BM Madhok*,1, S Yeluri1, SL Perry1, TA Hughes2 y DG Jayne1
1 Section of Translational Anaesthesia & Surgery, University of Leeds, Level 7 Clinical Sciences Building, St. James’s University Hospital, Leeds, UK
2 Leeds Institute of Molecular Medicine, University of Leeds, St. James’s University Hospital, Leeds, UK
Correspondencia: Dr BM Madhok; E-mail: [email protected]
Revisado: 23 de marzo de 2010
Aceptado: 26 de abril de 2010
Publicado: 18 de mayo de 2010
Resumen
Antecedentes
Las células cancerosas dependen en gran medida de la glucólisis. Nuestro objetivo era determinar si el cambio de metabolismo de la glucólisis hacia la respiración mitocondrial reduciría el crecimiento preferentemente en células de cáncer colorrectal sobre las células normales, y examinar los mecanismos subyacentes.
Métodos
Se trataron líneas celulares representativas de cáncer colorrectal y no cancerosas con dicloroacetato (DCA), un inhibidor de la piruvato deshidrogenasa cinasa.
Resultados
El dicloroacetato (20 mM) no redujo el crecimiento de las células no cancerosas, pero provocó una disminución significativa de la proliferación de las células cancerosas (P=0,009), que se asoció a la apoptosis y a la detención del ciclo celular en fase G2. El mayor efecto apoptótico fue evidente en las células metastásicas LoVo, en las que el DCA indujo un aumento de hasta diez veces en el recuento de células apoptóticas después de 48 h. La detención G2 más sorprendente fue evidente en las células HT29 bien diferenciadas, en las que el DCA causó un aumento de ocho veces en las células en fase G2 después de 48 h. El dicloroacetato redujo los niveles de lactato en los medios de crecimiento e indujo la desfosforilación de la subunidad E1α del complejo piruvato deshidrogenasa en todas las líneas celulares, pero el potencial de membrana mitocondrial intrínseco sólo se redujo en las células cancerosas (P=0,04).
Conclusiones
La inhibición de la piruvato deshidrogenasa cinasa atenúa la glucólisis y facilita la fosforilación oxidativa mitocondrial, lo que conduce a una reducción del crecimiento de las células de cáncer colorrectal, pero no de las células no cancerosas.
Palabras clave: dicloroacetato, cáncer colorrectal, piruvato deshidrogenasa, piruvato deshidrogenasa cinasa
British Journal of Cancer (2010) 102, 1746 – 1752, www.bjcancer.com
doi: 10.1038/sj.bjc.6605701
© 2010 Cancer Research UK
INTRODUCCIÓN
El cáncer colorrectal es el tercer cáncer más frecuente en el mundo y la cuarta causa de muerte relacionada con el cáncer (Shike et al, 1990). En 2007, el cáncer colorrectal representó 17,1 muertes por cada 100 000 personas en el Reino Unido (UK Bowel Cancer Statistics, 2009). A pesar de los avances recientes, el pronóstico de los pacientes con cáncer colorrectal avanzado y metastásico sigue siendo malo. La terapia del cáncer dirigida al metabolismo tumoral es un campo en rápido desarrollo (Pan y Mak, 2007). Las primeras observaciones sobre las diferencias metabólicas entre el cáncer y las células normales fueron realizadas por Otto Warburg, quien demostró que las células cancerosas dependen intrínsecamente de la glucólisis para la producción de energía química (Warburg, 1956). Ahora hay cada vez más pruebas de que este aumento de la glucólisis es el resultado de la influencia de múltiples vías moleculares, incluidas las respuestas adaptativas al microambiente tumoral hipóxico, la señalización oncogénica y la disfunción mitocondrial (Gatenby y Gillies, 2004; Gillies y Gatenby, 2007; Wu et al, 2007). El fenotipo glucolítico ofrece ventajas de crecimiento a las células cancerosas al resistirse a la apoptosis y facilitar la propagación tumoral y la metástasis (Yeluri et al, 2009).
Un regulador clave del metabolismo celular es la piruvato deshidrogenasa (PDH). La piruvato deshidrogenasa convierte el piruvato, producido a partir de la glucólisis, en acetil-CoA, que se oxida en el ciclo de los ácidos tricarboxílicos dentro de las mitocondrias. La actividad de la piruvato deshidrogenasa está estrechamente regulada por la fosforilación inhibitoria de la piruvato deshidrogenasa cinasa (PDK). La fosforilación se produce en la subunidad E1α de la PDH (PDHE1α) en tres sitios: Ser232, Ser293 y Ser300 (Rardin et al, 2009). El dicloroacetato (DCA) es un inhibidor de las cuatro isoenzimas de PDK(1-4) (Stacpoole, 1989), y recientemente se ha demostrado que reduce el crecimiento de líneas celulares de cáncer de pulmón, endometrio y mama (Bonnet et al, 2007; Wong et al, 2008; Sun et al, 2009). Se ha informado de que reduce el crecimiento de estas células cancerosas principalmente mediante la reducción de la fosforilación inhibitoria de la PDH, promoviendo así la fosforilación oxidativa mitocondrial e induciendo la apoptosis a través de vías mediadas por la mitocondria, NFAT-Kv 1.5 y el modulador de la apoptosis regulado por p53 (PUMA).
