BM Madhok*,1, S Yeluri1, SL Perry1, TA Hughes2 en DG Jayne1
1 Section of Translational Anaesthesia & Surgery, University of Leeds, Level 7 Clinical Sciences Building, St. James’s University Hospital, Leeds, UK
2 Leeds Institute of Molecular Medicine, University of Leeds, St. James’s University Hospital, Leeds, UK
Correspondentie: Dr BM Madhok; E-mail: [email protected]
Herzien: 23 maart 2010
Aanvaard: 26 april 2010
Gepubliceerd: 18 mei 2010
Abstract
Achtergrond
Kankercellen zijn sterk afhankelijk van glycolyse. Ons doel was om te bepalen of het omschakelen van het metabolisme van glycolyse naar mitochondriale ademhaling de groei van dikkedarmkankercellen preferentieel zou verminderen ten opzichte van normale cellen, en om de onderliggende mechanismen te onderzoeken.
Methoden
Representatieve colorectale kanker en niet-kankerachtige cellijnen werden behandeld met dichlooracetaat (DCA), een remmer van pyruvaat dehydrogenase kinase.
Resultaten
Dichlooracetaat (20 mM) verminderde de groei van niet-kankercellen niet, maar veroorzaakte een significante afname van de proliferatie van kankercellen (P=0,009), die gepaard ging met apoptose en celcyclusstilstand in de G2-fase. Het grootste apoptotische effect was duidelijk bij metastatische LoVo-cellen, waar DCA tot een tienvoudige toename van het aantal apoptotische cellen veroorzaakte na 48 uur. De meest opvallende G2-stilstand was duidelijk bij goed gedifferentieerde HT29-cellen, waar DCA een achtvoudige toename van cellen in de G2-fase veroorzaakte na 48 uur. Dichlooracetaat verlaagde het lactaatgehalte in de groeimedia en induceerde de defosforylering van de E1α-subeenheid van het pyruvaatdehydrogenasecomplex in alle cellijnen, maar het intrinsieke mitochondriale membraanpotentieel werd alleen in kankercellen verlaagd (P=0,04).
Conclusies
Inhibitie van pyruvaatdehydrogenase kinase vermindert de glycolyse en vergemakkelijkt mitochondriale oxidatieve fosforylering, hetgeen leidt tot verminderde groei van colorectale kankercellen, maar niet van niet-kankercellen.
Trefwoorden: dichlooracetaat, colorectale kanker, pyruvaat dehydrogenase, pyruvaat dehydrogenase kinase
British Journal of Cancer (2010) 102, 1746 – 1752, www.bjcancer.com
doi: 10.1038/sj.bjc.6605701
© 2010 Cancer Research UK
INLEIDING
Darmkanker is de derde meest voorkomende vorm van kanker ter wereld en de vierde belangrijkste doodsoorzaak door kanker (Shike et al, 1990). In 2007 was colorectale kanker verantwoordelijk voor 17,1 sterfgevallen per 100 000 personen in het Verenigd Koninkrijk (UK Bowel Cancer Statistics, 2009). Ondanks recente vooruitgang blijft de prognose van patiënten met gevorderde en uitgezaaide colorectale kanker slecht. Het richten van het tumormetabolisme op kankertherapie is een snel ontwikkelend gebied (Pan en Mak, 2007). Vroege waarnemingen betreffende de metabole verschillen tussen kankercellen en normale cellen werden gedaan door Otto Warburg, die aantoonde dat kankercellen inherent afhankelijk zijn van glycolyse voor de productie van chemische energie (Warburg, 1956). Er is nu steeds meer bewijs dat deze verhoogde glycolyse het gevolg is van de invloed van meerdere moleculaire routes, waaronder adaptieve reacties op de hypoxische tumormicro-omgeving, oncogene signalering en mitochondriale disfunctie (Gatenby en Gillies, 2004; Gillies en Gatenby, 2007; Wu et al, 2007). Het glycolytische fenotype biedt kankercellen groeivoordelen doordat het apoptose weerstaat en de verspreiding en uitzaaiing van de tumor vergemakkelijkt (Yeluri et al, 2009).
Een belangrijke regulator van het celmetabolisme is pyruvaatdehydrogenase (PDH). Pyruvaatdehydrogenase zet pyruvaat, afkomstig uit de glycolyse, om in acetyl-CoA, dat wordt geoxideerd in de tricarbonzuurcyclus in de mitochondriën. De activiteit van pyruvaatdehydrogenase wordt strak gereguleerd door remmende fosforylering door pyruvaatdehydrogenase kinase (PDK). Fosforylering vindt plaats op de E1α-subeenheid van PDH (PDHE1α) op drie plaatsen: Ser232, Ser293 en Ser300 (Rardin et al, 2009). Dichlooracetaat (DCA) is een remmer van alle vier de iso-enzymen van PDK(1-4) (Stacpoole, 1989), en recentelijk is aangetoond dat het de groei van long-, endometrium- en borstkankercellijnen vermindert (Bonnet et al, 2007; Wong et al, 2008; Sun et al, 2009). Er is gemeld dat het de groei van deze kankercellen vermindert, voornamelijk door de remmende fosforylering van PDH te verminderen en daardoor de mitochondriale oxidatieve fosforylering te bevorderen en apoptose te induceren via mitochondriale, NFAT-Kv 1.5 en p53 upregulated modulator of apoptosis (PUMA)-gemedieerde routes.
