E. D. Michelakis,1 * G. Sutendra,1 P . Dromparis,1 L. Webster,1 A. Haromy,1 E. Niven,2 C. Maguire,2 T.-L. Gammer,1 J. R. Mackey,3 D. Fulton,3 B. Abdulkarim,3 M. S. McMurtry,1 K. C. Petruk4
1Departamentode Medicina, Universidad de Alberta, Edmonton, Alberta, Canadá T6G 2B7.
2Departamentode Ingeniería Biomédica e Imagen Diagnóstica, Universidad de Alberta, Edmonton, Alberta, Canadá T6G 2B7.
3Departamentode Oncología, Universidad de Alberta, Edmonton, Alberta, Canadá T6G 2B7.
4Departamentode Neurocirugía, Universidad de Alberta, Edmonton, Alberta, Canadá T6G 2B7.
*A quien debe dirigirse la correspondencia. Correo electrónico: [email protected]
Volumen 2 Número 31 31ra34
Enviado: 11 de noviembre de 2009
Aceptado: 23 de abril de 2010
Publicado: 12 de mayo de 2010
Los tumores sólidos, incluido el agresivo cáncer cerebral primario glioblastoma multiforme, desarrollan resistencia a la muerte celular, en parte como resultado de un cambio de la fosforilación oxidativa mitocondrial a la glucólisis citoplasmática. Esta remodelación metabólica va acompañada de una hiperpolarización mitocondrial. Hemos comprobado si la pequeña molécula y fármaco huérfano dicloroacetato (DCA) puede revertir esta remodelación metabólica y mitocondrial específica del cáncer en el glioblastoma. Glioblastomas recién aislados de 49 pacientes mostraron hiperpolarización mitocondrial, que fue rápidamente revertida por el DCA. En un experimento separado con cinco pacientes que tenían glioblastoma, obtuvimos prospectivamente tejido tumoral basal y seriado, desarrollamos líneas celulares de glioblastoma específicas para cada paciente y células madre putativas de glioblastoma (células CD133+, nestin+ ), y tratamos a cada paciente con DCA oral durante un máximo de 15 meses. El DCA despolarizó las mitocondrias, aumentó las especies reactivas de oxígeno mitocondriales e indujo la apoptosis en las células de GBM, así como en las células madre putativas de GBM, tanto in vitro como in vivo. El tratamiento con DCA también inhibió el factor inducible por hipoxia-1α, promovió la activación de p53 y suprimió la angiogénesis tanto in vivo como in vitro. La toxicidad limitada por la dosis fue una neuropatía periférica reversible dependiente de la dosis, y no hubo toxicidad hematológica, hepática, renal o cardiaca. Se observaron indicios de eficacia clínica a una dosis que no causaba neuropatía periférica y a concentraciones séricas de DCA suficientes para inhibir la enzima diana del DCA, la piruvato deshidrogenasa quinasa II, que se expresaba en alto grado en todos los glioblastomas. La modulación metabólica puede ser un enfoque terapéutico viable en el tratamiento del glioblastoma.
INTRODUCCIÓN
El glioblastoma multiforme (GBM) es un tumor cerebral primario agresivo que presenta respuestas extremadamente pobres a las terapias aprobadas (1). La quimioterapia con temozolomida (TMZ) más radioterapia (RT), administrada tras la cirugía de citorreducción, aumenta la mediana de supervivencia de 12,1 meses con RT sola a 14,6 meses (1). La mediana de tiempo hasta la progresión del tumor tras RT y TMZ es de sólo 6,9 meses (1 ). En los gliomas recurrentes, la supervivencia libre de progresión y la respuesta a la TMZ son mucho peores (2). Los GBM son tumores muy vasculares con una notable heterogeneidad molecular y genética (1). Un tratamiento ideal debería aumentar la apoptosis de los GBM, superar la heterogeneidad molecular, inhibir la angiogénesis y atravesar la barrera hematoencefálica con una toxicidad sistémica mínima. Basándonos en nuestros recientes descubrimientos en modelos animales (3, 4), planteamos la hipótesis de que la pequeña molécula huérfana dicloroacetato (DCA) cumple estos criterios y puede ser eficaz en el tratamiento del GBM en humanos.
El DCA inhibe la enzima mitocondrial piruvato deshidrogenasa cinasa (PDK) (5). Al inhibir la PDK, el DCA activa la piruvato deshidrogenasa (PDH), una enzima portera que regula el flujo de carbohidratos (piruvato) a la mitocondria, aumentando la proporción de oxidación de glucosa respecto a la glucólisis (3-5). Si el piruvato permanece en el citoplasma, puede completar la glucólisis, produciendo ácido láctico y generando 2 moles de ATP por molécula de glucosa. Alternativamente, el piruvato puede entrar en varias vías anapleróticas y de biosíntesis de aminoácidos. Sin embargo, al activarse la PDH, el piruvato puede descarboxilarse en acetil-coenzima A, entrar en el ciclo de Krebs y completar la oxidación de la glucosa en la matriz mitocondrial, generando hasta 36 moles de ATP por molécula de glucosa en presencia de oxígeno. La oxidación de la glucosa no tiene lugar cuando el piruvato no entra en la mitocondria (por ejemplo, en mitocondrias enfermas o si la PDH está inhibida) o en ausencia de oxígeno.
Warburg (6) demostró por primera vez que el metabolismo de las células cancerosas, incluso en normoxia, se caracteriza por un aumento de la relación entre la glucólisis citoplasmática y la oxidación mitocondrial de la glucosa. Aunque se desconoce el mecanismo de este «efecto Warburg», y sigue sin demostrarse si está relacionado etiológicamente con la carcinogénesis (7), existe un interés creciente en el metabolismo como diana para las terapias contra el cáncer (8-11). El cambio energético de la oxidación mitocondrial de la glucosa a la glucólisis citoplasmática puede ofrecer una ventaja proliferativa a las células cancerosas (11). Por ejemplo, la mayoría de las enzimas glucolíticas también tienen acciones antiapoptóticas directas (12); el ácido láctico promueve la angiogénesis y la descomposición de la matriz intersticial, facilitando la metástasis (11); y la disminución de la función mitocondrial se asocia con la inhibición de la apoptosis dependiente de la mitocondria (3). El GBM tiene un fuerte fenotipo glucolítico, y se sabe que varias de las anomalías moleculares que ocurren en el GBM suprimen la oxidación mitocondrial de la glucosa y promueven la glucólisis citoplasmática (1), incluyendo la activación de las vías fosfatidilinositol 3-quinasa-AKT o myc o la supresión de la vía p53 (9, 10).