Se ha observado que las células de cáncer colorrectal sufren un aumento de la glucólisis (Bi et al, 2006) y que el microambiente tumoral es hipóxico y acidótico, debido principalmente a un suministro sanguíneo deficiente (Dewhirst et al, 1989; Milosevic et al, 2004). Anteriormente hemos demostrado que esto es especialmente cierto para el fenotipo más agresivo (Thorn et al, 2009), y la expresión de los marcadores importantes de la hipoxia se incrementa en el cáncer colorrectal, especialmente en el margen invasivo (Rajaganeshan et al, 2008, 2009). El propósito de este estudio fue investigar los efectos del DCA en el crecimiento de células de cáncer colorrectal en un intento de examinar la inhibición de PDK como una nueva estrategia terapéutica contra el cáncer colorrectal.
Materiales y métodos
Cultivos celulares
Todas las líneas celulares se adquirieron en la American Type Culture Collection (Manassas, VA, EE.UU.) o en la European Collection of Cell Cultures (Salisbury, Wiltshire, Reino Unido): HB2 (células epiteliales mamarias de origen no canceroso), 293 (células epiteliales de riñón de embrión humano), HT29 (adenocarcinoma colorrectal primario bien diferenciado), SW480 (adenocarcinoma colorrectal primario poco diferenciado) y LoVo (ganglio linfático supraclavicular izquierdo metastásico de adenocarcinoma colorrectal). las células 293 y HB2 se mantuvieron en medio DMEM, las HT29 y SW480 en medio RPMI 1640, y las LoVo en medio F12 (todos ellos de Invitrogen, Carlsbad, CA, EE.UU.), suplementado con un 10% de suero fetal de ternera, en un incubador humidificado a 37 °C y 5% deCO2. Para los experimentos en condiciones hipóxicas, incubamos las células en un incubador hipóxico humidificado (1% O2, 5%CO2, 94% N2, 37°C). El dicloroacetato sódico (Specials Lab, Prudhoe, Reino Unido) fue donado por el Departamento de Farmacia del Hospital Universitario St. James, Leeds, Reino Unido.
Ensayos MTT
Se sembraroncélulas(1 ×104) por pocillo en placas de cultivo tisular de 96 pocillos. Tras una incubación de una noche, se sustituyó el medio por medio fresco con dosis crecientes de DCA (0, 10, 15, 20, 30, 50 y 100 mM). Tras 24 y 48 h de incubación, se realizó el ensayo MTT sustituyendo el medio por 50 μlde solución de MTT de 1 mg ml-1 y las placas se incubaron en la oscuridad durante 3 h. A continuación, se retiró la solución de MTT y los precipitados de formazán azul oscuro se disolvieron en 100 μlde propan-1-ol. La densidad óptica se midió con un lector de microplacas (Opsys MR; Dynex Technologies Ltd, Worthing, West Sussex, Reino Unido) a 570 nm.
Annexin V and 7-AAD assays
Las célulasse sembraron en frascos de cultivo tisular de 25 cm2 y se incubaron durante la noche en condiciones estándar. El medio se sustituyó por medio fresco que contenía una serie de dosis de DCA (0, 10, 20 y 50 mM). Se realizaron análisis de citometría de flujo tras 24 y 48 horas de incubación. Las células se lavaron dos veces con PBS frío y se resuspendieron en 1 × tampón de unión (BD Bioscience, Franklin Lakes, NJ, EE.UU.) a 5 ×106 células por ml. se transfirieron 100 μlde solución (5 ×105 células) a tubos de cultivo de 5 ml. Estas células se tiñeron con 5 μlde anexina V-FITC y 10 μlde 7-AAD (BD Bioscience), se agitaron suavemente en vórtex y se incubaron a temperatura ambiente durante 15 min en la oscuridad. A continuación, se añadieron 400 μlde tampón de unión 1 × a cada tubo y se analizaron en una hora en el citómetro de flujo LSR II (BD Bioscience).
Ensayos con yoduro de propidio
Las célulasse propagaron como se ha mencionado para el ensayo de apoptosis. Se utilizó dicloroacetato (50 mM) y se comparó con el control del vehículo. Tras la recolección, se resuspendieron las células en 350 μlde PBS a una concentración de 0,5-1,0 ×106 células por ml. se añadieron 100 μlde 0,25 mg ml-1 de yoduro de propidio (PI)/5% de Tritón (Sigma, St Louis, MO, EE.UU.) a la suspensión celular. a continuación se añadieron 50 μlde 1 mg ml-1 de ribonucleasa A (Sigma). Los tubos de muestras se agitaron a fondo en vórtex y se incubaron durante 10 min en la oscuridad a temperatura ambiente. La citometría de flujo se realizó en un citómetro de flujo LSR II (BD Bioscience) y los datos se analizaron con el software FlowJo (FlowJo, Ashland, OR, EE.UU.).