Gebleken is dat colorectale kankercellen een verhoogde glycolyse ondergaan (Bi et al, 2006), en dat de tumormicro-omgeving hypoxisch en acidotisch is, voornamelijk als gevolg van een slecht ontwikkelde bloedtoevoer (Dewhirst et al, 1989; Milosevic et al, 2004). Wij hebben eerder aangetoond dat dit vooral geldt voor het agressievere fenotype (Thorn et al, 2009), en dat de expressie van de belangrijke markers van hypoxie verhoogd is bij colorectale kanker, vooral bij de invasieve marge (Rajaganeshan et al, 2008, 2009). Het doel van deze studie was de effecten van DCA op de groei van colorectale kankercellen te onderzoeken in een poging PDK-inhibitie als nieuwe therapeutische strategie tegen colorectale kanker te onderzoeken.
Materialen en methoden
Celculturen
Alle cellijnen werden gekocht van American Type Culture Collection (Manassas, VA, USA) of European Collection of Cell Cultures (Salisbury, Wiltshire, UK): HB2 (borstepitheelcellen van niet-kanker oorsprong), 293 (epitheelcellen van menselijke embryonale nieren), HT29 (goed gedifferentieerd primair colorectaal adenocarcinoom), SW480 (slecht gedifferentieerd primair colorectaal adenocarcinoom), en LoVo (metastatische linker supraclaviculaire lymfeklier van colorectaal adenocarcinoom). 293 en HB2 cellen werden onderhouden in DMEM medium, HT29 en SW480 in RPMI 1640 medium, en LoVo in F12 medium (alle van Invitrogen, Carlsbad, CA, USA), aangevuld met 10% foetaal kalfsserum, in een 37°C, 5%CO2 bevochtigde incubator. Voor experimenten onder hypoxische omstandigheden incubeerden we de cellen in een bevochtigde, hypoxische incubator (1% O2, 5%CO2, 94% N2, 37°C). Natriumdichlooracetaat (Specials Lab, Prudhoe, UK) werd geschonken door de apotheekafdeling van het St. James’s University Hospital, Leeds, UK.
MTT-tests
Cellen(1 ×104) per well werden uitgezaaid in 96-wells weefselkweekplaten. Na een nacht incuberen werd de media vervangen door verse media met toenemende doses DCA (0, 10, 15, 20, 30, 50 en 100 mM). Na 24 en 48 uur incubatie voerden we MTT-test uit door de media te vervangen door 50ulvan 1 mg ml-1 MTT-oplossing en de platen werden gedurende 3 uur in het donker geïncubeerd. MTT-oplossing werd vervolgens verwijderd en de donkerblauwe formazanneerslag werd opgelost in 100ulpropaan-1-ol. De optische dichtheid werd gemeten met een microplaatlezer (Opsys MR; Dynex Technologies Ltd, Worthing, West Sussex, UK) bij 570 nm.
Annexine V en 7-AAD assays
De cellenwerden gezaaid in 25 cm2 weefselkweekflessen en overnacht geïncubeerd onder standaardomstandigheden. De media werden vervangen door verse media met een reeks doses DCA (0, 10, 20 en 50 mM). Flowcytometrische analyse werd uitgevoerd na 24 en 48 uur incubatie. De cellen werden tweemaal gewassen met koude PBS en geresuspendeerd in 1 × bindingsbuffer (BD Bioscience, Franklin Lakes, NJ, USA) bij 5 ×106 cellen per ml. 100μloplossing (5 ×105 cellen) werd overgebracht naar 5 ml kweekbuisjes. Deze cellen werden gekleurd met 5μlannexine V-FITC en 10μl7-AAD (BD Bioscience), voorzichtig geveegd, en geïncubeerd bij omgevingstemperatuur gedurende 15 minuten in het donker. Vervolgens werd aan elke buis 400μl1 × bindingsbuffer toegevoegd en binnen een uur geanalyseerd op de LSR II-flowcytometer (BD Bioscience).
Propidiumjodidetests
De cellenwerden vermeerderd zoals vermeld voor de apoptosetest. Dichlooracetaat (50 mM) werd gebruikt en vergeleken met voertuigcontrole. Na het oogsten resuspendeerden we de cellen in 350μlPBS bij een concentratie van 0,5-1,0 ×106 cellen per ml. 100ulvan 0,25 mg ml-1 propidiumjodide (PI)/5% Triton (Sigma, St Louis, MO, USA) werd toegevoegd aan de celsuspensie. vervolgens werd 50μl1 mg ml-1 ribonuclease A (Sigma) toegevoegd. De monsterbuizen werden grondig geveerd en gedurende 10 minuten in het donker bij kamertemperatuur geïncubeerd. Flowcytometrie werd uitgevoerd op de LSR II flowcytometer (BD Bioscience) en de gegevens werden geanalyseerd met de FlowJo-software (FlowJo, Ashland, OR, USA).
Lactaatmetingen
Lactaatmetingenin groeimedia werden uitgevoerd door de afdeling chemische pathologie van de General Infirmary, Leeds Teaching Hospitals NHS Trust. De cellen werden geïncubeerd in kolven van 25 cm2 gedurende een nacht in normoxie. De volgende dag werd het medium vervangen door een reeks doses DCA (0, 10, 20 en 50 mM). Na 48 uur incubatie verzamelden we 2 ml media in fluoridebuisjes en brachten die onmiddellijk over naar het chemisch pathologisch laboratorium. De buisjes werden tijdens de overdracht op ijs gehouden. Het lactaatgehalte werd gemeten met een automatische analyser (Advia 1200 Chemistry system; Siemens Healthcare Diagnostics, Camberley, Surrey, UK).
