Mao-fa Zheng1, Si-yu Shen, Wei-da Huang
1Dipartimentodi Biochimica, Scuola di Scienze della Vita, Fudan University, Handan Road 220, Shanghai, 200433, Cina.
Email: e-mail: [email protected]
Ricevuto: 16 maggio 2013
Accettato: 27 agosto 2013
Pubblicato: 17 settembre 2013
Abstract
Scopo: La capecitabina è uno dei pochi farmaci chemioterapici con un’elevata disponibilità orale. Recentemente, il dicloroacetato di sodio (DCA) ha mostrato un grande potenziale come agente antitumorale. Nel presente studio, abbiamo valutato l’effetto antitumorale del DCA in combinazione con la capecitabina per i tumori che esprimevano modestamente TP.
Metodi: Un allotrapianto di melanoma B16 di topo e uno xenotrapianto di carcinoma polmonare umano non a piccole cellule A549 sono stati utilizzati per valutare l’effetto del trattamento combinato di DCA e capecitabina. L’istologia e l’immunoistochimica sono state utilizzate per rilevare l’apoptosi e la proliferazione delle cellule tumorali. La PCR in tempo reale e il Western blot sono stati eseguiti per rilevare l’espressione di TP e delle caspasi, rispettivamente.
Risultati: Per la prima volta, riportiamo che il DCA ha aumentato gli effetti antitumorali della capecitabina in un allotrapianto di topo B16 e in uno xenotrapianto umano A549, promuovendo l’apoptosi delle cellule tumorali. Il DCA ha un effetto limitato sull’espressione di TP.
Conclusioni: I nostri risultati suggeriscono che il DCA in combinazione con la capecitabina potrebbe essere un potenziale nuovo regime terapeutico contro alcuni tipi di cancro.
Parole chiave: DCA, Capecitabina, Combinazione, Effetto antitumorale
© Springer-Verlag Berlin Heidelberg 2013
Cancer Chemother Pharmacol (2013) 72:1031-1041
DOI: 10.1007/s00280-013-2281-z
INTRODUZIONE
Il dicloroacetato di sodio (DCA) è un piccolo sale molecolare dell’acido dicloroacetico con un peso molecolare di 150 Da. Il DCA inibisce l’attività della piruvato deidrogenasi chinasi, attivando così il complesso enzimatico mitocondriale piruvato deidrogenasi [1] e convertendo la via metabolica glicolitica in fosforilazione ossidativa. Negli ultimi 40 anni, il DCA è stato utilizzato come farmaco orfano nel trattamento dell’acidosi lattica congenita dei bambini e dell’acidosi lattica complicata da altre malattie [2] e ha dimostrato un’elevata efficacia e una bassa tossicità sia negli studi preclinici che in quelli clinici [3]. Recentemente, il DCA ha mostrato un grande potenziale come agente antitumorale, grazie alla somiglianza del rimodellamento metabolico di alcune cellule tumorali con quello che si verifica durante l’acidosi lattica [4]. Le cellule tumorali, in particolare le cellule staminali tumorali (CSC), resistono all’apoptosi producendo energia attraverso la glicolisi e la fermentazione dell’acido lattico, piuttosto che attraverso la fosforilazione ossidativa, a causa della natura ipossica del microambiente tumorale, un fenomeno noto come effetto Warburg [5,6]. Dopo la somministrazione orale, è stato dimostrato che il DCA ripristina la funzione mitocondriale e promuove selettivamente l’apoptosi delle cellule tumorali in un percorso mitocondriale dipendente [7,8]. Le attività terapeutiche del DCA contro il glioblastoma sono state testate in studi clinici (NCT00540176) e hanno mostrato alcuni risultati positivi [9]. Tuttavia, lo studio di fase II NCT01029925, volto a determinare il tasso di risposta del dicloroacetato orale in pazienti con cancro al seno e al polmone non a piccole cellule ricorrente e/o metastatico e pretrattato, è stato interrotto a causa di rischi e problemi di sicurezza superiori al previsto. Pertanto, l’utilità clinica del DCA per il controllo del cancro deve essere valutata con maggiore attenzione.
Come sensibilizzatore dell’apoptosi, il DCA è stato utilizzato anche in combinazione con altre terapie antitumorali. Cao et al. [10] hanno riportato che il DCA ha sensibilizzato le cellule di cancro alla prostata alle radiazioni in vitro. Xiao et al. [11] hanno determinato che il DCA ha aumentato la morte delle cellule tumorali quando è stato combinato con un adenovirus oncolitico che esprime il soppressore tumorale MDA-7/IL-24. Recentemente, la terapia a bersaglio metabolico con DCA è stata dimostrata come una nuova strategia di trattamento per migliorare i risultati della terapia fotodinamica [12]. Tong et al. [13] hanno scoperto che il DCA e il 5-fluorouracile hanno mostrato un effetto antitumorale sinergico nelle cellule di cancro colorettale in vitro. Tuttavia, ci sono ancora risultati controversi e dubbi sull’uso del DCA da solo o in combinazione con altri farmaci. Shahrzad et al. [14] hanno dimostrato che il DCA riduce l’apoptosi delle cellule tumorali in condizioni di ipossia sia in vitro che in vivo. Heshe et al. [15] hanno avvertito che il DCA ha ridotto la citotossicità di alcuni farmaci antitumorali standard, come il cisplatino e la doxorubicina, ma non ha influenzato l’attività della temozolomide in 7 delle 10 linee cellulari del loro studio. Questi risultati contrastanti implicano che l’uso del DCA da solo o in combinazione con altre terapie può essere specifico per il tipo di cancro e per l’agente.
La capecitabina è uno dei pochi farmaci chemioterapici con un’elevata disponibilità orale ed è autorizzata come trattamento di prima linea per il carcinoma rettale metastatico o come trattamento alternativo per il carcinoma mammario metastatico in combinazione con il docetaxel [16,17]. La capecitabina è un prodrug di 5-fluorouracile (5-FU) e richiede 3 reazioni enzimatiche per la conversione finale in 5-FU nelle cellule tumorali. La reazione finale è catalizzata dalla timidina fosforilasi (TP), che è espressa in misura maggiore in alcuni tumori rispetto ai tessuti normali [18]. Pertanto, il 5-FU citotossico viene generato in misura maggiore nelle cellule tumorali rispetto ai tessuti fuori bersaglio, il che rende la capecitabina un farmaco chemioterapico a bassa tossicità [18]. I livelli di espressione del TP sono diversi nei vari tipi di tumore [18]; questo limita l’uso della capecitabina solo ad alcuni tipi di cancro. Nel presente studio, abbiamo valutato l’effetto antitumorale del DCA in combinazione con la capecitabina per i tumori che esprimono modestamente TP. Abbiamo ipotizzato che il DCA aumenti gli effetti antitumorali e riduca la dose efficace di capecitabina. La combinazione di DCA e capecitabina potrebbe produrre un buon regime di trattamento perché entrambi gli agenti possono essere assunti per via orale con una buona aderenza da parte del paziente. Inoltre, le forme generiche di DCA possono ridurre la dose efficace di capecitabina, diminuendo così gli effetti collaterali e il costo del trattamento del cancro.