Las mitocondrias de las células cancerosas están hiperpolarizadas con respecto a las de las células no cancerosas (3, 13), una condición asociada a la supresión de la función mitocondrial. Aunque controvertido [revisado en (14)], el eflujo de mediadores proapoptóticos a través del poro de transición mitocondrial (PTM) depende en parte del potencial de membrana mitocondrial (ΔΨm) y, por tanto, la hiperpolarización mitocondrial puede marcar un estado de resistencia a la apoptosis (3, 15). Hemos demostrado que este estado puede ser revertido en células cancerosas por el DCA, que al inhibir la PDK promueve la entrada de piruvato en la mitocondria, revirtiendo el aumento de la relación glucólisis-oxidación de glucosa, mejorando la función mitocondrial y revirtiendo la hiperpolarización mitocondrial (3). Por lo tanto, el DCA disminuye el crecimiento tumoral in vitro e in vivo, sin afectar a las mitocondrias y tejidos no cancerosos (3, 16-20). El aumento de la respiración mitocondrial se asocia con un aumento de la producción de especies reactivas del oxígeno mitocondrial (mROS), predominantemente superóxido. El superóxido puede ser dismutado a H2O2, una mROS relativamente estable que puede alcanzar otras estructuras celulares más allá de las mitocondrias. Por ejemplo, el H2O2 puede activar canales de potasio dependientes de voltaje sensibles al redox en la membrana plasmática y, al menos en algunos tejidos, promover una disminución del calcio intracelular (3, 4). Otras dianas sensibles al redox pueden ser p53, que se activa cuando se oxida (21, 22). El eje p53 está inhibido en el GBM, lo que contribuye al aumento del estado proliferativo de las células del GBM (1). El p53 también reprime la transcripción estimulada por el factor inducible por hipoxia-1α (HIF-1α) porque el p53 y el HIF-1α compiten por el mismo factor de cotranscripción (23, 24). HIF-1α aumenta la expresión de los transportadores de glucosa y de varias enzimas glucolíticas, así como de PDK, manteniendo así el fenotipo glucolítico (25, 26). Además, HIF-1α aumenta la expresión del factor de crecimiento endotelial vascular (VEGF), potenciando la angiogénesis. La angiogénesis también puede verse favorecida por la activación de HIF-1α en condiciones de normoxia. Dado que las mitocondrias son importantes sensores de oxígeno (27), las mitocondrias inhibidas pueden transmitir señales redox pseudohipóxicas y activar HIF-1α incluso durante la normoxia (28-30). Además, una disminución del α-cetoglutarato, un producto directo del ciclo de Krebs, también puede promover la activación de HIF porque es un cofactor para la reacción de hidroxilación de prolilo que degrada HIF-1α (30).
Nuestra hipótesis era que el DCA administrado por vía oral, que atraviesa la barrera hematoencefálica, disminuiría el crecimiento del GBM in vivo. Además, sugerimos que esto podría ocurrir mediante (i) la inversión del fenotipo glucolítico y la normalización de ΔΨm, lo que promovería la apoptosis dependiente de mitocondrias; (ii) el aumento de mROS y la promoción de la activación de p53; y (iii) el aumento de las concentraciones de α-cetoglutarato. Los dos últimos efectos conducirían a la inhibición de HIF-1α, la disminución de VEGF y la inhibición de la angiogénesis.
Resultados
Efectos del DCA sobre las mitocondrias de 49 tumores GBM reciénaislados
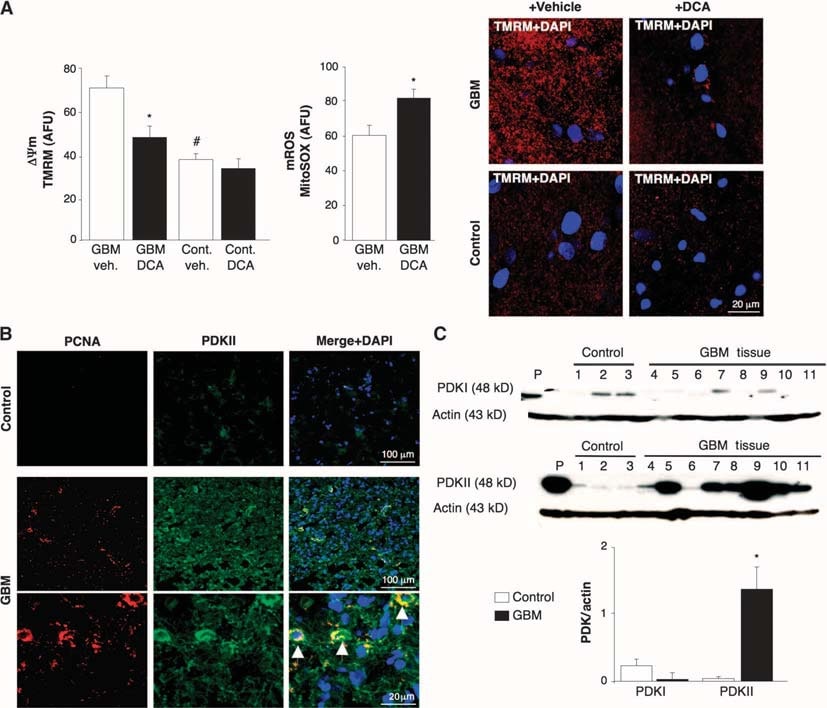
Para determinar si el GBM humano podría ser una diana para la terapia metabólica con DCA, estudiamos 49 GBM primarios consecutivos recién extirpados (60% varones, 48 ± 11 años). Además de los informes clínicos y neuropatológicos, confirmamos la identidad del GBM con inmunohistoquímica, que mostró expresión de proteína ácida fibrilar glial (GFAP) pero no de bIII-tubulina ni de marcadores oligodendrocitarios (fig. S1). El ΔΨm aumentó en los GBM recién aislados en comparación con los tejidos cerebrales no cancerosos obtenidos en la cirugía de la epilepsia (n = 3) (Fig. 1A). El DCA, pero no el vehículo (solución salina normal), causó despolarización mitocondrial en GBM pero no en tejido cerebral normal. El DCA también aumentó la mROS del GBM (Fig. 1A). Esto sugiere que la remodelación metabólica y mitocondrial en GBM es parcialmente reversible y que esta remodelación está regulada, al menos en parte, por PDK. La respuesta al DCA es consistente con una mayor concentración de PDKII [la isoforma más ubicuamente expresada y la que tiene elKi más bajo para el DCA (31)] en el GBM que en el tejido cerebral no canceroso, como se observa con inmunohistoquímica e inmunoblots (Fig. 1, B y C). Las células que mostraban las concentraciones más altas de PDKII también contenían antígeno nuclear de células proliferantes (PCNA), lo que sugiere que estas células estaban proliferando (Fig. 1B). Estos datos, recogidos durante un periodo de 2 años, reforzaron la justificación para administrar posteriormente DCA a pacientes con GBM (4).
