E. D. Michelakis,1 * G. Sutendra,1 P . Dromparis,1 L. Webster,1 A. Haromy,1 E. Niven,2 C. Maguire,2 T.-L. Gammer,1 J. R. Mackey,3 D. Fulton,3 B. Abdulkarim,3 M. S. McMurtry,1 K. C. Petruk4
1Dipartimentodi Medicina, Università di Alberta, Edmonton, Alberta, Canada T6G 2B7.
2Dipartimentodi Ingegneria biomedica e diagnostica per immagini, Università di Alberta, Edmonton, Alberta, Canada T6G 2B7.
3Dipartimentodi Oncologia, Università di Alberta, Edmonton, Alberta, Canada T6G 2B7.
4Dipartimentodi Neurochirurgia, Università di Alberta, Edmonton, Alberta, Canada T6G 2B7.
*A chi va indirizzata la corrispondenza. E-mail: [email protected]
Volume 2 Issue 31 31ra34
Inviato: 11 novembre 2009
Accettato: 23 aprile 2010
Pubblicato: 12 maggio 2010
I tumori solidi, compreso l’aggressivo tumore cerebrale primario glioblastoma multiforme, sviluppano una resistenza alla morte cellulare, in parte come risultato di un passaggio dalla fosforilazione ossidativa mitocondriale alla glicolisi citoplasmatica. Questo rimodellamento metabolico è accompagnato da iperpolarizzazione mitocondriale. Abbiamo testato se la piccola molecola e il farmaco orfano dicloroacetato (DCA) possano invertire questo rimodellamento metabolico e mitocondriale specifico del cancro nel glioblastoma. I glioblastomi appena isolati di 49 pazienti hanno mostrato un’iperpolarizzazione mitocondriale, che è stata rapidamente invertita dal DCA. In un esperimento separato con cinque pazienti affetti da glioblastoma, abbiamo assicurato in modo prospettico il tessuto tumorale di base e seriale, abbiamo sviluppato linee cellulari paziente-specifiche di glioblastoma e di cellule staminali putative di glioblastoma (cellule CD133+, nestin+ ) e abbiamo trattato ciascun paziente con DCA orale per un massimo di 15 mesi. Il DCA ha depolarizzato i mitocondri, aumentato le specie reattive dell’ossigeno mitocondriale e indotto l’apoptosi nelle cellule di GBM e nelle cellule staminali putative di GBM, sia in vitro che in vivo. La terapia con DCA ha anche inibito l’hypoxia-inducible factor-1α, promosso l’attivazione di p53 e soppresso l’angiogenesi sia in vivo che in vitro. La tossicità dose-limitante è stata una neuropatia periferica reversibile e dose-dipendente, mentre non è stata riscontrata alcuna tossicità ematologica, epatica, renale o cardiaca. Indicazioni di efficacia clinica erano presenti a una dose che non causava neuropatia periferica e a concentrazioni sieriche di DCA sufficienti a inibire l’enzima bersaglio del DCA, la piruvato deidrogenasi chinasi II, altamente espressa in tutti i glioblastomi. La modulazione metabolica può essere un approccio terapeutico valido per il trattamento del glioblastoma.
INTRODUZIONE
Il glioblastoma multiforme (GBM) è un tumore cerebrale primario aggressivo che presenta risposte estremamente scarse alle terapie approvate (1). La chemioterapia con temozolomide (TMZ) più radioterapia (RT), somministrata dopo l’intervento chirurgico di debulking, aumenta la sopravvivenza mediana da 12,1 mesi con la sola RT a 14,6 mesi (1). Il tempo mediano alla progressione del tumore dopo RT e TMZ è di soli 6,9 mesi (1). Nei gliomi ricorrenti, la sopravvivenza libera da progressione e la risposta alla TMZ sono molto peggiori (2). I GBM sono tumori molto vascolari con una notevole eterogeneità molecolare e genetica (1). Una terapia ideale dovrebbe aumentare l’apoptosi del GBM, superare l’eterogeneità molecolare, inibire l’angiogenesi e attraversare la barriera emato-encefalica con una tossicità sistemica minima. Sulla base dei nostri recenti risultati in modelli animali (3, 4), abbiamo ipotizzato che la piccola molecola orfana dicloroacetato (DCA) soddisfi questi criteri e possa essere efficace nel trattamento del GBM nell’uomo.
Il DCA inibisce l’enzima mitocondriale piruvato deidrogenasi chinasi (PDK) (5). Inibendo la PDK, il DCA attiva la piruvato deidrogenasi (PDH), un enzima che regola il flusso di carboidrati (piruvato) nei mitocondri, aumentando il rapporto tra ossidazione del glucosio e glicolisi (3-5). Se il piruvato rimane nel citoplasma, può completare la glicolisi, producendo acido lattico e generando 2 moli di ATP per molecola di glucosio. In alternativa, il piruvato può entrare in diverse vie anaplerotiche e di biosintesi degli aminoacidi. Tuttavia, su attivazione della PDH, il piruvato può essere decarbossilato ad acetil-coenzima A, entrare nel ciclo di Krebs e completare l’ossidazione del glucosio nella matrice mitocondriale, generando fino a 36 moli di ATP per molecola di glucosio in presenza di ossigeno. L’ossidazione del glucosio non avviene quando il piruvato non entra nei mitocondri (ad esempio, in mitocondri malati o se la PDH è inibita) o in assenza di ossigeno.
Warburg (6) ha dimostrato per primo che il metabolismo delle cellule tumorali, anche in condizioni di normossia, è caratterizzato da un aumento del rapporto tra glicolisi citoplasmatica e ossidazione mitocondriale del glucosio. Sebbene non si conosca il meccanismo di questo “effetto Warburg” e non si sappia se sia eziologicamente legato alla carcinogenesi (7), vi è un crescente interesse per il metabolismo come bersaglio per le terapie antitumorali (8-11). Il passaggio energetico dall’ossidazione mitocondriale del glucosio alla glicolisi citoplasmatica può offrire un vantaggio proliferativo alle cellule tumorali (11). Ad esempio, la maggior parte degli enzimi glicolitici ha anche un’azione antiapoptotica diretta (12); l’acido lattico promuove l’angiogenesi e la disgregazione della matrice interstiziale, facilitando le metastasi (11); la diminuzione della funzione mitocondriale è associata all’inibizione dell’apoptosi mitocondriale dipendente ( 3). Il GBM ha un forte fenotipo glicolitico e sono note alcune anomalie molecolari che si verificano nel GBM e che sopprimono l’ossidazione mitocondriale del glucosio e promuovono la glicolisi citoplasmatica (1), tra cui l’attivazione delle vie della fosfatidilinositolo 3-chinasi-AKT o myc o la soppressione della via p53 (9, 10).