Materiali e metodi
Materiali
Il dicloroacetatodi sodio(DCA, CSA:2156-56-1), con una purezza del 99%, è stato ottenuto da Shanghai Jieshi Chemical Co. ltd. (Cina). il 5-fluorouracile (5-FU) e la 5′-deossi-fluorouridina (5-DFUR) sono stati ottenuti da Sigma-Aldrich (USA). L’MTT è stato ottenuto da Shanghai Biological Engineering Co., Ltd. (Cina). ltd. (Cina). Le compresse di capecitabina (Xeloda) sono state ottenute da Roche (USA). Il melanoma B16 del topo e la linea cellulare A549 del carcinoma polmonare umano non a piccole cellule sono stati ottenuti dall’American Type Cell Culture Collection (ATCC, USA).
Studi su modelli animali
Modello di allotrapianto
Topi C57BL/6, femmine, di 6-8 settimane di età e del peso di circa 18-20 g, sono stati acquistati dallo Shanghai Laboratory Animal Center (SLAC, Cina) e lasciati acclimatare per 1 settimana. Un milione di cellule singole B16 sono state inoculate per via sottocutanea (s.c.) sul fianco destro dei topi C57BL/6. I topi sono stati raggruppati in modo casuale, con 6 topi per gruppo in una gabbia. Vi erano due gruppi di topi. DCA e capecitabina sono stati somministrati a questi due gruppi di topi rispettivamente a 3 e 10 giorni dall’inoculazione. Il DCA è stato aggiunto all’acqua potabile sterile a una concentrazione finale di 1,4 g/L. La misurazione del volume di acqua consumato ha dimostrato che la quantità di DCA somministrata a ciascun topo era approssimativamente pari a 100 mg/kg/die. Le compresse di capecitabina sono state macinate e sospese in acqua sterile con il 4% di carbossimetilcellulosa per ottenere diverse concentrazioni. Duecento microlitri di sospensioni di capecitabina sono stati somministrati per via intragastrica a ciascun topo. Ogni 2 giorni sono stati misurati i diametri lungo (a) e corto (b) dei tumori con un calibro a corsoio ed è stato registrato il peso corporeo. Il volume del tumore è stato calcolato con la formula V = 0,5ab2. Ventidue giorni dopo l’inoculazione, i topi sono stati uccisi e i tumori sono stati rimossi e pesati.
Modello di xenotrapianto
Topi BALB/c-nu, maschi, di 5-6 settimane di età e del peso di circa 18-20 g, sono stati acquistati dallo Shanghai Laboratory Animal Center (SLAC, Cina) e lasciati acclimatare per 1 settimana. Sezioni di circa 2 × 2 mm di tessuti tumorali A549 appena tritati, provenienti da topi BALB/c-nu precedentemente inoculati con cellule A549, sono state inoculate s.c. nella regione del fianco destro di topi maschi BALB/c-nu. DCA e capecitabina sono stati somministrati ai topi quando il loro volume tumorale ha raggiunto ~0,2 cm3. Da trenta a trentacinque giorni dopo il trattamento, i topi sono stati uccisi e i tumori sono stati rimossi e pesati. Gli altri metodi sono stati gli stessi descritti nell’esperimento di allotrapianto.
Gli studi sugli animali sono stati approvati dal Gruppo per il Benessere e l’Etica degli Animali del Dipartimento di Scienze degli Animali da Laboratorio dell’Università di Fudan.
Istologia e immunoistochimica
L’esameistologicodei noduli tumorali è stato eseguito utilizzando altri animali (3 topi per gruppo) non considerati per il monitoraggio della crescita tumorale. I tessuti sono stati fissati in paraformaldeide al 4% (w/v) e, dopo la fissazione notturna a temperatura ambiente, i campioni sono stati disidratati in etanolo graduato e incorporati in paraffina. Successivamente, diverse parti del tumore sono state tagliate in modo casuale per ottenere sezioni di 4μm al microtomo Leica. Dopo la deparaffinizzazione e la reidratazione, sono state scelte tre sezioni da parti diverse di ciascun campione per le operazioni di follow-up. Le colorazioni TUNEL (Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling) e DAPI (4′6-diamidino-2-phenylindole) sono state eseguite secondo le istruzioni del produttore dei kit TUNEL e DAPI (Beyotime, Cina). Le sezioni sono state analizzate con un microscopio a fluorescenza invertito (Olympus, Giappone). La rilevazione dell’antigene nucleare delle cellule proliferanti (PCNA) è stata effettuata dopo la deparaffinizzazione della sezione e l’incubazione a 96-100 °C per 20 minuti. L’attività della perossidasi endogena è stata spenta con perossido di idrogeno allo 0,3% (v/v) in metanolo al 60% (v/v) per 30 minuti. L’adsorbimento non specifico è stato minimizzato incubando le sezioni con il 2 % (v/v) di siero normale di capra in PBS per 20 minuti. Le sezioni di tessuto sono state incubate per una notte con l’anticorpo policlonale di coniglio anti-PCNA (Abcam; 1:100 in PBS), lavate con PBS 3 volte, 30 minuti per volta, e incubate con un IgG di capra antirabbit coniugato con biotina per 2 ore a 37 °C e con il complesso avidina-biotina-perossidasi per 1 h a 37 °C. Le sezioni sono state controcolorate con ematossilina (Sigma-Aldrich) e analizzate al microscopio ottico (Olympus, Giappone). Tre tumori per gruppo sono stati utilizzati per l’analisi immunoistochimica. Una o due sezioni per tumore sono state selezionate in cieco per misurare le cellule PCNA- o TUNEL-positive. Cinque campi casuali per vetrino con ingrandimento 400× sono stati misurati in cieco (n = 250 cellule per gruppo). Durante l’analisi quantitativa delle cellule TUNEL-positive, le possibili cellule necrotiche sono state escluse osservando la morfologia nucleare con la colorazione DAPI. Sono stati eseguiti controlli positivi e negativi per garantire risultati accurati.
Estrazione delle proteine e Western blot
I tessuti tumorali di ciascun gruppo (3 topi aggiuntivi per gruppo) sono stati riuniti e macinati sotto azoto liquido, quindi lisati in 150 μL di tampone di lisi tissutale (Beyotime, Cina). Le provette sono state agitate vigorosamente per 1 minuto, poste in ghiaccio per 20 minuti e centrifugate a 5.000g per 5 minuti a 4 °C. La concentrazione di proteine totali è stata determinata con un kit di determinazione delle proteine BCA (BioRad). Trenta microgrammi di proteine totali di ciascun campione sono stati separati mediante SDS-PAGE su gel al 10%, trasferiti su membrana PVDF, bloccati, incubati per una notte con l’anticorpo primario, incubati per 1 ora con l’anticorpo secondario e sviluppati con il substrato cromogenico NBT/BCIP. L’anticorpo monoclonale di topo anti-caspasi 3 (1:500), l’anticorpo anti-caspasi 9 (1:1.000), l’anticorpo anti-β-actina (1:2.000) e l’anticorpo policlonale di coniglio anti-caspasi 8 (1:1.000) sono stati ottenuti da Beyotime (Nanjing, Cina). L’anticorpo monoclonale di topo anti-TP (1:2.000) è stato fornito da Abcam (Regno Unito). la β-actina è stata utilizzata come controllo interno. La densità delle bande del Western blot è stata analizzata con Clinx Gel Analysis V2.02 (Clinx Science, Cina).