Mediciones de lactato
Las mediciones delactatoen los medios de crecimiento fueron realizadas por el departamento de patología química del General Infirmary, Leeds Teaching Hospitals NHS Trust. Las células se incubaron en matraces de 25 cm2 durante la noche en normoxia. Al día siguiente se sustituyó el medio por una serie de dosis de DCA (0, 10, 20 y 50 mM). Tras 48 h de incubación, se recogieron 2 ml de medio en tubos de fluoruro y se transfirieron inmediatamente al laboratorio de patología química. Los tubos se mantuvieron en hielo durante el traslado. Los niveles de lactato se midieron con un analizador automático (sistema Advia 1200 Chemistry; Siemens Healthcare Diagnostics, Camberley, Surrey, Reino Unido).
Ensayos TMRM
Las célulasse trataron con DCA como se describe para el ensayo de apoptosis. Tras 24 y 48 h de incubación, se lavaron las células en PBS y se suspendieron 1 ×106 células por ml en solución salina tamponada de Hank con 50 nM de éster metílico de tetrametilrhodamina (TMRM) (Invitrogen). se transfirieron 100 μlde la suspensión celular (1 ×105 células por pocillo) a placas opacas de 96 pocillos, se incubaron durante 30 min y se midió la fluorescencia a 530/620 nm a 37 °C utilizando un lector de placas (Mithras LB 40; Berthold Technologies, Bad, Wildbad, Alemania).
Western blotting
Las célulasse trataron con DCA como se ha descrito anteriormente. Tras 8 h de tratamiento, se extrajeron proteínas de las células en tampón Laemmli (2% SDS, 10% glicerol, 0,7% 2-mercaptoetanol, 0,05% azul de bromofenol y 0,5 M Tris-HCl). Los lisados se resolvieron por electroforesis en geles NuPAGE Novex 12% Bis-Tris (Invitrogen) en tampón MOPS-SDS (Invitrogen). Las proteínas se transfirieron a una membrana de fluoruro de polivinilideno (GE Healthcare, Chalford St Giles, Bucks, Reino Unido). La membrana se bloqueó durante 1 hora a temperatura ambiente en leche desnatada al 5% en TBS-T (solución salina tamponada con Tris y 0,1% de Tween). A continuación, se sondeó la membrana con anticuerpos primarios en leche desnatada al 1% en TBS-T durante 90 minutos, se lavó en TBS-T y se sondeó con el anticuerpo secundario adecuado conjugado con peroxidasa de rábano picante (HRP) durante 60 minutos. Anticuerpos primarios policlonales de conejo fosfodetectados anti-PDH-E1α (pSer293), 1 :500 (AP1062; EMD Chemicals, Darmstadt, Alemania), y monoclonal de ratón anti-PDHE1α, 1 :500 (459400; Invitrogen). Anticuerpos secundarios conjugados HRP anti-conejo o anti-ratón, 1 :1000 (Dako, Glostrup, Dinamarca). Las proteínas se visualizaron con sustrato quimioluminiscente Supersignal West Pico o Femto (Pierce Biotechnology, Rockford, IL, EE.UU.) y el sistema Chemidoc XRS (Bio-Rad, Hercules, CA, EE.UU.). la β-actina se utilizó como control de carga.
Análisis estadísticos
Los datos de citometría de flujo se adquirieron utilizando software específico, BD FACSDiva 6.0 y software FlowJo. Los análisis estadísticos se realizaron con SPSS para Windows (SPSS versión 15.0, Chicago, IL, EE.UU.). Las diferencias entre los grupos tratados con DCA y los de control con vehículo se evaluaron mediante la prueba U de Mann-Whitney y los intervalos de confianza del 95% de la diferencia de medias entre los dos grupos. Un valor P inferior a 0,05 se consideró estadísticamente significativo. Los datos se representan como la media de al menos tres experimentos independientes y las barras de error representan la desviación estándar de la media.
Resultados
El DCA reduce la proliferación de células cancerosas y el efecto es similar en normoxia e hipoxia
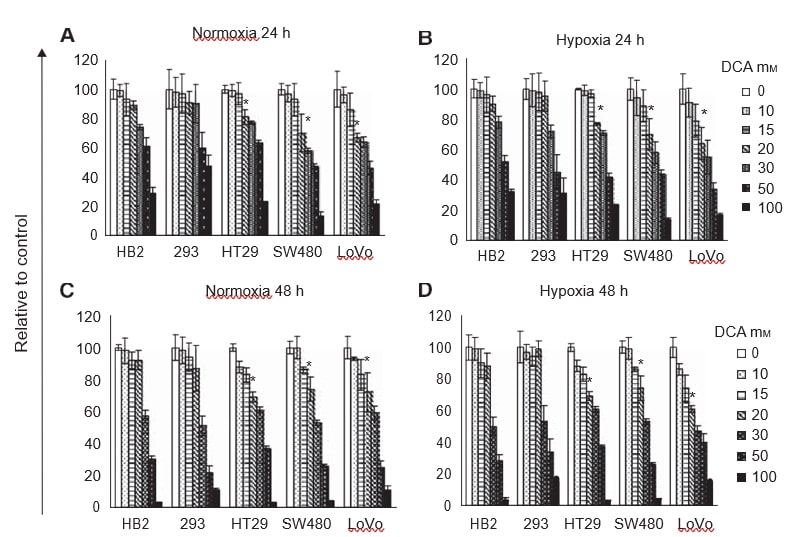
En primer lugar, queríamos determinar si el tratamiento con DCA inhibía la proliferación celular y si habría una respuesta diferencial en células cancerosas y no cancerosas en condiciones de normoxia e hipoxia. Con respecto a la hipoxia, nuestra hipótesis era que la influencia del DCA sería particularmente potente con niveles de oxígeno insuficientes para soportar una fosforilación oxidativa adicional. Todas las líneas celulares (HB2, 293, HT29, SW480 y LoVo) fueron tratadas con una serie de dosis de DCA durante 24-48 h en condiciones de normoxia e hipoxia. El número relativo de células se evaluó mediante ensayos MTT.
