E. D. Michelakis,1 * G. Sutendra,1 P . Dromparis,1 L. Webster,1 A. Haromy,1 E. Niven,2 C. Maguire,2 T.-L. Gammer,1 J. R. Mackey,3 D. Fulton,3 B. Abdulkarim,3 M. S. McMurtry,1 K. C. Petruk4
1Département demédecine, Université de l’Alberta, Edmonton, Alberta, Canada T6G 2B7.
2Départementdegénie biomédical et d’imagerie diagnostique, Université de l’Alberta, Edmonton, Alberta, Canada T6G 2B7.
3Départementd‘oncologie, Université de l’Alberta, Edmonton, Alberta, Canada T6G 2B7.
4Départementdeneurochirurgie, Université de l’Alberta, Edmonton, Alberta, Canada T6G 2B7.
*À qui la correspondance doit être adressée. Courriel : [email protected]
Volume 2 Numéro 31 31ra34
Soumis : 11 novembre 2009
Accepté : 23 avril 2010
Publié : 12 mai 2010
Les tumeurs solides, y compris le glioblastome multiforme, un cancer du cerveau primaire agressif, développent une résistance à la mort cellulaire, en partie à cause du passage de la phosphorylation oxydative mitochondriale à la glycolyse cytoplasmique. Ce remodelage métabolique s’accompagne d’une hyperpolarisation mitochondriale. Nous avons testé si la petite molécule et le médicament orphelin dichloroacétate (DCA) peuvent inverser ce remodelage métabolique et mitochondrial spécifique au cancer dans le glioblastome. Des glioblastomes fraîchement isolés provenant de 49 patients ont montré une hyperpolarisation mitochondriale, qui a été rapidement inversée par le DCA. Dans une expérience séparée avec cinq patients atteints de glioblastome, nous avons obtenu de manière prospective des tissus tumoraux de base et en série, développé des lignées cellulaires de glioblastome spécifiques au patient et des cellules souches putatives de glioblastome (cellules CD133+, nestin+ ), et traité chaque patient avec du DCA par voie orale pendant 15 mois. Le DCA a dépolarisé les mitochondries, augmenté les espèces réactives de l’oxygène mitochondrial et induit l’apoptose dans les cellules de GBM, ainsi que dans les cellules souches putatives de GBM, à la fois in vitro et in vivo. Le traitement par le DCA a également inhibé le facteur inductible de l’hypoxie-1α, favorisé l’activation de p53 et supprimé l’angiogenèse à la fois in vivo et in vitro. La toxicité limitant la dose était une neuropathie périphérique dose-dépendante et réversible, et il n’y a pas eu de toxicité hématologique, hépatique, rénale ou cardiaque. Des indications d’efficacité clinique étaient présentes à une dose qui ne provoquait pas de neuropathie périphérique et à des concentrations sériques de DCA suffisantes pour inhiber l’enzyme cible du DCA, la pyruvate déshydrogénase kinase II, qui était fortement exprimée dans tous les glioblastomes. La modulation métabolique pourrait être une approche thérapeutique viable dans le traitement du glioblastome.
INTRODUCTION
Le glioblastome multiforme (GBM) est une tumeur cérébrale primaire agressive qui répond extrêmement mal aux thérapies approuvées (1). La chimiothérapie au témozolomide (TMZ) associée à la radiothérapie (RT), administrée après une chirurgie de désobstruction, fait passer la survie médiane de 12,1 mois avec la RT seule à 14,6 mois (1). Le temps médian jusqu’à la progression de la tumeur après la RT et le TMZ n’est que de 6,9 mois (1). Dans les gliomes récurrents, la survie sans progression et la réponse au TMZ sont bien pires (2). Les GBM sont des tumeurs très vasculaires avec une hétérogénéité moléculaire et génétique remarquable (1). Une thérapie idéale devrait augmenter l’apoptose des GBM, surmonter l’hétérogénéité moléculaire, inhiber l’angiogenèse et traverser la barrière hémato-encéphalique tout en ayant une toxicité systémique minimale. Sur la base de nos récentes découvertes dans des modèles animaux (3, 4), nous avons émis l’hypothèse que la petite molécule orpheline dichloroacétate (DCA) remplit ces critères et pourrait être efficace dans le traitement des GBM chez l’homme.
Le DCA inhibe l’enzyme mitochondriale pyruvate déshydrogénase kinase (PDK) (5). En inhibant la PDK, le DCA active la pyruvate déshydrogénase (PDH), une enzyme gardienne qui régule le flux de glucides (pyruvate) dans la mitochondrie, augmentant ainsi le rapport entre l’oxydation du glucose et la glycolyse (3-5). Si le pyruvate reste dans le cytoplasme, il peut achever la glycolyse, produisant de l’acide lactique et générant 2 moles d’ATP par molécule de glucose. Sinon, le pyruvate peut entrer dans plusieurs voies anaplérotiques et de biosynthèse des acides aminés. Cependant, lors de l’activation de la PDH, le pyruvate peut être décarboxylé en acétyl-coenzyme A, entrer dans le cycle de Krebs et compléter l’oxydation du glucose dans la matrice mitochondriale, générant jusqu’à 36 moles d’ATP par molécule de glucose en présence d’oxygène. L’oxydation du glucose n’a pas lieu lorsque le pyruvate ne pénètre pas dans les mitochondries (par exemple, dans des mitochondries malades ou si la PDH est inhibée) ou en l’absence d’oxygène.
Warburg (6) a été le premier à montrer que le métabolisme des cellules cancéreuses, même en normoxie, est caractérisé par une augmentation du rapport entre la glycolyse cytoplasmique et l’oxydation mitochondriale du glucose. Bien que le mécanisme de cet « effet Warburg » soit inconnu et que son lien étiologique avec la cancérogenèse reste à prouver (7), on s’intéresse de plus en plus au métabolisme en tant que cible des thérapies anticancéreuses (8-11). Le passage énergétique de l’oxydation mitochondriale du glucose à la glycolyse cytoplasmique peut offrir un avantage prolifératif aux cellules cancéreuses (11). Par exemple, la plupart des enzymes glycolytiques ont également des actions antiapoptotiques directes (12); l’acide lactique favorise l’angiogenèse et la dégradation de la matrice interstitielle, facilitant ainsi les métastases (11); et une diminution de la fonction mitochondriale est associée à l’inhibition de l’apoptose mitochondriale dépendante (3). Le GBM présente un fort phénotype glycolytique, et un certain nombre d’anomalies moléculaires présentes dans le GBM sont connues pour supprimer l’oxydation mitochondriale du glucose et favoriser la glycolyse cytoplasmique (1), notamment l’activation des voies phosphatidylinositol 3-kinase-AKT ou myc ou la suppression de la voie p53 (9, 10).