TMRM-tests
De cellenwerden behandeld met DCA zoals beschreven voor de apoptosetest. Na 24 en 48 uur incubatie werden de cellen gewassen in PBS en werden 1 ×106 cellen per ml gesuspendeerd in Hank’s gebufferde zoutoplossing met 50 nM tetramethylrhodamine-methylester (TMRM) (Invitrogen). 100ulvan de celsuspensie (1 ×105 cellen per well) werd overgebracht naar ondoorzichtige 96-wells platen, geïncubeerd gedurende 30 min, en de fluorescentie werd gemeten bij 530/620 nm bij 37°C met behulp van een plaatlezer (Mithras LB 40; Berthold Technologies, Bad, Wildbad, Duitsland).
Western blotting
Cellenwerden behandeld met DCA zoals hierboven beschreven. Na 8 uur behandeling extraheerden we eiwitten uit de cellen in Laemmli buffer (2% SDS, 10% glycerol, 0,7% 2-mercaptoethanol, 0,05% broomfenolblauw en 0,5 M Tris-HCl). De lysaten werden opgelost door elektroforese op NuPAGE Novex 12% Bis-Tris-gels (Invitrogen) in MOPS-SDS-buffer (Invitrogen). Eiwitten werden overgebracht op een polyvinylideenfluoridemembraan (GE Healthcare, Chalford St Giles, Bucks, UK). Het membraan werd gedurende 1 uur bij kamertemperatuur geblokkeerd in 5% magere melk in TBS-T (Tris gebufferde zoutoplossing met 0,1% Tween). Het membraan werd vervolgens gedurende 90 minuten geprofileerd met primaire antilichamen in 1% magere melk in TBS-T, gewassen in TBS-T en vervolgens gedurende 60 minuten geprofileerd met het juiste mierikswortelperoxidase (HRP)-geconjugeerde secundaire antilichaam. Primaire antilichamen rabbit polyclonal phosphodetect anti-PDH-E1α (pSer293), 1 :500 (AP1062; EMD Chemicals, Darmstadt, Duitsland), en muismonoklonaal anti-PDHE1α, 1 :500 (459400; Invitrogen). Secundaire antilichamen anti-rabbit of anti-muis HRP-conjugaten, 1 :1000 (Dako, Glostrup, Denemarken). Eiwitten werden gevisualiseerd met Supersignal West Pico of Femto chemiluminescent substraat (Pierce Biotechnology, Rockford, IL, USA) en het Chemidoc XRS systeem (Bio-Rad, Hercules, CA, USA). β-Actine werd gebruikt als laadcontrole.
Statistische analyses
Flowcytometriegegevens werden verkregen met specifieke software, BD FACSDiva 6.0 en FlowJo-software. Statistische analyses werden uitgevoerd met SPSS voor Windows (SPSS versie 15.0, Chicago, IL, USA). Verschillen tussen met DCA behandelde en voertuigcontrolegroepen werden beoordeeld met de Mann-Whitney U-test en de 95%-betrouwbaarheidsintervallen van het verschil in gemiddelden tussen beide groepen. Een P-waarde van minder dan 0,05 werd beschouwd als statistisch significant. De gegevens worden weergegeven als gemiddelde van ten minste drie onafhankelijke experimenten en de foutbalkjes geven de standaardafwijking van het gemiddelde weer.
Resultaten
DCA vermindert de proliferatie van kankercellen en het effect is vergelijkbaar bij normoxie en hypoxie
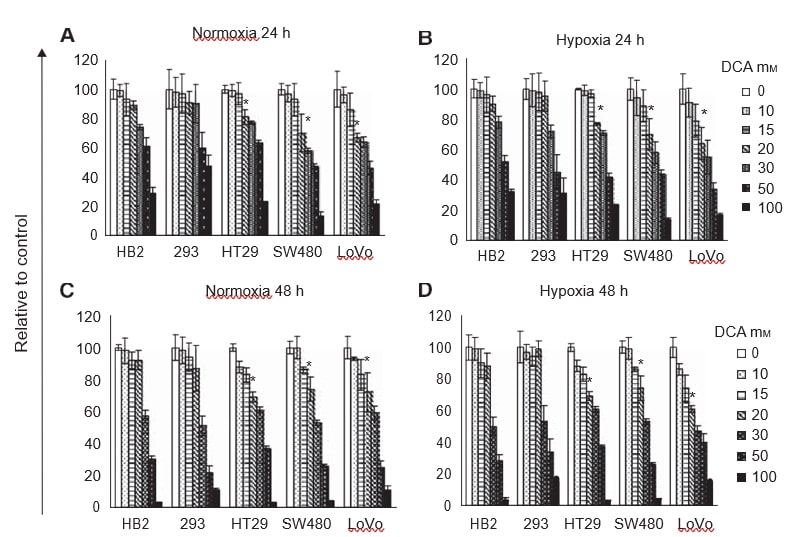
Allereerst wilden wij nagaan of behandeling met DCA de celproliferatie remt en of er een verschillende respons zou zijn bij kankercellen en niet-kankercellen in normoxische en hypoxische omstandigheden. Met betrekking tot hypoxie was onze hypothese dat de invloed van DCA bijzonder krachtig zou zijn bij zuurstofniveaus die onvoldoende zijn om extra oxidatieve fosforylering te ondersteunen. Alle cellijnen (HB2, 293, HT29, SW480 en LoVo) werden gedurende 24-48 uur in normoxische en hypoxische omstandigheden behandeld met een reeks doses DCA. Het relatieve aantal cellen werd beoordeeld met behulp van MTT-tests.