I mitocondri delle cellule tumorali sono iperpolarizzati rispetto a quelli delle cellule non tumorali (3, 13), una condizione associata alla soppressione della funzione mitocondriale. Anche se controverso [rivisto in (14)], l’efflusso di mediatori pro-apoptotici attraverso il poro di transizione mitocondriale (MTP) dipende in parte dal potenziale di membrana mitocondriale (ΔΨm) e quindi l’iperpolarizzazione mitocondriale può segnare uno stato di resistenza all’apoptosi (3, 15). Abbiamo dimostrato che questo stato può essere invertito nelle cellule tumorali dal DCA, che inibendo la PDK promuove l’ingresso del piruvato nei mitocondri, invertendo l’aumento del rapporto glicolisi/ossidazione del glucosio, migliorando la funzione mitocondriale e invertendo l’iperpolarizzazione mitocondriale (3). Il DCA riduce quindi la crescita tumorale in vitro e in vivo, senza influenzare i mitocondri e i tessuti non tumorali ( 3, 16-20). L’aumento della respirazione mitocondriale è associato a un aumento della produzione di specie reattive mitocondriali dell’ossigeno (mROS), prevalentemente superossido. Il superossido può essere dismesso in H2O2, un mROS relativamente stabile che può raggiungere altre strutture cellulari oltre i mitocondri. Ad esempio, l’H2O2 può attivare canali del potassio voltaggio-dipendenti sensibili al redox nella membrana plasmatica e, almeno in alcuni tessuti, promuovere una diminuzione del calcio intracellulare (3, 4). Altri bersagli sensibili al redox possono includere la p53, che viene attivata quando è ossidata (21, 22). L’asse p53 è inibito nel GBM, contribuendo all’aumento dello stato proliferativo delle cellule di GBM (1). p53 reprime anche la trascrizione stimolata dall’hypoxia-inducible factor-1α (HIF-1α) perché p53 e HIF-1α competono per lo stesso fattore di cotrascrizione (23, 24). HIF-1α aumenta l’espressione dei trasportatori di glucosio e di diversi enzimi glicolitici, nonché della PDK, sostenendo così il fenotipo glicolitico (25, 26). Inoltre, HIF-1α aumenta l’espressione del fattore di crescita endoteliale vascolare (VEGF), potenziando l’angiogenesi. L’angiogenesi può essere potenziata anche dall’attivazione di HIF-1α in condizioni di normossia. Poiché i mitocondri sono importanti sensori di ossigeno (27), i mitocondri inibiti possono trasmettere segnali redox pseudo-ipossici e attivare HIF-1α anche durante la normossia (28-30). Inoltre, una diminuzione dell’α-chetoglutarato, un prodotto diretto del ciclo di Krebs, può anche promuovere l’attivazione di HIF perché è un cofattore per la reazione di idrossilazione prolilica che degrada HIF-1α (30).
Abbiamo ipotizzato che il DCA somministrato per via orale, che attraversa la barriera emato-encefalica, possa ridurre la crescita del GBM in vivo. Abbiamo inoltre ipotizzato che ciò possa avvenire (i) invertendo il fenotipo glicolitico e normalizzando il ΔΨm, che promuoverebbe l’apoptosi mitocondri-dipendente; (ii) aumentando l’mROS e promuovendo l’attivazione di p53; e (iii) aumentando le concentrazioni di α-chetoglutarato. Gli ultimi due effetti porterebbero all’inibizione di HIF-1α, a una diminuzione del VEGF e all’inibizione dell’angiogenesi.
Risultati
Effetti del DCA sui mitocondri di 49 tumori GBM appena isolati
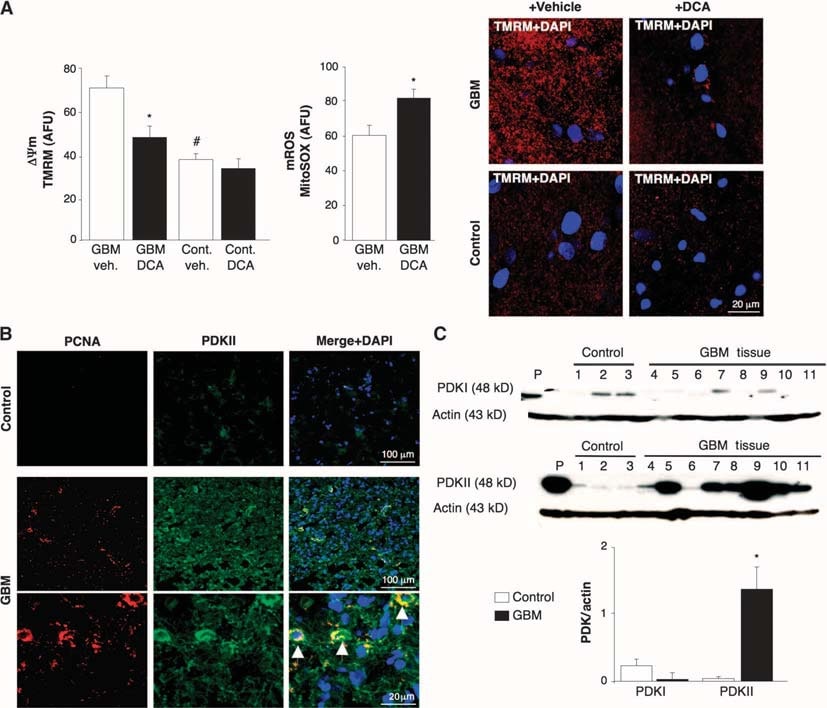
Per determinare se il GBM umano potesse essere un bersaglio per la terapia metabolica con il DCA, abbiamo studiato 49 GBM primari appena escissi (60% maschi, 48 ± 11 anni). Oltre ai referti clinici e neuropatologici, abbiamo confermato l’identità del GBM con l’immunoistochimica, che ha mostrato l’espressione della proteina gliale fibrillare acida (GFAP) ma non della bIII-tubulina o dei marcatori oligodendrocitari (fig. S1). Il ΔΨm era aumentato nei GBM appena isolati rispetto ai tessuti cerebrali non tumorali ottenuti durante la chirurgia dell’epilessia (n = 3) (Fig. 1A). Il DCA, ma non il veicolo (soluzione fisiologica normale), ha causato una depolarizzazione mitocondriale nel GBM ma non nel tessuto cerebrale normale. Il DCA ha anche aumentato il mROS del GBM (Fig. 1A). Ciò suggerisce che il rimodellamento metabolico e mitocondriale nel GBM è parzialmente reversibile e che questo rimodellamento è almeno in parte regolato dalla PDK. La risposta al DCA è coerente con una maggiore concentrazione di PDKII [l’isoforma più ubiquitariamente espressa e quella con ilKi più basso per il DCA (31)] nel GBM rispetto al tessuto cerebrale non tumorale, come si è visto con l’immunoistochimica e gli immunoblots (Fig. 1, B e C). Le cellule con le più alte concentrazioni di PDKII contenevano anche l’antigene nucleare delle cellule proliferanti (PCNA), suggerendo che queste cellule stavano proliferando (Fig. 1B). Questi dati, raccolti in un periodo di 2 anni, hanno rafforzato il razionale per la successiva somministrazione di DCA ai pazienti con GBM (4).