Les mitochondries des cellules cancéreuses sont hyperpolarisées par rapport à celles des cellules non cancéreuses (3, 13), un état associé à la suppression de la fonction mitochondriale. Bien que controversée [revue dans (14)], l’efflux des médiateurs proapoptotiques à travers le pore de transition mitochondrial (MTP) dépend en partie du potentiel de la membrane mitochondriale (ΔΨm), et ainsi, l’hyperpolarisation mitochondriale peut marquer un état de résistance à l’apoptose (3, 15). Nous avons montré que cet état peut être inversé dans les cellules cancéreuses par le DCA, qui en inhibant la PDK favorise l’entrée du pyruvate dans les mitochondries, inversant l’augmentation du rapport glycolyse/oxydation du glucose, améliorant la fonction mitochondriale et inversant l’hyperpolarisation mitochondriale (3). Le DCA diminue donc la croissance tumorale in vitro et in vivo, sans affecter les mitochondries et les tissus non cancéreux (3, 16-20). L’augmentation de la respiration mitochondriale est associée à une augmentation de la production d’espèces réactives de l’oxygène mitochondrial (mROS), principalement du superoxyde. Le superoxyde peut être dismuté en H2O2, une espèce d’oxygène réactif relativement stable qui peut atteindre d’autres structures cellulaires que les mitochondries. Par exemple, H2O2 peut activer les canaux potassiques voltage-dépendants sensibles à l’oxydoréduction dans la membrane plasmique et, au moins dans certains tissus, favoriser une diminution du calcium intracellulaire (3, 4). D’autres cibles redox-sensibles peuvent inclure p53, qui est activé lorsqu’il est oxydé (21, 22). L’axe p53 est inhibé dans le GBM, ce qui contribue à l’état prolifératif accru des cellules du GBM (1). p53 réprime également la transcription stimulée par le facteur inductible de l’hypoxie-1α (HIF-1α) car p53 et HIF-1α sont en compétition pour le même facteur de cotranscription (23, 24). HIF-1α augmente l’expression des transporteurs de glucose et de plusieurs enzymes glycolytiques ainsi que de la PDK, soutenant ainsi le phénotype glycolytique (25, 26). En outre, HIF-1α augmente l’expression du facteur de croissance endothélial vasculaire (VEGF), ce qui renforce l’angiogenèse. L’angiogenèse peut également être renforcée par l’activation normoxique de HIF-1α. Les mitochondries étant d’importants capteurs d’oxygène (27), les mitochondries inhibées peuvent transmettre des signaux redox pseudohypoxiques et activer HIF-1α même pendant la normoxie (28-30). En outre, une diminution de l’α-cétoglutarate, un produit direct du cycle de Krebs, peut également favoriser l’activation de HIF car il s’agit d’un cofacteur pour la réaction d’hydroxylation prolylique qui dégrade HIF-1α (30).
Nous avons émis l’hypothèse que le DCA administré par voie orale, qui traverse la barrière hémato-encéphalique, diminuerait la croissance des GBM in vivo. Nous avons en outre suggéré que cela pourrait se produire (i) en inversant le phénotype glycolytique et en normalisant le ΔΨm, ce qui favoriserait l’apoptose dépendante des mitochondries ; (ii) en augmentant le mROS et en favorisant l’activation de p53 ; et (iii) en augmentant les concentrations d’α-cétoglutarate. Ces deux derniers effets conduiraient à une inhibition de HIF-1α, une diminution du VEGF et une inhibition de l’angiogenèse.
Résultats
Effets du DCA sur les mitochondries de 49 tumeurs GBM fraîchement isolées
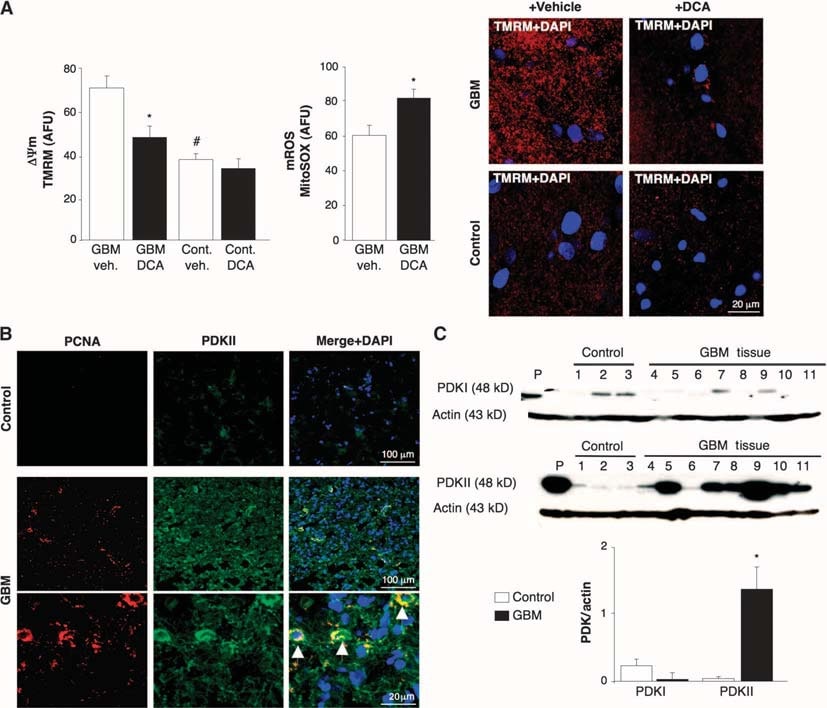
Pour déterminer si les GBM humains pouvaient être une cible pour une thérapie métabolique avec le DCA, nous avons étudié 49 GBM primaires consécutifs fraîchement excisés (60% d’hommes, 48 ± 11 ans). En plus des rapports cliniques et neuropathologiques, nous avons confirmé l’identité des GBM par immunohistochimie, qui a montré l’expression de la protéine acide fibrillaire gliale (GFAP) mais pas de bIII-tubuline ou de marqueurs oligodendrocytaires (fig. S1). Le ΔΨm était augmenté dans les GBM fraîchement isolés par rapport aux tissus cérébraux non cancéreux obtenus lors d’une chirurgie de l’épilepsie (n = 3) (fig. 1A). Le DCA, mais pas le véhicule (solution saline normale), a provoqué une dépolarisation mitochondriale dans les GBM mais pas dans le tissu cérébral normal. Le DCA a également augmenté le mROS des GBM (Fig. 1A). Cela suggère que le remodelage métabolique et mitochondrial dans le GBM est partiellement réversible et que ce remodelage est au moins en partie régulé par la PDK. La réponse au DCA est cohérente avec une concentration plus élevée de PDKII [l’isoforme la plus ubiquitairement exprimée et celle dontleKi pour le DCA est le plus faible (31)] dans le GBM que dans le tissu cérébral non cancéreux, comme le montrent l’immunohistochimie et les immunoblots (Fig. 1, B et C). Les cellules présentant les concentrations les plus élevées de PDKII contenaient également de l’antigène nucléaire des cellules en prolifération (PCNA), ce qui suggère que ces cellules étaient en prolifération (Fig. 1B). Ces données, recueillies sur une période de 2 ans, ont renforcé les arguments en faveur de l’administration ultérieure de DCA aux patients atteints de GBM (4).
