Mao-fa Zheng1, Si-yu Shen, Wei-da Huang
1Départementde biochimie, École des sciences de la vie, Université Fudan, Handan Road 220, Shanghai, 200433, Chine.
Courriel : courriel : [email protected]
Reçu : 16 mai 2013
Accepté : 27 août 2013
Publié : 17 septembre 2013
Résumé
Objectif : La capécitabine est l’un des rares médicaments de chimiothérapie dont la disponibilité orale est élevée. Récemment, le dichloroacétate (DCA) de sodium a montré un grand potentiel comme agent anticancéreux. Dans la présente étude, nous avons évalué l’effet anticancéreux du DCA en association avec la capécitabine pour les cancers exprimant modestement le TP.
Méthodes : Une allogreffe de mélanome B16 de souris et une xénogreffe de cancer du poumon non à petites cellules A549 humain ont été utilisées pour évaluer l’effet du traitement combiné DCA et capécitabine. L’histologie et l’immunohistochimie ont été utilisées pour détecter l’apoptose et la prolifération des cellules cancéreuses. La PCR en temps réel et le Western blot ont été réalisés pour détecter l’expression de TP et des caspases, respectivement.
Résultats : Pour la première fois, nous rapportons que le DCA augmente les effets antitumoraux de la capécitabine dans une allogreffe de souris B16 et une xénogreffe humaine A549 en favorisant l’apoptose des cellules tumorales. Le DCA a peu d’effet sur l’expression de TP.
Conclusions : Nos résultats suggèrent que le DCA en combinaison avec la capécitabine pourrait être un nouveau régime thérapeutique potentiel contre certains cancers.
Mots-clés : DCA, Capécitabine, Combinaison, Effet antitumoral
© Springer-Verlag Berlin Heidelberg 2013
Cancer Chemother Pharmacol (2013) 72:1031-1041
DOI: 10.1007/s00280-013-2281-z
INTRODUCTION
Le dichloroacétate (DCA) de sodium est un petit sel moléculaire de l’acide dichloroacétique dont le poids moléculaire est de 150 Da. Le DCA inhibe l’activité de la pyruvate déshydrogénase kinase, activant ainsi le complexe enzymatique mitochondrial pyruvate déshydrogénase [1] et convertissant la voie métabolique glycolytique en phosphorylation oxydative. Au cours des 40 dernières années, le DCA a été utilisé comme médicament orphelin dans le traitement de l’acidose lactique congénitale de l’enfant et d’autres acidoses lactiques compliquées par d’autres maladies [2] et a montré une grande efficacité et une faible toxicité dans les essais précliniques et cliniques [3]. Récemment, le DCA a montré un grand potentiel en tant qu’agent anticancéreux en raison des similitudes entre le remodelage métabolique de certaines cellules tumorales et celui qui se produit pendant l’acidose lactique [4]. Les cellules cancéreuses, notamment les cellules souches cancéreuses (CSC), résistent à l’apoptose en produisant de l’énergie par glycolyse et fermentation lactique, plutôt que par phosphorylation oxydative, en raison de la nature hypoxique du microenvironnement tumoral, un phénomène connu sous le nom d’effet Warburg [5,6]. Après administration orale, il a été démontré que le DCA restaure la fonction mitochondriale et favorise sélectivement l’apoptose des cellules tumorales par une voie dépendante des mitochondries [7,8]. Les activités thérapeutiques du DCA contre le glioblastome ont été testées dans des essais cliniques (NCT00540176) et ont donné quelques résultats positifs [9]. Cependant, l’essai de phase II NCT01029925 visant à déterminer le taux de réponse du dichloroacétate oral chez des patients atteints d’un cancer du sein ou d’un cancer du poumon non à petites cellules récurrent et/ou métastatique et prétraité a été interrompu en raison de risques et de problèmes de sécurité plus élevés que prévu. L’utilité clinique du DCA dans la lutte contre le cancer doit donc être évaluée plus soigneusement.
En tant que sensibilisateur à l’apoptose, le DCA a également été utilisé en association avec d’autres thérapies anticancéreuses. Cao et al. [10] ont signalé que le DCA sensibilisait les cellules cancéreuses de la prostate aux radiations in vitro. Xiao et al. [11] ont déterminé que le DCA augmentait la mort des cellules tumorales lorsqu’il était associé à un adénovirus oncolytique exprimant le suppresseur de tumeur MDA-7/IL-24. Récemment, la thérapie ciblant le métabolisme avec le DCA a été démontrée comme une nouvelle stratégie de traitement pour améliorer les résultats de la thérapie photodynamique [12]. Tong et al [13] ont découvert que le DCA et le 5-fluorouracile avaient un effet antitumoral synergique sur les cellules cancéreuses colorectales in vitro. Cependant, des résultats controversés et des doutes subsistent quant à l’utilisation du DCA seul ou en association avec d’autres médicaments. Shahrzad et al. [14] ont montré que le DCA réduisait l’apoptose des cellules cancéreuses dans des conditions hypoxiques à la fois in vitro et in vivo. Heshe et al. [15] ont indiqué que le DCA réduisait la cytotoxicité de certains médicaments anticancéreux standard, tels que le cisplatine et la doxorubicine, mais qu’il n’affectait pas l’activité du témozolomide dans 7 des 10 lignées cellulaires de leur étude. Ces résultats contradictoires impliquent que l’utilisation du DCA seul ou en association avec d’autres thérapies peut être spécifique au type de cancer et à l’agent.
La capécitabine est l’un des rares médicaments chimiothérapeutiques très disponibles par voie orale. Elle est autorisée comme traitement de première intention du cancer du rectum métastatique ou comme traitement alternatif du cancer du sein métastatique lorsqu’elle est associée au docétaxel [16,17]. La capécitabine est un promédicament du 5-fluorouracile (5-FU) et nécessite 3 réactions enzymatiques pour une conversion finale en 5-FU dans les cellules tumorales. La réaction finale est catalysée par la thymidine phosphorylase (TP), qui est exprimée dans une plus grande mesure dans certaines tumeurs que dans les tissus normaux [18]. Par conséquent, le 5-FU cytotoxique est généré à un degré plus élevé dans les cellules tumorales que dans les tissus non ciblés, ce qui fait de la capécitabine un médicament chimiothérapeutique à faible toxicité [18]. Les niveaux d’expression du TP sont différents selon les types de tumeurs [18], ce qui limite l’utilisation de la capécitabine à quelques types de cancers seulement. Dans la présente étude, nous avons évalué l’effet anticancéreux du DCA en association avec la capécitabine pour les cancers qui expriment modestement le TP. Nous avons émis l’hypothèse que le DCA augmente les effets anticancéreux et réduit la dose efficace de la capécitabine. L’association du DCA à la capécitabine pourrait produire un bon schéma thérapeutique car les deux agents peuvent être pris par voie orale avec une bonne adhésion du patient. En outre, les formes génériques de DCA peuvent réduire la dose efficace de capécitabine, diminuant ainsi les effets secondaires et le coût du traitement du cancer.