Effetti clinici del DCA su cinque pazienti con GBM
Abbiamo poi trattato con il DCA cinque pazienti consecutivi con GBM primario, inviati dal nostro programma di tumori cerebrali e dai quali era disponibile il tessuto dell’ultimo intervento di debulking. Tre pazienti (pazienti da 1 a 3) avevano GBM ricorrente con progressione della malattia dopo diverse chemioterapie (oltre al trattamento standard con chirurgia, RT e TMZ) e sono stati considerati idonei per una terapia palliativa. Altri due pazienti (pazienti 4 e 5) sono stati diagnosticati di recente e, dopo l’intervento iniziale di debulking, è stato somministrato DCA in aggiunta al trattamento standard con RT e TMZ. Nel paziente 4, un pretrattamento di 3 mesi con DCA è stato seguito dall’aggiunta di RT e TMZ, mentre nel paziente 5 il DCA è stato iniziato contemporaneamente a RT e TMZ, dopo l’intervento di debulking. Se i pazienti hanno richiesto un nuovo intervento o un’autopsia, il tessuto dell’ultimo intervento di debulking (prima della somministrazione di DCA) è stato confrontato con il tessuto successivo al trattamento con DCA. Le informazioni cliniche sono riassunte nella tabella S1. Il DCA viene somministrato ai pazienti da oltre 30 anni, principalmente nel trattamento degli errori congeniti del metabolismo mitocondriale, e sono disponibili dati farmacocinetici e farmacodinamici (5, 32-34). Abbiamo trattato i pazienti con una dose iniziale di 12,5 mg/kg per via orale due volte al giorno per 1 mese, dopodiché la dose è stata aumentata a 25 mg/kg per via orale due volte al giorno. Abbiamo poi seguito un protocollo di de-escalation della dose, diminuendo la dose del 50% quando si verificava una tossicità dose-limitante. I pazienti sono stati seguiti clinicamente fino a 15 mesi. Nessuno dei pazienti ha manifestato tossicità ematologica, epatica, renale o cardiaca (tabella S1). La neuropatia periferica è stata l’unica tossicità apparente. I pazienti hanno manifestato un grado variabile di neuropatia periferica in funzione della dose, che è risultata reversibile, confermando studi precedenti (35-37). Quando la dose è stata ridotta a 6,25 mg/kg per via orale due volte al giorno, nessuno dei pazienti ha presentato una neuropatia periferica clinicamente significativa (tabella S1). Inizialmente, l’emivita del DCA è di <1 ora. Il DCA inibisce il proprio metabolismo e le concentrazioni sieriche aumentano, raggiungendo infine un plateau (34). Le concentrazioni plasmatiche di DCA nei nostri pazienti sono rimaste non rilevabili per i primi 2 o 3 mesi, ma in seguito hanno raggiunto concentrazioni terapeutiche. Alla dose di 6,25 mg/kg per via orale due volte al giorno per almeno 3 mesi, le concentrazioni trogolo di DCA erano di 0,44 ± 0,16 mM (media ± SD; n = 4) (tabella S1). Questi valori sono simili a quelli osservati nel trattamento cronico con DCA di adulti con difetti mitocondriali (34) e sono nello stesso intervallo delKi di DCA per PDKII (0,2 mM) (31). I pazienti 1, 4 e 5 hanno mostrato alcune evidenze di regressione radiologica alla risonanza magnetica (MRI) (Fig. 2A e figg. da S2 a S4). Il paziente 3 aveva un tumore molto grande con edema cerebrale al basale (fig. S5), nonostante la somministrazione di alte dosi di steroidi, e un basso punteggio Karnofsky e ha continuato a peggiorare. È morto per le complicazioni dell’edema cerebrale 3 mesi dopo l’inizio della terapia con DCA. Il paziente 2 ha richiesto il drenaggio di una cisti e il debulking al mese 11 di terapia con DCA. Il paziente 4 ha mostrato una progressione radiologica al mese 3 della terapia con DCA; a questo punto è stato eseguito un ulteriore debulking ed è stata somministrata RT più TMZ in aggiunta al DCA. Tutti, tranne il paziente 3, erano clinicamente stabili al mese 15 della terapia con DCA e vivi al mese 18 (follow-up telefonico). Ulteriori dettagli clinici sono descritti nel Materiale supplementare.
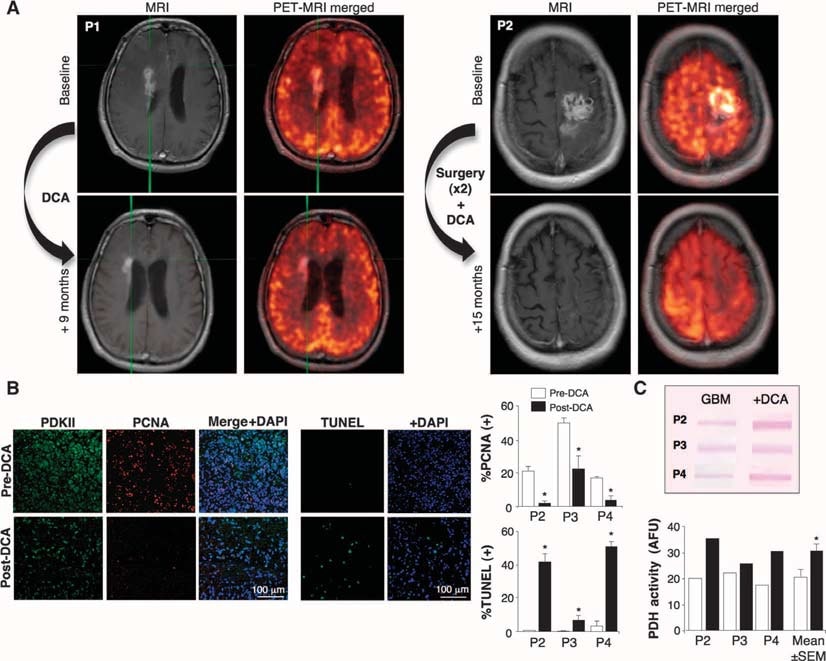
Effetti del DCA sui tumori GBM in vivo, sulle linee cellulari GBM primarie e sulle putative GBM-SC derivate dai pazienti trattati con DCA
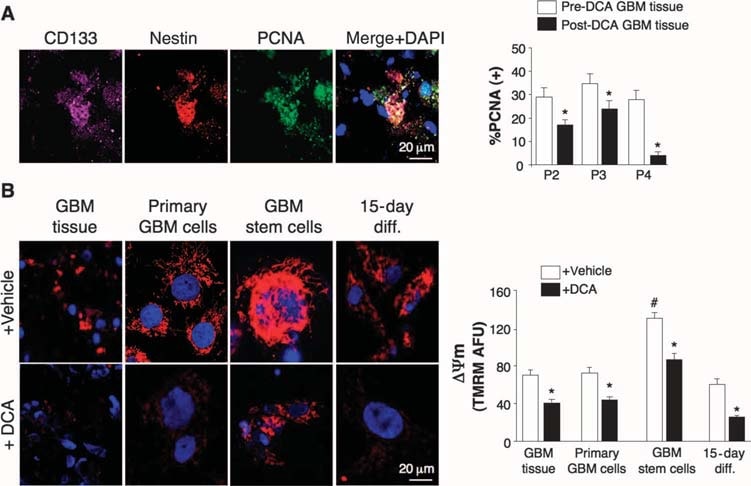