Behandeling met toenemende doses DCA verminderde de celproliferatie op dosisafhankelijke wijze (figuur 1A-D). In tegenstelling tot onze verwachting waren de profielen van verminderde celgroei vergelijkbaar in hypoxie en normoxie. Bij 24 en 48 uur had maximaal 20 mM DCA geen invloed op de groei van kweken van de niet-kankercellen HB2 en 293. Echter, 20 mM DCA verminderde de groei van kweken van alle drie de colorectale kankercellijnen aanzienlijk (P⩽0,009). Het effect van DCA was groter op de slecht gedifferentieerde SW480-cellen en de metastatische LoVo-cellen dan op de goed gedifferentieerde HT29-cellen. De groei van kweken van LoVo-cellen behandeld met 20 mM DCA werd tot 40% verminderd in vergelijking met cellen behandeld met voertuigcontrole. Omdat er relatief weinig verschil was in de vermindering van de groei van met DCA behandelde kweken in hypoxische en normoxische omstandigheden, werden verdere experimenten alleen in normoxie uitgevoerd.
DCA bevordert apoptose in kankercellen en spaart niet-kankercellen
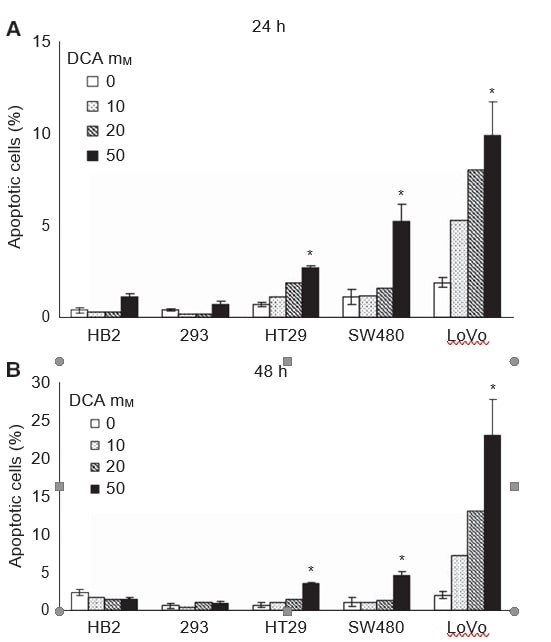
Vervolgens wilden wij onderzoeken of de verminderde groei van de kweken na behandeling met DCA gepaard ging met inductie van apoptose. De cellen werden behandeld met verschillende doses DCA (0, 10, 20 en 50 mM) gedurende 24 en 48 uur, en het percentage cellen dat apoptose onderging werd bepaald door membraanfosfatidylserine te detecteren met annexine V-FITC. De cellen werden gekleurd met annexine V-FITC en de vitale kleurstof 7-AAD, en geanalyseerd met flowcytometrie. Er was een dosisafhankelijke inductie van apoptose in de kankercellijnen na 24 en 48 uur behandeling, en weinig of geen apoptose in de niet-kankercellen (figuur 2A en B). Het grootste effect werd waargenomen bij de metastatische LoVo-cellen; 50 mM DCA veroorzaakte een tienvoudige toename van het percentage apoptotische cellen na 48 uur, terwijl er een zeven- en vijfvoudige toename was bij HT29- en SW480-cellen. De toename van het gemiddelde percentage totale apoptotische cellen met 50 mM DCA was: 2,8 (95% CI: 2-3) in HT29-cellen, 3,5 (95% CI: 2-5) in SW480-cellen, en 21 (95% CI: 8-34) in LoVo-cellen. Er was minimale apoptose geïnduceerd in de 293 cellen, zelfs met 50 mM DCA, 0,2 (95% CI: -0,2 tot 0,6). In HB2-cellen was er een niet-significante afname van het percentage apoptotische cellen bij behandeling met 50 mM DCA, -0,9 (95% CI: -2,2 tot 0,4).
DCA induceert G2-fase-stilstand in colorectale kankercellen, maar heeft geen effect op het celcyclusprofiel van niet-kankerachtige 293 cellen
Wij wilden ook nagaan of de groeivermindering van de kweken na behandeling met DCA samenhing met inductie van groeistilstand. De cellen werden behandeld met 50 mM DCA gedurende 24 of 48 uur, en de celcyclusprofielen werden geanalyseerd met behulp van flowcytometrische bepaling van het DNA-gehalte na PI-kleuring. Behandeling met dichlooracetaat veroorzaakte veranderingen in de celcyclusprofielen van alle kankercellen, maar had geen effect op de niet-kankercellen. De veranderingen in het celcyclusprofiel waren na 24 uur behandeling aantoonbaar en bleven na 48 uur bestaan (figuur 3A en B).