Matériel et méthodes
Matériaux
Le dichloroacétate desodium(DCA, CSA:2156-56-1), pureté 99 %, a été obtenu auprès de Shanghai Jieshi Chemical Co., Ltd. (Chine). le 5-fluorouracil (5-FU) et la 5′-déoxy-fluorouridine (5-DFUR) ont été obtenus auprès de Sigma-Aldrich (USA). Le MTT a été obtenu auprès de Shanghai Biological Engineering Co., Ltd. (Chine). Les comprimés de capécitabine (Xeloda) ont été obtenus auprès de Roche (USA). Le mélanome B16 de la souris et la lignée cellulaire A549 du cancer du poumon non à petites cellules humain ont été obtenus auprès de l’American Type Cell Culture Collection (ATCC, États-Unis).
Études sur des modèles animaux
Modèle d’allogreffe
Des souris C57BL/6, femelles, âgées de 6 à 8 semaines et pesant environ 18 à 20 g, ont été achetées au Shanghai Laboratory Animal Center (SLAC, Chine) et laissées s’acclimater pendant une semaine. Un million de cellules uniques B16 ont été inoculées par voie sous-cutanée (s.c.) dans le flanc droit des souris C57BL/6. Les souris ont été regroupées de manière aléatoire, avec 6 souris par groupe dans une cage. Il y avait deux groupes de souris. Le DCA et la capécitabine ont été administrés à ces deux ensembles de souris, respectivement 3 et 10 jours après l’inoculation. Le DCA a été ajouté à de l’eau potable stérile jusqu’à une concentration finale de 1,4 g/L. La mesure du volume d’eau consommé a montré que la quantité de DCA administrée à chaque souris était approximativement égale à 100 mg/kg/jour. Les comprimés de capécitabine ont été broyés et mis en suspension dans de l’eau stérile avec 4 % de carboxyméthylcellulose pour obtenir différentes concentrations. Deux cents microlitres des suspensions de capécitabine ont été administrés par voie intragastrique à chaque souris. Tous les 2 jours, les diamètres long (a) et court (b) des tumeurs ont été mesurés à l’aide d’un pied à coulisse, et le poids corporel a été enregistré. Le volume de la tumeur a été calculé par la formule V = 0,5ab2. Vingt-deux jours après l’inoculation, les souris ont été tuées et les tumeurs ont été retirées et pesées.
Modèle de xénogreffe
Des souris BALB/c-nu, de sexe masculin, âgées de 5 à 6 semaines et pesant environ 18 à 20 g, ont été achetées au Shanghai Laboratory Animal Center (SLAC, Chine) et laissées s’acclimater pendant une semaine. Des sections d’environ 2 × 2 mm de tissus tumoraux A549 récemment hachés, provenant de souris BALB/c-nu préalablement inoculées avec des cellules A549, ont été inoculées s.c. dans la région du flanc droit de souris BALB/c-nu mâles. Le DCA et la capécitabine ont été administrés aux souris lorsque le volume de leur tumeur atteignait ~0,2 cm3. Trente à trente-cinq jours après le traitement, les souris ont été tuées et les tumeurs ont été retirées et pesées. Les autres méthodes étaient les mêmes que celles décrites dans l’expérience d’allogreffe.
Les études sur les animaux ont été approuvées par le Groupe d’éthique et de bien-être des animaux du Département de science des animaux de laboratoire de l’Université Fudan.
Histologie et immunohistochimie
L’examen histologique des nodules tumoraux a été réalisé sur des animaux supplémentaires (3 souris par groupe) qui n’ont pas été pris en compte pour le suivi de la croissance tumorale. Les tissus ont été fixés dans du paraformaldéhyde à 4 % (p/v), et après fixation pendant une nuit à température ambiante, les échantillons ont été déshydratés dans de l’éthanol gradué et inclus dans de la paraffine. Par la suite, différentes parties de la tumeur ont été coupées au hasard pour réaliser des sections de 4μm sur le microtome Leica. Après déparaffinisation et réhydratation, trois sections de différentes parties de chaque échantillon ont été choisies pour les opérations de suivi. Le marquage à l’aide de la désoxynucléotidyl transférase terminale médiée par le dUTP nick end-labeling (TUNEL) et la coloration au 4′6-diamidino-2-phénylindole (DAPI) ont été réalisés conformément aux instructions du fabricant des kits TUNEL et DAPI (Beyotime, Chine). Les coupes ont été analysées à l’aide d’un microscope inversé à fluorescence (Olympus, Japon). La détection de l’antigène nucléaire des cellules proliférantes (PCNA) a été effectuée après déparaffinisation des sections et incubation à 96-100 °C pendant 20 minutes. L’activité peroxydase endogène a été éteinte avec du peroxyde d’hydrogène à 0,3 % (v/v) dans du méthanol à 60 % (v/v) pendant 30 minutes. L’adsorption non spécifique a été minimisée en incubant les sections dans du sérum de chèvre normal à 2 % (v/v) dans du PBS pendant 20 minutes. Les coupes de tissu ont été incubées pendant une nuit avec un anticorps polyclonal de lapin anti-PCNA (Abcam ; 1:100 dans du PBS), lavées avec du PBS 3 fois, 30 min par fois, et incubées avec une IgG de chèvre anti-lapin conjuguée à de la biotine pendant 2 h à 37 °C et un complexe avidine-biotine-peroxydase pendant 1 h à 37 °C. Les sections ont été contre-colorées avec de l’hématoxyline (Sigma-Aldrich) et analysées par microscopie optique (Olympus, Japon). Trois tumeurs par groupe ont été utilisées pour l’analyse immunohistochimique. Une ou deux sections par tumeur ont été sélectionnées en aveugle pour mesurer les cellules positives au PCNA ou au TUNEL. Cinq champs aléatoires par lame en grossissement 400× ont été mesurés en aveugle (n = 250 cellules par groupe). Lors de l’analyse quantitative des cellules TUNEL-positives, les éventuelles cellules nécrotiques ont été exclues en observant la morphologie nucléaire avec la coloration DAPI. Des contrôles positifs et négatifs ont été effectués pour garantir la précision des résultats.
Extraction des protéines et Western blot
Les tissustumorauxde chaque groupe (3 souris supplémentaires par groupe) ont été regroupés et broyés sous azote liquide, puis lysés dans 150 μL de tampon de lyse tissulaire (Beyotime, Chine). Les tubes ont été agités vigoureusement pendant 1 min, placés sur la glace pendant 20 min et centrifugés à 5 000g pendant 5 min à 4 °C. La concentration en protéines totales a été déterminée à l’aide d’un kit de détermination des protéines BCA (BioRad). Trente microgrammes de protéines totales de chaque échantillon ont été séparés par SDS-PAGE sur un gel à 10 %, transférés sur une membrane PVDF, bloqués, incubés pendant une nuit avec l’anticorps primaire, incubés pendant 1 h avec l’anticorps secondaire, et développés avec le substrat chromogène NBT/BCIP. L’anticorps monoclonal de souris anticaspase 3 (1:500), anticaspase 9 (1:1 000), anticorps anti-β-actine (1:2 000) et anticorps polyclonal de lapin anticaspase 8 (1:1 000) ont été obtenus auprès de Beyotime (Nanjing, Chine). L’anticorps monoclonal de souris anti-TP (1:2,000) provient d’Abcam (UK). la β-Actine a été utilisée comme contrôle interne. La densité des bandes du Western blot a été analysée avec Clinx Gel Analysis V2.02 (Clinx Science, Chine).