PCR in tempo reale
I tumoriB16e i tessuti epatici sono stati prelevati dallo stesso topo C57BL/6 precedentemente inoculato con cellule B16 (sono stati utilizzati 3 topi, dono di Wenlong Ren dell’Istituto di Industria Farmaceutica di Shanghai). I tessuti di cellule Colo205/A549 e di fegato sono stati prelevati dallo stesso topo BALB/c-nu precedentemente inoculato con cellule Colo205/A549 (sono stati utilizzati rispettivamente 3 topi, dono di Wenlong Ren dell’Istituto di Industria Farmaceutica di Shanghai). Per l’analisi dell’espressione di TP dopo il trattamento, i campioni tumorali sono stati raggruppati da 3 tessuti tumorali di peso uguale per ogni gruppo. L’RNA totale è stato isolato con il reagente Trizol (Invitrogen, USA) e trascritto inversamente con il kit PrimeScript® RT reagent (Takara, Giappone). Il cDNA è stato normalizzato con la β-actina. La PCR in tempo reale è stata eseguita con metodi a tre fasi utilizzando il kit SYBR® Premix Ex Taq™ II (Takata, Giappone) con temperatura di annealing di 55 °C e 40 cicli di amplificazione. I singoli test sono stati eseguiti in triplo. la β-actina è stata utilizzata come controllo interno. La quantità relativa di ciascun cDNA è stata analizzata mediante2-△△Ct. Primers per la real-time PCR: β-actina F: 5′-TCAAGATCATTGCTC CTCCTG-3′ e β-actina R: 5′-CTGCTTGCTGATCCATCTG-3′, hTP F: 5′-TGGCTCAGTCGACAGCAG-3′ e hTP R: 5′-TCCGCTGATCATTG GCACCT-3′, mTP F: 5′-GCCTAGCTAAAGCATTGTCTC-3′ e mTP R: 5′-AAGGGTGCT CGATCTGATAGCA-3′.
Statistiche
Abbiamo utilizzato l’ANOVA a confronti multipli con analisi post hoc (test di Tukey), utilizzando il software SPSS 16.0 (SPSS Inc., USA). I dati sono presentati come media ± SEM e P < 0,05 è stato considerato significativo.
Risultati
Espressione di TP nel melanoma B16 di topo e nei tumori NSCLC A549 umani
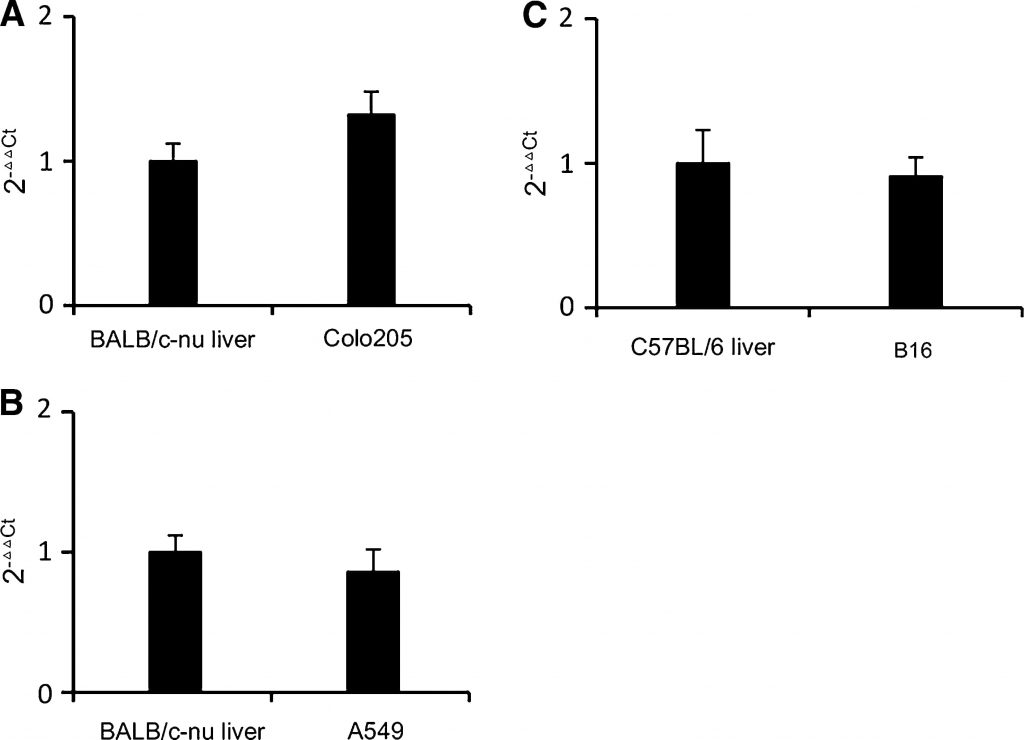
L’espressione di TP nei tumori B16 e A549 prelevati dai topi è stata analizzata mediante PCR in tempo reale. Il fegato umano esprime una quantità relativamente maggiore di TP rispetto ad altri tessuti normali [18]. Nei tipici tipi di cancro adatti al trattamento con capecitabina, l’espressione di TP nel tumore è vicina o superiore a quella del fegato, ad esempio nei tumori del colon-retto e della mammella [18,19]. Negli xenotrapianti di cancro umano, i tumori con elevata attività di TP sono risultati più sensibili al trattamento con capecitabina rispetto a quelli con bassa attività di TP [20]. La linea cellulare di cancro colorettale Colo205 è stata scelta come riferimento in base alla sua moderata espressione di TP e alla moderata sensibilità alla capecitabina [21]. Come mostrato nella Fig. 1a, i livelli di trascrizione di TP in Colo205 erano leggermente superiori a quelli del fegato del topo BALB/c-nu. Come mostrato nelle Fig. 1b, c, i livelli di trascrizione di TP nei tumori B16 e A549 erano vicini a quelli dei fegati di topo. Pertanto, anche le B16 e le A549 erano linee cellulari moderatamente esprimenti TP. Possiamo dedurre che B16 e A549 risponderanno in qualche misura al trattamento con capecitabina senza coprire l’effetto del DCA. Pertanto, i modelli di allotrapianto B16 e di xenotrapianto A549 erano adatti per studiare l’effetto antitumorale di DCA e capecitabina in combinazione.
IlDCA aumenta l’effetto antitumorale della capecitabina in un allotrapianto di melanoma B16 di topo senza tossicità aggiuntiva
Poiché la natura ipossica del microambiente tumorale è fondamentale per un’attività ottimale del DCA, l’effetto antitumorale del DCA più la capecitabina è stato testato in modelli animali anziché in linee cellulari. Trentasei topi C57BL/6 sono stati inoculati con 1 ×106 cellule di melanoma B16 e separati casualmente in 6 gruppi (n = 6): un gruppo di controllo che non ha ricevuto alcun farmaco, un gruppo di solo DCA, un gruppo di sola capecitabina 10 mg/die e 3 gruppi di DCA più capecitabina 5, 10 o 20 mg/die. Tre giorni dopo l’inoculazione delle cellule tumorali, sono stati somministrati ai topi DCA in acqua potabile e capecitabina per via orale (p.o.) come agenti singoli o in combinazione con concentrazioni crescenti di capecitabina. Come mostrato nel pannello superiore della Fig. 2a, sia i trattamenti con DCA che con capecitabina 10 mg/die da soli hanno inibito modestamente la crescita dei tumori di melanoma B16 rispetto al gruppo di controllo. Al contrario, DCA più 10 mg/die di capecitabina hanno aumentato significativamente l’inibizione della crescita tumorale (P < 0,05). L’effetto antitumorale di DCA più 20 mg/die di capecitabina è stato simile a quello osservato con DCA più 10 mg/die di capecitabina; la crescita tumorale è stata quasi completamente inibita. È da notare che i topi trattati con DCA più 20 mg/die di capecitabina hanno subito una perdita acuta di peso corporeo (Fig. 2a, pannello inferiore), mentre DCA più 10 mg/die di capecitabina ha avuto un effetto minimo sul peso corporeo rispetto al controllo.