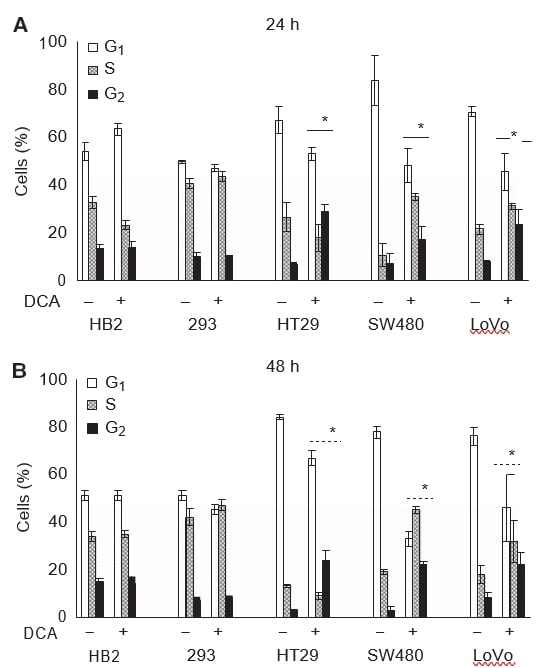
Na 48 uur behandeling met 50 mM DCA was er een achtvoudige toename van cellen in G2-fase in HT29- en SW480-cellen, en een drievoudige toename in LoVo-cellen. De toename van het gemiddelde percentage van alle kankercellen in G2-fase was: 21 (95% CI: 13-30) voor HT29, 19 (95% CI: 13-24) voor SW480-cellen, en 14 (95% CI: 10-21) voor LoVo-cellen; terwijl er geen verschil was in de 293-cellen, 1 (95% CI: -4 tot 7), en HB2-cellen, -0,3 (95% CI: -9 tot 9). Er was een overeenkomstige afname van cellen in G0/G1-fase in alle kankercellijnen. Intrigerend genoeg was er bij HT29-cellen een kleine afname, maar bij SW480- en LoVo-cellen was er een significante toename van het aandeel cellen dat geacht werd zich in de S-fase te bevinden (zie Discussie). Het celcyclusprofiel van 293 en HB2 cellen veranderde minimaal bij behandeling met DCA.
DCA verlaagt het extracellulaire lactaatgehalte in de groeimedia
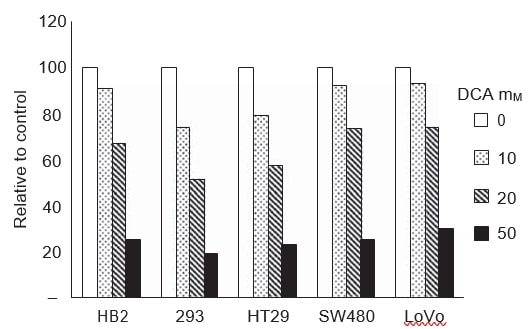
Om vast te stellen of de veranderingen in groei en apoptose geïnduceerd door DCA samenhingen met verminderde glycolyse, maten we het lactaatgehalte in de groeimedia. Melkzuur is het eindproduct van glycolyse. Als DCA de mitochondriale oxidatieve fosforylering induceert, wordt pyruvaat gedecarboxyleerd tot acetyl-CoA en niet gereduceerd tot lactaat. Het lactaatgehalte in de groeimedia van alle cellijnen werd gemeten na 48 uur behandeling met een reeks doses DCA (figuur 4). Het lactaatgehalte werd bepaald met een auto-analysator die routinematig wordt gebruikt voor biochemische metingen van het lactaatgehalte; de tests zijn gebaseerd op een colorimetrische reactie die wordt gekatalyseerd door lactaatoxidase. Behandeling met DCA verminderde het extracellulaire lactaatgehalte in de groeimedia op dosisafhankelijke wijze in alle kankercellijnen en niet-kankercellijnen.
DCA depolariseert de intrinsieke mitochondriale membraan in colorectale kankercellen maar niet in niet-kankercellen
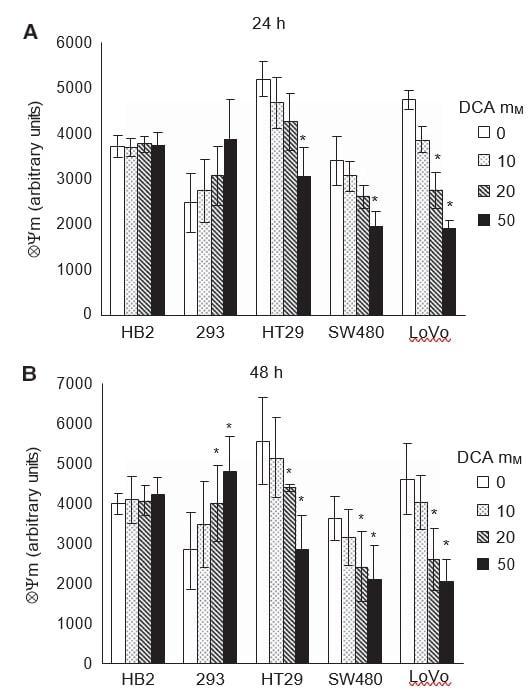
Om na te gaan of de inductie van apoptose in kankercellen bij behandeling met DCA gepaard ging met bevordering van de mitochondriale oxidatieve fosforylering, maten we de intrinsieke mitochondriale membraanpotentiaal (ΔΨm). Escalatie van de mitochondriale ademhaling zou de elektronentransportketen reactiveren en de gehyperpolariseerde ΔΨm in kankercellen verminderen. Cellen werden gedurende 24 en 48 uur behandeld met doses DCA en gekleurd met de kleurstof TMRM, waarmee ΔΨm fluorescent gemeten kan worden.
