Rev Cancer 4 : 891-899
6 Gillies RJ, Gatenby RA (2007) Adaptive landscapes and emergent phenotypes : why do cancers have high glycolysis ? J Bioenerg Biomembr 39 : 251-257
7 Klucar J, Al-Rubeai M (1997) G2 cell cycle arrest and apoptosis are induced in Burkitt’s lymphoma cells by the anticancer agent oracin. FEBS Lett 400 : 127-130
8 Ko L, Allalunis-Turner J (2009) Investigation on the mechanism of dichloroacetate (DCA) induced apoptosis in breast cancer. J Clin Oncol 27 (Suppl 15) : e14637
9 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99 : 989-994
10 Milosevic M, Fyles A, Hedley D, Hill R (2004) The human tumor microenvironment : invasive (needle) measurement of oxygen and interstitial fluid pressure. Semin Radiat Oncol 14 : 249-258
11 Pan JG, Mak TW (2007) Metabolic targeting as an anticancer strategy : dawn of a new era ? Sci STKE 2007 : e14
12 Rajaganeshan R, Prasad R, Guillou PJ, Poston G, Scott N, Jayne DG (2008) The role of hypoxia in recurrence following resection of Dukes’ B colorectal cancer. Int J Colorectal Dis 23 : 1049-1055
13 Rajaganeshan R, Prasad R, Guillou PJ, Scott N, Poston G, Jayne DG (2009) Expression patterns of hypoxic markers at the invasive margin of colorectal cancers and liver metastases. Eur J Surg Oncol 35 (12) : 1286-1294
14 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE (2009) Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 389 : 157-164
15 Shike M, Winawer SJ, Greenwald PH, Bloch A, Hill MJ, Swaroop SV (1990) Primary prevention of colorectal cancer. Le centre collaborateur de l’OMS pour la prévention du cancer colorectal. Bull World Health Organ 68 : 377-385
16 Shin YK, Yoo BC, Hong YS, Chang HJ, Jung KH, Jeong SY, Park JG (2009) Upregulation of glycolytic enzymes in proteins secreted from human colon cancer cells with 5-fluorouracil resistance. Electrophorèse 30 : 2182-2192
17 Stacpoole PW (1989) The pharmacology of dichloroacetate. Metabolism 38 : 1124-1144 (en anglais seulement)
18 Stacpoole PW, Henderson GN, Yan Z, James MO (1998) Clinical pharmacology and toxicology of dichloroacetate. Environ Health Perspect 106 (Suppl 4) : 989-994
19 Stacpoole PW, Nagaraja NV, Hutson AD (2003) Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol 43 : 683-691
20SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC (2009) Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 120 (1) : 253-260
21 Thorn CC, Freeman TC, Scott N, Guillou PJ, Jayne DG (2009) Laser microdissection expression profiling of marginal edges of colorectal tumours reveals evidence of increased lactate metabolism in the aggressive phenotype. Gut 58 : 404-412
22 Statistiques britanniques sur le cancer de l’intestin (2009) http://info.cancerresearchuk.org/cancerstats/types/bowel/
23 Warburg O (1956) On the origin of cancer cells. Science 123 : 309-314
24 Whitehouse S, Randle PJ (1973) Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate. Biochem J 134 : 651-653
25 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Le dichloroacétate induit l’apoptose dans les cellules cancéreuses de l’endomètre. Gynecol Oncol 109 : 394-402
26 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA (2007) Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 292 : C125-C136
27 Yeluri S, Madhok B, Prasad KR, Quirke P, Jayne DG (2009) Cancer’s craving for sugar : an opportunity for clinical exploitation. J Cancer Res Clin Oncol 135 : 867-877