El tratamiento con dosis crecientes de DCA redujo la proliferación celular de forma dependiente de la dosis (Figura 1A-D). Contrariamente a lo que esperábamos, los perfiles de reducción del crecimiento celular fueron similares en hipoxia y normoxia. A las 24 y 48 h, hasta 20 mM de DCA no afectó al crecimiento de los cultivos de las células no cancerosas, HB2 y 293. Sin embargo, 20 mM de DCA afectó significativamente al crecimiento de las células no cancerosas, HB2 y 293. Sin embargo, 20 mM de DCA redujo significativamente el crecimiento de los cultivos de las tres líneas celulares de cáncer colorrectal (P⩽0,009). El efecto del DCA fue mayor en las células SW480 poco diferenciadas y en las células LoVo metastásicas que en las células HT29 bien diferenciadas. El crecimiento de los cultivos de células LoVo tratadas con 20 mM de DCA se redujo hasta en un 40% en comparación con las células tratadas con el vehículo de control. Dado que había relativamente poca diferencia en la reducción del crecimiento de los cultivos tratados con DCA en condiciones hipóxicas y normoxicas, se realizaron más experimentos sólo en normoxia.
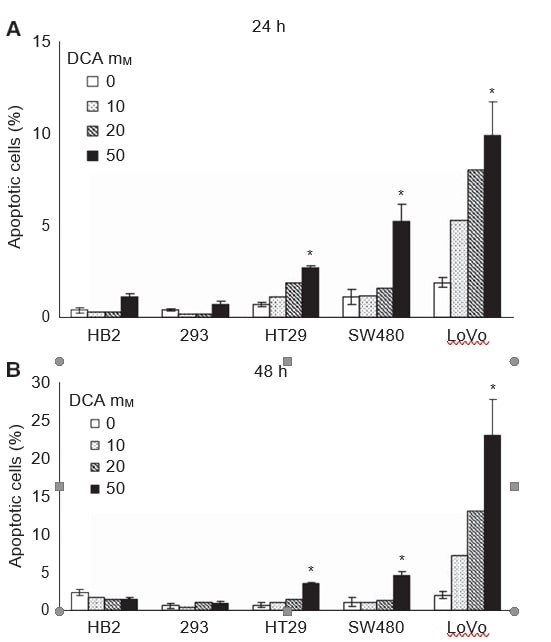
El DCA promueve la apoptosis en las células cancerosas, preservando las células no cancerosas
A continuación, quisimos investigar si la reducción del crecimiento de los cultivos en el tratamiento con DCA estaba asociada con la inducción de la apoptosis. Las células se trataron con distintas dosis de DCA (0, 10, 20 y 50 mM) durante 24 y 48 h, y la proporción de células que sufrían apoptosis se evaluó mediante la detección de fosfatidilserina de membrana con anexina V-FITC. Las células se tiñeron con anexina V-FITC y colorante vital 7-AAD, y se analizaron mediante citometría de flujo. Se produjo una inducción de la apoptosis dependiente de la dosis en las líneas celulares cancerosas tras 24 y 48 h de tratamiento, mientras que la apoptosis inducida en las células no cancerosas fue escasa o nula (Figura 2A y B). El mayor efecto se observó en las células metastásicas LoVo; 50 mM de DCA causó un aumento de diez veces en la proporción de células apoptóticas después de 48 h, mientras que hubo un aumento de siete y cinco veces en las células HT29 y SW480, respectivamente. El aumento del porcentaje medio de células apoptóticas totales con 50 mM de DCA fue: 2,8 (IC 95%: 2-3) en células HT29, 3,5 (IC 95%: 2-5) en células SW480 y 21 (IC 95%: 8-34) en células LoVo. La apoptosis inducida en las células 293 fue mínima incluso con 50 mM de DCA, 0,2 (IC 95%: -0,2 a 0,6). En las células HB2, se produjo una disminución no significativa del porcentaje de células apoptóticas en el tratamiento con 50 mM de DCA, -0,9 (IC del 95%: -2,2 a 0,4).
El DCAinduce la detención de la fase G2 en células de cáncer colorrectal pero no tiene ningún efecto sobre el perfil del ciclo celular de las células 293 no cancerosas
También quisimos examinar si la reducción del crecimiento de los cultivos en el tratamiento con DCA estaba asociada con la inducción de la detención del crecimiento. Las células se trataron con 50 mM de DCA durante 24 o 48 h, y los perfiles del ciclo celular se analizaron mediante citometría de flujo del contenido de ADN tras la tinción con PI. El tratamiento con dicloroacetato provocó cambios en los perfiles del ciclo celular de todas las células cancerosas, pero no afectó a las células no cancerosas. Los cambios en el perfil del ciclo celular fueron detectables tras 24 h de tratamiento y persistieron a las 48 h (Figura 3A y B).