Net als bij eerdere experimenten was het effect van DCA zichtbaar na 24 uur behandeling en hield het aan na 48 uur (figuur 5A en B). Behandeling met dichlooracetaat verminderde de gehyperpolariseerde ΔΨm in alle kankercellen op dosis-afhankelijke wijze. Dichlooracetaat had geen effect op ΔΨm van de niet-kankercellen van HB2, terwijl verrassend genoeg het ΔΨm van de niet-kankercellen van 293 op een dosis-afhankelijke manier toenam. Na 24 uur behandeling verminderde 50 mM DCA ΔΨm aanzienlijk in alle kankercellen; in LoVo-cellen was er echter zelfs met 20 mM DCA een significante vermindering (figuur 5A, P=0,02). In de niet-kankercellen van 293 was er een trend naar toename van ΔΨm bij DCA-behandeling, hoewel dit niet statistisch significant was (P=0,08). Na 48 uur behandeling was er een significante vermindering van ΔΨm in alle kankercellen en een toename in de 293 cellen, met 20-50 mM DCA (figuur 5B, P⩽0,04).
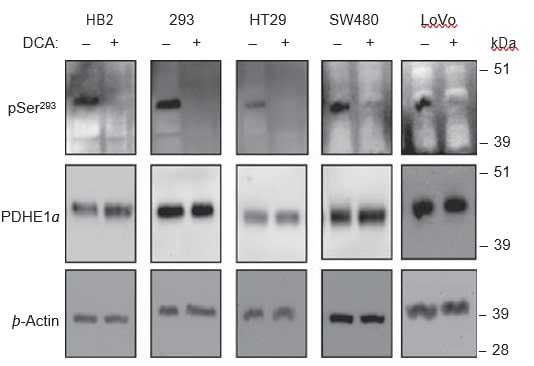
DCA-behandeling leidt tot defosforylering van de PDHE1α-subeenheid
DCA zou alle vier de iso-enzymen van PDK remmen, en daardoor de fosforylering van de PDHE1α-subeenheid verminderen, wat op zijn beurt leidt tot activering van het PDH-complex. Om na te gaan of de fosforylering van PDHE1α optrad bij behandeling met DCA in de gebruikte cellijnen, gebruikten we western blot analyses op lysaten van met DCA behandelde en onbehandelde cellen. In alle cellijnen veroorzaakte behandeling met 20 mM DCA gedurende 8 uur een dramatische vermindering van het signaal voor fosforylering op de pSer293-plaats, maar er werd geen verandering waargenomen in de niveaus van totaal PDHE1α (figuur 6). Fosfosesspecifieke antilichamen voor de andere twee fosforyleringsplaatsen, Ser232 en Ser300, zijn nog niet in de handel verkrijgbaar.
Bespreking
Wij hebben aangetoond dat DCA een dosis-afhankelijke vermindering van de groei van in vitro culturen van colorectale kankercellen en niet-kankercellen induceert. De kankercellen waren echter gevoeliger voor DCA, waarbij een dosis van 20 mM een significante remming van de kankercelgroei veroorzaakte, maar weinig effect had op de niet-kankercellen. Wij hebben aangetoond dat de componenten van dit differentiële effect de volgende zijn: een krachtige inductie van apoptose en celcyclusstilstand in de kankercellen, maar niet in de niet-kankercellen.
Deze conclusies ondersteunen een eenvoudig model van differentiële gevoeligheid voor DCA. Enkele gegevens vereisen echter nadere bespreking. Ten eerste verminderde 50 mM DCA de groei van kweken van de niet-kankercellen 293 en HB2, maar er werd geen toename van apoptotische cellen of verandering in het celcyclusprofiel van deze cellen waargenomen. Een mogelijke verklaring voor deze bevindingen zou kunnen zijn dat deze dosis DCA leidde tot een tragere doorgang van deze niet-kankerachtige cellen door alle stadia van de celcyclus, zonder de relatieve verhoudingen binnen elk stadium te veranderen. Ten tweede wijzen onze resultaten erop dat DCA G2-stilstand induceerde in colorectale kankercellen. Dit staat in contrast met eerdere studies, die G1-arrest of geen verandering in het celcyclusprofiel bij DCA-behandeling aantoonden (Cao e.a., 2008; Wong e.a., 2008). Wong e.a. (2008) toonden verhoogde expressie van PUMA in alle endometriumkankercellijnen die een apoptotische reactie op DCA vertoonden, en concludeerden dat deze p53 activering leidde tot G1-stilstand. De colorectale kankercellen in onze studie arresteerden echter in de G2-fase na behandeling met DCA, en wij vonden geen inductie van p53 door DCA in onze colorectale kankercellijnen (gegevens niet getoond). Merkwaardig genoeg vonden Cao e.a. (2008) dat de combinatie van DCA en radiotherapie prostaatkankercellen in de G2-fase arresteerde, hoewel DCA op zichzelf het celcyclusprofiel niet beïnvloedde. Ten derde, in SW480 en LoVo cellen resulteerde DCA behandeling in een toename van het percentage cellen dat geacht werd zich in de S fase te bevinden. Dit wijst op een toename van de proliferatie en de inductie van apoptose. Een soortgelijke bevinding werd gerapporteerd door Wong et al. (2008) in een van de geteste endometriumkankercellen. Een alternatieve verklaring is dat een deel van de cellen die na DCA-behandeling van de kankercellijnen in “S-fase” verkeren, in werkelijkheid apoptotische cellen in het “sub-G2“-gebied zijn, zoals eerder is gerapporteerd bij lymfoomcellen (Klucar en Al-Rubeai, 1997).