Efectos clínicos del DCA en cinco pacientes con GBM
A continuación, tratamos con DCA a cinco pacientes consecutivos con GBM primario, remitidos por nuestro programa de cáncer cerebral y de los que disponíamos de tejido de la última cirugía de citorreducción. Tres pacientes (pacientes 1 a 3) tenían GBM recurrente con progresión de la enfermedad después de varias quimioterapias (además del tratamiento estándar con cirugía, RT y TMZ) y se consideraron apropiados para terapia paliativa. Otros dos pacientes (pacientes 4 y 5) fueron diagnosticados recientemente y, tras la cirugía de citorreducción inicial, se administró DCA además del tratamiento estándar de RT y TMZ. En el paciente 4, a un tratamiento previo de 3 meses con DCA le siguió la adición de RT y TMZ, mientras que en el paciente 5 el DCA se inició simultáneamente con RT y TMZ, tras la cirugía de citorreducción. Si los pacientes requerían reintervención o autopsia, se comparó el tejido de la última cirugía de citorreducción (antes de la administración de DCA) con el tejido posterior al tratamiento con DCA. Su información clínica se resume en la tabla S1. El DCA se ha administrado a pacientes durante más de 30 años, principalmente en el tratamiento de errores congénitos del metabolismo mitocondrial, y se dispone de datos farmacocinéticos y farmacodinámicos (5, 32-34). Tratamos a los pacientes con una dosis inicial de 12,5 mg/kg por vía oral dos veces al día durante 1 mes, momento en el que se aumentó la dosis a 25 mg/kg por vía oral dos veces al día. A continuación, se siguió un protocolo de desescalada de la dosis, disminuyéndola en un 50% cuando se producía toxicidad limitante de la dosis. Se realizó un seguimiento clínico de los pacientes durante 15 meses. Ninguno de los pacientes presentó toxicidad hematológica, hepática, renal o cardiaca (tabla S1). La neuropatía periférica fue la única toxicidad aparente. Los pacientes presentaron grados variables de neuropatía periférica en función de la dosis, que fue reversible, lo que confirma estudios anteriores (35-37). Cuando se disminuyó la dosis a 6,25 mg/kg por vía oral dos veces al día, ninguno de los pacientes presentó neuropatía periférica clínicamente significativa (tabla S1). Inicialmente, la semivida del DCA es <1 hora. El DCA inhibe su propio metabolismo y las concentraciones séricas aumentan, alcanzando finalmente una meseta (34). Las concentraciones plasmáticas mínimas de DCA en nuestros pacientes permanecieron indetectables durante los primeros 2 a 3 meses, pero posteriormente alcanzaron concentraciones terapéuticas. Con una dosis de 6,25 mg/kg por vía oral dos veces al día durante al menos 3 meses, las concentraciones mínimas de DCA fueron de 0,44 ± 0,16 mM (media ± DE; n = 4) (tabla S1). Estos valores son similares a los observados en el tratamiento crónico con DCA de adultos con defectos mitocondriales (34) y están en el mismo rango que elKi del DCA para la PDKII (0,2 mM) (31). Los pacientes 1, 4 y 5 mostraron cierta evidencia de regresión radiológica en la resonancia magnética (RM) (Fig. 2A y figs. S2 a S4). El paciente 3 tenía un tumor muy grande con edema cerebral al inicio (fig. S5), a pesar de estar recibiendo altas dosis de esteroides, y una puntuación de Karnofsky baja y continuó deteriorándose. Falleció por complicaciones del edema cerebral 3 meses después del inicio del tratamiento con DCA. El paciente 2 requirió drenaje de un quiste y citorreducción en el mes 11 de tratamiento con DCA. El paciente 4 mostró progresión radiológica en el mes 3 del tratamiento con DCA, momento en el que se realizó una citorreducción adicional y se administró RT más TMZ además de DCA. Todos, excepto el paciente 3, estaban clínicamente estables en el mes 15 del tratamiento con DCA y vivos en el mes 18 (seguimiento telefónico). En el Material suplementario se describen más detalles clínicos.