Abbiamo condotto esperimenti su tessuti derivati da questi cinque pazienti e siamo stati in grado di fare confronti tra i tessuti prima e dopo il trattamento con DCA nei pazienti da 2 a 4; abbiamo avuto a disposizione solo tessuti “prima” nei pazienti 1 e 5. Rispetto al tessuto pre-DCA, il tessuto GBM post-DCA di tutti e tre i pazienti ha mostrato una riduzione del numero di cellule per unità di volume, una diminuzione della proliferazione e un aumento dell’apoptosi (Fig. 2B), nonché un aumento dell’attività enzimatica tissutale della PDH, suggerendo un’efficace inibizione della PDK in vivo (Fig. 2C). Le cellule staminali putative del cancro del GBM (GBM-SCs) possono essere responsabili della resistenza post-trattamento e della recidiva del GBM (38-43). Queste cellule sono caratterizzate come GBM-SC CD133+ / nestin+ e formano nicchie intorno ai capillari (41). In queste unità vascolari GBM-SC, le GBM-SC possono indurre angiogenesi, mentre il loro fenotipo di cellule staminali molecolari è mantenuto dalla loro accessibilità ai fattori di crescita circolanti (44). La proliferazione delle GBM-SC è associata a un esito clinico particolarmente sfavorevole (42). Le GBM-SC CD133+ / nestin+ esprimevano PCNA in vivo in tutti i tumori pre-DCA, indicando che si stavano dividendo, ma la percentuale di cellule CD133+ / nestin+ che esprimevano PCNA era significativamente diminuita dopo la terapia con DCA nei pazienti da 2 a 4 (Fig. 3A). La colorazione simultanea con un anticorpo CD133 e con l’estere metilico della tetrametil rodamina (TMRM) ha mostrato che le cellule CD133+ avevano il ΔΨm più alto rispetto alle cellule non-GBM-SC vicine in vivo (fig. S6). Nelle linee cellulari primarie derivate dal tumore, circa il 10% delle cellule esprimeva sia il CD133 che la nestina, mentre >90% delle cellule esprimeva il marcatore maturo GFAP (ma non la bIII-tubulina o l’oligodendrocita) (fig. S7), simile all’istopatologia del GBM (fig. S1). Abbiamo isolato le GBM-SC putative da tumori GBM e le abbiamo coltivate con i fattori di crescita appropriati (fattore di crescita dei fibroblasti umani, 20 ng/ml; fattore di crescita epidermico umano, 20 ng/ml). Queste cellule avevano un’espressione molto elevata di CD133 e nestina, un’espressione molto bassa di marcatori gliali maturi (fig. S7) e formavano le caratteristiche neurosfere (fig. 4 e fig. S7), un fattore predittivo indipendente di esito clinico sfavorevole (43). Abbiamo misurato il DYm in tumori appena escissi, in linee cellulari primarie e in GBM-SC isolati da tali tumori, nonché in cellule differenziate derivate da GBM-SC (Fig. 3B). Il potenziale più elevato è stato riscontrato nelle GBM-SC putative. Sia le cellule GBM primarie che quelle secondarie derivate da GBM-SC (differenziazione di 15 giorni) avevano potenziali mitocondriali simili a quelli dei tumori parentali. Il DCA (0,5 mM per 24 ore) ha ridotto il potenziale in tutti i gruppi di cellule. Sebbene la causa dell’aumento del ΔΨm nel cancro (3, 13) non sia ancora stata completamente definita, è stato proposto che sia causato in parte da una traslocazione dell’esochinasi II (HXKII), un enzima glicolitico chiave, dal citoplasma alla membrana mitocondriale esterna (45, 46). Lì, HXKII può legarsi e inibire il canale anionico voltaggio-dipendente (un componente del MTP), aumentando il ΔΨm e la soglia apoptotica. L’inibizione di questa traslocazione diminuisce il ΔΨm del tumore e inverte la resistenza all’apoptosi (45, 46). Le nostre linee cellulari primarie generate da tumori pre-DCA hanno mostrato una traslocazione mitocondriale sostenuta di HXKII, spiegando potenzialmente l’aumento del ΔΨm. La traslocazione di HXKII non era presente nelle linee cellulari primarie dei tumori dopo il trattamento con DCA (fig. S8), compatibile con l’idea che il DCA abbia indotto la soppressione della glicolisi e diminuito il ΔΨm. Come nei tumori, la PDKII era presente ad alte concentrazioni nelle linee cellulari di GBM generate dai pazienti da 2 a 4, sebbene fossero espressi anche gli altri isoenzimi noti (fig. S9A). Quando le GBM-SC sono state autorizzate a differenziarsi in linee cellulari GBM secondarie, la percentuale di cellule con marcatori GBM-SC è diminuita a un valore simile a quello delle linee cellulari primarie (~10%). Quando è stato permesso di differenziarsi in presenza di DCA (0,5 mM), tuttavia, la percentuale di cellule con marcatori GBM-SC è diminuita ulteriormente fino a ~5% (fig. S7). In effetti, il DCA ha indotto l’apoptosi nelle GBM-SC in vitro (fig. 4 e fig. S9B) e nelle linee cellulari primarie di GBM (fig. S9C). L’apoptosi è stata ulteriormente aumentata nelle GBM-SC dalla combinazione di DCA e TMZ (Fig. 4 e fig. S9B), fornendo un razionale per la terapia combinata. L’apoptosi delle GBM-SC si è verificata anche in vivo nei tumori post-trattamento con DCA, come dimostrato dalla colocalizzazione di nestina, CD133 e dalla colorazione TUNEL (terminal deoxynucleotidyl transferase-mediated deoxyuridine triphosphate nick end labeling) (Fig. 4 e fig. S9D).