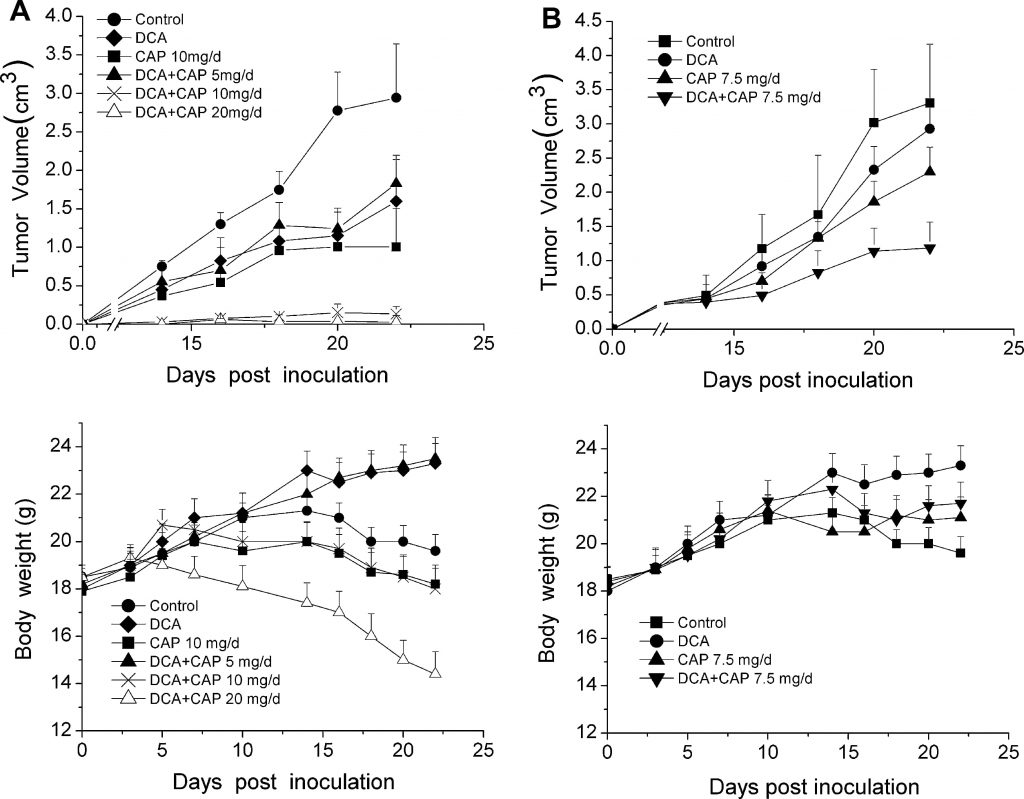
Per valutare l’effetto antitumorale di DCA più capecitabina contro i tumori palpabili e rilevabili, 10 giorni dopo l’inoculazione delle cellule tumorali, DCA e capecitabina da soli o in combinazione sono stati somministrati a un secondo gruppo di 36 topi C57BL/6 inoculati con 1 ×106 cellule di melanoma B16. I topi con tumori palpabili sono stati separati casualmente in 4 gruppi (n = 9, 3 topi sono stati utilizzati per l’analisi immunoistochimica), controllo, DCA da solo, capecitabina da sola a 7,5 mg/die e DCA più capecitabina a 7,5 mg/die. Come mostrato nel pannello superiore della Fig. 2b, DCA più capecitabina a 7,5 mg/die ha inibito significativamente la crescita tumorale rispetto a DCA o capecitabina da soli (P < 0,05). Ventidue giorni dopo l’inoculazione, DCA più 7,5 mg/die di capecitabina hanno inibito la crescita tumorale del 75% (P < 0,05), mentre DCA e capecitabina da soli hanno inibito la crescita solo del 25% e del 35%, rispettivamente (P < 0,05). Il DCA non ha causato alcuna perdita acuta di peso corporeo rispetto al trattamento con la sola capecitabina (Fig. 2b, pannello inferiore). Questi risultati dimostrano che DCA e capecitabina possono avere un effetto antitumorale sinergico nei tumori del melanoma B16.
IlDCA aumenta l’effetto antitumorale della capecitabina in un modello di xenotrapianto di NSCLC umano A549 senza tossicità aggiuntiva
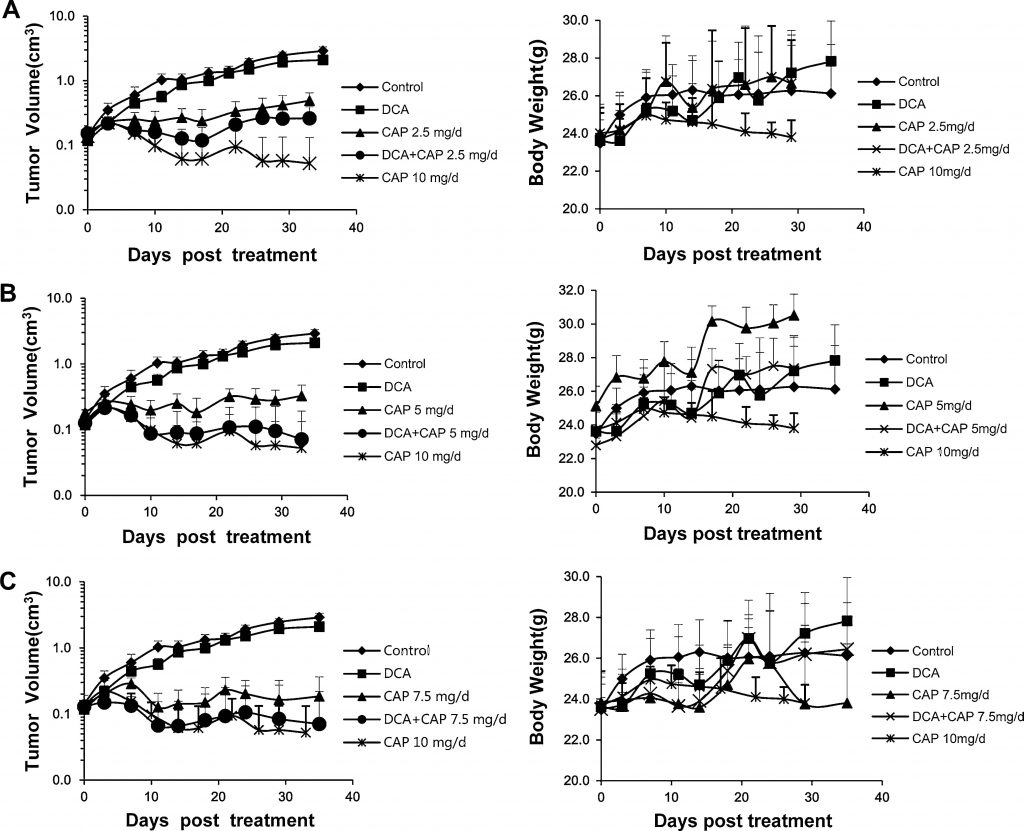
In precedenza è stato riportato che il NSCLC umano può essere trattato sia con il DCA [7] sia con la capecitabina [22-24]. Nel presente studio, abbiamo analizzato l’effetto antitumorale di DCA più capecitabina in un modello di xenotrapianto di NSCLC umano A549. Sessantasei topi maschi BALB/c-nu con tumori NSCLC A549 umani (~2 × 2 mm) inoculati s.c. nel fianco destro sono stati randomizzati in 9 gruppi: controllo; DCA da solo; 2,5, 5, 7,5, o 10 mg/die di capecitabina da sola; o DCA più 2,5, 5, o 7.5 mg/die di capecitabina (n = 6, tranne controllo, DCA da solo, 7,5 mg/die di capecitabina e DCA più 7,5 mg/die di capecitabina; in questi gruppi, n = 9, 3 topi sono stati utilizzati per l’analisi immunoistochimica e l’analisi Western blot o PCR in tempo reale). La capecitabina a 10 mg/die è stata utilizzata come controllo ad alte dosi. Quando il volume del tumore ha raggiunto 0,15-0,2 cm3, i farmaci sono stati somministrati ai topi. La capecitabina è stata somministrata per via parenterale secondo uno schema di 14 giorni sì e 7 no. Come mostrato nei pannelli di sinistra delle Fig. 3a, b e c, il DCA da solo ha avuto un leggero effetto di inibizione della crescita sui tumori A549; questo risultato è in contrasto con quelli di precedenti studi [7] in cui il DCA da solo aveva mostrato un maggiore effetto antitumorale. La capecitabina da sola a 10 mg/die ha inibito fortemente la crescita dei tumori A549, ma si è notata una drastica perdita di peso corporeo, che implica una grave tossicità (Fig. 3a, b, c, pannelli di destra). Come mostrato nel pannello sinistro della Fig. 3a, 