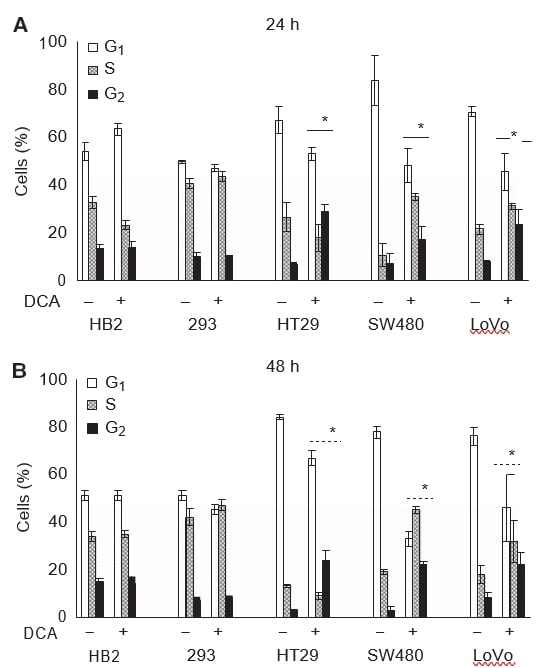
Tras 48 h de tratamiento con 50 mM de DCA, se produjo un aumento de ocho veces de las células en fase G2 en las células HT29 y SW480, y de tres veces en las células LoVo. El aumento del porcentaje medio de todas las células cancerosas en fase G2 fue: 21 (IC 95%: 13-30) para las células HT29, 19 (IC 95%: 13-24) para las células SW480, y 14 (IC 95%: 10-21) para las células LoVo; mientras que no hubo diferencias en las células 293, 1 (IC 95%: -4 a 7), y las células HB2, -0,3 (IC 95%: -9 a 9). En todas las líneas celulares cancerosas se produjo una disminución correspondiente de las células en fase G0/G1. Curiosamente, en las células HT29 se produjo una pequeña disminución, pero en las células SW480 y LoVo se produjo un aumento significativo de la proporción de células consideradas en fase S (véase la sección Discusión). El perfil del ciclo celular de las células 293 y HB2 cambió mínimamente con el tratamiento con DCA.
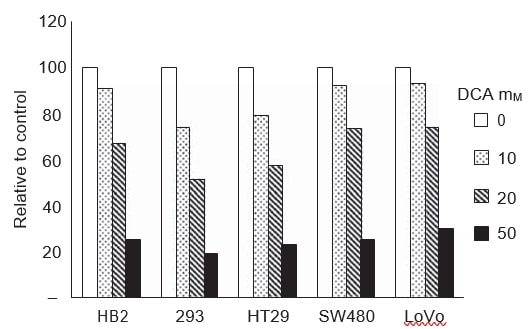
El DCA reduce los niveles de lactato extracelular en los medios de crecimiento
Para establecer si los cambios en el crecimiento y la apoptosis inducidos por el DCA se correlacionaban con una reducción de la glucólisis, medimos los niveles de lactato en los medios de crecimiento. El ácido láctico es el producto final de la glucólisis. Si el DCA estuviera induciendo la fosforilación oxidativa mitocondrial, el piruvato se descarboxilaría a acetil-CoA y no se reduciría a lactato, por lo que los niveles de lactato en los medios de crecimiento disminuirían. Los niveles de lactato en los medios de crecimiento de todas las líneas celulares se midieron tras 48 h de tratamiento con una serie de dosis de DCA (Figura 4). Los niveles de lactato se determinaron con un autoanalizador que se utiliza habitualmente para la medición bioquímica de los niveles de lactato; los ensayos se basan en una reacción colorimétrica catalizada por la lactato oxidasa. El tratamiento con DCA redujo los niveles de lactato extracelular en los medios de crecimiento de forma dependiente de la dosis en todas las líneas celulares cancerosas y no cancerosas.
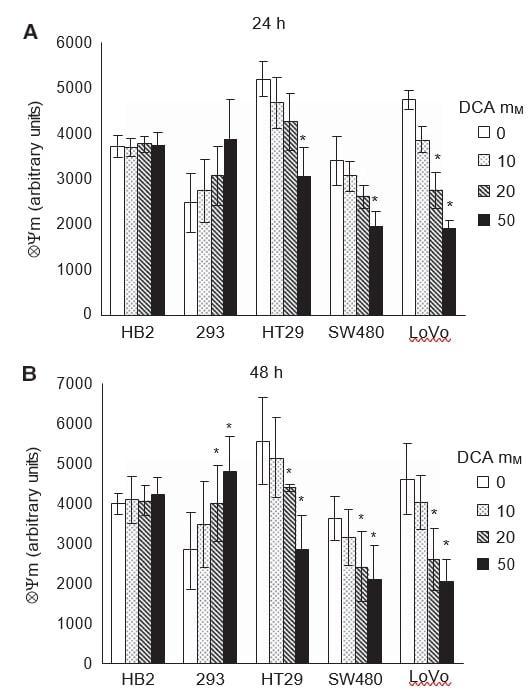
ElDCA despolariza la membrana mitocondrial intrínseca en células de cáncer colorrectal pero no en células no cancerosas
Para verificar si la inducción de la apoptosis en células cancerosas en tratamiento con DCA estaba asociada con la promoción de la fosforilación oxidativa mitocondrial, medimos el potencial de membrana mitocondrial intrínseca (ΔΨm). La intensificación de la respiración mitocondrial reactivaría la cadena de transporte de electrones y reduciría el ΔΨm hiperpolarizado en las células cancerosas. Las células se trataron con dosis de DCA durante 24 y 48 h y se tiñeron con el colorante TMRM, que permite la medición fluorescente de ΔΨm.