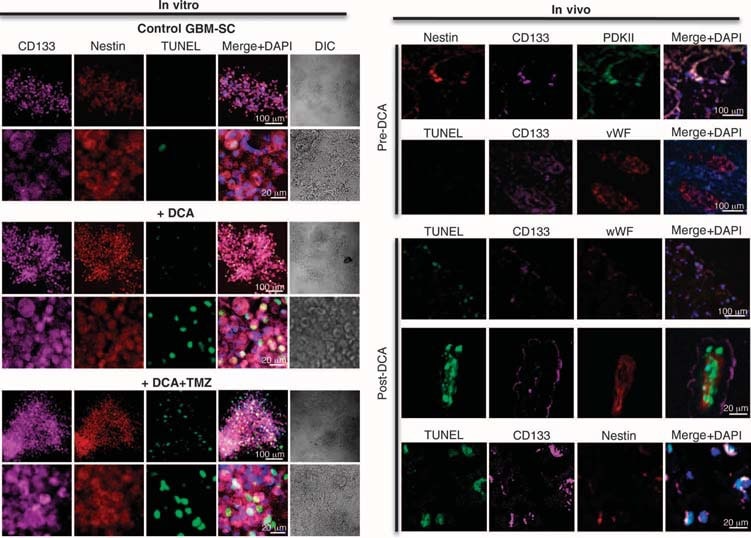
Effetti del DCA sull’unità microvessuale GBM-SC e sull’angiogenesi in vivo e in vitro
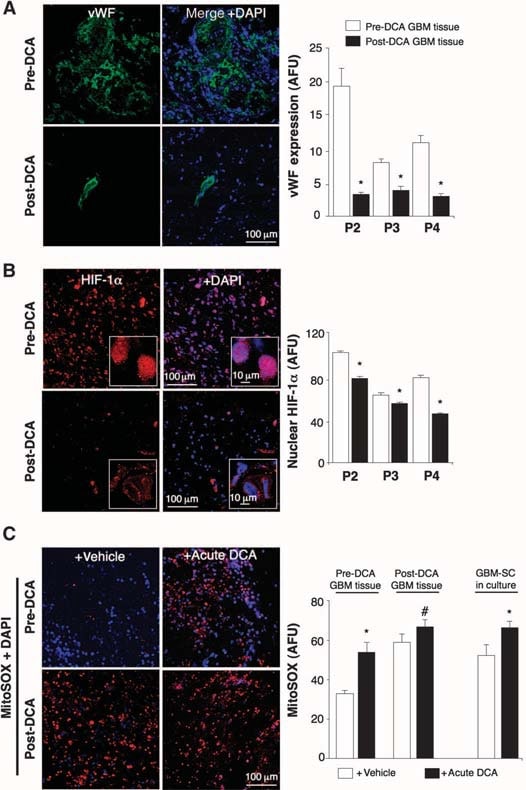
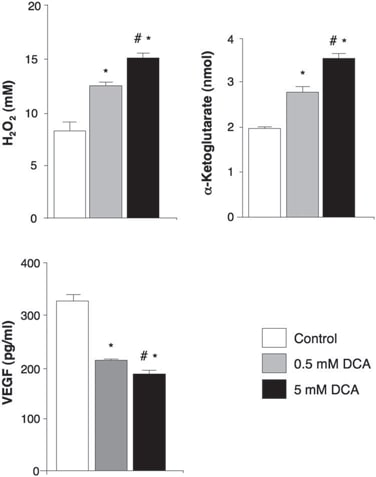
Nei tessuti non trattati, prima del DCA, sono state trovate nicchie di GBM-SC intorno a letti di microvasi [colorazione del fattore von Willebrand (vWF)], come riportato (41). Questa unità di microvasi GBM-SC (44) è stata distrutta dal trattamento con DCA perché, oltre alle GBM-SC, l’apoptosi è aumentata anche nelle cellule endoteliali microvascolari (Fig. 4 e fig. S9D), suggerendo una potenziale inibizione dell’angiogenesi. Infatti, la diminuzione della colorazione vWF nei tumori post-trattamento con DCA ha suggerito una riduzione della vascolarizzazione (Fig. 5A). HIF-1α era altamente espresso o attivato (localizzazione nucleare) nei tessuti tumorali pre-DCA e inibito in quelli post-DCA (Fig. 5B). I tumori post-trattamento con DCA dei pazienti da 2 a 4 hanno mostrato un aumento significativo di mROS in vivo (superossido misurato mediante mitoSOX) rispetto ai tumori pre-DCA degli stessi pazienti (Fig. 5C). Nei tumori pre-DCA, il DCA acuto ha aumentato l’mROS fino ai valori osservati nei tumori trattati dopo il DCA. Al contrario, nei tumori post-DCA, il DCA acuto ha aumentato solo minimamente l’mROS, suggerendo un effetto quasi massimo in vivo. Il DCA ha aumentato l’mROS anche nelle GBM-SC (Fig. 5C). Le basse concentrazioni di ROS nelle cellule staminali tumorali possono riflettere la resistenza all’apoptosi e si ritiene che le terapie che aumentano i ROS delle cellule staminali tumorali siano più efficaci (47). Sebbene abbiamo studiato il mROS (superossido mitocondriale), è controverso se l’attivazione di HIF-1α nel cancro sia associata a un aumento o a una diminuzione complessiva dei ROS [rivisto in (28)]. Utilizzando una tecnica diversa, abbiamo misurato anche l’H2O2 a cellula intera. Il DCA ha aumentato l’H2O2 delle cellule di GBM in modo dose-dipendente (Fig. 6). Inoltre, il DCA ha anche aumentato le concentrazioni intracellulari di α-chetoglutarato in modo dose-dipendente (Fig. 6). Ciò è compatibile con l’aumento dell’ossidazione del glucosio che segue l’attivazione della PDH (3) , poiché l’α-chetoglutarato è un prodotto del ciclo di Krebs. Il ciclo di Krebs produce i donatori di elettroni che alimentano la catena di trasporto degli elettroni durante la respirazione. Di conseguenza, il DCA ha aumentato i tassi di respirazione del 44 ± 4% nelle cellule di GBM (media ± SEM; n = 3; P < 0,05), sostenendo un aumento complessivo dell’attività mitocondriale. L’aumento di H2O2 e α-chetoglutarato può spiegare la diminuzione dell’attività di HIF-1α (Fig. 5B), confermata anche dalla diminuzione dose-dipendente della produzione di VEGF da parte delle cellule di GBM (Fig. 6). Questi dati sono coerenti con la diminuzione dell’angiogenesi in vivo (Figg. 4 e 5) e suggeriscono che le cellule di GBM segnalano alle cellule endoteliali in modo paracrino. Per studiare se il DCA possa sopprimere direttamente l’angiogenesi, abbiamo utilizzato la tecnica standard di formazione di tubi di cellule endoteliali umane in Matrigel. In condizioni di ipossia moderata fisiologica, il DCA ha causato un’inibizione diretta dose-dipendente dell’angiogenesi in vitro (fig. S10). I tumori post-trattamento con DCA dei pazienti da 2 a 4 hanno mostrato un aumento dell’attività della p53 sensibile all’mROS (traslocazione nucleare), confermato anche dall’aumento dell’attività e dell’abbondanza del suo bersaglio a valle p21 (fig. S11). Questi effetti su p53 o p21 possono spiegare anche la diminuzione della trascrizione guidata da HIF e sono coerenti con gli effetti antiproliferativi, oltre che proapoptotici, del DCA nel GBM (fig. S12).