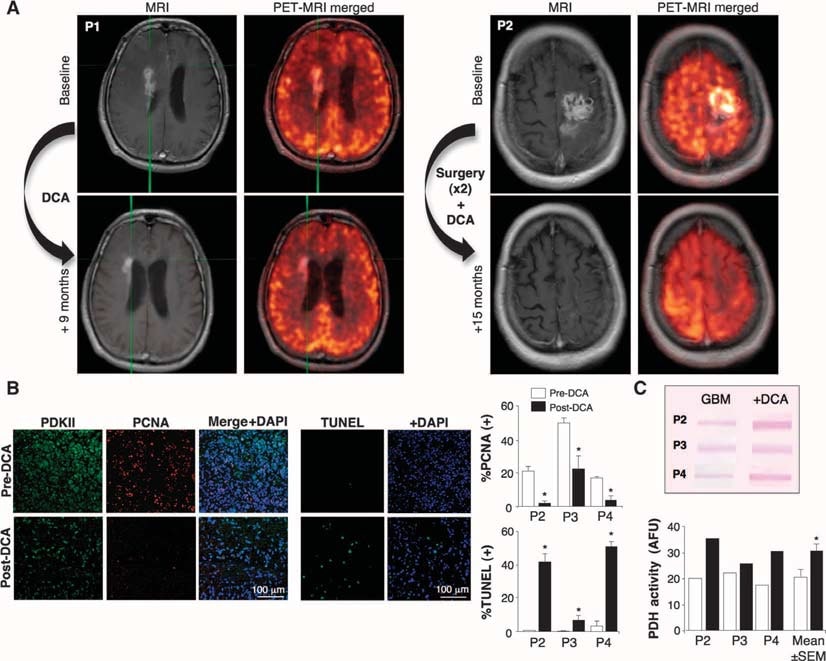
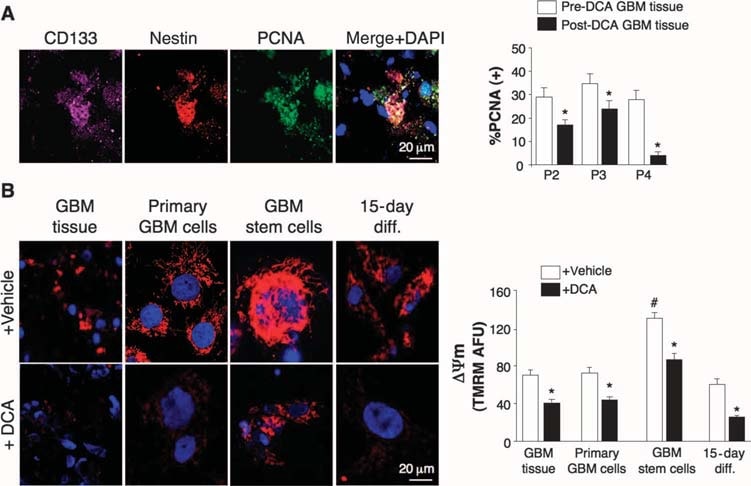
Efectos del DCA en tumores de GBM in vivo, líneas celulares primarias de GBM, y putativas GBM-SC derivadas de los pacientes tratados con DCA
Llevamos a cabo experimentos en tejidos derivados de estos cinco pacientes y pudimos hacer comparaciones en tejidos antes y después del tratamiento con DCA en los pacientes 2 a 4; sólo teníamos tejidos «antes» en los pacientes 1 y 5. En comparación con los tejidos pre-DCA, los tejidos antes y después del tratamiento con DCA en los pacientes 1 y 5 eran más similares. Comparado con el tejido pre-DCA, el tejido GBM post-DCA de los tres pacientes mostró una disminución del número de células por unidad de volumen, una disminución de la proliferación y un aumento de la apoptosis (Fig. 2B), así como un aumento de la actividad enzimática tisular de la PDH, lo que sugiere una inhibición efectiva de la PDK in vivo (Fig. 2C). Las células madre del cáncer de GBM (GBM-SCs) pueden ser responsables de la resistencia post-tratamiento y de la recurrencia del GBM (38-43). Estas células se caracterizan como CD133+ / nestin+ GBM-SC y forman nichos alrededor de los capilares (41). En estas unidades vasculares GBM-SC pueden inducir angiogénesis, mientras que su fenotipo molecular de células madre se mantiene por su accesibilidad a factores de crecimiento circulantes (44). La proliferación de GBM-SC se asocia con un resultado clínico particularmente malo (42). CD133+ /nestin+ GBM-SC expresaron PCNA in vivo en todos los tumores pre-DCA, indicando que se están dividiendo, pero el porcentaje de células CD133+ / nestin+ que expresaron PCNA disminuyó significativamente después de la terapia DCA en los pacientes 2 a 4 (Fig. 3A). La tinción simultánea con un anticuerpo CD133 y éster metílico de tetrametil rodamina (TMRM) mostró que las células CD133+ tenían el ΔΨm más alto en comparación con las células vecinas no-GBM-SC in vivo (fig. S6). En las líneas celulares primarias derivadas de tumores, ~10% de las células expresaban tanto CD133 como nestina, mientras que >90% de las células expresaban el marcador maduro GFAP (pero no bIII-tubulina u oligodendrocitos) (fig. S7), similar a la histopatología del GBM (fig. S1). Aislamos supuestas GBM-SC de tumores GBM y las cultivamos con los factores de crecimiento apropiados (factor de crecimiento de fibroblastos humanos, 20 ng/ml; factor de crecimiento epidérmico humano, 20 ng/ml). Estas células tenían una expresión muy alta tanto de CD133 como de nestina, tenían una expresión muy baja de marcadores gliales maduros (fig. S7) y formaban neuroesferas características (fig. 4 y fig. S7), un predictor independiente de mal resultado clínico (43). Medimos el DYm en tumores recién extirpados, en líneas celulares primarias y en GBM-SC aisladas de esos tumores, así como en células diferenciadas derivadas de GBM-SC (Fig. 3B). El potencial más alto se encontró en las GBM-SC putativas. Tanto las células GBM primarias como las secundarias derivadas de GBM-SC (diferenciación de 15 días) tenían potenciales mitocondriales similares a los de los tumores parentales. El DCA (0,5 mM durante 24 horas) disminuyó el potencial en todos los grupos de células. Aunque la causa del aumento de ΔΨm en el cáncer (3, 13) aún no está totalmente definida, se ha propuesto que se debe en parte a una translocación de la hexoquinasa II (HXKII), una enzima glucolítica clave, del citoplasma a la membrana mitocondrial externa (45, 46). Allí, la HXKII puede unirse e inhibir el canal aniónico dependiente de voltaje (un componente de la MTP), aumentando el ΔΨm y el umbral apoptótico. La inhibición de esta translocación disminuye el ΔΨm del cáncer e invierte la resistencia a la apoptosis (45, 46). Nuestras líneas celulares primarias generadas a partir de tumores pre-DCA mostraron una translocación mitocondrial sostenida de HXKII, explicando potencialmente el aumento de ΔΨm. La translocación de HXKII no estaba presente en las líneas celulares primarias de los tumores tras el tratamiento con DCA (fig. S8), lo que es compatible con la idea de que el DCA indujo la supresión de la glucólisis y disminuyó la ΔΨm. Al igual que en los tumores, la PDKII estaba presente en altas concentraciones en las líneas celulares de GBM generadas a partir de los pacientes 2 a 4, aunque las otras isoenzimas conocidas también se expresaban (fig. S9A). Cuando se permitió que las GBM-SC se diferenciaran en líneas celulares secundarias de GBM, la proporción de células con marcadores GBM-SC disminuyó a un valor similar al de las líneas celulares primarias (~10%). Sin embargo, cuando se les permitió diferenciarse en presencia de DCA (0,5 mM), la proporción de células con marcadores GBM-SC disminuyó aún más, hasta ~5% (fig. S7). De hecho, el DCA indujo apoptosis en GBM-SC in vitro (Fig. 4 y fig. S9B) así como en líneas celulares primarias de GBM (fig. S9C). La apoptosis se incrementó aún más en GBM-SCs por la combinación de DCA más TMZ (Fig. 4 y fig. S9B), proporcionando una justificación para la terapia de combinación. La apoptosis de las GBM-SC también tuvo lugar in vivo en los tumores tratados con DCA, como muestra la colocalización de nestina, CD133 y la tinción TUNEL mediada por la desoxinucleotidil transferasa terminal (Fig. 4 y fig. S9D).