Veranderingen in het cellulaire metabolisme bij behandeling met DCA
DCA bleek de melkzuurproductie uit pyruvaat in zowel kankercellen als niet-kankercellen te onderdrukken. Bovendien leidde behandeling met DCA tot defosforylering van PDHE1α, en daarmee tot activering van PDH in alle onderzochte cellijnen. De basis van het verschillende effect van DCA op kankercellen en niet-kankercellen kan dus liggen in de invloed ervan op de mitochondriale functie. Behandeling met DCA verminderde het hoge ΔΨm van alle kankercellen, maar niet van de niet-kankercellen. Dit wijst erop dat DCA, door remming van PDK en dus activering van PDH, de mitochondriale ademhaling bevordert die leidt tot depolarisatie van het intrinsieke mitochondriale membraan, en apoptose induceert via de proximale mitochondriale route, zoals beschreven in eerdere studies (Bonnet et al, 2007; Cao et al, 2008; Wong et al, 2008). Inductie van apoptose en veranderingen in de mitochondriale functie waren het meest uitgesproken in de zeer invasieve en metastatische LoVo-cellen dan in de minder invasieve HT29- en SW480-cellen. Dit zou klinische implicaties kunnen hebben voor de behandeling van uitgezaaide colorectale kanker, aangezien het gewoonlijk de zeer invasieve uitgezaaide kankers zijn die het meest resistent zijn tegen conventionele chemotherapie, en die wellicht het gevoeligst zijn voor PDK-remming. Ter ondersteuning hiervan is in een recente studie gemeld dat de colorectale tumoren die resistent zijn tegen 5-fluorouracil vaker een verhoogde glycolyse hebben, en dus meer vatbaar zijn voor therapie gericht op het kankermetabolisme (Shin et al, 2009). In dit opzicht contrasteren onze resultaten met de bevindingen van Wong e.a. (2008), die vonden dat zeer invasieve endometriumkankercellen het meest resistent zijn tegen behandeling met DCA.
PDK-remming als kankertherapie tegen dikkedarmkanker
Wij vonden dat doses van 20-50 mM DCA verschillende reacties gaven tussen kankercellen en niet-kankercellen. Potentiële therapeutische DCA-doses liggen dus tussen 20 en 50 mM. Bovendien meldde een recente studie dat de IC50 van DCA voor borstkankercellen tussen 20 en 30 mM ligt (Ko en Allalunis-Turner, 2009). Dit staat in contrast met eerdere studies die meldden dat DCA de proliferatie vermindert en apoptose in kankercellen induceert met doses van slechts 0,5-10 mM (Bonnet et al, 2007; Wong et al, 2008; Sun et al, 2009). Dichlooracetaat is bij mensen relatief veilig gebleken bij gebruik voor de behandeling van melkziekte (Stacpoole et al, 2003). De belangrijkste bijwerkingen met maximaal 100 mg kg-1 DCA hebben betrekking op het zenuwstelsel en de lever, en veroorzaken lichte sedatie of slaperigheid, omkeerbare perifere neuropathie, en lichte asymptomatische verhoging van serumtransaminasen die wijzen op hepatocellulaire schade (Stacpoole et al, 1998). Bovendien meldden recente studies dat DCA zowel in vitro als in vivo in klinisch bereikbare doses de tumorgroei doeltreffend vermindert (Bonnet et al, 2007; Sun et al, 2009). Er werd gesuggereerd dat DCA snel zou kunnen leiden tot klinische proeven met kanker in een vroeg stadium (Michelakis et al, 2008). Het is echter onwaarschijnlijk dat de dosis DCA die nodig is om de groei van colorectale kankercellen in onze studie te remmen, klinisch kan worden bereikt zonder aanzienlijke bijwerkingen te veroorzaken. De dosis DCA die nodig is om de equivalente plasmaconcentraties in vivo te bereiken, zou ongeveer vijf tot tien keer zo hoog zijn als de dosis die in klinische proeven tegen melkziekte wordt gebruikt. Het lijkt erop dat de in onze studie gebruikte colorectale kankercellen resistenter zijn tegen DCA dan long-, endometrium- en borstkankercellen. Intrigerend is dat Sun et al. (2009) in hun studie over borstkankercellen vonden dat DCA de proliferatie van kankercellen remde, maar geen apoptose of celdood induceerde. Deze resultaten verschillen duidelijk van de effecten van DCA die in onze studie werden waargenomen op long- (Bonnet et al, 2007), endometrium- (Wong et al, 2008) en colorectale kankercellen. Hoewel DCA dus de groei van verschillende kankercellen remt, lijken het effect en de onderliggende mechanismen afhankelijk te zijn van het celtype. Een waarschijnlijke verklaring voor deze verschillende effecten zou het verschil in expressie van de PDK-isoenzymen in de onderzochte kankercellen kunnen zijn. Dichlooracetaat is een niet-specifieke remmer van PDK (Whitehouse en Randle, 1973), en heeft een verschillendeKi voor elk van de vier PDK-isoenzymen (Bowker-Kinley et al, 1998). Bovendien is bekend dat de vier PDK-isoenzymen in verschillende weefsels verschillend tot expressie komen. Er is dus behoefte aan de ontwikkeling van remmers voor de afzonderlijke PDK-isoenzymen die metabole manipulatie per kankerceltype mogelijk moeten maken.