PCR en temps réel
Les tumeursB16et les tissus hépatiques ont été prélevés sur la même souris C57BL/6 préalablement inoculée avec des cellules B16 (3 souris ont été utilisées, don de Wenlong Ren de l’Institut d’industrie pharmaceutique de Shanghai). Les tissus de Colo205/A549 et du foie ont été prélevés sur la même souris BALB/c-nu à laquelle on avait précédemment inoculé des cellules Colo205/A549 (3 souris ont été utilisées, respectivement, don de Wenlong Ren à l’Institut d’industrie pharmaceutique de Shanghai). Pour l’analyse de l’expression de TP après traitement, les échantillons de tumeurs ont été regroupés à partir de 3 tissus tumoraux de poids égal pour chaque groupe. L’ARN total a été isolé avec le réactif Trizol (Invitrogen, USA) et transcrit de manière inverse avec le kit de réactif PrimeScript® RT (Takara, Japon). L’ADNc a été normalisé avec la β-actine. La PCR en temps réel a été réalisée par des méthodes en trois étapes en utilisant le kit SYBR® Premix Ex Taq™ II (Takata, Japon) avec une température d’hybridation de 55 °C et 40 cycles d’amplification. Les tests individuels ont été réalisés en triplicata. la β-actine a été utilisée comme contrôle interne. La quantité relative de chaque ADNc a été analysée au moyen du2-△△Ct. Amorces pour la PCR en temps réel : β-actine F : 5′-TCAAGATCATTGCTC CTCCTG-3′ et β-actine R : 5′-CTGCTTGCTGATCCACATCTG-3′, hTP F : 5′-TGGCTCAGTCGGGACAGCAG-3′ et hTP R : 5′-TCCGCTGATCATTG GCACCT-3′, mTP F : 5′-GCCTAGCTAAAGCATTGTGCTC-3′ et mTP R : 5′-AAGGGTGCT CGATCTGATAGCA-3′.
Statistiques
Nous avons utilisé l’ANOVA à comparaisons multiples avec analyse post hoc (test de Tukey), en utilisant le logiciel SPSS 16.0 (SPSS Inc., USA). Les données sont présentées sous forme de moyenne ± SEM, et P < 0,05 a été considéré comme significatif.
Résultats
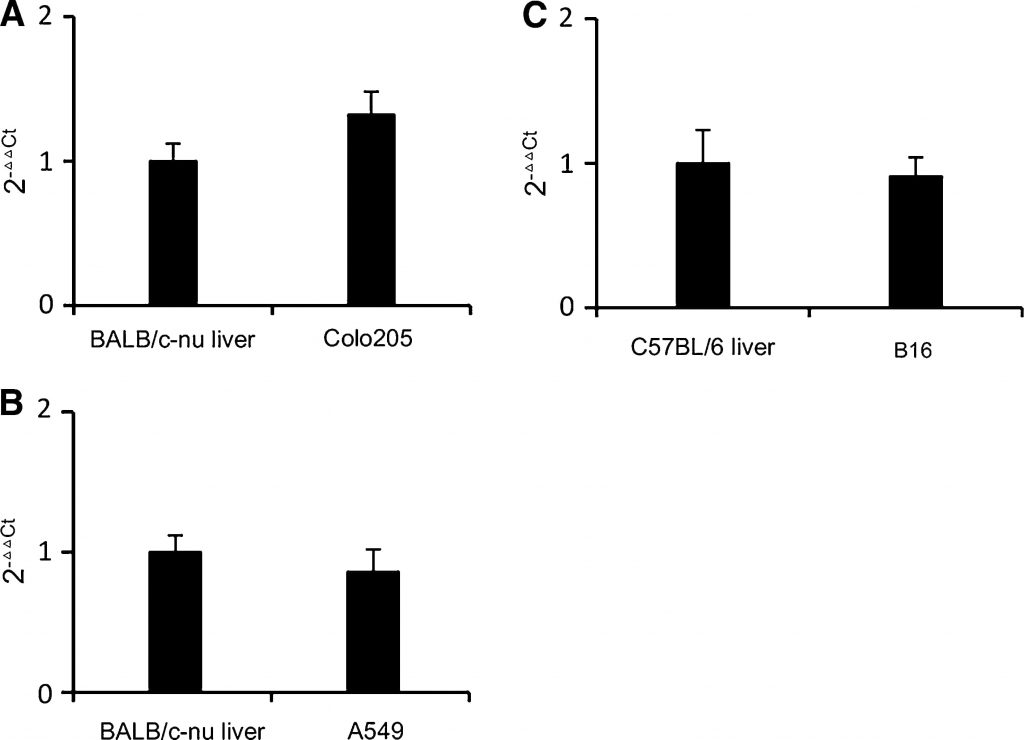
Expression de la TP dans les mélanomes B16 de souris et les tumeurs humaines A549 du NSCLC
L’expression de la TP dans les tumeurs B16 et A549 prélevées sur des souris a été analysée par PCR en temps réel. Le foie humain exprime relativement plus de TP que les autres tissus normaux [18]. Dans un type de cancer typique qui convient au traitement par la capécitabine, l’expression de la TP dans la tumeur est proche ou supérieure à celle du foie, par exemple dans les cancers colorectaux et du sein [18,19]. Dans les xénogreffes de cancers humains, les cancers à forte activité TP sont plus sensibles au traitement par capécitabine que ceux à faible activité TP [20]. La lignée cellulaire de cancer colorectal Colo205 a été choisie comme référence en raison de son expression TP modérée et de sa sensibilité modérée à la capécitabine [21]. Comme le montre la figure 1a, les niveaux de transcription de la TP dans Colo205 étaient un peu plus élevés que dans le foie de la souris BALB/c-nu. Comme le montrent les figures 1bet 1c, les niveaux de transcription du TP dans les tumeurs B16 et A549 étaient proches de ceux du foie des souris. Par conséquent, B16 et A549 étaient également des lignées cellulaires exprimant modérément le TP. Nous pouvons en déduire que les B16 et A549 répondront dans une certaine mesure au traitement par capécitabine sans couvrir l’effet du DCA. Ainsi, les modèles d’allogreffe B16 et de xénogreffe A549 étaient appropriés pour étudier l’effet antitumoral du DCA et de la capécitabine en association.
LeDCA augmente l’effet antitumoral de la capécitabine dans une allogreffe de mélanome B16 de souris sans toxicité supplémentaire
Comme la nature hypoxique du microenvironnement tumoral est essentielle pour une activité optimale du DCA, l’effet antitumoral du DCA plus la capécitabine a été testé sur des modèles animaux plutôt que sur des lignées cellulaires. Trente-six souris C57BL/6 ont été inoculées avec 1 ×106 cellules de mélanome B16 et séparées au hasard en 6 groupes (n = 6) : un groupe témoin qui n’a reçu aucun médicament, un groupe DCA seul, un groupe capécitabine seule à 10 mg/jour, et 3 groupes DCA plus 5, 10 ou 20 mg/jour de capécitabine. Trois jours après l’inoculation des cellules tumorales, on a administré à des souris du DCA dans l’eau potable et de la capécitabine par voie orale (p.o.) en tant qu’agents uniques ou en association avec des concentrations croissantes de capécitabine. Comme le montre le panneau supérieur de la figure 2a, les traitements par DCA et par 10 mg/jour de capécitabine seuls ont inhibé modestement la croissance des tumeurs du mélanome B16 par rapport au groupe témoin. En revanche, l’association de DCA et de 10 mg/jour de capécitabine a considérablement augmenté l’inhibition de la croissance tumorale (P < 0,05). L’effet antitumoral du DCA plus 20 mg/jour de capécitabine a été similaire à celui observé avec le DCA plus 10 mg/jour de capécitabine ; la croissance tumorale a été presque totalement inhibée. Il convient de noter que les souris traitées par DCA plus 20 mg/jour de capécitabine ont subi une perte aiguë de poids corporel (figure 2a, panneau inférieur), alors que DCA plus 10 mg/jour de capécitabine a eu peu d’effet sur le poids corporel par rapport au contrôle.