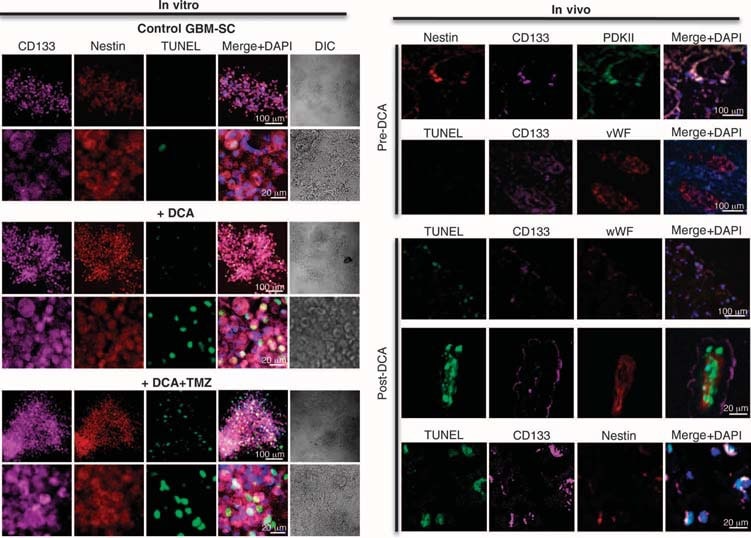
Efectos del DCA en la unidad de microvasos de GBM-SC y angiogénesis in vivo e in vitro
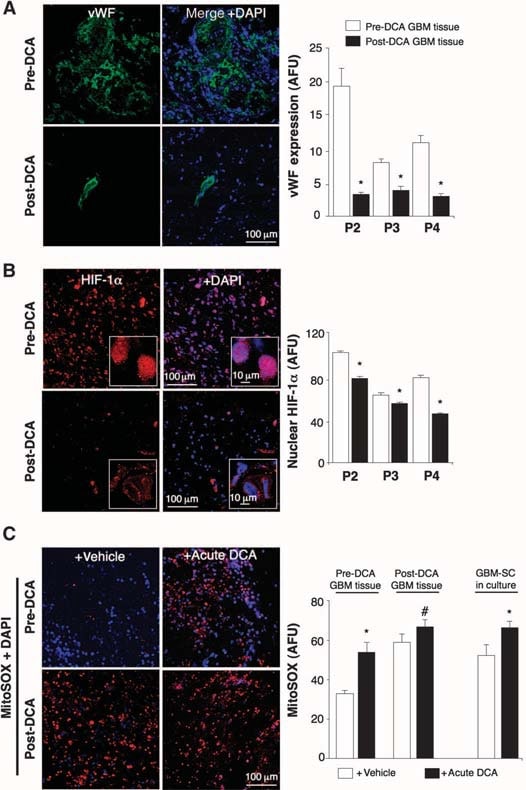
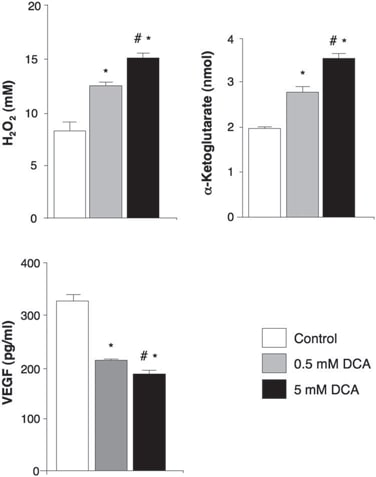
En los tejidos no tratados, pre-DCA, se encontraron nichos de GBM-SC alrededor de lechos de microvasos [tinción de factor von Willebrand (vWF)], como se ha informado (41). Esta unidad de microvasos GBM-SC (44) fue destruida por el tratamiento con DCA porque, además de GBM-SC, la apoptosis también aumentó en las células endoteliales microvasculares (Fig. 4 y fig. S9D), lo que sugiere una posible inhibición de la angiogénesis. De hecho, la disminución de la tinción de vWF en los tumores tratados con DCA sugiere una disminución de la vascularidad (Fig. 5A). HIF-1α estaba altamente expresado o activado (localización nuclear) en los tejidos tumorales pre-DCA e inhibido en los tejidos tumorales post-DCA (Fig. 5B). Los tumores post-tratamiento con DCA de los pacientes 2 a 4 mostraron un aumento significativo de mROS in vivo (superóxido medido por mitoSOX) en comparación con los tumores pre-DCA de los mismos pacientes (Fig. 5C). En los tumores pre-DCA, el DCA agudo incrementó la mROS hasta valores observados en los tumores post-tratamiento con DCA. En contraste, en los tumores post-DCA, el DCA agudo sólo aumentó mínimamente mROS, sugiriendo un efecto casi máximo in vivo. El DCA también aumentó mROS en GBM-SC (Fig. 5C). Las bajas concentraciones de ROS en células madre cancerosas pueden reflejar resistencia a la apoptosis, y se sugiere que las terapias que aumentan las ROS en células madre cancerosas son más efectivas (47). Aunque estudiamos mROS (superóxido mitocondrial), existe controversia sobre si la activación de HIF-1α en el cáncer se asocia con un aumento o disminución general de ROS [revisado en (28)]. Utilizando una técnica diferente, también medimos el H2O2 de células enteras. El DCA incrementó el H2O2 de las células GBM de forma dosis-dependiente (Fig. 6). Además, el DCA también incrementó las concentraciones intracelulares de α-cetoglutarato de forma dosis-dependiente (Fig. 6). Esto es compatible con el incremento en la oxidación de la glucosa que sigue a la activación de la PDH (3) porque el α-cetoglutarato es un producto del ciclo de Krebs. El ciclo de Krebs produce los donadores de electrones que alimentan la cadena de transporte de electrones durante la respiración. En consecuencia, el DCA aumentó las tasas de respiración en un 44 ± 4% en las células GBM (media ± SEM; n = 3; P < 0,05), lo que apoya un aumento general de la actividad mitocondrial. El aumento de H2O2 y α-cetoglutarato puede explicar la disminución de la actividad de HIF-1α (Fig. 5B), que también se confirma por la disminución dependiente de la dosis en la producción de VEGF por las células GBM (Fig. 6). Estos datos son consistentes con la disminución de la angiogénesis in vivo (Figs. 4 y 5) y sugieren que las células GBM señalan a las células endoteliales de forma paracrina. Para estudiar si el DCA puede suprimir directamente la angiogénesis, utilizamos la técnica estándar de formación de tubos de células endoteliales humanas en Matrigel. En condiciones fisiológicas de hipoxia moderada, el DCA causó una inhibición directa de la angiogénesis in vitro dependiente de la dosis (fig. S10). Los tumores post-tratamiento con DCA de los pacientes 2 a 4 mostraron un aumento de la actividad de la mROS-sensible p53 (translocación nuclear), también confirmado por el aumento de la actividad y abundancia de su diana descendente p21 (fig. S11). Estos efectos sobre p53 o p21 también pueden explicar la disminución de la transcripción impulsada por HIF y son consistentes con los efectos antiproliferativos, además de proapoptóticos, del DCA en el GBM (fig. S12).