Effets cliniques du DCA sur cinq patients atteints de GBM
Nous avons ensuite traité au DCA cinq patients consécutifs atteints de GBM primaire, envoyés par notre programme de lutte contre le cancer du cerveau et dont le tissu était disponible lors de la dernière chirurgie de débulking. Trois patients (patients 1 à 3) présentaient un GBM récurrent avec une progression de la maladie après plusieurs chimiothérapies (en plus du traitement standard avec chirurgie, RT et TMZ) et ont été considérés comme appropriés pour un traitement palliatif. Deux autres patients (patients 4 et 5) ont été diagnostiqués récemment et, après la chirurgie initiale de débulking, le DCA a été administré en plus du traitement standard de RT et TMZ. Chez le patient 4, un prétraitement de 3 mois par DCA a été suivi de l’ajout de la RT et du TMZ, tandis que chez le patient 5, le DCA a été initié simultanément à la RT et au TMZ, après la chirurgie de débulking. Si les patients ont dû être réopérés ou autopsiés, les tissus de la dernière chirurgie de débulking (avant l’administration du DCA) ont été comparés aux tissus du traitement post-DCA. Leurs informations cliniques sont résumées dans le tableau S1. Le DCA est administré aux patients depuis plus de 30 ans, principalement dans le traitement des erreurs innées du métabolisme mitochondrial, et des données pharmacocinétiques et pharmacodynamiques sont disponibles (5, 32-34). Nous avons traité les patients avec une dose initiale de 12,5 mg/kg par voie orale deux fois par jour pendant 1 mois, puis nous avons augmenté la dose à 25 mg/kg par voie orale deux fois par jour. Nous avons ensuite suivi un protocole de désescalade de la dose, en diminuant la dose de 50 % en cas de toxicité limitant la dose. Les patients ont été suivis cliniquement jusqu’à 15 mois. Aucun des patients n’a présenté de toxicité hématologique, hépatique, rénale ou cardiaque (tableau S1). La neuropathie périphérique a été la seule toxicité apparente. Les patients ont présenté des degrés variables de neuropathie périphérique en fonction de la dose, qui était réversible, ce qui confirme les études précédentes (35-37). Lorsque la dose a été réduite à 6,25 mg/kg par voie orale deux fois par jour, aucun des patients n’a présenté de neuropathie périphérique cliniquement significative (tableau S1). Initialement, la demi-vie du DCA est < 1 heure. Le DCA inhibe son propre métabolisme et les concentrations sériques augmentent pour finalement atteindre un plateau (34). Les concentrations plasmatiques minimales de DCA chez nos patients sont restées indétectables pendant les 2 à 3 premiers mois, mais ont ensuite atteint des concentrations thérapeutiques. À une dose de 6,25 mg/kg par voie orale deux fois par jour pendant au moins 3 mois, les concentrations minimales de DCA étaient de 0,44 ± 0,16 mM (moyenne ± SD ; n = 4) (tableau S1). Ces valeurs sont semblables à celles observées dans le traitement chronique par le DCA d’adultes présentant des anomalies mitochondriales (34) et se situent dans la même fourchette queleKi du DCA pour la PDKII (0,2 mM) (31). Les patients 1, 4 et 5 ont montré des signes de régression radiologique à l’imagerie par résonance magnétique (IRM) (figure 2A et figures S2 à S4). Le patient 3 présentait une très grosse tumeur avec un œdème cérébral au départ (fig. S5), bien qu’il ait été traité par de fortes doses de stéroïdes, et un faible score de Karnofsky ; son état a continué à se détériorer. Il est décédé de complications liées à l’œdème cérébral 3 mois après le début du traitement par DCA. Le patient 2 a nécessité le drainage d’un kyste et un débulking au 11ème mois du traitement par DCA. Le patient 4 a montré une progression radiologique au troisième mois du traitement par DCA, et à ce moment-là, un débulking supplémentaire a été effectué et une RT plus TMZ a été administrée en plus du DCA. Tous les patients, à l’exception du patient 3, étaient cliniquement stables au mois 15 du traitement DCA et vivants au mois 18 (suivi téléphonique). D’autres détails cliniques sont décrits dans le matériel supplémentaire.