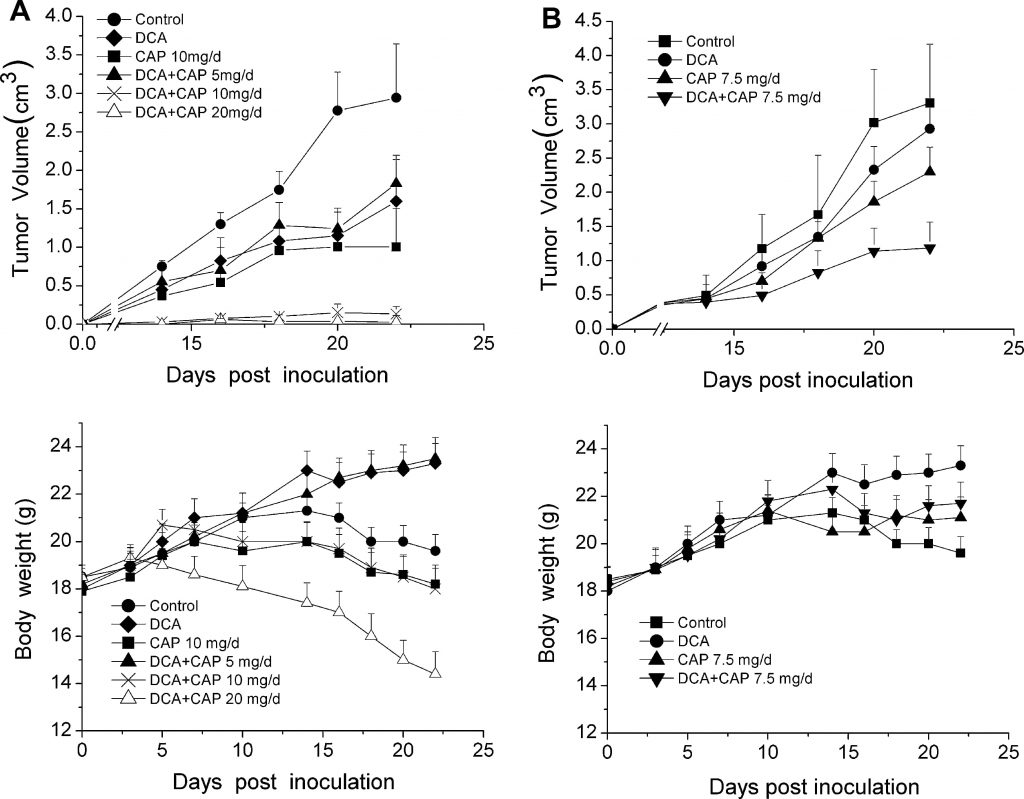
Pour évaluer l’effet antitumoral du DCA et de la capécitabine sur les tumeurs palpables et détectables, 10 jours après l’inoculation des cellules tumorales, le DCA et la capécitabine, seuls ou en association, ont été administrés à un second groupe de 36 souris C57BL/6 inoculées avec 1 ×106 cellules de mélanome B16. Les souris présentant des tumeurs palpables ont été séparées au hasard en 4 groupes (n = 9, 3 souris ont été utilisées pour l’analyse immunohistochimique), contrôle, DCA seul, capécitabine seule à 7,5 mg/jour, et DCA plus capécitabine à 7,5 mg/jour. Comme le montre le panneau supérieur de la figure 2b, le DCA plus la capécitabine à 7,5 mg/jour ont considérablement inhibé la croissance tumorale par rapport au DCA ou à la capécitabine seuls (P < 0,05). Vingt-deux jours après l’inoculation, le DCA plus 7,5 mg/jour de capécitabine ont inhibé la croissance tumorale de 75 % (P < 0,05), alors que le DCA et la capécitabine seuls n’ont inhibé la croissance que de 25 % et 35 %, respectivement (P < 0,05). Le DCA n’a pas entraîné de perte aiguë de poids corporel par rapport au traitement par la capécitabine seule (figure 2b, panneau inférieur). Ces résultats montrent que le DCA et la capécitabine peuvent avoir un effet antitumoral synergique sur les tumeurs du mélanome B16.
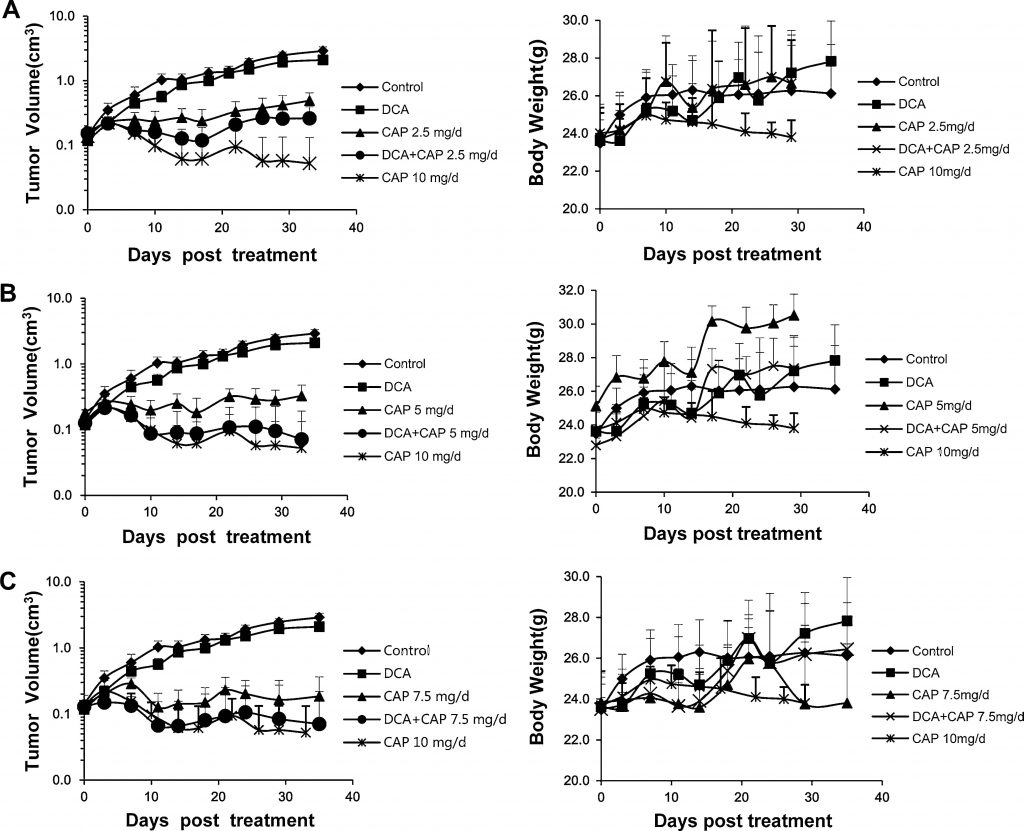
LeDCA augmente l’effet antitumoral de la capécitabine dans un modèle de xénogreffe A549 de NSCLC humain sans toxicité supplémentaire
Il a déjà été signalé que le NSCLC humain pouvait être traité soit par le DCA [7] soit par la capécitabine [22-24]. Dans la présente étude, nous avons examiné l’effet antitumoral du DCA associé à la capécitabine dans un modèle de xénogreffe A549 de CPNPC humain. Soixante-six souris mâles BALB/c-nu avec des tumeurs A549 de CPNPC humain (~2 × 2 mm) inoculées par voie s.c. dans le flanc droit ont été réparties de manière aléatoire en 9 groupes : contrôle ; DCA seul ; 2,5, 5, 7,5 ou 10 mg/jour de capécitabine seule ; ou DCA plus 2,5, 5 ou 7.5 mg/jour de capécitabine (n = 6, sauf le groupe témoin, le DCA seul, la capécitabine à 7,5 mg/jour et le DCA plus 7,5 mg/jour de capécitabine ; dans ces groupes, n = 9, 3 souris ont été utilisées pour l’analyse immunohistochimique et l’analyse Western blot ou PCR en temps réel). La capécitabine à 10 mg/jour a servi de témoin à forte dose. Lorsque le volume des tumeurs atteignait 0,15-0,2 cm3, les médicaments étaient administrés aux souris. La capécitabine a été administrée par voie orale selon un schéma de 14 jours de marche et de 7 jours d’arrêt. Comme le montrent les panneaux de gauche des figures 3a, b et c, le DCA seul a eu un léger effet d’inhibition de la croissance sur les tumeurs A549 ; cette constatation ne correspond pas à celle des rapports précédents [7], dans lesquels le DCA seul avait un effet antitumoral plus important. La capécitabine seule à 10 mg/jour a fortement inhibé la croissance des tumeurs A549, mais une perte spectaculaire de poids corporel a été observée, ce qui implique une toxicité grave (Fig. 3a, b, c, panneaux de droite). Comme le montre le panneau de gauche