2,5 mg/die di capecitabina da sola hanno ridotto significativamente la crescita dei tumori A549; la combinazione di DCA più 2,5 mg/die di capecitabina ha aumentato l’inibizione della crescita. La curva crescente del volume tumorale del trattamento con il solo DCA suggerisce che la capecitabina esercita un effetto antitumorale dominante nel trattamento combinato. L’effetto di DCA più 5 mg/die di capecitabina è stato leggermente migliore rispetto a DCA più 2,5 mg/die di capecitabina, anche se peggiore rispetto a 10 mg/die di capecitabina da sola, e non è stata osservata alcuna perdita significativa del peso corporeo (Fig. 3b, pannello destro). È da notare che DCA più 5 mg/die di capecitabina ha aumentato significativamente l’inibizione della crescita tumorale rispetto alla sola capecitabina 5 mg/die (Fig. 3b, pannello di sinistra). L’effetto antitumorale della combinazione era simile a quello del trattamento con 10 mg/die di capecitabina, ma senza una perdita significativa del peso corporeo (Fig. 3b, pannello destro). L’effetto di 7,5 mg/die di capecitabina da sola (Fig. 3c) è stato leggermente migliore di quello di 5 mg/die di capecitabina da sola; tuttavia, quando è stata combinata con DCA, non c’è stata alcuna differenza significativa tra DCA più 7,5 mg/die di capecitabina e DCA più 5 mg/die di capecitabina. Questi risultati implicano che il DCA può ridurre la dose di capecitabina senza perdere gli effetti antitumorali o aumentare la tossicità.
IlDCA aumenta l’effetto apoptotico della capecitabina sulle cellule B16 e A549 in vivo
L’esameistologicodei tumori di melanoma B16 è stato eseguito 7 giorni dopo l’inizio del trattamento. La colorazione TUNEL ha rivelato che il trattamento dei tumori di melanoma B16 con DCA o 7,5 mg/die di capecitabina da soli ha indotto l’apoptosi cellulare rispettivamente all’8 e al 17%. In combinazione, DCA più 7,5 mg/die di capecitabina hanno indotto un’apoptosi di circa il 30%, superiore alla somma dei singoli trattamenti (Fig. 4a, c, pannello sinistro). La colorazione del PCNA nei tumori del melanoma B16 ha rivelato che il DCA da solo ha avuto un impatto minimo sulla proliferazione, mentre la capecitabina 7,5 mg/die da sola ha ridotto significativamente la proliferazione. DCA più 7,5 mg/die di capecitabina non ha aumentato l’inibizione della proliferazione della sola capecitabina nelle cellule tumorali B16 (Fig. 4b, c, pannello destro).
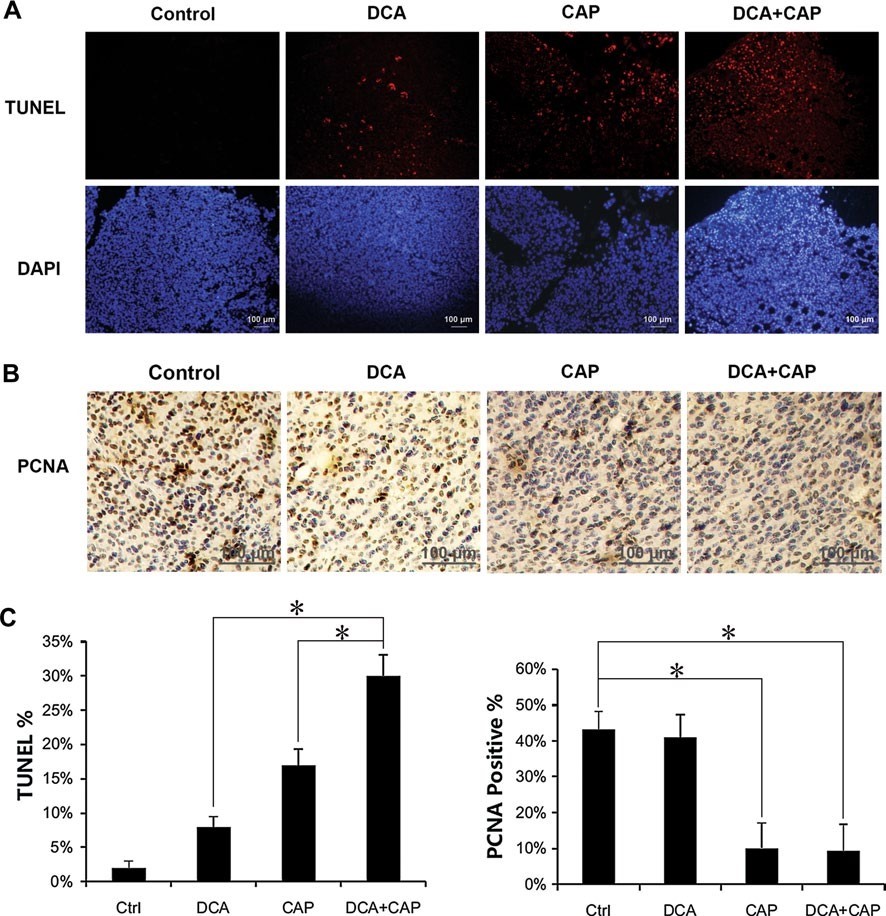
Analogamente ai tumori del melanoma B16, il trattamento dei tumori NSCLC A549 con DCA o capecitabina 7,5 mg/die da soli ha indotto l’apoptosi rispettivamente al 15 e al 30%. Se combinati insieme, DCA più 7,5 mg/die di capecitabina hanno indotto un’apoptosi del 50%, superiore alla somma dei trattamenti con i singoli agenti (Fig. 5a, b). Questi risultati suggeriscono che DCA e capecitabina hanno un effetto sinergico sull’apoptosi delle cellule tumorali NSCLC A549. Il DCA non ha aumentato l’inibizione della proliferazione della capecitabina nelle cellule tumorali NSCLC A549 (dati non mostrati).