DISCUSSIONE
Il modulatore metabolico DCA esercita effetti antitumorali in cellule in coltura e nei roditori (3, 16-20). Abbiamo ora dimostrato che il DCA può essere utilizzato nei pazienti affetti da GBM. In alcuni pazienti affetti da GBM, il trattamento con DCA è stato associato a una prolungata stabilizzazione radiologica o regressione del tumore e, in generale, ha mostrato un profilo di sicurezza complessivamente buono. Questa prima relazione nell’uomo fornisce un razionale per studi più ampi con questa piccola molecola generica in pazienti affetti da GBM. I nostri risultati indicano che il GBM è un buon candidato per un intervento metabolico. Il bersaglio del DCA, PDKII, è altamente espresso nei tumori e nelle linee cellulari di GBM e il DCA può inibire la sua attività in vivo. Il GBM è caratterizzato da iperpolarizzazione mitocondriale, in linea con il rimodellamento metabolico (effetto Warburg) e la relativa resistenza all’apoptosi che caratterizzano il GBM e la maggior parte dei tumori solidi (11). Mutazioni nei geni per le isocitrato deidrogenasi citoplasmatiche e mitocondriali sono state descritte in GBM derivanti da gliomi di grado inferiore (GBM secondari) (48), ma il meccanismo con cui queste mutazioni sono correlate alla carcinogenesi rimane poco chiaro (49, 50). I nostri pazienti avevano GBM primari e il rimodellamento mitocondriale era almeno parzialmente reversibile con DCA, suggerendo che non era dovuto a una disfunzione irreversibile. Inoltre, dimostriamo che le GBM-SC putative possono subire lo stesso rimodellamento metabolico e mitocondriale, ma in misura maggiore, perché le GBM-SC presentano i mitocondri più iperpolarizzati sia in vivo che in vitro. L’inversione di questo rimodellamento mitocondriale da parte del DCA ha indotto l’apoptosi in GBM-SC sia in vitro che in vivo. Sebbene l’entità dell’induzione dell’apoptosi da parte del DCA non sia elevata (rispetto agli agenti citotossici), è relativamente selettiva, risparmiando le cellule non tumorali (3) e, poiché coinvolge le GBM-SC, può determinare un effetto clinico più duraturo. Il paziente 3 è deceduto e il paziente 4 ha avuto una recidiva entro i primi 3 mesi, quando il DCA non aveva raggiunto valori terapeutici sostenuti (come dimostrato dal fatto che le concentrazioni plasmatiche trough erano non rilevabili). Pertanto, i pazienti possono essere sottotrattati con la somministrazione iniziale di DCA e sono a rischio di progressione iniziale della malattia. Diminuendo la resistenza all’apoptosi mitocondriale, il DCA può essere in grado di potenziare gli effetti delle terapie standard. In effetti, gli effetti del DCA più TMZ sull’apoptosi delle GBMSC potrebbero essere alla base dei benefici a lungo termine riscontrati dal paziente 5. Gli effetti del DCA sulle cellule tumorali sono imitati dal PDKII small interfering RNA e il DCA non ha effetti superiori a quelli osservati dopo l’eliminazione del PDKII (3). Ciò suggerisce che il meccanismo degli effetti antitumorali del DCA sia l’inibizione di PDKII, un enzima che si trova in concentrazioni elevate nel GBM. Inibendo la PDKII, il DCA normalizza l’aumento del rapporto tra glicolisi e ossidazione del glucosio nelle cellule tumorali, aumentando la funzione mitocondriale (3). Questo meccanismo è supportato dai risultati attuali, secondo cui il DCA aumenta sia la respirazione delle cellule di GBM sia la concentrazione di a-chetoglutarato. Tuttavia, a differenza delle condizioni controllate della coltura cellulare, altri meccanismi possono contribuire agli effetti del DCA sul metabolismo delle cellule tumorali e sull’apoptosi in vivo. Oltre agli effetti precedentemente descritti del DCA sulle cellule tumorali (3), i nostri dati su HXKII, mROS, HIF-1α, p53 e p21 sono coerenti con l’inibizione dell’angiogenesi, l’induzione dell’apoptosi e la soppressione della proliferazione indotte dal DCA sia nel GBM che nel GBM-SC, come riassunto nel Materiale supplementare e nella fig. S12. S12. Il DCA non ha presentato alcuna tossicità apparente, se non una neurotossicità reversibile non demielinizzante già descritta in precedenza (32, 34), che era minima o assente alla dose orale di 6,25 mg/kg, due volte al giorno. Questa dose ha mostrato efficacia biologica e clinica e ha raggiunto concentrazioni plasmatiche ai valori richiesti per l’inibizione della PDK (31). Dato l’esiguo numero di pazienti trattati nel nostro studio, non è possibile trarre conclusioni definitive sul DCA come terapia per il GBM. Il nostro lavoro sostiene la necessità di ulteriori studi con il DCA nel GBM, con particolare attenzione ai protocolli di terapia combinata. Il GBM potrebbe essere vulnerabile anche ad altri farmaci della famiglia emergente dei modulatori metabolici, indicando un nuovo approccio nella gestione di questo tumore incurabile.
MATERIALE SUPPLEMENTARE
www.sciencetranslationalmedicine.org/cgi/content/full/2/31/31ra34/DC1
Materiali e metodi
Risultati
Discussione
Fig. S1. Caratterizzazione molecolare dei tumori GBM.
Fig. S2. Evoluzione della risposta tumorale nel paziente 1.
Fig. S3. Evoluzione della risposta tumorale nel paziente 4.
Fig. S4. Evoluzione della risposta tumorale nel paziente 5.
Fig. S5. Risonanza magnetica del GBM del paziente 3.
Fig. S6. Potenziale di membrana mitocondriale in GBM-SC da tessuto GBM appena escisso.
Fig. S7. Caratterizzazione di cellule GBM primarie e GBM-SC.
Fig. S8. HXKII in cellule GBM derivate da pazienti prima e dopo il trattamento cronico con DCA.
Fig. S9 Effetti della terapia con DCA su GBM-SC e apoptosi vascolare.
Fig. S10. Effetti del DCA sull’angiogenesi in vitro.
Fig. S11. Effetti del trattamento con DCA sull’attività di p53 e p21 in vivo.
Fig. S12. Meccanismo completo proposto per gli effetti antitumorali del DCA nel GBM (vedi
Supplementary Discussion).