de la figure 3a, 2,5 mg/jour de capécitabine seule a réduit de manière significative la croissance des tumeurs A549 ; l’association DCA plus 2,5 mg/jour de capécitabine a augmenté l’inhibition de la croissance. La courbe du volume tumoral croissant du traitement par DCA seul suggère que la capécitabine exerce un effet antitumoral dominant dans le traitement combiné. L’effet du DCA plus 5 mg/jour de capécitabine était légèrement meilleur que celui du DCA plus 2,5 mg/jour de capécitabine, mais moins bon que celui de la capécitabine seule à 10 mg/jour, et aucune perte significative de poids corporel n’a été observée (figure 3b, panneau de droite). Il convient de noter que l’association DCA plus 5 mg/jour de capécitabine a augmenté de manière significative l’inhibition de la croissance tumorale par rapport à la capécitabine 5 mg/jour seule (Fig. 3b, panneau de gauche). L’effet antitumoral de l’association était proche de celui du traitement par 10 mg/jour de capécitabine, mais sans perte significative de poids corporel (Fig. 3b, panneau de droite). L’effet de la capécitabine 7,5 mg/jour seule (Fig. 3c) était légèrement meilleur que celui de la capécitabine 5 mg/jour seule ; cependant, lorsqu’elle était associée au DCA, il n’y avait pas de différence significative entre le DCA plus 7,5 mg/jour de capécitabine et le DCA plus 5 mg/jour de capécitabine. Ces résultats impliquent que le DCA peut réduire la dose de capécitabine sans perdre les effets antitumoraux ni augmenter la toxicité.
LeDCA augmente l’effet apoptotique de la capécitabine sur les cellules B16 et A549 in vivo
Un examen histologique des tumeurs de mélanome B16 a été effectué 7 jours après le début du traitement. La coloration TUNEL a révélé que le traitement des tumeurs de mélanome B16 par le DCA ou la capécitabine à 7,5 mg/jour seuls induisait une apoptose cellulaire de 8 et 17 %, respectivement. L’association du DCA et de 7,5 mg/jour de capécitabine a provoqué une apoptose d’environ 30 %, soit plus que la somme des traitements individuels combinés (figure 4a, c, panneau de gauche). La coloration du PCNA dans les tumeurs de mélanome B16 a révélé que le DCA seul avait peu d’impact sur la prolifération, tandis que la capécitabine seule à 7,5 mg/jour réduisait significativement la prolifération. L’association du DCA et de 7,5 mg/jour de capécitabine n’a pas augmenté l’inhibition de la prolifération par la capécitabine seule dans les cellules tumorales B16 (Fig. 4b, c, panneau de droite).
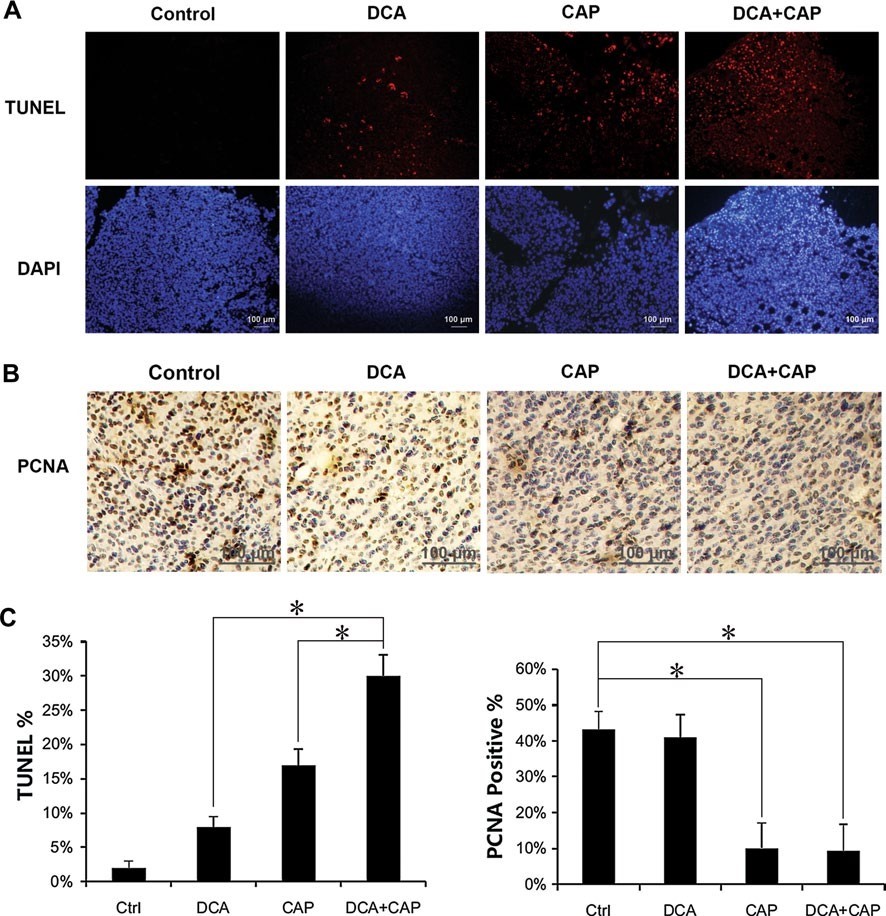
Comme pour les tumeurs du mélanome B16, le traitement des tumeurs du CPNPC A549 par le DCA ou la capécitabine à 7,5 mg/jour seuls a induit une apoptose de 15 et 30 %, respectivement. Lorsqu’ils ont été combinés, le DCA et la capécitabine à 7,5 mg/jour ont induit une apoptose de 50 %, soit plus que la somme des traitements à agent unique combinés (Fig. 5a, b). Ces résultats suggèrent que le DCA et la capécitabine ont un effet synergique sur l’apoptose des cellules tumorales A549 du NSCLC. Le DCA n’a pas augmenté l’inhibition de la prolifération des cellules tumorales A549 du NSCLC par la capécitabine (données non présentées).