Al igual que en experimentos anteriores, el efecto del DCA fue evidente tras 24 h de tratamiento y persistió a las 48 h (Figura 5A y B). El tratamiento con dicloroacetato redujo la ΔΨm hiperpolarizada en todas las células cancerosas de forma dependiente de la dosis. El dicloroacetato no tuvo ningún efecto sobre el ΔΨm de las células HB2 no cancerosas, mientras que, sorprendentemente, el ΔΨm de las células 293 no cancerosas aumentó de forma dependiente de la dosis. A las 24 h de tratamiento, 50 mM de DCA redujeron significativamente el ΔΨm en todas las células cancerosas; sin embargo, en las células LoVo se produjo una reducción significativa incluso con 20 mM de DCA (Figura 5A, P=0,02). En las células 293 no cancerosas, se observó una tendencia al aumento de ΔΨm en el tratamiento con DCA, aunque no fue estadísticamente significativa (P=0,08). A las 48 h de tratamiento, se produjo una reducción significativa de ΔΨm en todas las células cancerosas y un aumento en las células 293, con 20-50 mM de DCA (Figura 5B, P⩽0,04).
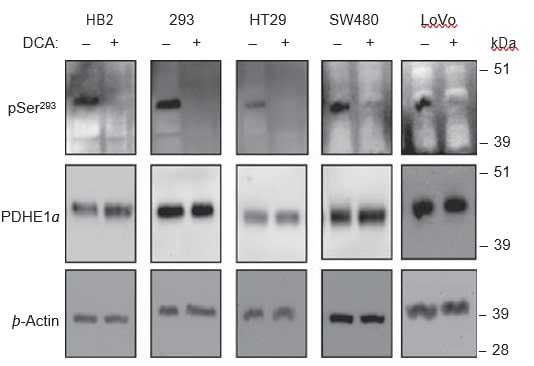
Eltratamiento con DCA conduce a la desfosforilación de lasubunidadPDHE1α
Se cree queel DCAinhibe las cuatro isoenzimas de PDK y, por tanto, reduce la fosforilación de la subunidad PDHE1α, lo que conduce, a su vez, a la activación del complejo PDH. Para verificar si se producía la desfosforilación de PDHE1α con el tratamiento con DCA en las líneas celulares utilizadas, utilizamos análisis de Western blot en lisados de células tratadas y no tratadas con DCA. En todas las líneas celulares, el tratamiento con 20 mM de DCA durante 8 h provocó una drástica reducción de la señal de fosforilación en el sitio pSer293, pero no se detectaron cambios en los niveles de PDHE1α total (Figura 6). Los anticuerpos fosfoespecíficos para los otros dos sitios de fosforilación, Ser232 y Ser300, aún no están disponibles comercialmente.
Discusión
Hemos demostrado que el DCA induce una reducción dependiente de la dosis en el crecimiento de cultivos in vitro de células de cáncer colorrectal y de células no cancerosas. Sin embargo, las células cancerosas fueron más sensibles al DCA, con una dosis de 20 mM causando una inhibición significativa del crecimiento de las células cancerosas, pero teniendo poco efecto sobre las células no cancerosas. Hemos demostrado que los componentes de este efecto diferencial son los siguientes: una potente inducción de la apoptosis y la detención del ciclo celular en las células cancerosas, pero no en las células no cancerosas.