DISCUSIÓN
El modulador metabólico DCA ejerce efectos anticancerígenos en células cultivadas y roedores (3, 16-20). Ahora hemos demostrado que el DCA puede utilizarse en pacientes que padecen GBM. El tratamiento con DCA se asoció en algunos pacientes con GBM a una estabilización radiológica prolongada o a la regresión del tumor y, en general, mostró un buen perfil de seguridad global. Este primer informe en humanos proporciona una justificación para ampliar los estudios con esta pequeña molécula genérica en pacientes con GBM. Nuestros resultados indican que el GBM es un buen candidato para la intervención metabólica. La diana del DCA, PDKII, está altamente expresada en tumores y líneas celulares de GBM, y el DCA puede inhibir su actividad in vivo. El GBM se caracteriza por la hiperpolarización mitocondrial, en consonancia con la remodelación metabólica (efecto Warburg) y la resistencia a la apoptosis relacionada que caracteriza al GBM y a la mayoría de los tumores sólidos (11). Se han descrito mutaciones en los genes de las isocitrato deshidrogenasas citoplasmáticas y mitocondriales en GBM que surgen de gliomas de grado inferior (GBM secundarios) (48), pero el mecanismo por el que estas mutaciones se relacionan con la carcinogénesis sigue sin estar claro (49, 50). Nuestros pacientes tenían GBM primarios y la remodelación mitocondrial fue al menos parcialmente reversible con DCA, lo que sugiere que no se debió a una disfunción irreversible. Además, mostramos que los putativos GBM-SC pueden sufrir la misma remodelación metabólica y mitocondrial, pero en mayor grado, porque los GBM-SC tenían las mitocondrias más hiperpolarizadas tanto in vivo como in vitro. La reversión de esta remodelación mitocondrial mediante DCA indujo apoptosis en GBM-SC tanto in vitro como in vivo. Aunque la magnitud de la inducción de la apoptosis por el DCA no es alta (en comparación con los agentes citotóxicos), es relativamente selectiva, ya que no afecta a las células no cancerosas (3), y, dado que afecta a GBM-SC, puede dar lugar a un efecto clínico más sostenido. El paciente 3 falleció y el paciente 4 sufrió una recidiva en los 3 primeros meses, cuando el DCA no había alcanzado valores terapéuticos sostenidos (como demuestra el hecho de que las concentraciones plasmáticas mínimas fueran indetectables). Por lo tanto, los pacientes pueden estar infratratados en la administración inicial de DCA y en riesgo de progresión inicial de la enfermedad. Al disminuir la resistencia a la apoptosis dependiente de las mitocondrias, el DCA puede potenciar los efectos de las terapias estándar. De hecho, los efectos del DCA más TMZ sobre la apoptosis de GBMSC podrían ser la base de los beneficios a largo plazo experimentados por el paciente 5. Los efectos del DCA en las células cancerosas son imitados por el ARN de interferencia pequeño PDKII, y el DCA no tiene efectos más allá de los observados tras el knockdown de PDKII (3). Esto sugiere que el mecanismo de los efectos anticancerígenos del DCA es la inhibición de la PDKII, una enzima que se encuentra en concentraciones elevadas en el GBM. Al inhibir la PDKII, el DCA normaliza el aumento de la proporción entre glucólisis y oxidación de glucosa en las células cancerosas, aumentando la función mitocondrial (3). Este mecanismo está respaldado por nuestros hallazgos actuales de que el DCA aumenta tanto la respiración de las células GBM como la concentración de a-cetoglutarato. Sin embargo, a diferencia de las condiciones controladas del cultivo celular, otros mecanismos pueden contribuir a los efectos del DCA sobre el metabolismo de las células cancerosas y la apoptosis in vivo. Además de los efectos descritos anteriormente del DCA en las células cancerosas (3), nuestros datos sobre HXKII, mROS, HIF-1α, p53 y p21 son coherentes con la inhibición de la angiogénesis inducida por el DCA, la inducción de la apoptosis y la supresión de la proliferación tanto en GBM como en GBM-SC, como se resume en el Material suplementario y en la fig. S12. El DCA no presentó toxicidad aparente, salvo una neurotoxicidad no mielinizante reversible y dependiente de la dosis descrita previamente (32, 34), que fue mínima o inexistente a la dosis oral de 6,25 mg/kg, dos veces al día. Esta dosis mostró eficacia biológica y clínica y alcanzó concentraciones plasmáticas en los valores requeridos para la inhibición de la PDK (31). Con el pequeño número de pacientes tratados en nuestro estudio, no se pueden sacar conclusiones firmes con respecto al DCA como terapia para el GBM. Nuestro trabajo apoya la necesidad de más estudios con DCA en GBM, con énfasis en protocolos de terapia combinada. El GBM también puede ser vulnerable a otros fármacos de la familia emergente de moduladores metabólicos, lo que apunta a un nuevo enfoque en el tratamiento de este cáncer incurable.
MATERIAL SUPLEMENTARIO
www.sciencetranslationalmedicine.org/cgi/content/full/2/31/31ra34/DC1
Materiales y Métodos
Resultados
Discusión
Fig. S1. Caracterización molecular de los tumores GBM.
Fig. S2. Evolución de la respuesta tumoral en el paciente 1.
Fig. S3. Evolución de la respuesta tumoral en el paciente
Fig. S4. Evolución de la respuesta tumoral en el paciente
Fig. S5. RMN de GBM del paciente 3.
Fig. S6. Potencial de membrana mitocondrial en GBM-SC de tejido GBM recién extirpado.
Fig. S7. Caracterización de células GBM primarias y GBM-SC.
Fig. S8. HXKII en células GBM derivadas de pacientes antes y después del tratamiento crónico con DCA.