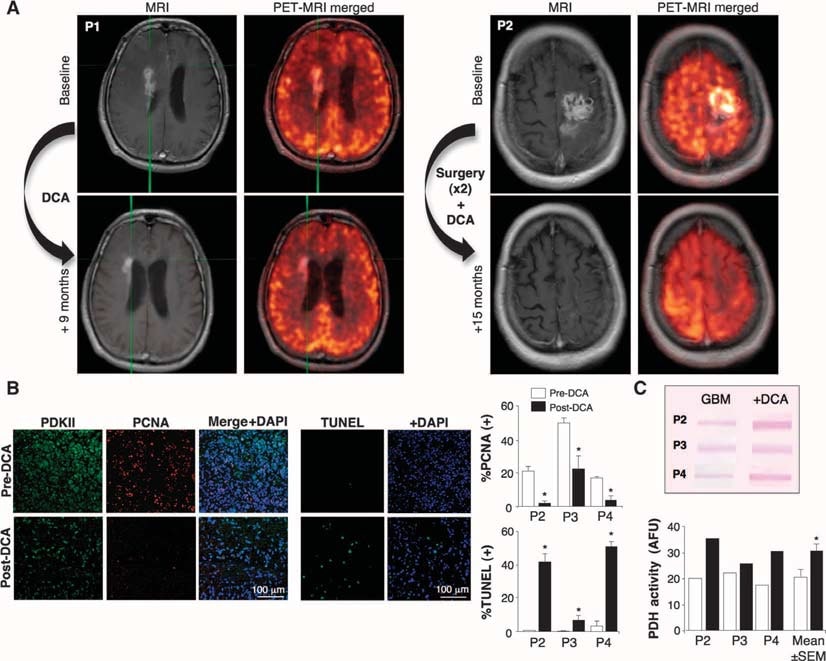
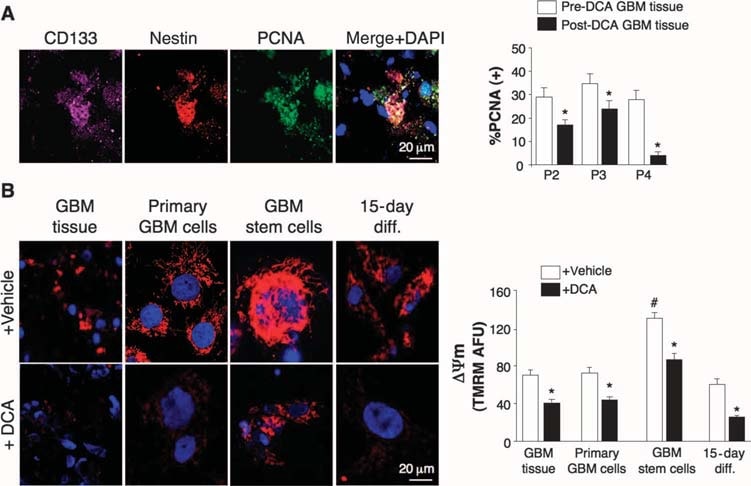
Effets du DCA sur les tumeurs GBM in vivo, les lignées cellulaires GBM primaires et les GBM-SC putatifs dérivés des patients traités au DCA
Nous avons réalisé des expériences sur des tissus dérivés de ces cinq patients et avons pu faire des comparaisons entre les tissus avant et après le traitement au DCA chez les patients 2 à 4 ; nous ne disposions que des tissus » avant » chez les patients 1 et 5. Par rapport aux tissus pré-DCA, les tissus de GBM post-DCA des trois patients présentaient une diminution du nombre de cellules par unité de volume, une diminution de la prolifération et une augmentation de l’apoptose (Fig. 2B), ainsi qu’une augmentation de l’activité enzymatique tissulaire de la PDH, ce qui suggère une inhibition efficace de la PDK in vivo (Fig. 2C). Les cellules souches cancéreuses putatives de GBM (GBM-SC) peuvent être responsables de la résistance post-traitement et de la récurrence des GBM (38-43). Ces cellules sont caractérisées comme des GBM-SC CD133+ / nestin+ et forment des niches autour des capillaires (41). Dans ces unités vasculaires, les GBM-SC peuvent induire l’angiogenèse, tandis que leur phénotype de cellules souches moléculaires est maintenu par leur accessibilité aux facteurs de croissance circulants (44). La prolifération des GBM-SC est associée à un résultat clinique particulièrement mauvais (42). Les GBM-SC CD133+ /nestin+ exprimaient le PCNA in vivo dans toutes les tumeurs pré-DCA, ce qui indique qu’elles se divisent, mais le pourcentage de cellules CD133+ /nestin+ qui exprimaient le PCNA était significativement réduit après le traitement DCA chez les patients 2 à 4 (Fig. 3A). La coloration simultanée avec un anticorps CD133 et de l’ester méthylique de tétraméthyl rhodamine (TMRM) a montré que les cellules CD133+ avaient le ΔΨm le plus élevé par rapport aux cellules non GBM-SC voisines in vivo (fig. S6). Dans les lignées cellulaires primaires dérivées des tumeurs, ~10% des cellules exprimaient à la fois CD133 et nestin, tandis que >90% des cellules exprimaient le marqueur mature GFAP (mais pas la bIII-tubuline ou l’oligodendrocyte) (fig. S7), ce qui est similaire à l’histopathologie des GBM (fig. S1). Nous avons isolé des cellules GBM-SC putatives à partir de tumeurs GBM et les avons cultivées avec les facteurs de croissance appropriés (facteur de croissance des fibroblastes humains, 20 ng/ml ; facteur de croissance épidermique humain, 20 ng/ml). Ces cellules avaient une très forte expression de CD133 et de nestin, une très faible expression des marqueurs gliaux matures (fig. S7) et formaient des neurosphères caractéristiques (fig. 4 et fig. S7), un prédicteur indépendant de mauvais résultats cliniques (43). Nous avons mesuré le DYm dans des tumeurs fraîchement excisées, dans des lignées cellulaires primaires et dans des GBM-SC isolés de ces tumeurs ainsi que dans des cellules différenciées dérivées de GBM-SC (Fig. 3B). Le potentiel le plus élevé a été trouvé dans le GBM-SC putatif. Les cellules de GBM primaires et secondaires dérivées de GBM-SC (différenciation de 15 jours) avaient des potentiels mitochondriaux similaires à ceux des tumeurs mères. Le DCA (0,5 mM pendant 24 heures) a diminué le potentiel dans tous les groupes de cellules. Bien que la cause de l’augmentation du ΔΨm dans le cancer (3, 13) reste à définir complètement, il a été proposé qu’elle soit causée en partie par une translocation de l’hexokinase II (HXKII), une enzyme glycolytique clé, du cytoplasme à la membrane mitochondriale externe (45, 46). Là, la HXKII peut se lier au canal anionique dépendant du voltage (un composant du MTP) et l’inhiber, ce qui augmente le ΔΨm et le seuil apoptotique. L’inhibition de cette translocation diminue le ΔΨm du cancer et inverse la résistance à l’apoptose (45, 46). Nos lignées cellulaires primaires générées à partir de tumeurs pré-DCA ont montré une translocation mitochondriale soutenue de HXKII, expliquant potentiellement l’augmentation du ΔΨm. La translocation de HXKII n’était pas présente dans les lignées cellulaires primaires des tumeurs après le traitement au DCA (fig. S8), ce qui est compatible avec l’idée que le DCA a induit une suppression de la glycolyse et une diminution du ΔΨm. Comme dans les tumeurs, PDKII était présent à des concentrations élevées dans les lignées cellulaires de GBM générées par les patients 2 à 4, bien que les autres isoenzymes connues aient également été exprimées (fig. S9A). Lorsqu’on a laissé les GBM-SC se différencier en lignées cellulaires secondaires de GBM, la proportion de cellules avec des marqueurs GBM-SC a diminué jusqu’à une valeur similaire à celle des lignées cellulaires primaires (~10%). Cependant, lorsqu’on les laissait se différencier en présence de DCA (0,5 mM), la proportion de cellules présentant des marqueurs GBM-SC diminuait encore plus pour atteindre ~5 % (fig. S7). En effet, le DCA a induit l’apoptose dans les GBM-SC in vitro (fig. 4 et fig. S9B) ainsi que dans les lignées cellulaires primaires de GBM (fig. S9C). L’apoptose a encore été augmentée dans les cellules de GBM-SC par l’association du DCA et du TMZ (fig. 4 et fig. S9B), ce qui justifie la thérapie combinée. L’apoptose des GBM-SC a également eu lieu in vivo dans les tumeurs traitées par le DCA, comme le montre la colocalisation de la nestine, du CD133 et de la coloration TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling) (fig. 4 et fig. S9D).