Estas conclusiones apoyan un modelo simple de sensibilidad diferencial al DCA. Sin embargo, algunos datos requieren una mayor discusión. En primer lugar, 50 mM de DCA redujo el crecimiento de los cultivos de las células no cancerosas 293 y HB2, pero no se observó ningún aumento de células apoptóticas ni ningún cambio en el perfil del ciclo celular de estas células. Una posible explicación de estos hallazgos podría ser que esta dosis de DCA condujo a un tránsito más lento de estas células no cancerosas a través de todas las etapas del ciclo celular, sin cambiar las proporciones relativas dentro de cada etapa. En segundo lugar, nuestros resultados indican que el DCA indujo la detención G2 en las células de cáncer colorrectal. Esto contrasta con estudios anteriores, que han mostrado una detención G1 o ningún cambio en el perfil del ciclo celular con el tratamiento con DCA (Cao et al, 2008; Wong et al, 2008). Wong et al (2008) mostraron un aumento de la expresión de PUMA en todas las líneas celulares de cáncer de endometrio que presentaban una respuesta apoptótica al DCA, y concluyeron que esta activación de p53 conducía a la detención en G1. Sin embargo, las células de cáncer colorrectal de nuestro estudio se detuvieron en la fase G2 en el tratamiento con DCA, y no encontramos ninguna inducción de p53 por DCA en nuestras líneas celulares de cáncer colorrectal (datos no mostrados). Curiosamente, Cao et al (2008) descubrieron que la combinación de DCA y radioterapia detenía las células de cáncer de próstata en fase G2, aunque el DCA por sí solo no afectaba al perfil del ciclo celular. En tercer lugar, en las células SW480 y LoVo, el tratamiento con DCA dio lugar a un aumento de la proporción de células consideradas en fase S. Esto sugiere un aumento de la proliferación a medida que las células se desarrollan. Esto sugiere un aumento de la proliferación, así como la inducción de la apoptosis. Un hallazgo similar fue comunicado por Wong et al (2008) en una de varias células de cáncer de endometrio analizadas. Una explicación alternativa es que una proporción de las células observadas en «fase S» tras el tratamiento con DCA de las líneas celulares cancerosas representen en realidad células apoptóticas en la región «sub-G2«, como se ha señalado anteriormente en células de linfoma (Klucar y Al-Rubeai, 1997).
Cambios en el metabolismo celular con el tratamiento con DCA
El DCApareció suprimir la producción de ácido láctico a partir de piruvato tanto en células cancerosas como no cancerosas. Además, el tratamiento con DCA provocó la desfosforilación de PDHE1α y, por tanto, la activación de PDH en todas las líneas celulares investigadas. Por lo tanto, la base del efecto diferencial del DCA sobre las células cancerosas y no cancerosas puede residir en su influencia sobre la función mitocondrial. El tratamiento con DCA redujo la elevada ΔΨm de todas las células cancerosas, pero no de las no cancerosas. Esto sugiere que el DCA, al inhibir la PDK y por lo tanto activar la PDH, promueve la respiración mitocondrial que conduce a la despolarización de la membrana mitocondrial intrínseca, e induce la apoptosis por la vía mitocondrial proximal como se describe en los estudios anteriores (Bonnet et al, 2007; Cao et al, 2008; Wong et al, 2008). La inducción de la apoptosis y los cambios en la función mitocondrial fueron más pronunciados en las células LoVo altamente invasivas y metastásicas que en las células HT29 y SW480, menos invasivas. Esto podría tener implicaciones clínicas para el tratamiento del cáncer colorrectal metastásico, ya que suelen ser los cánceres metastásicos altamente invasivos los más resistentes a la quimioterapia convencional, y los que podrían ser más sensibles a la inhibición de la PDK. En apoyo de esta idea, un estudio reciente ha informado de que los tumores colorrectales resistentes al 5-fluorouracilo tienen más probabilidades de presentar una regulación al alza de la glucólisis y, por tanto, son más susceptibles de terapias dirigidas al metabolismo del cáncer (Shin et al, 2009). En este sentido, nuestros resultados contrastan con los de Wong et al (2008), que hallaron que las células de cáncer de endometrio altamente invasivo eran las más resistentes al tratamiento con DCA.
Inhibición de PDK como terapia contra el cáncer colorrectal
Encontramos que dosis de 20-50 mM de DCA daban respuestas diferenciales entre células cancerosas y no cancerosas. Por lo tanto, las dosis terapéuticas potenciales de DCA estarían entre 20 y 50 mM. Además, un estudio reciente informó de que el IC50 del DCA para las células de cáncer de mama se sitúa entre 20 y 30 mM (Ko y Allalunis-Turner, 2009). Esto contrasta con estudios anteriores que han informado de que el DCA reduce la proliferación e induce la apoptosis en células cancerosas con dosis tan bajas como 0,5-10 mM (Bonnet et al, 2007; Wong et al, 2008; Sun et al, 2009). Se ha comprobado que el dicloroacetato es relativamente seguro en humanos cuando se utiliza para el tratamiento de la acidosis láctica (Stacpoole et al, 2003). Los principales efectos secundarios con hasta 100 mg kg-1 de DCA son sobre el sistema nervioso y el hígado, causando sedación o somnolencia leve, neuropatía periférica reversible y elevación asintomática leve de las transaminasas séricas que refleja daño hepatocelular (Stacpoole et al, 1998). Además, estudios recientes informaron de que el DCA reducía eficazmente el crecimiento tumoral en dosis clínicamente alcanzables tanto in vitro como in vivo (Bonnet et al, 2007; Sun et al, 2009). Se sugirió que el DCA podría trasladarse rápidamente a ensayos clínicos de cáncer en fase inicial (Michelakis et al, 2008). Sin embargo, es improbable que la dosis de DCA necesaria para inhibir el crecimiento de células de cáncer colorrectal en nuestro estudio pueda alcanzarse clínicamente sin causar efectos secundarios significativos. La dosis de DCA necesaria para alcanzar las concentraciones plasmáticas equivalentes in vivo sería entre cinco y diez veces superior a la utilizada en los ensayos clínicos contra la acidosis láctica. Parece que las células de cáncer colorrectal utilizadas en nuestro estudio son más resistentes al DCA que las células de cáncer de pulmón, endometrio y mama. Curiosamente, Sun et al (2009), en su estudio sobre células de cáncer de mama, descubrieron que el DCA inhibía la proliferación de las células cancerosas, pero no inducía la apoptosis ni la muerte celular. Estos resultados fueron notablemente diferentes a los efectos del DCA observados en células de cáncer de pulmón (Bonnet et al, 2007), endometrio (Wong et al, 2008) y colorrectal en nuestro estudio. Así pues, aunque el DCA inhibe el crecimiento de diversas células cancerosas, el efecto y los mecanismos subyacentes parecen depender del tipo celular. Una explicación probable de estos efectos diferenciales podría ser la diferencia en la expresión de las isoenzimas PDK en las células cancerosas examinadas. El dicloroacetato es un inhibidor inespecífico de la PDK (Whitehouse y Randle, 1973), y tiene unKi diferente para cada una de las cuatro isoenzimas PDK (Bowker-Kinley et al, 1998). Además, se sabe que las cuatro isoenzimas PDK se expresan de forma diferencial en diversos tejidos. Por lo tanto, es necesario desarrollar inhibidores de las isoenzimas PDK individuales que permitan una manipulación metabólica específica para cada tipo de célula cancerosa.