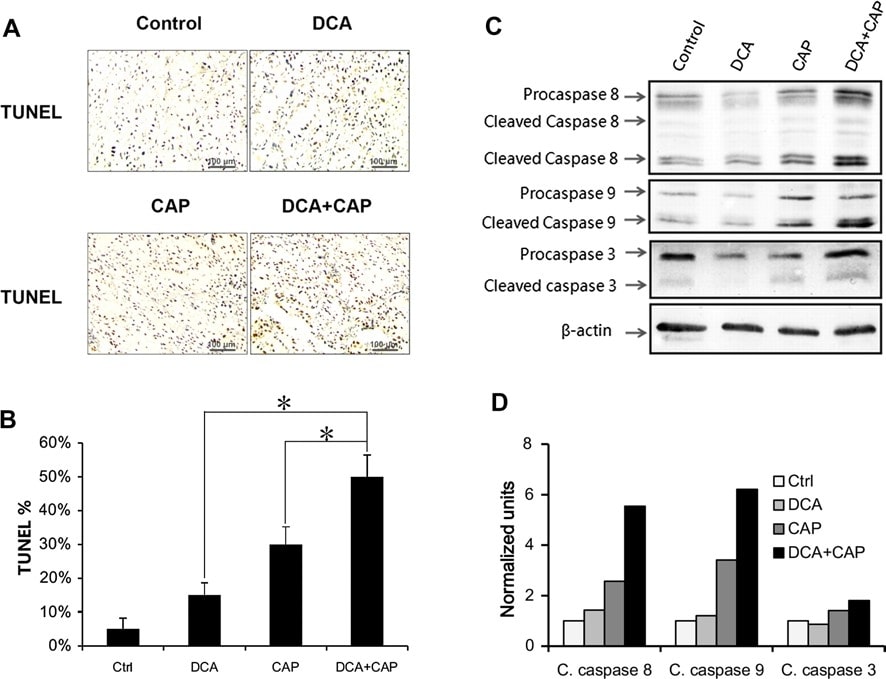
Le Western blot a montré que le DCA avait peu d’effet sur l’expression et l’activation de la procaspase 8, de la procaspase 9 et de la procaspase 3 dans les tumeurs A549 du NSCLC par rapport au contrôle au jour 7 après le traitement (Fig. 5c, d). La capécitabine seule à 7,5 mg/jour a augmenté l’activation des 3 procaspases (Fig. 5c, d). Fait intéressant, bien que le DCA seul ait eu peu d’effet sur l’expression et l’activation des 3 procaspases, le DCA plus la capécitabine ont favorisé l’expression de la procaspase 8 et de la procaspase 3 par rapport au traitement par la capécitabine seule et ont augmenté l’activation de la procaspase 8, de la procaspase 9 et de la procaspase 3.
Le DCA a peu d’effet sur l’expression de la TP dans la tumeur
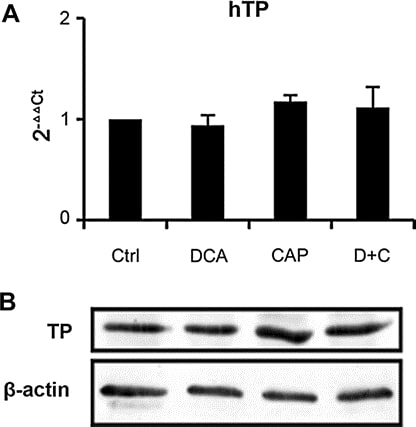
Nous avons analysé l’expression de la TP dans des échantillons de tumeurs provenant de xénogreffes A549 au jour 7 après le traitement. La PCR en temps réel (Fig. 6a) et le Western blot (Fig. 6b) ont montré que le DCA, seul ou en association avec la capécitabine, a peu d’effet sur l’expression de TP, ce qui signifie que le mécanisme du DCA et de la capécitabine en association est différent de celui des autres synergistes de la capécitabine rapportés précédemment [19,25-27]. La PCR en temps réel a également montré que le DCA n’a pas affecté l’expression d’autres enzymes métaboliques de la capécitabine (thymidylate synthase, orotate phosphoribosyl transférase, dihydropyrimidine déshydrogénase, thymidine kinase 1 et cytidine désaminase, données non présentées). Les résultats suggèrent que le DCA a peu d’effet sur le métabolisme de la capécitabine, ce qui n’augmenterait pas la toxicité de la capécitabine.
Discussion
Le DCA, seul ou en association avec d’autres thérapies, a été testé dans des essais cliniques ; cependant, aucun rapport sur les effets antitumoraux du DCA en association avec la capécitabine n’est disponible. Nous avons émis l’hypothèse que l’effet de promotion de l’apoptose du DCA sur les cellules tumorales solides les rendrait plus sensibles à la capécitabine. Dans la présente étude, le DCA à 1,4 g/L seul a eu peu d’effet sur la croissance de la tumeur A549 du CPNPC chez la souris. Cependant, l’administration combinée de DCA et de capécitabine a permis de réduire de 50 % la dose efficace de capécitabine. Le DCA a augmenté l’effet antitumoral de la capécitabine in vivo via la sensibilisation à l’apoptose. Le DCA a peu d’effet sur le métabolisme de la capécitabine, ce qui n’augmenterait pas la toxicité de la capécitabine.
Comme des résultats positifs et négatifs ont été rapportés (comme indiqué dans la section Introduction), l’utilisation du DCA comme agent anticancéreux fait toujours l’objet de controverses. Cette divergence peut être due à des conditions expérimentales différentes, notamment entre les résultats in vivo et in vitro. Les essais cellulaires utilisés dans la présente étude ont montré que les cellules tumorales sont insensibles au DCA lorsqu’elles sont cultivées in vitro, la CI50 étant supérieure à 50 mM. (données non présentées). L’effet du DCA sur les cellules cancéreuses n’est pas dû à une cytotoxicité directe mais dépend du modèle métabolique des cellules cancéreuses [7, 28]. Les lignées cellulaires cultivées in vitro ne peuvent pas reproduire le microenvironnement tumoral et peuvent perdre l' »effet Warburg ». Par conséquent, les essais in vitro basés sur les cellules ne sont peut-être pas les systèmes modèles les plus pertinents pour déterminer l’utilisation du DCA. C’est pourquoi l’effet antitumoral combiné du DCA et de la capécitabine a d’abord été évalué dans des modèles de tumeurs de souris in vivo dans la présente étude. Nous avons également constaté que l’effet du DCA sur la xénogreffe A549 de NSCLC dans la présente étude était inférieur à celui rapporté par Bonnet et al [7], même si une dose plus importante de DCA a été utilisée. Cela peut être dû à la différence de capacité métabolique du DCA entre les souris et les rats. Les résultats suggèrent que l’effet du DCA peut être affecté par l’état des cellules cancéreuses et des patients, ce qui nécessite une étude attentive de l’utilisation clinique du DCA dans le traitement du cancer.