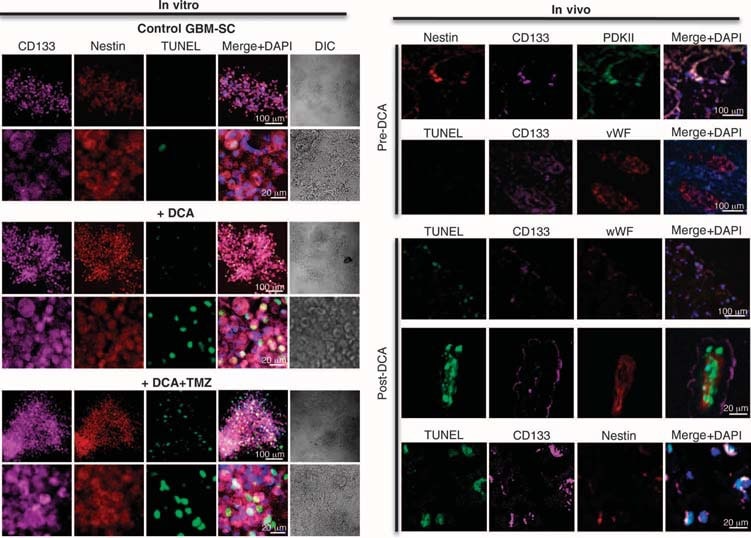
Effets du DCA sur l’unité de microvaisseaux du GBM-SC et sur l’angiogenèse in vivo et in vitro
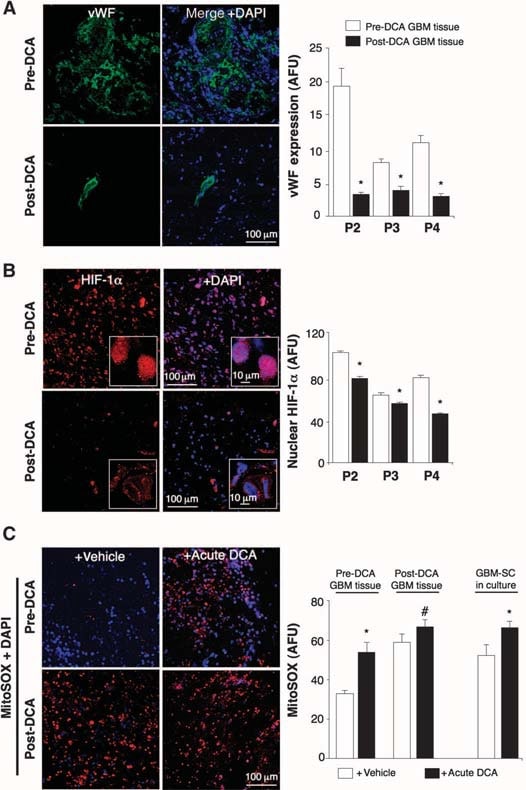
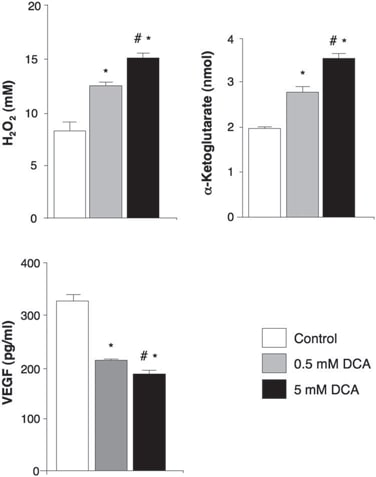
Dans les tissus pré-DCA non traités, des niches de GBM-SC ont été trouvées autour des lits de microvaisseaux [coloration du facteur de von Willebrand (vWF)], comme indiqué (41). Cette unité de microvaisseaux GBM-SC (44) a été détruite par le traitement au DCA car, outre les GBM-SC, l’apoptose a également augmenté dans les cellules endothéliales microvasculaires (fig. 4 et fig. S9D), ce qui suggère une inhibition potentielle de l’angiogenèse. En effet, la diminution de la coloration du vWF dans les tumeurs traitées après le DCA suggère une diminution de la vascularisation (Fig. 5A). HIF-1α était fortement exprimé ou activé (localisation nucléaire) dans les tissus tumoraux pré-DCA et inhibé dans les tissus post-DCA (Fig. 5B). Les tumeurs des patients 2 à 4 traitées après le DCA ont montré une augmentation significative du mROS in vivo (superoxyde mesuré par mitoSOX) par rapport aux tumeurs pré-DCA des mêmes patients (Fig. 5C). Dans les tumeurs pré-DCA, le DCA aigu a augmenté la mROS jusqu’aux valeurs observées dans les tumeurs traitées après le DCA. En revanche, dans les tumeurs post-DCA, le DCA aigu n’a que très peu augmenté la mROS, ce qui suggère un effet presque maximal in vivo. Le DCA a également augmenté la mROS dans les GBM-SC (Fig. 5C). Les faibles concentrations de ROS dans les cellules souches cancéreuses peuvent refléter une résistance à l’apoptose, et les thérapies qui augmentent les ROS des cellules souches cancéreuses sont censées être plus efficaces (47). Bien que nous ayons étudié le mROS (superoxyde mitochondrial), il existe une controverse quant à savoir si l’activation de HIF-1α dans le cancer est associée à une augmentation ou à une diminution globale des ROS [revue dans (28)]. En utilisant une technique différente, nous avons également mesuré le H2O2 dans les cellules entières. Le DCA a augmenté le H2O2 des cellules de GBM de manière dose-dépendante (Fig. 6). En outre, le DCA a également augmenté les concentrations intracellulaires d’α-cétoglutarate de manière dose-dépendante (Fig. 6). Ceci est compatible avec l’augmentation de l’oxydation du glucose qui suit l’activation de la PDH (3) car l’α-cétoglutarate est un produit du cycle de Krebs. Le cycle de Krebs produit les donneurs d’électrons qui alimentent la chaîne de transport d’électrons pendant la respiration. En conséquence, le DCA a augmenté les taux de respiration de 44 ± 4 % dans les cellules de GBM (moyenne ± SEM ; n = 3 ; P < 0,05), soutenant une augmentation globale de l’activité mitochondriale. L’augmentation de H2O2 et d’α-cétoglutarate peut expliquer la diminution de l’activité de HIF-1α (Fig. 5B), ce qui est également confirmé par la diminution dose-dépendante de la production de VEGF par les cellules de GBM (Fig. 6). Ces données sont cohérentes avec la diminution de l’angiogenèse in vivo (Figs. 4 et 5) et suggèrent que les cellules GBM envoient des signaux aux cellules endothéliales de manière paracrine. Pour étudier si le DCA peut supprimer directement l’angiogenèse, nous avons utilisé la technique standard de formation de tubes de cellules endothéliales humaines dans du Matrigel. Sous hypoxie physiologique modérée, le DCA a provoqué une inhibition directe dose-dépendante de l’angiogenèse in vitro (fig. S10). Les tumeurs des patients 2 à 4 traitées au DCA ont montré une augmentation de l’activité de p53 sensible au mROS (translocation nucléaire), également confirmée par l’augmentation de l’activité et de l’abondance de sa cible en aval p21 (fig. S11). Ces effets sur p53 ou p21 peuvent également expliquer la diminution de la transcription induite par HIF et sont cohérents avec les effets antiprolifératifs, en plus des effets proapoptotiques, du DCA dans les GBM (fig. S12).