REFERENCIAS
1 Bi X, Lin Q, Foo TW, Joshi S, You T, Shen HM, Ong CN, Cheah PY, Eu KW, Hew CL (2006) Proteomic analysis of colorectal cancer reveals alterations in metabolic pathways: mechanism of tumorigenesis. Mol Cell Proteomics 5: 1119-1130
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11: 37-51
3 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 329 (Parte 1): 191-196
4 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 68: 1223-1231
4 Dewhirst MW, Tso CY, Oliver R, Gustafson CS, Secomb TW, Gross JF (1989) Morphologic and hemodynamic comparison of tumor and healing normal tissue microvasculature. Int J Radiat Oncol Biol Phys 17: 91-99
5 Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis? Nat Rev Cancer 4: 891-899
6 Gillies RJ, Gatenby RA (2007) Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? J Bioenerg Biomembr 39: 251-257
7 Klucar J, Al-Rubeai M (1997) G2 cell cycle arrest and apoptosis are induced in Burkitt’s lymphoma cells by the anticancer agent oracin. FEBS Lett 400: 127-130
8 Ko L, Allalunis-Turner J (2009) Investigation on the mechanism of dichloroacetate (DCA) induced apoptosis in breast cancer. J Clin Oncol 27 (Suppl 15): e14637
9 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99: 989-994
10 Milosevic M, Fyles A, Hedley D, Hill R (2004) The human tumor microenvironment: invasive (needle) measurement of oxygen and interstitial fluid pressure. Semin Radiat Oncol 14: 249-258
11 Pan JG, Mak TW (2007) Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE 2007: e14
12 Rajaganeshan R, Prasad R, Guillou PJ, Poston G, Scott N, Jayne DG (2008) The role of hypoxia in recurrence following resection of Dukes’ B colorectal cancer. Int J Colorectal Dis 23: 1049-1055
13 Rajaganeshan R, Prasad R, Guillou PJ, Scott N, Poston G, Jayne DG (2009) Expression patterns of hypoxic markers at the invasive margin of colorectal cancers and liver metastases. Eur J Surg Oncol 35 (12): 1286-1294
14 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE (2009) Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 389: 157-164
15 Shike M, Winawer SJ, Greenwald PH, Bloch A, Hill MJ, Swaroop SV (1990) Primary prevention of colorectal cancer. El Centro Colaborador de la OMS para la Prevención del Cáncer Colorrectal. Bull World Health Organ 68: 377-385
16 Shin YK, Yoo BC, Hong YS, Chang HJ, Jung KH, Jeong SY, Park JG (2009) Upregulation of glycolytic enzymes in proteins secreted from human colon cancer cells with 5-fluorouracil resistance. Electroforesis 30: 2182-2192
17 Stacpoole PW (1989) The pharmacology of dichloroacetate. Metabolism 38: 1124-1144
18 Stacpoole PW, Henderson GN, Yan Z, James MO (1998) Clinical pharmacology and toxicology of dichloroacetate. Environ Health Perspect 106 (Suppl 4): 989-994
19 Stacpoole PW, Nagaraja NV, Hutson AD (2003) Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol 43: 683-691
20SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC (2009) Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 120 (1): 253-260
21 Thorn CC, Freeman TC, Scott N, Guillou PJ, Jayne DG (2009) Laser microdissection expression profiling of marginal edges of colorectal tumours reveals evidence of increased lactate metabolism in the aggressive phenotype. Gut 58: 404-412
22 UK Bowel Cancer Statistics (2009) http://info.cancerresearchuk.org/cancerstats/types/bowel/
23 Warburg O (1956) On the origin of cancer cells (Sobre el origen de las células cancerosas). Science 123: 309-314
24 Whitehouse S, Randle PJ (1973) Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate. Biochem J 134: 651-653
25 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 109: 394-402
26 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA (2007) Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol 292: C125-C136
27 Yeluri S, Madhok B, Prasad KR, Quirke P, Jayne DG (2009) Cancer’s craving for sugar: an opportunity for clinical exploitation. J Cancer Res Clin Oncol 135: 867-877