Tabella S1. Parametri clinici e di laboratorio di cinque pazienti affetti da GBM prima e dopo il trattamento con DCA.
Referenze
RIFERIMENTI
1 1. P. Y. Wen, S. Kesari, Malignant gliomas in adults. N. Engl. J. Med. 359, 492-507 (2008).
2 W. K. Yung, R. E. Albright, J. Olson, R. Fredericks, K. Fink, M. D. Prados, M. Brada, A. Spence, R. J. Hohl, W. Shapiro, M. Glantz, H. Greenberg, R. G. Selker, N. A. Vick, R. Rampling, H. Friedman, P. Phillips, J. Bruner, N. Yue, D. Osoba, S. Zaknoen, V. A. Levin, A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br. J. Cancer 83, 588-593 (2000).
3 S. Bonnet, S. L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C. T. Lee, G. D. Lopaschuk, L. Puttagunta, S. Bonnet, G. Harry, K. Hashimoto, C. J. Porter, M. A. Andrade, B. Thebaud, E. D. Michelakis, Un asse mitocondriale-K+ channel è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita del cancro. Cancer Cell 11, 37-51 (2007).
4 E. D. Michelakis, L. Webster, J. R. Mackey, Il dicloroacetato (DCA) come potenziale terapia mirata al metabolismo per il cancro. Br. J. Cancer 99, 989-994 (2008).
5 P. W. Stacpoole, La farmacologia del dicloroacetato. Metabolism 38, 1124-1144 (1989).
6 O. Warburg, Ueber den Stoffwechsel der Tumoren (Constable, Londra, 1930).
7 S. Weinhouse, L’ipotesi Warburg cinquant’anni dopo. Z. Krebsforsch. Klin. Onkol. Cancer Res. Clin. Oncol. 87, 115-126 (1976).
8 M. G. Vander Heiden, L. C. Cantley, C. B. Thompson, Understanding the Warburg effect: I requisiti metabolici della proliferazione cellulare. Science 324, 1029-1033 (2009).
9 J. G. Pan, T. W. Mak, Metabolic targeting as an anticancer strategy: L’alba di una nuova era? Sci. STKE 2007, pe14 (2007).
10 J. W. Kim, C. V. Dang, Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 66, 8927-8930 (2006).
11 R. A. Gatenby, R. J. Gillies, Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891-899 (2004).
12 J. W. Kim, C. V. Dang, Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 30, 142-150 (2005).
13 L. B. Chen, Potenziale di membrana mitocondriale nelle cellule viventi. Annu. Rev. Cell Biol. 4, 155-181 (1988).
14 G. Kroemer, L. Galluzzi, C. Brenner, Permeabilizzazione della membrana mitocondriale nella morte cellulare. Physiol. Rev. 87, 99-163 (2007).
15. N. Zamzami, G. Kroemer, Il mitocondrio nell’apoptosi: come si apre il vaso di Pandora. Nat. Rev. Mol. Cell Biol. 2, 67-71 (2001).
16 R. A. Cairns, I. Papandreou, P. D. Sutphin, N. C. Denko, Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc. Natl. Acad. Sci. U.S.A. 104, 9445-9450 (2007).
17 W. Cao, S. Yacoub, K. T. Shiverick, K. Namiki, Y. Sakai, S. Porvasnik, C. Urbanek, C. J. Rosser, Il dicloroacetato (DCA) sensibilizza alle radiazioni sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2 in vitro. Prostate 68, 1223-1231 (2008).
18 S. Dhar, S. J. Lippard, Mitaplatin, una potente fusione di cisplatino e del farmaco orfano dicloroacetato. Proc. Natl. Acad. Sci. U.S.A. 106, 22199-22204 (2009).
19 R. C. Sun, M. Fadia, J. E. Dahlstrom, C. R. Parish, P. G. Board, A. C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer growth in vitro and in vivo. Breast Cancer Res. Treat. 120, 253-260 (2010).
20 J. Y. Wong, G. S. Huggins, M. Debidda, N. C. Munshi, I. De Vivo, Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol. Oncol. 109, 394-402 (2008).
21 S. Wang, S. S. Leonard, J. Ye, M. Ding, X. Shi, Il ruolo del radicale idrossile come messaggero nell’attivazione di p53 indotta dal Cr(VI). Am. J. Physiol. Cell Physiol. 279, C868-C875 (2000).
22 C. Huang, Z. Zhang, M. Ding, J. Li, J. Ye, S. S. Leonard, H. M. Shen, L. Butterworth, Y. Lu, M. Costa, Y. Rojanasakul, V. Castranova, V. Vallyathan, X. Shi, Vanadate induce la transattivazione di p53 attraverso il perossido di idrogeno e causa apoptosi. J. Biol. Chem. 275, 32516-32522 (2000).
23 T. Schmid, J. Zhou, R. Köhl, B. Brüne, p300 allevia la repressione trascrizionale di hypoxia-inducible factor-1 (HIF-1) indotta da p53. Biochem. J. 380, 289-295 (2004).
24 M. V. Blagosklonny, W. G. An, L. Y. Romanova, J. Trepel, T. Fojo, L. Neckers, p53 inibisce la trascrizione stimolata dall’hypoxia-inducible factor. J. Biol. Chem. 273, 11995-11998 (1998).
25 J. W. Kim, I. Tchernyshyov, G. L. Semenza, C. V. Dang, HIF-1-mediated expression of pyruvate dehydrogenase kinase: Un interruttore metabolico necessario per l’adattamento cellulare all’ipossia. Cell Metab. 3, 177-185 (2006).
26 G. L. Semenza, D. Artemov, A. Bedi, Z. Bhujwalla, K. Chiles, D. Feldser, E. Laughner, R. Ravi, J. Simons, P. Taghavi, H. Zhong, “The metabolism of tumours”: 70 anni dopo. Novartis Found. Symp. 240, 251-260 (2001).
27 E. K. Weir, J. López-Barneo, K. J. Buckler, S. L. Archer, Acute oxygen-sensing mechanisms. N. Engl. J. Med. 353, 2042-2055 (2005).
28 N. C. Denko, Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705-713 (2008).
29 G. L. Semenza, Targeting HIF-1 per la terapia del cancro. Nat. Rev. Cancer 3, 721-732 (2003).
30 E. D. MacKenzie, M. A. Selak, D. A. Tennant, L. J. Payne, S. Crosby, C. M. Frederiksen, D. G. Watson, E. Gottlieb, Cell-permeating a-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell. Biol. 27, 3282-3289 (2007).
31 M. M. Bowker-Kinley, W. I. Davis, P. Wu, R. A. Harris, K. M. Popov, Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329 (Pt 1), 191-196 (1998).