DISCUSSION
Le modulateur métabolique DCA exerce des effets anticancéreux dans des cellules cultivées et chez les rongeurs (3, 16-20). Nous avons maintenant montré que le DCA peut être utilisé chez les patients souffrant de GBM. Le traitement au DCA a été associé chez certains patients atteints de GBM à une stabilisation radiologique prolongée ou à une régression tumorale et, en général, il a présenté un bon profil de sécurité global. Ce rapport précoce, premier chez l’homme, justifie la poursuite des études avec cette petite molécule générique chez les patients atteints de GBM. Nos résultats indiquent que le GBM est un bon candidat pour une intervention métabolique. La cible du DCA, PDKII, est fortement exprimée dans les tumeurs et les lignées cellulaires de GBM, et le DCA peut inhiber son activité in vivo. Le GBM est caractérisé par une hyperpolarisation mitochondriale, ce qui correspond au remodelage métabolique (effet Warburg) et à la résistance à l’apoptose qui caractérisent le GBM et la plupart des tumeurs solides (11). Des mutations dans les gènes des isocitrates déshydrogénases cytoplasmiques et mitochondriales ont été décrites dans des GBM issus de gliomes de moindre importance (GBM secondaires) (48), mais le mécanisme par lequel ces mutations sont liées à la carcinogenèse reste obscur (49, 50). Nos patients étaient atteints de GBM primaires et le remodelage mitochondrial était au moins partiellement réversible avec le DCA, ce qui suggère qu’il n’était pas dû à un dysfonctionnement irréversible. De plus, nous montrons que les GBM-SC putatifs peuvent subir le même remodelage métabolique et mitochondrial, mais à un degré plus élevé, car les GBM-SC avaient les mitochondries les plus hyperpolarisées à la fois in vivo et in vitro. L’inversion de ce remodelage mitochondrial par le DCA a provoqué l’apoptose des GBM-SC à la fois in vitro et in vivo. Bien que l’ampleur de l’induction de l’apoptose par le DCA ne soit pas élevée (par rapport aux agents cytotoxiques), elle est relativement sélective, épargnant les cellules non cancéreuses (3), et, parce qu’elle concerne les GBM-SC, elle peut entraîner un effet clinique plus durable. Le patient 3 est décédé et le patient 4 a connu une récidive dans les 3 premiers mois, alors que le DCA n’avait pas atteint des valeurs thérapeutiques durables (comme le montre le fait que les concentrations plasmatiques minimales étaient indétectables). Ainsi, les patients peuvent être sous-traités lors de l’administration initiale de DCA et risquer une progression initiale de la maladie. En diminuant la résistance à l’apoptose dépendante des mitochondries, le DCA pourrait potentialiser les effets des thérapies standard. En effet, les effets du DCA et du TMZ sur l’apoptose des GBMSC pourraient être à l’origine des avantages à long terme dont a bénéficié le patient 5. Les effets du DCA sur les cellules cancéreuses sont imités par le petit ARN interférent de PDKII, et le DCA n’a pas d’autres effets que ceux observés après le knockdown de PDKII (3). Cela suggère que le mécanisme des effets anticancéreux du DCA est l’inhibition de la PDKII, une enzyme qui se trouve à des concentrations élevées dans les GBM. En inhibant PDKII, le DCA normalise l’augmentation du rapport glycolyse/oxydation du glucose dans les cellules cancéreuses, augmentant ainsi la fonction mitochondriale (3). Ce mécanisme est corroboré par nos observations actuelles selon lesquelles le DCA augmente à la fois la respiration des cellules de GBM et la concentration d’a-cétoglutarate. Cependant, contrairement aux conditions contrôlées de la culture cellulaire, d’autres mécanismes peuvent contribuer aux effets du DCA sur le métabolisme et l’apoptose des cellules cancéreuses in vivo. Outre les effets du DCA sur les cellules cancéreuses décrits précédemment (3), nos données sur HXKII, mROS, HIF-1α, p53 et p21 sont cohérentes avec l’inhibition de l’angiogenèse, l’induction de l’apoptose et la suppression de la prolifération induites par le DCA dans les GBM et les GBM-SC, comme le résument le matériel supplémentaire et la fig. S12. Le DCA n’a présenté aucune toxicité apparente, à l’exception d’une neurotoxicité non démyélinisante, réversible et dépendante de la dose, décrite précédemment (32, 34), qui était minime ou absente à la dose orale de 6,25 mg/kg, deux fois par jour. Cette dose a montré une efficacité biologique et clinique et a permis d’atteindre des concentrations plasmatiques aux valeurs requises pour l’inhibition de la PDK (31). Le petit nombre de patients traités dans notre étude ne permet pas de tirer des conclusions définitives sur le DCA en tant que traitement du GBM. Notre travail confirme la nécessité de poursuivre les études sur le DCA dans le GBM, en mettant l’accent sur les protocoles de thérapie combinée. Le GBM pourrait également être vulnérable à d’autres médicaments de la famille émergente des modulateurs métaboliques, ce qui laisse entrevoir une nouvelle approche dans la gestion de ce cancer incurable.
MATÉRIEL SUPPLÉMENTAIRE
www.sciencetranslationalmedicine.org/cgi/content/full/2/31/31ra34/DC1
Matériaux et méthodes
Résultats
Discussion
Fig. S1. Caractérisation moléculaire des tumeurs GBM.
Fig. S2. Évolution de la réponse tumorale chez le patient 1.
Fig. S3. Evolution de la réponse tumorale chez le patient 4.
Fig. S4. Evolution de la réponse tumorale chez le patient 5.
Fig. S5. IRM du GBM du patient 3.
Fig. S6. Potentiel de la membrane mitochondriale dans des GBM-SC provenant de tissus de GBM fraîchement excisés.
Fig. S7. Caractérisation des cellules primaires de GBM et des GBM-SC.
Fig. S8. HXKII dans les cellules de GBM dérivées de patients avant et après un traitement chronique au DCA.
Fig. S9. Effets du traitement au DCA sur les GBM-SC et l’apoptose vasculaire.
Fig. S10. Effets du DCA sur l’angiogenèse in vitro.
Fig. S11. Effets du traitement au DCA sur l’activité de p53 et p21 in vivo.
Fig. S12. Proposition d’un mécanisme complet pour les effets anticancéreux du DCA sur les GBM (voir
Discussion supplémentaire).
Tableau S1. Paramètres de laboratoire et cliniques de cinq patients atteints de GBM avant et après le traitement au DCA.
Références
RÉFÉRENCES
1 1. P. Y. Wen, S. Kesari, Malignant gliomas in adults. N. Engl. J. Med. 359, 492-507 (2008).
2 W. K. Yung, R. E. Albright, J. Olson, R. Fredericks, K. Fink, M. D. Prados, M. Brada, A. Spence, R. J. Hohl, W. Shapiro, M. Glantz, H. Greenberg, R. G. Selker, N. A. Vick, R. Rampling, H. Friedman, P. Phillips, J. Bruner, N. Yue, D. Osoba, S. Zaknoen, V. A. Levin, A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br. J. Cancer 83, 588-593 (2000).
3 S. Bonnet, S. L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C. T. Lee, G. D. Lopaschuk, L. Puttagunta, S. Bonnet, G. Harry, K. Hashimoto, C. J.. Porter, M. A. Andrade, B. Thebaud, E. D. Michelakis, A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37-51 (2007).
4 E. D. Michelakis, L. Webster, J. R. Mackey, Dichloroacetate (DCA) as a potential metabolictargeting therapy for cancer. Br. J. Cancer 99, 989-994 (2008).
5 P. W. Stacpoole, The pharmacology of dichloroacetate. Metabolism 38, 1124-1144 (1989).
6 O. Warburg, Ueber den Stoffwechsel der Tumoren (Constable, Londres, 1930).
7 S. Weinhouse, The Warburg hypothesis fifty years later. Z. Krebsforsch. Klin. Onkol. Cancer Res. Clin. Oncol. 87, 115-126 (1976).
8 M. G. Vander Heiden, L. C. Cantley, C. B. Thompson, Understanding the Warburg effect : Les exigences métaboliques de la prolifération cellulaire. Science 324, 1029-1033 (2009).
9 J. G. Pan, T. W. Mak, Metabolic targeting as an anticancer strategy : Dawn of a new era ? Sci. STKE 2007, pe14 (2007).
10 J. W. Kim, C. V. Dang, Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 66, 8927-8930 (2006).
11 R. A. Gatenby, R. J. Gillies, Why do cancers have high aerobic glycolysis ? Nat. Rev. Cancer 4, 891-899 (2004).
12 J. W. Kim, C. V. Dang, Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 30, 142-150 (2005).
13 L. B. Chen, Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 4, 155-181 (1988).
14 G. Kroemer, L. Galluzzi, C. Brenner, Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99-163 (2007).
15. N. Zamzami, G. Kroemer, The mitochondrion in apoptosis : How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2, 67-71 (2001).
16 R. A. Cairns, I. Papandreou, P. D. Sutphin, N. C. Denko, Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc. Natl. Acad. Sci. U.S.A. 104, 9445-9450 (2007).
17 W. Cao, S. Yacoub, K. T. Shiverick, K. Namiki, Y. Sakai, S. Porvasnik, C. Urbanek, C. J. Rosser, Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 68, 1223-1231 (2008).
18 S. Dhar, S. J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. U.S.A. 106, 22199-22204 (2009).
19 R. C. Sun, M. Fadia, J. E. Dahlstrom, C. R. Parish, P. G. Board, A. C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 120, 253-260 (2010).
20 J. Y. Wong, G. S. Huggins, M. Debidda, N. C. Munshi, I. De Vivo, Dichloroacetate induit l’apoptose dans les cellules cancéreuses de l’endomètre. Gynecol. Oncol. 109, 394-402 (2008).
21 S. Wang, S. S. Leonard, J. Ye, M. Ding, X. Shi, The role of hydroxyl radical as a messenger in Cr(VI)-induced p53 activation. Am. J. Physiol. Cell Physiol. 279, C868-C875 (2000).
22 C. Huang, Z. Zhang, M. Ding, J. Li, J. Ye, S. S. Leonard, H. M. Shen, L. Butterworth, Y. Lu, M. Costa, Y. Rojanasakul, V. Castranova, V. Vallyathan, X. Shi, Vanadate induit la transactivation de p53 par le peroxyde d’hydrogène et provoque l’apoptose. J. Biol. Chem. 275, 32516-32522 (2000).
23 T. Schmid, J. Zhou, R. Köhl, B. Brüne, p300 relie la répression transcriptionnelle du facteur-1 inductible par l’hypoxie (HIF-1) provoquée par p53. Biochem. J. 380, 289-295 (2004).
24 M. V. Blagosklonny, W. G. An, L. Y. Romanova, J. Trepel, T. Fojo, L. Neckers, p53 inhibe la transcription stimulée par le facteur inductible de l’hypoxie. J. Biol. Chem. 273, 11995-11998 (1998).
25 J. W. Kim, I. Tchernyshyov, G. L. Semenza, C. V. Dang, HIF-1-mediated expression of pyruvate dehydrogenase kinase : A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177-185 (2006).
26 G. L. Semenza, D. Artemov, A. Bedi, Z. Bhujwalla, K. Chiles, D. Feldser, E. Laughner, R. Ravi, J. Simons, P. Taghavi, H. Zhong, ‘The metabolism of tumours’ : 70 years later. Novartis Found. Symp. 240, 251-260 (2001).
27 E. K. Weir, J. López-Barneo, K. J. Buckler, S. L. Archer, Acute oxygen-sensing mechanisms. N. Engl. J. Med. 353, 2042-2055 (2005).
28 N. C. Denko, Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705-713 (2008).
29 G. L. Semenza, Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721-732 (2003).
30 E. D. MacKenzie, M. A. Selak, D. A. Tennant, L. J. Payne, S. Crosby, C. M. Frederiksen, D. G. Watson, E. Gottlieb, Cell-permeating a-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell. Biol. 27, 3282-3289 (2007).
31 M. M. Bowker-Kinley, W. I. Davis, P. Wu, R. A. Harris, K. M. Popov, Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329 (Pt 1), 191-196 (1998).