VERWIJZINGEN
1 Bi X, Lin Q, Foo TW, Joshi S, You T, Shen HM, Ong CN, Cheah PY, Eu KW, Hew CL (2006) Proteomic analysis of colorectal cancer reveals alterations in metabolic pathways: mechanism of tumorigenesis. Mol Cell Proteomics 5: 1119-1130
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) Een mitochondria-K+ kanaal as wordt onderdrukt in kanker en de normalisatie ervan bevordert apoptose en remt kanker groei. Kankercel 11: 37-51
3 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM (1998) Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 329 (Deel 1): 191-196
4 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostaat 68: 1223-1231
4 Dewhirst MW, Tso CY, Oliver R, Gustafson CS, Secomb TW, Gross JF (1989) Morfologische en hemodynamische vergelijking van tumor en genezend normaal weefsel microvasculatuur. Int J Radiat Oncol Biol Phys 17: 91-99
5 Gatenby RA, Gillies RJ (2004) Waarom hebben kankers een hoge aerobe glycolyse? Nat Rev Cancer 4: 891-899
6 Gillies RJ, Gatenby RA (2007) Adaptive landscapes and emergent phenotypes: why do cancers have high glycolysis? J Bioenerg Biomembr 39: 251-257
7 Klucar J, Al-Rubeai M (1997) G2 cell cycle arrest and apoptosis are induced in Burkitt’s lymphoma cells by the anticancer agent oracin. FEBS Lett 400: 127-130
8 Ko L, Allalunis-Turner J (2009) Onderzoek naar het mechanisme van door dichlooracetaat (DCA) geïnduceerde apoptose bij borstkanker. J Clin Oncol 27 (Suppl 15): e14637
9 Michelakis ED, Webster L, Mackey JR (2008) Dichlooracetaat (DCA) als een potentiële metabool-gerichte therapie voor kanker. Br J Cancer 99: 989-994
10 Milosevic M, Fyles A, Hedley D, Hill R (2004) The human tumor microenvironment: invasive (needle) measurement of oxygen and interstitial fluid pressure. Seminar Radiat Oncol 14: 249-258
11 Pan JG, Mak TW (2007) Metabolic targeting als antikankerstrategie: dageraad van een nieuw tijdperk? Sci STKE 2007: e14
12 Rajaganeshan R, Prasad R, Guillou PJ, Poston G, Scott N, Jayne DG (2008) The role of hypoxia in recurrence following resection of Dukes’ B colorectal cancer. Int J Colorectal Dis 23: 1049-1055
13 Rajaganeshan R, Prasad R, Guillou PJ, Scott N, Poston G, Jayne DG (2009) Expressiepatronen van hypoxische markers bij de invasieve marge van colorectale kankers en levermetastasen. Eur J Surg Oncol 35 (12): 1286-1294
14 Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE (2009) Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem 389: 157-164
15 Shike M, Winawer SJ, Greenwald PH, Bloch A, Hill MJ, Swaroop SV (1990) Primary prevention of colorectal cancer. Het samenwerkingscentrum van de WHO voor de preventie van colorectale kanker. Bull World Health Organ 68: 377-385
16 Shin YK, Yoo BC, Hong YS, Chang HJ, Jung KH, Jeong SY, Park JG (2009) Upregulation of glycolytic enzymes in proteins secreted from human colon cancer cells with 5-fluorouracil resistance. Elektroforese 30: 2182-2192
17 Stacpoole PW (1989) De farmacologie van dichlooracetaat. Metabolisme 38: 1124-1144
18 Stacpoole PW, Henderson GN, Yan Z, James MO (1998) Clinical pharmacology and toxicology of dichloroacetate. Environ Health Perspect 106 (Suppl. 4): 989-994
19 Stacpoole PW, Nagaraja NV, Hutson AD (2003) Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol 43: 683-691
20SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC (2009) Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 120 (1): 253-260
21 Thorn CC, Freeman TC, Scott N, Guillou PJ, Jayne DG (2009) Laser microdissection expression profiling of marginal edges of colorectal tumors reveals evidence of increased lactate metabolism in the aggressive phenotype. Darm 58: 404-412
22 UK Darmkankerstatistieken (2009) http://info.cancerresearchuk.org/cancerstats/types/bowel/
23 Warburg O (1956) Over het ontstaan van kankercellen. Science 123: 309-314
24 Whitehouse S, Randle PJ (1973) Activation of pyruvate dehydrogenase in perfused rat heart by dichloroacetate. Biochem J 134: 651-653
25 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I (2008) Dichlooracetaat induceert apoptose in endometriumkankercellen. Gynecol Oncol 109: 394-402
26 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA (2007) Multiparameter metabole analyse onthult een nauw verband tussen verzwakte mitochondriale bio-energetische functie en verhoogde glycolyse afhankelijkheid in menselijke tumorcellen. Am J Physiol Cell Physiol 292: C125-C136
27 Yeluri S, Madhok B, Prasad KR, Quirke P, Jayne DG (2009) Cancer’s craving for sugar: an opportunity for clinical exploitation. J Cancer Res Clin Oncol 135: 867-877