32 P. W. Stacpoole, D. S. Kerr, C. Barnes, S. T. Bunch, P. R. Carney, E. M. Fennell, N. M. Felitsyn, R. L. Gilmore, M. Greer, G. N. Henderson, A. D. Hutson, R. E. Neiberger, R. G. O’Brien, L. A. Perkins, R. G. Quisling, A. L. Shroads, J. J. Shuster, J. H. Silverstein, D. W. Theriaque, E. Valenstein, Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 117, 1519-1531 (2006).
33 P. W. Stacpoole, A. C. Lorenz, R. G. Thomas, E. M. Harman, Il dicloroacetato nel trattamento dell’acidosi lattica. Ann. Intern. Med. 108, 58-63 (1988).
34 P. W. Stacpoole, T. L. Kurtz, Z. Han, T. Langaee, Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv. Drug Deliv. Rev. 60, 1478-1487 (2008).
35 P. Kaufmann, K. Engelstad, Y. Wei, S. Jhung, M. C. Sano, D. C. Shungu, W. S. Millar, X. Hong, C. L. Gooch, X. Mao, J. M. Pascual, M. Hirano, P. W. Stacpoole, S. DiMauro, D. C. De Vivo, Dichloroacetate causes toxic neuropathy in MELAS: A randomized, controlled clinical trial. Neurology 66, 324-330 (2006).
36 P. W. Stacpoole, L. R. Gilbert, R. E. Neiberger, P. R. Carney, E. Valenstein, D. W. Theriaque, J. J. Shuster, Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121, e1223-e1228 (2008).
37 P. W. Stacpoole, G. N. Henderson, Z. Yan, M. O. James, Farmacologia clinica e tossicologia del dicloroacetato. Environ. Health Perspect. 106 (Suppl. 4), 989-994 (1998).
38 N. Sanai, A. Alvarez-Buylla, M. S. Berger, Neural stem cells and the origin of gliomas. N. Engl. J. Med. 353, 811-822 (2005).
39 F. Zindy, T. Uziel, O. Ayrault, C. Calabrese, M. Valentine, J. E. Rehg, R. J. Gilbertson, C. J. Sherr, M. F. Roussel, Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 67, 2676-2684 (2007).
40 T. Hide, T. Takezaki, H. Nakamura, J. Kuratsu, T. Kondo, Brain tumor stem cells as research and treatment targets. Brain Tumor Pathol. 25, 67-72 (2008).
41 C. Calabrese, H. Poppleton, M. Kocak, T. L. Hogg, C. Fuller, B. Hamner, E. Y. Oh, M. W. Gaber, D. Finklestein, M. Allen, A. Frank, I. T. Bayazitov, S. S. Zakharenko, A. Gajjar, A. Davidoff, R. J. Gilbertson, A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69-82 (2007).
42 R. Pallini, L. Ricci-Vitiani, G. L. Banna, M. Signore, D. Lombardi, M. Todaro, G. Stassi, M. Martini, G. Maira, L. M. Larocca, R. De Maria, Analisi delle cellule staminali tumorali ed esito clinico in pazienti con glioblastoma multiforme. Clin. Cancer Res. 14, 8205-8212 (2008).
43 D. R. Laks, M. Masterman-Smith, K. Visnyei, B. Angenieux, N. M. Orozco, I. Foran, W. H. Yong, H. V. Vinters, L. M. Liau, J. A. Lazareff, P. S. Mischel, T. F. Cloughesy, S. Horvath, H. I. Kornblum, Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 27, 980-987 (2009).
44 R. J. Gilbertson, J. N. Rich, Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 7, 733-736 (2007).
45 J. G. Pastorino, J. B. Hoek, Hexokinase II: The integration of energy metabolism and control of apoptosis. Curr. Med. Chem. 10, 1535-1551 (2003).
46 J. G. Pastorino, J. B. Hoek, N. Shulga, Activation of glycogen synthase kinase 3b disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545-10554 (2005).
47M. Diehn, R. W. Cho, N. A. Lobo, T. Kalisky, M. J. Dorie, A. N. Kulp, D. Qian, J. S. Lam, L. E. Ailles, M. Wong, B. Joshua, M. J. Kaplan, I. Wapnir, F. M. Dirbas, G. Somlo, C. Garberoglio, B. Paz, J. Shen, S. K. Lau, S. R. Quake, J. M. Brown, I. L. Weissman, M. F. Clarke, Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780-783 (2009).
48 H. Yan, D. W. Parsons, G. Jin, R. McLendon, B. A. Rasheed, W. Yuan, I. Kos, I. Batinic-Haberle, S. Jones, G. J. Riggins, H. Friedman, A. Friedman, D. Reardon, J. Herndon, K. W. Kinzler, V. E. Velculescu, B. Vogelstein, D. D. Bigner, IDH1 e IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765-773 (2009).
49 C. B. Thompson, Metabolic enzymes as oncogenes or tumor suppressors. N. Engl. J. Med. 360, 813-815 (2009).
50 L. Dang, D. W. White, S. Gross, B. D. Bennett, M. A. Bittinger, E. M. Driggers, V. R. Fantin, H. G. Jang, S. Jin, M. C. Keenan, K. M. Marks, R. M. Prins, P. S. Ward, K. E. Yen, L. M. Liau, J. D. Rabinowitz, L. C. Cantley, C. B. Thompson, M. G. Vander Heiden, S. M. Su, Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739-744 (2009).
51 Finanziamenti: Questo studio è stato finanziato dalla Hecht Foundation (Vancouver, British Columbia, Canada; E.D.M.), dal Canada Institutes for Health Research e dal Canada Research Chairs Program (E.D.M.) e da donazioni pubbliche al programma DCA (ricevute e gestite dai Reggenti dell’Università di Alberta e dalla Facoltà di Medicina). Gli autori desiderano riconoscere il sostegno dell’Alberta Health Services (D. Gordon, Senior VicePresident, Major Tertiary Hospitals). Contributi degli autori: E.D.M. ha progettato gli studi, ha supervisionato gli studi meccanicistici, ha ottenuto i finanziamenti, ha analizzato i dati e ha scritto il manoscritto. K.C.P. ha co-progettato gli studi, supervisionato tutti gli studi clinici e co-scritto il manoscritto. G.S. e P.D. hanno eseguito tutti gli studi meccanici e hanno redatto il manoscritto. L.W. ha coordinato tutti gli studi, ha contribuito all’acquisizione dei dati, ha analizzato i dati clinici e ha redatto il manoscritto. A.H., E.N., C.M., T.-L.G. e M.S.M. hanno contribuito all’acquisizione e all’analisi dei dati e hanno redatto il manoscritto. J.R.M., D.F. e B.A. hanno co-progettato gli studi clinici, hanno contribuito all’acquisizione dei dati e hanno redatto il manoscritto. Interessi contrastanti: E.D.M. è comproprietario di un brevetto in corso di registrazione sull’uso del DCA come terapia antitumorale. Non c’è stata alcuna commercializzazione attiva o programmata di questo brevetto.