Dans la présente étude, les études sur les modèles animaux ont démontré que le DCA seul a montré un léger effet antitumoral contre les tumeurs établies. Mais lorsqu’il est associé à la capécitabine, le DCA augmente considérablement l’effet antitumoral de la capécitabine, comme le montrent les études sur les modèles animaux et les résultats de l’immunohistochimie de l’apoptose. Dans le même ordre d’idées, une analyse par transfert Western a montré que le DCA lui-même avait peu d’effet sur l’expression et le clivage des procaspases. Mais lorsqu’il est associé à la capécitabine, le DCA augmente remarquablement l’expression et le clivage des procaspases 8, 9 et 3. Il a été signalé que les agents cytotoxiques, comme le 5-Fu, peuvent entraîner une augmentation de l’expression et de l’activation de la caspase 8 [29,30]. La raison pour laquelle la capécitabine entraîne une augmentation de l’expression et de l’activation de la caspase 9 n’est pas encore claire. Elle pourrait être liée à l’effet antiangiogénétique de la capécitabine. Il a été signalé que le DCA peut normaliser l’axe mitochondrial du canal K+ et agir comme un sensibilisateur à l’apoptose [7]. Nous supposons que le DCA peut dépolariser le potentiel mitochondrial des cellules cancéreuses en normalisant l’axe, et donc augmenter l’activation de la caspase 8 et de la caspase 9 par la capécitabine. L’activation accrue de la caspase 8 et de la caspase 9 entraîne une activation accrue de la caspase 3.
Récemment, la contribution critique des CSC, avec leur capacité tumorigène accrue et leur résistance à la radiothérapie et à la chimiothérapie, au comportement malin a été mise en évidence [31]. Dans les tumeurs solides, les CSC joueraient un rôle important dans les caractéristiques anti-chimiothérapie et anti-radiothérapie des tumeurs [32, 33]. De nombreuses thérapies visant à cibler les CSC ont été discutées [34, 35], et les thérapies combinées visant à cibler à la fois les CSC et les cellules cancéreuses « normales » ont suscité un grand intérêt [36-38]. Il a été signalé que le DCA induisait l’apoptose des cellules souches putatives de glioblastome, à la fois in vitro et in vivo [9]. Dans notre étude, après 7 jours de traitement au DCA, le nombre de cellules CD133-positives dans les lames de la tumeur A549, déterminé par immunohistochimie, a été réduit à 0,5 % par rapport à 6 % dans le groupe témoin (données non présentées), ce qui nous amène à supposer que le DCA peut également avoir un effet sur l’induction de l’apoptose dans les CSC du cancer du poumon. Nous supposons que le DCA peut sensibiliser les cellules tumorales, en particulier les CSC, à la capécitabine. L’association du DCA et de la capécitabine agit de deux manières contre les cellules tumorales : la capécitabine cible les cellules cancéreuses » normales « , en épargnant les CSC, et le DCA cible les CSC et favorise l’apoptose à la capécitabine. Un autre scénario probable pour expliquer les effets combinés est que le DCA a supprimé l’angiogenèse du cancer in vivo [9], dans lequel le DCA n’a pas d’effet direct sur les cellules cancéreuses. En plus de son activité antitumorale classique, la capécitabine peut également agir en tant que molécule antiangiogénétique selon des études récentes [39]. Le DCA peut augmenter l’effet antiangiogénique de la capécitabine, ce qui explique l’effet antitumoral combiné. Des études approfondies seront menées pour déterminer le mécanisme détaillé.
En conclusion, nous avons utilisé des modèles de tumeurs greffées par syngénétique et de tumeurs xénogreffées pour étudier l’effet antitumoral combiné du DCA et de la capécitabine et nous avons constaté que le DCA potentialisait pour la première fois l’effet antitumoral de la capécitabine. Nous avons déterminé que le DCA avait la capacité de sensibiliser les cellules cancéreuses et d’augmenter les effets apoptotiques de la capécitabine. Administré en association, le DCA a permis de réduire la dose efficace de capécitabine sans augmenter la toxicité. Le DCA, générique et peu coûteux, administré par voie orale, en association avec la capécitabine par voie orale, pourrait constituer un bon schéma thérapeutique contre les cancers.
Remerciements
Nous sommes très reconnaissants à Wenlong Ren, de l’Institut de l’industrie pharmaceutique de Shanghai, pour son aide dans la préparation des modèles de tumeurs de souris.
Conflit d’intérêt
Aucun.
RÉFÉRENCES
1 Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL (2008) PDK-1 régule la production de lactate dans l’hypoxie et est associé à un mauvais pronostic dans le cancer squameux de la tête et du cou. Br J Cancer 98(12):1975-1984. doi:10.1038/sj.bjc.6604356
2 Cohen RD, Iles RA (1978) Dichloroacetate and the treatment of lactic acidosis. New Engl J Med 298(24):1364. doi:10.1056/NEJM197806152982413
3 Agbenyega T, Planche T, Bedu-Addo G, Ansong D, Owusu-Ofori A, Bhattaram VA, Nagaraja NV, Shroads AL, Henderson GN, Hutson AD, Derendorf H, Krishna S, Stacpoole PW (2003) Population kinetics, efficacy, and safety of dichloroacetate for lactic acidosis due to severe malaria in children. J Clin Pharmacol 43(4):386-396
4 Michelakis ED, Webster L, Mackey JR (2008) Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 99(7):989-994. doi:10.1038/sj.bjc.6604554
5 Warburg O, Wind F, Negelein E (1927) The metabolism of tumors in the body. J General Physiol 8(6):519-530
6 Gatenby RA, Gillies RJ (2004) Why do cancers have high aerobic glycolysis ? Nat Rev Cancer 4(11):891-899. doi:10.1038/nrc1478
7 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED (2007) A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11(1):37-51. doi:10.1016/j.ccr.2006.10.020
8 Pan JG, Mak TW (2007) Metabolic targeting as an anticancer strategy : dawn of a new era ? Science’s STKE : signal transduction knowledge environment 2007(381):pe14. doi:10.1126/stke.3812007pe14
9 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2(31):31ra34. doi:10.1126/scitranslmed.3000677
10 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ (2008) Le dichloroacétate (DCA) sensibilise in vitro les cellules cancéreuses de la prostate de type sauvage et sur-exprimant Bcl-2 aux radiations. Prostate 68(11):1223-1231. doi:10.1002/pros.20788
11 Xiao L, Li X, Niu N, Qian J, Xie G, Wang Y (2010) Le dichloroacétate (DCA) renforce la mort des cellules tumorales en combinaison avec un adénovirus oncolytique armé de MDA-7/IL-24. Mol Cell Biochem 340(1-2):31-40. doi:10.1007/s11010-010-0397-6