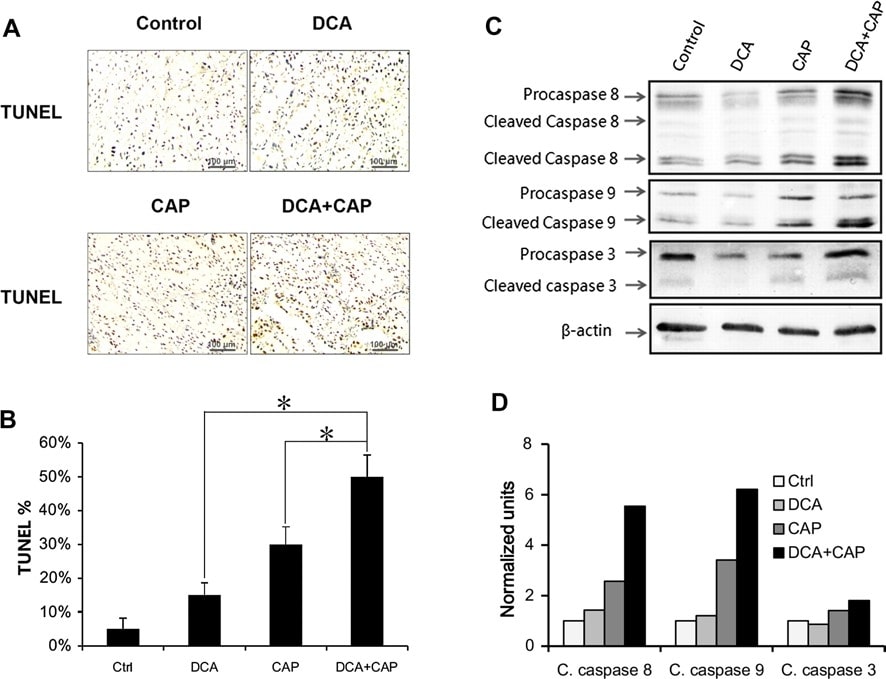
Il Western blot ha mostrato che il DCA ha avuto uno scarso effetto sull’espressione e sull’attivazione della procaspasi 8, della procaspasi 9 e della procaspasi 3 nei tumori NSCLC A549 rispetto al controllo al giorno 7 dopo il trattamento (Fig. 5c, d). La sola capecitabina a 7,5 mg/die ha aumentato l’attivazione di tutte e 3 le procaspasi (Fig. 5c, d). È interessante notare che, sebbene il DCA da solo abbia avuto uno scarso effetto sull’espressione e sull’attivazione delle 3 procaspasi, il DCA più la capecitabina ha promosso l’espressione della procaspasi 8 e della procaspasi 3 rispetto al trattamento con la sola capecitabina e ha aumentato l’attivazione della procaspasi 8, della procaspasi 9 e della procaspasi 3.
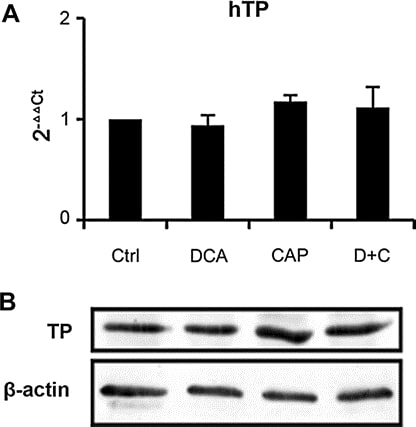
IlDCA ha uno scarso effetto sull’espressione di TP nel tumore
Abbiamo analizzato l’espressione di TP in campioni tumorali di xenotrapianti A549 al giorno 7 post-trattamento. Sia la PCR in tempo reale (Fig. 6a) che il Western blot (Fig. 6b) hanno mostrato che il DCA come agente singolo o in combinazione con la capecitabina ha uno scarso effetto sull’espressione di TP, il che significa che il meccanismo della combinazione di DCA e capecitabina è diverso da quello di altri sinergizzanti della capecitabina precedentemente riportati [19,25-27]. La PCR in tempo reale ha anche mostrato che il DCA non ha influenzato l’espressione di altri enzimi metabolici della capecitabina (timidilato sintasi, orotato fosforibosil transferasi, diidropirimidina deidrogenasi, timidina chinasi 1 e citidina deaminasi, dati non mostrati). I risultati suggeriscono che il DCA ha uno scarso effetto sul metabolismo della capecitabina, che non aumenterebbe la tossicità della capecitabina.
Discussione
Il DCA da solo o in combinazione con altre terapie è stato testato in studi clinici; tuttavia, non sono disponibili rapporti sugli effetti antitumorali del DCA in combinazione con la capecitabina. Abbiamo ipotizzato che l’effetto di promozione dell’apoptosi del DCA sulle cellule tumorali solide le rendesse più sensibili alla capecitabina. Nel presente studio, 1,4 g/L di DCA da solo ha avuto un effetto limitato sulla crescita del tumore NSCLC A549 nei topi. Tuttavia, la somministrazione combinata di DCA e capecitabina è stata in grado di ridurre la dose efficace di capecitabina del 50%. Il DCA ha aumentato l’effetto antitumorale della capecitabina in vivo attraverso la sensibilizzazione all’apoptosi. Il DCA ha uno scarso effetto sul metabolismo della capecitabina, il che non aumenta la tossicità della capecitabina.
Poiché sono stati riportati sia risultati positivi che negativi (come illustrato nella sezione Introduzione), vi sono ancora controversie sull’uso del DCA come agente antitumorale. Questa discrepanza può essere dovuta a condizioni sperimentali diverse, in particolare tra i risultati in vivo e in vitro. I saggi cellulari utilizzati nel presente studio hanno dimostrato che le cellule tumorali sono insensibili al DCA quando coltivate in vitro, con un IC50 superiore a 50 mM. (dati non mostrati). L’effetto del DCA sulle cellule tumorali non è dovuto alla citotossicità diretta, ma dipende dal modello metabolico delle cellule tumorali [7, 28]. Le linee cellulari coltivate in vitro non possono replicare il microambiente tumorale e possono perdere l'”effetto Warburg”. Pertanto, i saggi in vitro basati sulle cellule potrebbero non essere i sistemi modello più pertinenti per determinare l’uso del DCA. Per questo motivo, nel presente studio è stato valutato per la prima volta l’effetto antitumorale combinato di DCA e capecitabina in modelli tumorali murini in vivo. Abbiamo anche riscontrato che l’effetto del DCA sullo xenotrapianto di NSCLC A549 nel presente studio è stato inferiore a quello riportato da Bonnet et al. [7], anche se è stata utilizzata una dose maggiore di DCA. Ciò potrebbe essere dovuto alla diversa capacità metabolica del DCA tra topi e ratti. I risultati suggeriscono che l’effetto del DCA può essere influenzato dallo stato delle cellule tumorali e dei pazienti, il che richiede un attento studio nell’uso clinico del DCA nel trattamento del cancro.