32 P. W. Stacpoole, D. S. Kerr, C. Barnes, S. T. Bunch, P. R. Carney, E. M. Fennell, N. M. Felitsyn, R. L. Gilmore, M. Greer, G. N. Henderson, A. D. Hutson, R. E. Neiberger, R. G. O’Brien, L. A.. Perkins, R. G. Quisling, A. L. Shroads, J. J. Shuster, J. H. Silverstein, D. W. Theriaque, E. Valenstein, Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 117, 1519-1531 (2006).
33 P. W. Stacpoole, A. C. Lorenz, R. G. Thomas, E. M. Harman, Dichloroacetate in the treatment of lactic acidosis. Ann. Intern. Med. 108, 58-63 (1988).
34 P. W. Stacpoole, T. L. Kurtz, Z. Han, T. Langaee, Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv. Drug Deliv. Rev. 60, 1478-1487 (2008).
35 P. Kaufmann, K. Engelstad, Y. Wei, S. Jhung, M. C. Sano, D. C. Shungu, W. S. Millar, X. Hong, C. L. Gooch, X. Mao, J. M. Pascual, M. Hirano, P. W. Stacpoole, S. DiMauro, D. C. De Vivo, Dichloroacetate causes toxic neuropathy in MELAS : A randomized, controlled clinical trial. Neurology 66, 324-330 (2006).
36 P. W. Stacpoole, L. R. Gilbert, R. E. Neiberger, P. R. Carney, E. Valenstein, D. W. Theriaque, J. J. Shuster, Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121, e1223-e1228 (2008).
37 P. W. Stacpoole, G. N. Henderson, Z. Yan, M. O. James, Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 106 (Suppl. 4), 989-994 (1998).
38 N. Sanai, A. Alvarez-Buylla, M. S. Berger, Neural stem cells and the origin of gliomas. N. Engl. J. Med. 353, 811-822 (2005).
39 F. Zindy, T. Uziel, O. Ayrault, C. Calabrese, M. Valentine, J. E. Rehg, R. J. Gilbertson, C. J. Sherr, M. F. Roussel, Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 67, 2676-2684 (2007).
40 T. Hide, T. Takezaki, H. Nakamura, J. Kuratsu, T. Kondo, Brain tumor stem cells as research and treatment targets. Brain Tumor Patrol. 25, 67-72 (2008).
41 C. Calabrese, H. Poppleton, M. Kocak, T. L. Hogg, C. Fuller, B. Hamner, E. Y. Oh, M. W. Gaber, D. Finklestein, M. Allen, A. Frank, I. T. Bayazitov, S. S. Zakharenko, A. Gajjar, A. Davidoff, R. J. Gilbertson, A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69-82 (2007).
42 R. Pallini, L. Ricci-Vitiani, G. L. Banna, M. Signore, D. Lombardi, M. Todaro, G. Stassi, M. Martini, G. Maira, L. M. Larocca, R. De Maria, Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin. Cancer Res. 14, 8205-8212 (2008).
43 D. R. Laks, M. Masterman-Smith, K. Visnyei, B. Angenieux, N. M. Orozco, I. Foran, W. H. Yong, H. V. Vinters, L. M. Liau, J. A. Lazareff, P. S. Mischel, T. F. Cloughesy, S. Horvath, H. I. Kornblum, Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 27, 980-987 (2009).
44 R. J. Gilbertson, J. N. Rich, Making a tumour’s bed : Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 7, 733-736 (2007).
45 J. G. Pastorino, J. B. Hoek, Hexokinase II : The integration of energy metabolism and control of apoptosis. Curr. Med. Chem. 10, 1535-1551 (2003).
46 J. G. Pastorino, J. B. Hoek, N. Shulga, Activation of glycogen synthase kinase 3b disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545-10554 (2005).
47M. Diehn, R. W. Cho, N. A. Lobo, T. Kalisky, M. J. Dorie, A. N. Kulp, D. Qian, J. S. Lam, L. E. Ailles, M. Wong, B. Joshua, M. J. Kaplan, I. Wapnir, F. M. Dirbas, G. Somlo, C. Garberoglio, B. Paz, J. Shen, S. K. Lau, S. R. Quake, J. M. Brown, I. L. Weissman, M. F. Clarke, Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780-783 (2009).
48 H. Yan, D. W. Parsons, G. Jin, R. McLendon, B. A. Rasheed, W. Yuan, I. Kos, I. Batinic-Haberle, S. Jones, G. J. Riggins, H. Friedman, A. Friedman, D. Reardon, J. Herndon, K. W. Kinzler, V. E. Velculescu, B. Vogelstein, D. D. Bigner, IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765-773 (2009).
49 C. B. Thompson, Metabolic enzymes as oncogenes or tumor suppressors. N. Engl. J. Med. 360, 813-815 (2009).
50 L. Dang, D. W. White, S. Gross, B. D. Bennett, M. A. Bittinger, E. M. Driggers, V. R. Fantin, H. G. Jang, S. Jin, M. C. Keenan, K. M. Marks, R. M. Prins, P. S.. Ward, K. E. Yen, L. M. Liau, J. D. Rabinowitz, L. C. Cantley, C. B. Thompson, M. G. Vander Heiden, S. M. Su, Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739-744 (2009).
51 Financement : Cette étude a été financée par la Fondation Hecht (Vancouver, Colombie-Britannique, Canada ; E.D.M.), les Instituts de recherche en santé du Canada et le Programme des chaires de recherche du Canada (E.D.M.), ainsi que par des dons publics au programme DCA (reçus et gérés par les Régents de l’Université d’Alberta et la Faculté de médecine). Les auteurs tiennent à remercier les Services de santé de l’Alberta pour leur soutien (D. Gordon, premier vice-président, grands hôpitaux tertiaires). Contributions des auteurs : E.D.M. a conçu les études, supervisé les études mécanistiques, obtenu le financement, analysé les données et rédigé le manuscrit. K.C.P. a co-conçu les études, supervisé toutes les études cliniques et co-écrit le manuscrit. G.S. et P.D. ont réalisé toutes les études mécanistiques et ont édité le manuscrit. L.W. a coordonné toutes les études, a contribué à l’acquisition des données, a analysé les données cliniques et a édité le manuscrit. A.H., E.N., C.M., T.-L.G. et M.S.M. ont contribué à l’acquisition et à l’analyse des données et ont révisé le manuscrit. J.R.M., D.F. et B.A. ont conçu les études cliniques, contribué à l’acquisition des données et révisé le manuscrit. Intérêts concurrents : E.D.M. est le co-propriétaire d’un brevet d’utilisation en instance sur l’utilisation du DCA en tant que thérapie du cancer. Il n’y a pas eu de commercialisation active ou planifiée de ce brevet.