12 Kwitniewski M, Moan J, Juzeniene A (2011) Metabolic-targeted therapy with dichloroacetate (DCA) : a novel treatment strategy to improve the outcome of photodynamic therapy. Photochem Photobiol Sci Off J Eur Photochem Assoc Eur Soc Photobiol 10(1):25-28. doi:10.1039/c0pp00193g
13 Tong J, Xie G, He J, Li J, Pan F, Liang H (2011) Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer. J Biomed Biotechnol 2011:740564. doi:10.1155/2011/740564
14 Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL (2010) Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancer Lett 297(1):75-83. doi:10.1016/j.canlet.2010.04.027
15 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C (2011) La thérapie métabolique ciblée par le dichloroacétate déjoue la cytotoxicité des médicaments anticancéreux standard. Cancer Chemother Pharmacol 67(3):647-655. doi:10.1007/s00280-010-1361-6
16 Mandelblat J, Bashir T, Budman DR (2006) Capecitabine-docetaxel combination treatment. Expert Rev Anticancer Ther 6(9):1169-1178. doi:10.1586/14737140.6.9.1169
17 Budman DR (2000) Capecitabine. Invest New Drugs 18(4):355-363
18 Miwa M, Ura M, Nishida M, Sawada N, Ishikawa T, Mori K, Shimma N, Umeda I, Ishitsuka H (1998) Design of a novel oral fluoropyrimidine carbamate, capecitabine, which generates 5-fluorouracil selectively in tumours by enzymes concentrated in human liver and cancer tissue. Eur J Cancer 34(8):1274-1281
19 Endo M, Shinbori N, Fukase Y, Sawada N, Ishikawa T, Ishitsuka H, Tanaka Y (1999) Induction de l’expression de la thymidine phosphorylase et amélioration de l’efficacité de la capécitabine ou de la 5′-désoxy-5-fluorouridine par le cyclophosphamide dans des modèles de tumeurs mammaires. Int J cancer J int du cancer 83(1):127-134
20 Ishikawa T, Sekiguchi F, Fukase Y, Sawada N, Ishitsuka H (1998) Corrélation positive entre l’efficacité de la capécitabine et de la doxifluridine et le rapport entre les activités de la thymidine phosphorylase et de la dihydropyrimidine déshydrogénase dans les tumeurs des xénogreffes de cancer humain. Cancer Res 58(4):685-690
21 Kolinsky K, Shen BQ, Zhang YE, Kohles J, Dugan U, Zioncheck TF, Heimbrook D, Packman K, Higgins B (2009) In vivo activity of novel capecitabine regimens alone and with bevacizumab and oxaliplatin in colorectal cancer xenograft models. Mol Cancer Ther 8(1):75-82. doi:10.1158/1535-7163.MCT-08-0596
22 Lee DH, Han JY, Yoon SM, Lee JJ, Lee HG, Kim HY, Yoon SJ, Hong EK, Lee JS (2006) A pilot trial of gemcitabine and vinorelbine plus capecitabine in locally advanced or metastatic nonsmall cell lung cancer. Am J Clin Oncol 29(2):143-147. doi:10.1097/01.coc.0000203743.32845.40
23LeeJJ, Han JY, Lee DH, Kim HY, Chun JH, Lee HG, Yoon SM, Lee SY, Lee JS (2006) A phase II trial of docetaxel plus capecitabine in patients with previously treated non-small cell lung cancer. Jpn J Clin Oncol 36(12):761-767. doi:10.1093/jjco/hyl106
24 Kindwall-Keller T, Otterson GA, Young D, Neki A, Criswell T, Nuovo G, Soong R, Diasio R, Villalona-Calero MA (2005) Phase II evaluation of docetaxel-modulated capecitabine in previously treated patients with non-small cell lung cancer. Clin Cancer Res off J Am Assoc Cancer Res 11(5):1870-1876. doi:10.1158/1078-0432.CCR-04-1727
25 Sawada N, Ishikawa T, Fukase Y, Nishida M, Yoshikubo T, Ishitsuka H (1998) Induction of thymidine phosphorylase activity and enhancement of capecitabine efficacy by taxol/taxotere in human cancer xenografts. Clin Cancer Res Off J Am Assoc Cancer Res 4(4):1013-1019
26 Sawada N, Kondoh K, Mori K (2007) Enhancement of capecitabine efficacy by oxaliplatin in human colorectal and gastric cancer xenografts. Oncol Rep 18(4):775-778
27 Sawada N, Ishikawa T, Sekiguchi F, Tanaka Y, Ishitsuka H (1999) X-ray irradiation induces thymidine phosphorylase and enhance the efficacy of capecitabine (Xeloda) in human cancer xenografts. Clin Cancer Res Off J Am Assoc Cancer Res 5(10):2948-2953
28 Kumar A, Kant S, Singh SM (2012) Novel molecular mechanisms of antitumor action of dichloroacetate against T cell lymphoma : implication of altered glucose metabolism, pH homeostasis and cell survival regulation. Chem Biol Interact 199(1):29-37. doi:10.1016/j.cbi.2012.06.005
29 Milner AE, Palmer DH, Hodgkin EA, Eliopoulos AG, Knox PG, Poole CJ, Kerr DJ, Young LS (2002) Induction of apoptosis by chemotherapeutic drugs : the role of FADD in activation of caspase-8 and synergy with death receptor ligands in ovarian carcinoma cells. Cell Death Differ 9(3):287-300. doi:10.1038/sj.cdd.4400945
30 Ehrhardt H, Hacker S, Wittmann S, Maurer M, Borkhardt A, Toloczko A, Debatin KM, Fulda S, Jeremias I (2008) Cytotoxic drug-induced, p53-mediated upregulation of caspase-8 in tumor cells. Oncogene 27(6):783-793. doi:10.1038/sj.onc.1210666
31 Ghotra VP, Puigvert JC, Danen EH (2009) The cancer stem cell microenvironment and anti-cancer therapy. Int J Radiat Biol 85(11):955-962. doi:10.3109/09553000903242164
32 Kitamura H, Okudela K, Yazawa T, Sato H, Shimoyamada H (2009) Cancer stem cell : implications in cancer biology and therapy with special reference to lung cancer. Lung Cancer 66(3):275-281. doi:10.1016/j.lungcan.2009.07.019
33 Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J, Gurney AL (2008) Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One 3(6):e2428. doi:10.1371/journal.pone.0002428
34 Tao H, Zhu Y (2011) Colorectal cancer stem cell : a potential therapeutic target. Clin Transl Oncol Off Publ Fed Span Oncol Soc Natl Cancer Inst Mexico 13(12):833-838. doi:10.1007/s12094-011-0743-5
35 Tang C, Ang BT, Pervaiz S (2007) Cancer stem cell : target for anti-cancer therapy. FASEB J Off Publ Fed Am Soc Exp Biol 21(14):3777-3785. doi:10.1096/fj.07-8560rev
36 Sun XY, Nong J, Qin K, Warnock GL, Dai LJ (2011) Mesenchymal stem cell-mediated cancer therapy : a dual-targeted strategy of personalized medicine. World J Stem Cells 3(11):96-103. doi:10.4252/wjsc.v3.i11.96
37 Sapi E (2009) Novel cancer stem cell therapy on the horizon. Cancer Biol Ther 8(18):1754-1755
38 Shigdar S, Lin J, Li Y, Yang CJ, Wei M, Zhus Y, Liu H, Duan W (2012) Cancer stem cell targeting : the next generation of cancer therapy and molecular imaging. Ther Deliv 3(2):227-244
39RanieriG, Roccaro AM, Vacca A, Ribatti D (2006) Thymidine phosphorylase (facteur de croissance des cellules endothéliales dérivées des plaquettes) comme cible de la capécitabine : de la biologie au chevet du patient. Recent Pat Anti-Cancer Drug Discov 1(2):171-183
Contenu connexe :