Fig. S9 Efectos de la terapia con DCA en GBM-SC y apoptosis vascular.
Fig. S10. Efectos del DCA sobre la angiogénesis in vitro.
Fig. S11. Efectos del tratamiento con DCA sobre la actividad de p53 y p21 in vivo.
Fig. S12. Mecanismo global propuesto para los efectos anticancerígenos del DCA en GBM (véase
Supplementary Discussion).
Tabla S1. Parámetros clínicos y de laboratorio de cinco pacientes con GBM antes y después del tratamiento con DCA.
Referencias
REFERENCIAS
1 1. P. Y. Wen, S. Kesari, Gliomas malignos en adultos. N. Engl. J. Med. 359, 492-507 (2008).
2 W. K. Yung, R. E. Albright, J. Olson, R. Fredericks, K. Fink, M. D. Prados, M. Brada, A. Spence, R. J. Hohl, W. Shapiro, M. Glantz, H. Greenberg, R. G. Selker, N. A. Vick, R. Rampling, H. Friedman, P. Phillips, J. Bruner, N. Yue, D. Osoba, S. Zaknoen, V. A. Levin, A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br. J. Cancer 83, 588-593 (2000).
3 S. Bonnet, S. L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C. T. Lee, G. D. Lopaschuk, L. Puttagunta, S. Bonnet, G. Harry, K. Hashimoto, C. J. Porter, M. A. Andrade, B. Thebaud, E. D. Michelakis, A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37-51 (2007).
4 E. D. Michelakis, L. Webster, J. R. Mackey, Dichloroacetate (DCA) as a potential metabolictargeting therapy for cancer. Br. J. Cancer 99, 989-994 (2008).
5 P. W. Stacpoole, The pharmacology of dichloroacetate. Metabolism 38, 1124-1144 (1989).
6 O. Warburg, Ueber den Stoffwechsel der Tumoren (Constable, Londres, 1930).
7 S. Weinhouse, The Warburg hypothesis fifty years later. Z. Krebsforsch. Klin. Onkol. Cancer Res. Clin. Oncol. 87, 115-126 (1976).
8 M. G. Vander Heiden, L. C. Cantley, C. B. Thompson, Understanding the Warburg effect: Los requisitos metabólicos de la proliferación celular. Science 324, 1029-1033 (2009).
9 J. G. Pan, T. W. Mak, Metabolic targeting as an anticancer strategy: Dawn of a new era? Sci. STKE 2007, pe14 (2007).
10 J. W. Kim, C. V. Dang, Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 66, 8927-8930 (2006).
11 R. A. Gatenby, R. J. Gillies, Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891-899 (2004).
12 J. W. Kim, C. V. Dang, Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 30, 142-150 (2005).
13 L. B. Chen, Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 4, 155-181 (1988).
14 G. Kroemer, L. Galluzzi, C. Brenner, Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99-163 (2007).
15. N. Zamzami, G. Kroemer, The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2, 67-71 (2001).
16 R. A. Cairns, I. Papandreou, P. D. Sutphin, N. C. Denko, Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc. Natl. Acad. Sci. U.S.A. 104, 9445-9450 (2007).
17 W. Cao, S. Yacoub, K. T. Shiverick, K. Namiki, Y. Sakai, S. Porvasnik, C. Urbanek, C. J. Rosser, Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 68, 1223-1231 (2008).
18 S. Dhar, S. J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. U.S.A. 106, 22199-22204 (2009).
19 R. C. Sun, M. Fadia, J. E. Dahlstrom, C. R. Parish, P. G. Board, A. C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 120, 253-260 (2010).
20 J. Y. Wong, G. S. Huggins, M. Debidda, N. C. Munshi, I. De Vivo, Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol. Oncol. 109, 394-402 (2008).
21 S. Wang, S. S. Leonard, J. Ye, M. Ding, X. Shi, The role of hydroxyl radical as a messenger in Cr(VI)-induced p53 activation. Am. J. Physiol. Cell Physiol. 279, C868-C875 (2000).
22 C. Huang, Z. Zhang, M. Ding, J. Li, J. Ye, S. S. Leonard, H. M. Shen, L. Butterworth, Y. Lu, M. Costa, Y. Rojanasakul, V. Castranova, V. Vallyathan, X. Shi, Vanadate induces p53 transactivation through hydrogen peroxide and causes apoptosis. J. Biol. Chem. 275, 32516-32522 (2000).
23 T. Schmid, J. Zhou, R. Köhl, B. Brüne, p300 relieves p53-evoked transcriptional repression of hypoxia-inducible factor-1 (HIF-1). Biochem. J. 380, 289-295 (2004).
24 M. V. Blagosklonny, W. G. An, L. Y. Romanova, J. Trepel, T. Fojo, L. Neckers, p53 inhibits hypoxia-inducible factor-stimulated transcription. J. Biol. Chem. 273, 11995-11998 (1998).
25 J. W. Kim, I. Tchernyshyov, G. L. Semenza, C. V. Dang, HIF-1-mediated expression of pyruvate dehydrogenase kinase: Un interruptor metabólico necesario para la adaptación celular a la hipoxia. Cell Metab. 3, 177-185 (2006).
26 G. L. Semenza, D. Artemov, A. Bedi, Z. Bhujwalla, K. Chiles, D. Feldser, E. Laughner, R. Ravi, J. Simons, P. Taghavi, H. Zhong, ‘The metabolism of tumours’: 70 years later. Novartis Found. Symp. 240, 251-260 (2001).
27 E. K. Weir, J. López-Barneo, K. J. Buckler, S. L. Archer, Acute oxygen-sensing mechanisms. N. Engl. J. Med. 353, 2042-2055 (2005).
28 N. C. Denko, Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705-713 (2008).
29 G. L. Semenza, Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721-732 (2003).
30 E. D. MacKenzie, M. A. Selak, D. A. Tennant, L. J. Payne, S. Crosby, C. M. Frederiksen, D. G. Watson, E. Gottlieb, Cell-permeating a-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell. Biol. 27, 3282-3289 (2007).
31 M. M. Bowker-Kinley, W. I. Davis, P. Wu, R. A. Harris, K. M. Popov, Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329 (Pt 1), 191-196 (1998).
32 P. W. Stacpoole, D. S. Kerr, C. Barnes, S. T. Bunch, P. R. Carney, E. M. Fennell, N. M. Felitsyn, R. L. Gilmore, M. Greer, G. N. Henderson, A. D. Hutson, R. E. Neiberger, R. G. O’Brien, L. A. Perkins, R. G. Quisling, A. L. Shroads, J. J. Shuster, J. H. Silverstein, D. W. Theriaque, E. Valenstein, Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 117, 1519-1531 (2006).