Nel presente studio, gli studi su modelli animali hanno dimostrato che il DCA di per sé ha mostrato un lieve effetto antitumorale contro i tumori stabiliti. Ma quando è stato combinato con la capecitabina, il DCA ha aumentato drasticamente l’effetto antitumorale della capecitabina, come si evince sia dagli studi su modelli animali sia dai risultati dell’immunoistochimica dell’apoptosi. Coerentemente, l’analisi Western blot ha mostrato che il DCA di per sé ha avuto un effetto minimo sull’espressione e sul clivaggio delle procaspasi. Tuttavia, se combinato con la capecitabina, il DCA aumenta notevolmente l’espressione e il clivaggio delle procaspasi 8, 9 e 3. È stato riportato che gli agenti citotossici, come il 5-Fu, possono portare a un aumento dell’espressione e dell’attivazione della caspasi 8 [29,30]. Il motivo per cui la capecitabina porta a un aumento dell’espressione e dell’attivazione della caspasi 9 non è ancora chiaro. Potrebbe essere legato all’effetto antiangiogenetico della capecitabina. È stato riportato che il DCA può normalizzare l’asse dei canali K+ mitocondriali e agire come sensibilizzatore dell’apoptosi [7]. Si ipotizza che il DCA possa depolarizzare il potenziale mitocondriale delle cellule tumorali normalizzando l’asse, aumentando così l’attivazione della caspasi 8 e della caspasi 9 da parte della capecitabina. L’aumento dell’attivazione della caspasi 8 e della caspasi 9 porta a un’aumentata attivazione della caspasi 3.
Recentemente, è stato evidenziato il contributo critico delle CSC, con la loro maggiore capacità tumorigenica e resistenza alla radioterapia e alla chemioterapia, al comportamento maligno [31]. È stato riferito che le CSC nei tumori solidi svolgono un ruolo importante nelle caratteristiche antimoterapiche e antiradioterapiche dei tumori [32, 33]. Sono state discusse molte terapie per colpire le CSC [34, 35] e le terapie combinate per colpire sia le CSC che le cellule tumorali “normali” hanno suscitato grande interesse [36-38]. È stato riportato che il DCA ha indotto l’apoptosi nelle cellule staminali putative del glioblastoma, sia in vitro che in vivo [9]. Nel nostro studio, dopo 7 giorni di trattamento con DCA, il numero di cellule CD133-positive nei vetrini del tumore A549, determinato mediante immunoistochimica, si è ridotto allo 0,5% rispetto al 6% del gruppo di controllo (dati non mostrati), il che ci porta a ipotizzare che il DCA possa avere anche un effetto sull’induzione dell’apoptosi nelle CSC del cancro del polmone. Si ipotizza che il DCA possa sensibilizzare le cellule tumorali, soprattutto le CSC, alla capecitabina. La combinazione di DCA e capecitabina agisce in due modi contro le cellule tumorali: la capecitabina colpisce le cellule tumorali “normali”, risparmiando le CSC, mentre il DCA colpisce le CSC e promuove l’apoptosi della capecitabina. Un altro scenario probabile per spiegare gli effetti della combinazione è che il DCA sopprima l’angiogenesi del cancro in vivo [9], in cui il DCA non mostra un effetto diretto sulle cellule tumorali. Oltre alla sua classica attività antitumorale, secondo recenti studi la capecitabina potrebbe agire anche come molecola antiangiogenetica [ 39]. Il DCA potrebbe potenziare l’effetto antiangiogenetico della capecitabina, il che spiega l’effetto antitumorale combinato. Saranno effettuati studi approfonditi per determinare il meccanismo dettagliato.
In conclusione, abbiamo utilizzato modelli di tumore innestato per via singenetica e xenotrapianto per studiare l’effetto antitumorale combinato di DCA e capecitabina e abbiamo scoperto che il DCA potenzia per la prima volta l’effetto antitumorale della capecitabina. Abbiamo determinato che il DCA ha la capacità di sensibilizzare le cellule tumorali e di aumentare gli effetti apoptotici della capecitabina. Somministrato in combinazione, il DCA è stato in grado di ridurre la dose efficace di capecitabina senza aumentare la tossicità. Il DCA a basso costo, generico e orale, in combinazione con la capecitabina orale, può essere un buon regime terapeutico contro i tumori.
Ringraziamenti
Siamo molto grati a Wenlong Ren dello Shanghai Institute of Pharmaceutical Industry per l’assistenza nella preparazione dei modelli tumorali murini.
Conflitto di interessi
Nessuno.
RIFERIMENTI
1 Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL (2008) PDK-1 regola la produzione di lattato in ipossia ed è associato a una prognosi sfavorevole nel tumore squamoso della testa e del collo. Br J Cancer 98(12):1975-1984. doi:10.1038/sj.bjc.6604356
2 Cohen RD, Iles RA (1978) Il dicloroacetato e il trattamento dell’acidosi lattica. New Engl J Med 298(24):1364. doi:10.1056/NEJM197806152982413
3 Agbenyega T, Planche T, Bedu-Addo G, Ansong D, Owusu-Ofori A, Bhattaram VA, Nagaraja NV, Shroads AL, Henderson GN, Hutson AD, Derendorf H, Krishna S, Stacpoole PW (2003) Population kinetics, efficacy, and safety of dichloroacetate for lactic acidosis due to severe malaria in children. J Clin Pharmacol 43(4):386-396
4 Michelakis ED, Webster L, Mackey JR (2008) Il dicloroacetato (DCA) come potenziale terapia a bersaglio metabolico per il cancro. Br J Cancer 99(7):989-994. doi:10.1038/sj.bjc.6604554
5 Warburg O, Wind F, Negelein E (1927) Il metabolismo dei tumori nell’organismo. J General Physiol 8(6):519-530
6 Gatenby RA, Gillies RJ (2004) Perché i tumori hanno un’elevata glicolisi aerobica? Nat Rev Cancer 4(11):891-899. doi:10.1038/nrc1478
7 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11(1):37-51. doi:10.1016/j.ccr.2006.10.020
8 Pan JG, Mak TW (2007) Il targeting metabolico come strategia antitumorale: l’alba di una nuova era? Science’s STKE: signal transduction knowledge environment 2007(381):pe14. doi:10.1126/stke.3812007pe14
9 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2(31):31ra34. doi:10.1126/scitranslmed.3000677
10 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Il dicloroacetato (DCA) sensibilizza alle radiazioni in vitro sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2. Prostata 68(11):1223-1231. doi:10.1002/pros.20788
11 Xiao L, Li X, Niu N, Qian J, Xie G, Wang Y (2010) Il dicloroacetato (DCA) potenzia la morte delle cellule tumorali in combinazione con l’adenovirus oncolitico armato di MDA-7/IL-24. Mol Cell Biochem 340(1-2):31-40. doi:10.1007/s11010-010-0397-6