33 P. W. Stacpoole, A. C. Lorenz, R. G. Thomas, E. M. Harman, Dicloroacetato en el tratamiento de la acidosis láctica. Ann. Intern. Med. 108, 58-63 (1988).
34 P. W. Stacpoole, T. L. Kurtz, Z. Han, T. Langaee, Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv. Drug Deliv. Rev. 60, 1478-1487 (2008).
35 P. Kaufmann, K. Engelstad, Y. Wei, S. Jhung, M. C. Sano, D. C. Shungu, W. S. Millar, X. Hong, C. L. Gooch, X. Mao, J. M. Pascual, M. Hirano, P. W. Stacpoole, S. DiMauro, D. C. De Vivo, Dichloroacetate causes toxic neuropathy in MELAS: A randomized, controlled clinical trial. Neurology 66, 324-330 (2006).
36 P. W. Stacpoole, L. R. Gilbert, R. E. Neiberger, P. R. Carney, E. Valenstein, D. W. Theriaque, J. J. Shuster, Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121, e1223-e1228 (2008).
37 P. W. Stacpoole, G. N. Henderson, Z. Yan, M. O. James, Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 106 (Suppl. 4), 989-994 (1998).
38 N. Sanai, A. Alvarez-Buylla, M. S. Berger, Neural stem cells and the origin of gliomas. N. Engl. J. Med. 353, 811-822 (2005).
39 F. Zindy, T. Uziel, O. Ayrault, C. Calabrese, M. Valentine, J. E. Rehg, R. J. Gilbertson, C. J. Sherr, M. F. Roussel, Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 67, 2676-2684 (2007).
40 T. Hide, T. Takezaki, H. Nakamura, J. Kuratsu, T. Kondo, Brain tumor stem cells as research and treatment targets. Brain Tumor Pathol. 25, 67-72 (2008).
41 C. Calabrese, H. Poppleton, M. Kocak, T. L. Hogg, C. Fuller, B. Hamner, E. Y. Oh, M. W. Gaber, D. Finklestein, M. Allen, A. Frank, I. T. Bayazitov, S. S. Zakharenko, A. Gajjar, A. Davidoff, R. J. Gilbertson, A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69-82 (2007).
42 R. Pallini, L. Ricci-Vitiani, G. L. Banna, M. Signore, D. Lombardi, M. Todaro, G. Stassi, M. Martini, G. Maira, L. M. Larocca, R. De Maria, Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin. Cancer Res. 14, 8205-8212 (2008).
43 D. R. Laks, M. Masterman-Smith, K. Visnyei, B. Angenieux, N. M. Orozco, I. Foran, W. H. Yong, H. V. Vinters, L. M. Liau, J. A. Lazareff, P. S. Mischel, T. F. Cloughesy, S. Horvath, H. I. Kornblum, Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 27, 980-987 (2009).
44 R. J. Gilbertson, J. N. Rich, Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 7, 733-736 (2007).
45 J. G. Pastorino, J. B. Hoek, Hexokinase II: The integration of energy metabolism and control of apoptosis. Curr. Med. Chem. 10, 1535-1551 (2003).
46 J. G. Pastorino, J. B. Hoek, N. Shulga, Activation of glycogen synthase kinase 3b disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545-10554 (2005).
47M. Diehn, R. W. Cho, N. A. Lobo, T. Kalisky, M. J. Dorie, A. N. Kulp, D. Qian, J. S. Lam, L. E. Ailles, M. Wong, B. Joshua, M. J. Kaplan, I. Wapnir, F. M. Dirbas, G. Somlo, C. Garberoglio, B. Paz, J. Shen, S. K. Lau, S. R. Quake, J. M. Brown, I. L. Weissman, M. F. Clarke, Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780-783 (2009).
48 H. Yan, D. W. Parsons, G. Jin, R. McLendon, B. A. Rasheed, W. Yuan, I. Kos, I. Batinic-Haberle, S. Jones, G. J. Riggins, H. Friedman, A. Friedman, D. Reardon, J. Herndon, K. W. Kinzler, V. E. Velculescu, B. Vogelstein, D. D. Bigner, IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765-773 (2009).
49 C. B. Thompson, Enzimas metabólicas como oncogenes o supresores tumorales. N. Engl. J. Med. 360, 813-815 (2009).
50 L. Dang, D. W. White, S. Gross, B. D. Bennett, M. A. Bittinger, E. M. Driggers, V. R. Fantin, H. G. Jang, S. Jin, M. C. Keenan, K. M. Marks, R. M. Prins, P. S. Ward, K. E. Yen, L. M. Liau, J. D. Rabinowitz, L. C. Cantley, C. B. Thompson, M. G. Vander Heiden, S. M. Su, Cancer-associated IDH1 mutations produce 2-hidroxiglutarato. Nature 462, 739-744 (2009).
51 Financiación: Este estudio fue financiado por la Fundación Hecht (Vancouver, Columbia Británica, Canadá; E.D.M.), los Institutos Canadienses de Investigación Sanitaria y el Programa de Cátedras de Investigación de Canadá (E.D.M.), y por donaciones públicas al programa DCA (recibidas y gestionadas por los Regentes de la Universidad de Alberta y la Facultad de Medicina). Los autores desean agradecer el apoyo de los Servicios de Salud de Alberta (D. Gordon, Vicepresidente Senior, Grandes Hospitales Terciarios). Contribuciones de los autores: E.D.M. diseñó los estudios, supervisó los estudios mecanísticos, consiguió la financiación, analizó los datos y redactó el manuscrito. K.C.P. co-diseñó los estudios, supervisó todos los estudios clínicos, y co-escribió el manuscrito. G.S. y P.D. realizado todos los estudios mecanicistas y editado el manuscrito. L.W. coordinado todos los estudios, contribuyó a la adquisición de datos, analizó los datos clínicos, y editó el manuscrito. A.H., E.N., C.M., T.-L.G., y M.S.M. contribuido a la adquisición de datos y análisis de datos y editado el manuscrito. J.R.M., D.F. y B.A. co-diseñaron los estudios clínicos, contribuyeron a la adquisición de datos y editaron el manuscrito. Intereses en conflicto: E.D.M. es copropietario de una patente de uso pendiente sobre el uso del DCA como terapia contra el cáncer. No ha habido ninguna comercialización activa o planeada de esta patente.