12 Kwitniewski M, Moan J, Juzeniene A (2011) Metabolic-targeted therapy with dichloroacetate (DCA): a novel treatment strategy to improve the outcome of photodynamic therapy. Photochem Photobiol Sci Off J Eur Photochem Assoc Eur Soc Photobiol 10(1):25-28. doi:10.1039/c0pp00193g
13 Tong J, Xie G, He J, Li J, Pan F, Liang H (2011) Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. J Biomed Biotechnol 2011:740564. doi:10.1155/2011/740564
14 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL (2010) Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett 297(1):75-83. doi:10.1016/j.canlet.2010.04.027
15 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C (2011) La terapia metabolicamente mirata con dicloroacetato sconfigge la citotossicità dei farmaci antitumorali standard. Cancer Chemother Pharmacol 67(3):647-655. doi:10.1007/s00280-010-1361-6
16 Mandelblat J, Bashir T, Budman DR (2006) Trattamento di combinazione capecitabina-docetaxel. Expert Rev Anticancer Ther 6(9):1169-1178. doi:10.1586/14737140.6.9.1169
17 Budman DR (2000) Capecitabina. Invest New Drugs 18(4):355-363
18 Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K, Shimma N, Umeda I, Ishitsuka H (1998) Progettazione di un nuovo carbammato fluoropirimidinico orale, la capecitabina, che genera 5-fluorouracile in modo selettivo nei tumori grazie a enzimi concentrati nel fegato umano e nel tessuto canceroso. Eur J Cancer 34(8):1274-1281
19 Endo M, Shinbori N, Fukase Y, Sawada N, Ishikawa T, Ishitsuka H, Tanaka Y (1999) Induction of thymidine phosphorylase expression and enhancement of efficacy of capecitabine or 5′-deoxy-5-fluorouridine by cyclophosphamide in mammary tumor models. Int J cancer J int du cancer 83(1):127-134
20 Ishikawa T, Sekiguchi F, Fukase Y, Sawada N, Ishitsuka H (1998) Correlazione positiva tra l’efficacia della capecitabina e della doxifluridina e il rapporto tra le attività della timidina fosforilasi e della diidropirimidina deidrogenasi nei tumori in xenotrapianti di cancro umano. Cancer Res 58(4):685-690
21 Kolinsky K, Shen BQ, Zhang YE, Kohles J, Dugan U, Zioncheck TF, Heimbrook D, Packman K, Higgins B (2009) In vivo activity of novel capecitabine regimens alone and with bevacizumab and oxaliplatin in colorectal cancer xenograft models. Mol Cancer Ther 8(1):75-82. doi:10.1158/1535-7163.MCT-08-0596
22 Lee DH, Han JY, Yoon SM, Lee JJ, Lee HG, Kim HY, Yoon SJ, Hong EK, Lee JS (2006) A pilot trial of gemcitabine and vinorelbine plus capecitabine in locally advanced or metastatic nonsmall cell lung cancer. Am J Clin Oncol 29(2):143-147. doi:10.1097/01.coc.0000203743.32845.40
23LeeJJ, Han JY, Lee DH, Kim HY, Chun JH, Lee HG, Yoon SM, Lee SY, Lee JS (2006) A phase II trial of docetaxel plus capecitabine in patients with previously treated non-small cell lung cancer. Jpn J Clin Oncol 36(12):761-767. doi:10.1093/jjco/hyl106
24 Kindwall-Keller T, Otterson GA, Young D, Neki A, Criswell T, Nuovo G, Soong R, Diasio R, Villalona-Calero MA (2005) Phase II evaluation of docetaxel-modulated capecitabine in previously treated patients with non-small cell lung cancer. Clin Cancer Res off J Am Assoc Cancer Res 11(5):1870-1876. doi:10.1158/1078-0432.CCR-04-1727
25 Sawada N, Ishikawa T, Fukase Y, Nishida M, Yoshikubo T, Ishitsuka H (1998) Induction of thymidine phosphorylase activity and enhancement of capecitabine efficacy by taxol/taxotere in human cancer xenografts. Clin Cancer Res Off J Am Assoc Cancer Res 4(4):1013-1019
26 Sawada N, Kondoh K, Mori K (2007) Enhancement of capecitabine efficacy by oxaliplatin in human colorectal and gastric cancer xenografts. Oncol Rep 18(4):775-778
27 Sawada N, Ishikawa T, Sekiguchi F, Tanaka Y, Ishitsuka H (1999) L’irradiazione a raggi X induce la timidina fosforilasi e potenzia l’efficacia della capecitabina (Xeloda) in xenotrapianti di cancro umano. Clin Cancer Res Off J Am Assoc Cancer Res 5(10):2948-2953
28 Kumar A, Kant S, Singh SM (2012) Nuovi meccanismi molecolari dell’azione antitumorale del dicloroacetato contro il linfoma a cellule T: implicazione dell’alterazione del metabolismo del glucosio, dell’omeostasi del pH e della regolazione della sopravvivenza cellulare. Chem Biol Interact 199(1):29-37. doi:10.1016/j.cbi.2012.06.005
29 Milner AE, Palmer DH, Hodgkin EA, Eliopoulos AG, Knox PG, Poole CJ, Kerr DJ, Young LS (2002) Induction of apoptosis by chemotherapeutic drugs: the role of FADD in activation of caspase-8 and synergy with death receptor ligands in ovarian carcinoma cells. Cell Death Differ 9(3):287-300. doi:10.1038/sj.cdd.4400945
30 Ehrhardt H, Hacker S, Wittmann S, Maurer M, Borkhardt A, Toloczko A, Debatin KM, Fulda S, Jeremias I (2008) Cytotoxic drug-induced, p53-mediated upregulation of caspase-8 in tumor cells. Oncogene 27(6):783-793. doi:10.1038/sj.onc.1210666
31 Ghotra VP, Puigvert JC, Danen EH (2009) The cancer stem cell microenvironment and anti-cancer therapy. Int J Radiat Biol 85(11):955-962. doi:10.3109/09553000903242164
32 Kitamura H, Okudela K, Yazawa T, Sato H, Shimoyamada H (2009) Cancer stem cell: implications in cancer biology and therapy with special reference to lung cancer. Lung Cancer 66(3):275-281. doi:10.1016/j.lungcan.2009.07.019
33 Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J, Gurney AL (2008) Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One 3(6):e2428. doi:10.1371/journal.pone.0002428
34 Tao H, Zhu Y (2011) Le cellule staminali del cancro colorettale: un potenziale bersaglio terapeutico. Clin Transl Oncol Off Publ Fed Span Oncol Soc Natl Cancer Inst Mexico 13(12):833-838. doi:10.1007/s12094-011-0743-5
35 Tang C, Ang BT, Pervaiz S (2007) Cancer stem cell: target for anti-cancer therapy. FASEB J Off Publ Fed Am Soc Exp Biol 21(14):3777-3785. doi:10.1096/fj.07-8560rev
36 Sun XY, Nong J, Qin K, Warnock GL, Dai LJ (2011) Mesenchymal stem cell-mediated cancer therapy: a dual-targeted strategy of personalized medicine. World J Stem Cells 3(11):96-103. doi:10.4252/wjsc.v3.i11.96
37 Sapi E (2009) All’orizzonte una nuova terapia con le cellule staminali del cancro. Cancer Biol Ther 8(18):1754-1755
38 Shigdar S, Lin J, Li Y, Yang CJ, Wei M, Zhus Y, Liu H, Duan W (2012) Cancer stem cell targeting: the next generation of cancer therapy and molecular imaging. Ther Deliv 3(2):227-244
39RanieriG, Roccaro AM, Vacca A, Ribatti D (2006) La timidina fosforilasi (fattore di crescita delle cellule endoteliali di derivazione piastrinica) come bersaglio della capecitabina: dalla biologia al letto del malato. Recent Pat Anti-Cancer Drug Discov 1(2):171-183
Contenuti correlati: