E. D. Michelakis,1 * G. Sutendra,1 P . Dromparis,1 L. Webster,1 A. Haromy,1 E. Niven,2 C. Maguire,2 T.-L. Gammer,1 J. R. Mackey,3 D. Fulton,3 B. Abdulkarim,3 M. S. McMurtry,1 K. C. Petruk4
1Departementgeneeskunde, Universiteit van Alberta, Edmonton, Alberta, Canada T6G 2B7.
2Departmentof Biomedical Engineering and Diagnostic Imaging, Universiteit van Alberta, Edmonton, Alberta, Canada T6G 2B7.
3DepartementOncologie, Universiteit van Alberta, Edmonton, Alberta, Canada T6G 2B7.
4DepartementNeurochirurgie, Universiteit van Alberta, Edmonton, Alberta, Canada T6G 2B7.
*Aan wie de correspondentie moet worden gericht. E-mail: [email protected]
Volume 2 Issue 31 31ra34
Ingediend: 11 november 2009
Aanvaard: 23 april 2010
Gepubliceerd: 12 mei 2010
Vaste tumoren, waaronder de agressieve primaire hersenkanker glioblastoma multiforme, ontwikkelen resistentie tegen celdood, deels als gevolg van een omschakeling van mitochondriale oxidatieve fosforylering naar cytoplasmatische glycolyse. Deze metabole remodellering gaat gepaard met mitochondriale hyperpolarisatie. Wij testten of het kleine molecuul en weesgeneesmiddel dichlooracetaat (DCA) deze kankerspecifieke metabole en mitochondriale remodellering in glioblastoom kan omkeren. Vers geïsoleerde glioblastomen van 49 patiënten vertoonden mitochondriale hyperpolarisatie, die snel werd omgekeerd door DCA. In een afzonderlijk experiment met vijf patiënten die glioblastoom hadden, hebben we prospectief basislijn- en serieel tumorweefsel veiliggesteld, patiëntspecifieke cellijnen van glioblastoom en vermeende glioblastoomstamcellen (CD133+, nestin+ cellen) ontwikkeld, en elke patiënt gedurende 15 maanden oraal met DCA behandeld. DCA depolariseerde de mitochondriën, verhoogde de mitochondriale reactieve zuurstofspecies en induceerde apoptose in GBM-cellen en vermeende GBM-stamcellen, zowel in vitro als in vivo. DCA-therapie remde ook de hypoxia-induceerbare factor-1α, bevorderde de activering van p53 en onderdrukte angiogenese, zowel in vivo als in vitro. De dosisbeperkende toxiciteit was een dosisafhankelijke, reversibele perifere neuropathie, en er was geen hematologische, lever-, nier- of harttoxiciteit. Er waren aanwijzingen voor klinische werkzaamheid bij een dosis die geen perifere neuropathie veroorzaakte en bij serumconcentraties van DCA die voldoende waren om het doelenzym van DCA, pyruvaatdehydrogenase kinase II, te remmen, dat in alle glioblastomen sterk tot expressie kwam. Metabolische modulatie kan een haalbare therapeutische aanpak zijn bij de behandeling van glioblastomen.
INLEIDING
Glioblastoma multiforme (GBM) is een agressieve primaire hersentumor die zeer slecht reageert op goedgekeurde therapieën (1). Chemotherapie met temozolomide (TMZ) plus bestralingstherapie (RT), toegediend na debulkingschirurgie, verhoogt de mediane overleving van 12,1 maanden met RT alleen tot 14,6 maanden (1). De mediane tijd tot progressie van de tumor na RT en TMZ is slechts 6,9 maanden ( 1). Bij terugkerende gliomen zijn de progressievrije overleving en de respons op TMZ veel slechter (2). GBM’s zijn zeer vasculaire tumoren met een opmerkelijke moleculaire en genetische heterogeniteit (1). Een ideale therapie moet de GBM-apoptose verhogen, de moleculaire heterogeniteit overwinnen, angiogenese remmen, en de bloed-hersenbarrière passeren met minimale systemische toxiciteit. Op basis van onze recente bevindingen in diermodellen (3, 4) stelden wij de hypothese dat het weesgeneesmiddel dichlooracetaat (DCA) aan deze criteria voldoet en effectief kan zijn bij de behandeling van GBM bij de mens.
DCA remt het mitochondriale enzym pyruvaatdehydrogenase kinase (PDK) (5). Door PDK te remmen, activeert DCA pyruvaatdehydrogenase (PDH), een poortwachtersenzym dat de stroom koolhydraten (pyruvaat) naar de mitochondriën regelt, waardoor de verhouding tussen glucose-oxidatie en glycolyse toeneemt (3-5). Als pyruvaat in het cytoplasma blijft, kan het de glycolyse voltooien, waarbij melkzuur wordt geproduceerd en 2 mol ATP per glucosemolecuul wordt gegenereerd. Als alternatief kan pyruvaat in verschillende anaplerotische en aminozuurbiosynthesewegen terechtkomen. Bij activering van PDH kan pyruvaat echter worden gedecarboxyleerd tot acetyl-co-enzym A, in de Krebs-cyclus terechtkomen en de glucose-oxidatie in de mitochondriale matrix voltooien, waarbij in aanwezigheid van zuurstof tot 36 mol ATP per glucosemolecuul wordt gegenereerd. Glucose-oxidatie vindt niet plaats als pyruvaat niet in de mitochondriën komt (bijvoorbeeld in zieke mitochondriën of als PDH geremd is) of bij afwezigheid van zuurstof.
Warburg (6) toonde voor het eerst aan dat het metabolisme van kankercellen, zelfs onder normoxie, wordt gekenmerkt door een toename van de verhouding tussen cytoplasmatische glycolyse en mitochondriale glucose-oxidatie. Hoewel het mechanisme van dit “Warburg-effect” onbekend is, en of het etiologisch verband houdt met carcinogenese nog steeds niet bewezen is (7), is er toenemende belangstelling voor het metabolisme als doelwit voor kankertherapieën (8-11). De energetische omschakeling van mitochondriale glucose-oxidatie naar cytoplasmatische glycolyse kan kankercellen een proliferatief voordeel bieden (11). Zo hebben de meeste glycolytische enzymen ook een directe anti-apoptotische werking (12); melkzuur bevordert angiogenese en interstitiële matrixafbraak, wat metastase vergemakkelijkt (11); en een verminderde mitochondriale functie wordt in verband gebracht met remming van mitochondriale apoptose ( 3). GBM heeft een sterk glycolytisch fenotype, en van een aantal van de bij GBM voorkomende moleculaire afwijkingen is bekend dat zij de mitochondriale glucoseoxidatie onderdrukken en de cytoplasmatische glycolyse bevorderen (1), waaronder de activering van de fosfatidylinositol 3-kinase-AKT- of myc-route of de onderdrukking van de p53-route (9, 10).
De mitochondriën van kankercellen zijn hypergepolariseerd ten opzichte van die van niet-kankercellen (3, 13), een toestand die geassocieerd wordt met een onderdrukte mitochondriale functie. Hoewel controversieel [besproken in (14)], hangt de uitstroom van pro-apoptotische mediatoren door de mitochondriale overgangspore (MTP) gedeeltelijk af van het mitochondriale membraanpotentiaal (ΔΨm), en dus kan mitochondriale hyperpolarisatie een apoptose-resistentie toestand markeren (3, 15). Wij hebben aangetoond dat deze toestand in kankercellen kan worden omgekeerd door DCA, dat door remming van PDK de toetreding van pyruvaat tot de mitochondriën bevordert, de toename van de verhouding tussen glycolyse en glucose-oxidatie omkeert, de mitochondriale functie verbetert en de mitochondriale hyperpolarisatie omkeert (3). Daarom vermindert DCA de groei van tumoren in vitro en in vivo, zonder de mitochondriën en weefsels buiten de kanker aan te tasten (3, 16-20). De toename van de mitochondriale ademhaling gaat gepaard met een toename van de productie van mitochondriale reactieve zuurstofsoorten (mROS), voornamelijk superoxide. Superoxide kan worden gedesmuteerd tot H2O2, een relatief stabiel mROS dat andere cellulaire structuren buiten de mitochondriën kan bereiken. H2O2 kan bijvoorbeeld redoxgevoelige spanningsafhankelijke kaliumkanalen in de plasmamembraan activeren en, althans in sommige weefsels, een afname van het intracellulaire calcium bevorderen (3, 4). Andere redoxgevoelige doelen zijn onder meer p53, dat wordt geactiveerd bij oxidatie (21, 22). De p53-as is geremd in GBM, hetgeen bijdraagt tot de toegenomen proliferatie van GBM-cellen (1). p53 onderdrukt ook de door hypoxie geïnduceerde factor-1α (HIF-1α)-gestimuleerde transcriptie, omdat p53 en HIF-1α concurreren om dezelfde cotranscriptiefactor (23, 24). HIF-1α verhoogt de expressie van glucosetransporters en verscheidene glycolytische enzymen, alsook van PDK, en ondersteunt zo het glycolytische fenotype (25, 26). Bovendien verhoogt HIF-1α de expressie van vasculaire endotheliale groeifactor (VEGF), wat de angiogenese bevordert. Angiogenese kan ook worden bevorderd door normoxische activering van HIF-1α. Omdat mitochondriën belangrijke zuurstofsensoren zijn (27), kunnen geremde mitochondriën pseudohypoxische redoxsignalen doorgeven en HIF-1α zelfs tijdens normoxie activeren (28-30). Bovendien kan een afname van α-ketoglutaraat, een direct product van de Krebs-cyclus, ook de activering van HIF bevorderen, omdat het een cofactor is voor de prolyl-hydroxyleringsreactie die HIF-1α afbreekt (30).
Wij veronderstelden dat oraal toegediend DCA, dat de bloed-hersenbarrière passeert, de groei van GBM in vivo zou verminderen. Wij stelden verder dat dit zou kunnen gebeuren door (i) het omkeren van het glycolytische fenotype en het normaliseren van ΔΨm, wat mitochondria-afhankelijke apoptose zou bevorderen; (ii) het verhogen van mROS en het bevorderen van p53 activering; en (iii) het verhogen van α-ketoglutaraat concentraties. De laatste twee effecten zouden leiden tot remming van HIF-1α, afname van VEGF en remming van angiogenese.
Resultaten
Effecten van DCA op mitochondriën van 49 vers geïsoleerde GBM-tumoren
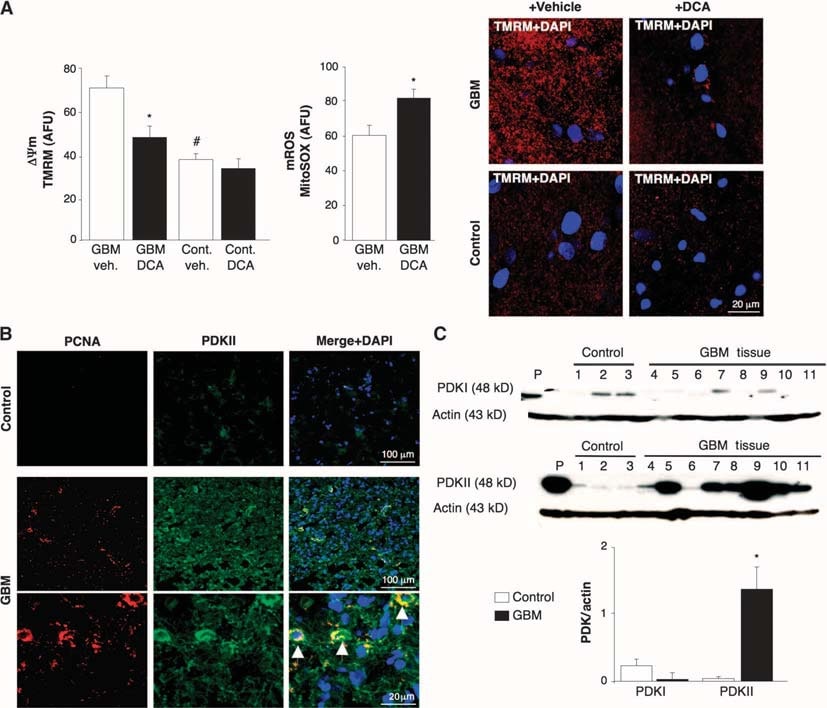
Om te bepalen of humane GBM een doelwit zouden kunnen zijn voor metabole therapie met DCA, bestudeerden wij 49 vers geëxcideerde opeenvolgende primaire GBM’s (60% man, 48 ± 11 jaar). In aanvulling op de klinische en neuropathologische rapporten, bevestigden wij de GBM identiteit met immunohistochemie, die expressie van gliaal fibrillair zuur proteïne (GFAP) liet zien, maar geen bIII-tubuline of oligodendrocyten markers (fig. S1). ΔΨm was verhoogd in de vers geïsoleerde GBM’s vergeleken met niet-kanker hersenweefsel verkregen bij epilepsiechirurgie (n = 3) (fig. 1A). DCA, maar niet het voertuig (normale zoutoplossing), veroorzaakte mitochondriale depolarisatie in GBM, maar niet in normaal hersenweefsel. DCA verhoogde ook GBM mROS (Fig. 1A). Dit suggereert dat de metabole en mitochondriale remodeling in GBM gedeeltelijk omkeerbaar is en dat deze remodeling ten minste gedeeltelijk wordt gereguleerd door PDK. De respons op DCA is consistent met een hogere concentratie van PDKII [de meest alomtegenwoordig uitgedrukte isovorm en degene met de laagsteKi voor DCA (31)] in GBM dan in hersenweefsel zonder kanker, zoals gezien met immunohistochemie en immunoblots (Fig. 1, B en C). Cellen met de hoogste PDKII-concentraties bevatten ook prolifererend celkernantigeen (PCNA), hetgeen suggereert dat deze cellen prolifereerden (fig. 1B). Deze gegevens, verzameld over een periode van 2 jaar, versterkten de reden om DCA toe te dienen aan patiënten met GBM (4).
Klinische effecten van DCA op vijf patiënten met GBM
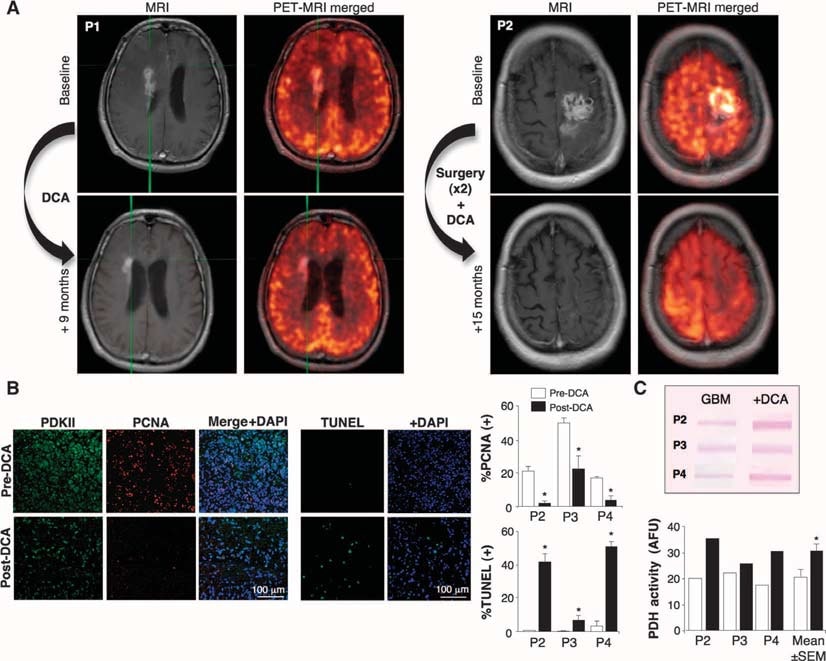
Vervolgens behandelden wij met DCA vijf opeenvolgende patiënten met primair GBM, verwezen uit ons hersenkankerprogramma en van wie weefsel beschikbaar was van de laatste debulkingsoperatie. Drie patiënten (patiënten 1 tot 3) hadden terugkerende GBM met ziekteprogressie na verschillende chemotherapieën (naast de standaardbehandeling met chirurgie, RT en TMZ) en werden geschikt geacht voor palliatieve therapie. Twee andere patiënten (patiënten 4 en 5) waren nieuw gediagnosticeerd, en na de eerste debulkingsoperatie werd DCA toegediend naast de standaardbehandeling met RT en TMZ. Bij patiënt 4 werd een voorbehandeling van 3 maanden met DCA gevolgd door toevoeging van RT en TMZ, terwijl bij patiënt 5 DCA gelijktijdig met RT en TMZ werd gestart, na de debulkingsoperatie. Indien de patiënten opnieuw moesten worden geopereerd of autopsie moesten ondergaan, werd weefsel van de laatste debulkingsoperatie (vóór toediening van DCA) vergeleken met het weefsel van na de DCA-behandeling. Hun klinische informatie is samengevat in tabel S1. DCA wordt al meer dan 30 jaar aan patiënten toegediend, voornamelijk voor de behandeling van aangeboren fouten in het mitochondriaal metabolisme, en er zijn farmacokinetische en farmacodynamische gegevens beschikbaar (5, 32-34). Wij behandelden patiënten met een startdosis van 12,5 mg/kg tweemaal daags oraal gedurende 1 maand, waarna de dosis werd verhoogd tot 25 mg/kg tweemaal daags oraal. Daarna volgden wij een de-escalatieprotocol, waarbij de dosis met 50% werd verlaagd wanneer er dosislimiterende toxiciteit optrad. De patiënten werden tot 15 maanden klinisch gevolgd. Geen van de patiënten had hematologische, lever-, nier- of harttoxiciteit (tabel S1). Perifere neuropathie was de enige duidelijke toxiciteit. De patiënten hadden variabele dosisafhankelijke graden van perifere neuropathie, die omkeerbaar was, hetgeen eerdere studies bevestigt (35-37). Toen de dosis werd verlaagd tot 6,25 mg/kg oraal tweemaal daags, had geen van de patiënten klinisch significante perifere neuropathie (tabel S1). Aanvankelijk is de halfwaardetijd van DCA <1 uur. DCA remt zijn eigen metabolisme en de serumconcentraties stijgen, om uiteindelijk een plateau te bereiken (34). De plasma-dalconcentraties van DCA bij onze patiënten bleven de eerste 2 tot 3 maanden ondetecteerbaar, maar bereikten daarna therapeutische concentraties. Bij een dosis van 6,25 mg/kg oraal tweemaal daags gedurende ten minste 3 maanden bedroegen de dalspiegelconcentraties van DCA 0,44 ± 0,16 mM (gemiddelde ± SD; n = 4) (tabel S1). Deze waarden zijn vergelijkbaar met die gezien bij chronische DCA-behandeling van volwassenen met mitochondriale defecten (34) en liggen in hetzelfde bereik als deKi van DCA voor PDKII (0,2 mM) (31). Patiënten 1, 4 en 5 vertoonden enig bewijs van radiologische regressie op magnetische resonantiebeeldvorming (MRI) (fig. 2A en fig. S2 tot S4). Patiënt 3 had bij aanvang een zeer grote tumor met hersenoedeem (fig. S5), ondanks hoge steroïdendoses, en een lage Karnofsky-score en bleef achteruitgaan. Hij overleed 3 maanden na aanvang van de DCA-therapie aan complicaties door hersenoedeem. Bij patiënt 2 was drainage van een cyste en debulking nodig in maand 11 van de DCA-therapie. Patiënt 4 vertoonde radiologische progressie in maand 3 van de DCA-therapie, waarop verdere debulking werd uitgevoerd en RT plus TMZ werd gegeven naast DCA. Allen, behalve patiënt 3, waren klinisch stabiel in maand 15 van de DCA-therapie en in leven in maand 18 (telefonische follow-up). Verdere klinische details worden beschreven in het aanvullend materiaal.
Effecten van DCA op GBM-tumoren in vivo, primaire GBM-cellijnen, en vermoedelijk GBM-SC afkomstig van de met DCA behandelde patiënten
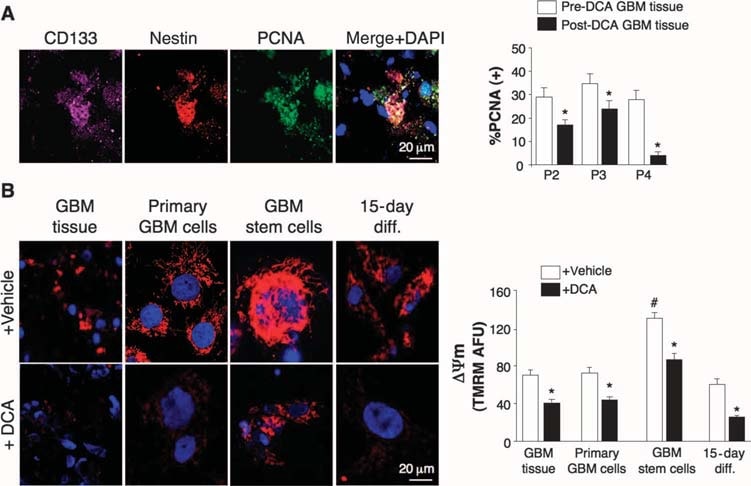
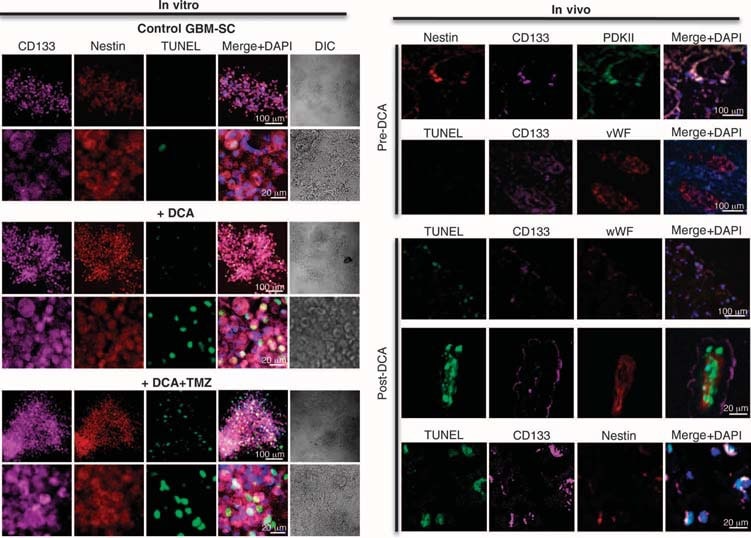
We hebben experimenten uitgevoerd op weefsels afkomstig van deze vijf patiënten en konden vergelijkingen maken in weefsels voor en na DCA-behandeling bij patiënten 2 tot en met 4; we hadden alleen “voor” weefsels bij patiënten 1 en 5. Vergeleken met pre-DCA weefsel vertoonde post-DCA GBM-weefsel van alle drie patiënten een verminderd aantal cellen per volume-eenheid, verminderde proliferatie en toegenomen apoptose (fig. 2B), alsmede een toegenomen enzymatische activiteit van PDH in het weefsel, hetgeen wijst op een effectieve remming van PDK in vivo (fig. 2C). Veronderstelde GBM-kanker stamcellen (GBM-SC’s) kunnen verantwoordelijk zijn voor resistentie na behandeling en recidief van GBM (38-43). Deze cellen worden gekenmerkt als CD133+/nestin+ GBM-SC en vormen niches rond haarvaten (41). In dergelijke vasculaire GBM-SC-eenheden kunnen GBM-SC angiogenese induceren, terwijl hun moleculaire stamcelfenotype in stand wordt gehouden door hun toegankelijkheid tot circulerende groeifactoren (44). Proliferatie van GBM-SC wordt in verband gebracht met een bijzonder slecht klinisch resultaat (42). CD133+ /nestin+ GBM-SC brachten PCNA tot expressie in vivo in alle pre-DCA tumoren, wat erop wijst dat zij zich delen, maar het percentage CD133+ /nestin+ cellen dat PCNA tot expressie bracht was significant afgenomen na DCA-therapie in patiënten 2 tot 4 (Fig. 3A). Gelijktijdige kleuring met een CD133 antilichaam en tetramethyl rhodamine methyl ester (TMRM) toonde aan dat CD133+ cellen de hoogste ΔΨm hadden in vergelijking met naburige niet-GBM-SC in vivo (fig. S6). In tumor-afgeleide primaire cellijnen, ~10% van de cellen uitgedrukt zowel CD133 en nestin, terwijl >90% van de cellen uitgedrukt de volwassen marker GFAP (maar niet bIII-tubuline of oligodendrocyt) (fig. S7), vergelijkbaar met de histopathologie van GBM (fig. S1). Wij isoleerden vermeende GBM-SC uit GBM-tumoren en kweekten ze met de juiste groeifactoren (menselijke fibroblastgroeifactor, 20 ng/ml; menselijke epidermale groeifactor, 20 ng/ml). Deze cellen hadden een zeer hoge expressie van zowel CD133 als nestine, hadden een zeer lage expressie van rijpe gliale markers (fig. S7), en vormden karakteristieke neurosferen (fig. 4 en fig. S7), een onafhankelijke voorspeller van slechte klinische uitkomst (43). Wij hebben DYm gemeten in vers uitgesneden tumoren, in primaire cellijnen en in GBM-SC geïsoleerd uit die tumoren, alsmede in gedifferentieerde cellen afgeleid van GBM-SC (fig. 3B). Het hoogste potentieel werd gevonden in het vermoedelijke GBM-SC. Zowel de primaire als de van GBM-SC afgeleide secundaire GBM-cellen (15 dagen differentiatie) hadden mitochondriale potentialen die vergelijkbaar waren met die van de ouderlijke tumoren. DCA (0,5 mM gedurende 24 uur) verminderde de potentiaal in alle groepen cellen. Hoewel de oorzaak van het verhoogde ΔΨm bij kanker (3, 13) nog niet volledig is vastgesteld, is voorgesteld dat het gedeeltelijk wordt veroorzaakt door een translocatie van hexokinase II (HXKII), een belangrijk glycolytisch enzym, van het cytoplasma naar het buitenste mitochondriale membraan (45, 46). Daar kan HXKII zich binden aan het spanningsafhankelijke anionkanaal (een onderdeel van de MTP) en dit remmen, waardoor de ΔΨm en de apoptotische drempel worden verhoogd. Inhibitie van deze translocatie verlaagt het ΔΨm van de kanker en keert de weerstand tegen apoptose om (45, 46). Onze primaire cellijnen gegenereerd uit pre-DCA tumoren vertoonden een aanhoudende mitochondriale translocatie van HXKII, wat mogelijk de toename van ΔΨm verklaart. HXKII translocatie was niet aanwezig in primaire cellijnen van tumoren na DCA behandeling (fig. S8), verenigbaar met het idee dat DCA onderdrukking van glycolyse en verlaging van ΔΨm induceerde. Evenals in de tumoren was PDKII in hoge concentraties aanwezig in de GBM-cellijnen van patiënten 2 t/m 4, hoewel ook de andere bekende isoenzymen tot expressie kwamen (fig. S9A). Wanneer GBM-SC’s mochten differentiëren tot secundaire GBM-cellijnen, nam het percentage cellen met GBM-SC-markers af tot een waarde die vergelijkbaar was met die van de primaire cellijnen (~10%). Bij differentiatie in aanwezigheid van DCA (0,5 mM) nam het percentage cellen met GBM-SC-markers echter nog verder af tot ~5% (fig. S7). DCA induceerde inderdaad apoptose in GBM-SC in vitro (fig. 4 en fig. S9B) en in primaire cellijnen van GBM (fig. S9C). Apoptose werd verder verhoogd in GBM-SC’s door de combinatie van DCA plus TMZ (fig. 4 en fig. S9B), hetgeen een reden is voor combinatietherapie. GBM-SC apoptose vond ook in vivo plaats in de tumoren na behandeling met DCA, zoals blijkt uit de colocatie van nestine, CD133, en terminal deoxynucleotidyl transferase-gemedieerde deoxyuridine triphosphate nick end labeling (TUNEL) kleuring (Fig. 4 en fig. S9D).
Effecten van DCA op de GBM-SC microvasculaire eenheid en angiogenese in vivo en in vitro
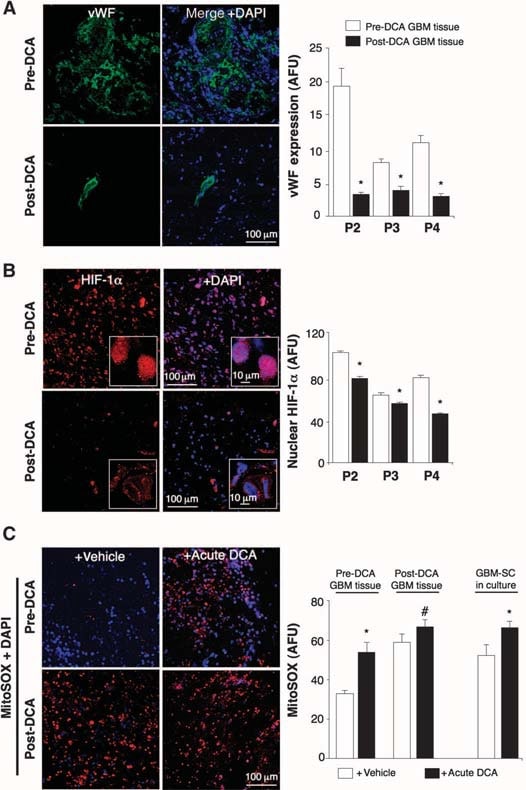
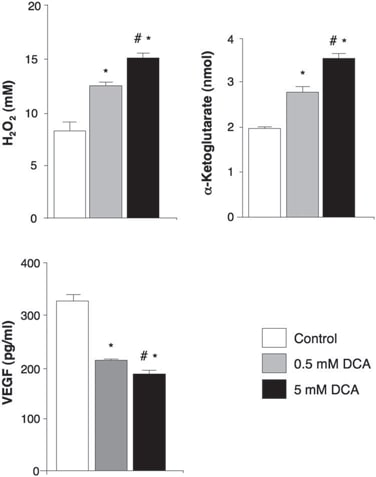
In de onbehandelde, pre-DCA weefsels werden niches van GBM-SC gevonden rond microvaatbedden [von Willebrand factor (vWF) kleuring], zoals gerapporteerd (41). Deze GBM-SC microvasculaire eenheid (44) werd vernietigd door DCA-behandeling omdat, naast GBM-SC, ook apoptose was toegenomen in microvasculaire endotheelcellen (fig. 4 en fig. S9D), hetgeen wijst op een mogelijke remming van angiogenese. Verminderde vWF-kleuring in tumoren na behandeling met DCA wees inderdaad op verminderde vasculariteit (fig. 5A). HIF-1α werd in hoge mate tot expressie gebracht of geactiveerd (nucleaire lokalisatie) in de pre-DCA en geremd in de post-DCA tumorweefsels (fig. 5B). Post-DCA behandelde tumoren van patiënten 2 tot 4 vertoonden een significante toename van mROS in vivo (superoxide gemeten met mitoSOX) in vergelijking met de pre-DCA tumoren van dezelfde patiënten (Fig. 5C). In de pre-DCA tumoren verhoogde acute DCA de mROS tot waarden gezien in de post-DCA behandelde tumoren. In de post-DCA-tumoren daarentegen verhoogde acute DCA de mROS slechts minimaal, wat duidt op een vrijwel maximaal effect in vivo. DCA verhoogde mROS ook in GBM-SC (fig. 5C). Lage ROS-concentraties in kankerstamcellen kunnen wijzen op resistentie tegen apoptose, en therapieën die de ROS in kankerstamcellen verhogen zouden effectiever zijn (47). Hoewel wij mROS (mitochondriaal superoxide) bestudeerden, bestaat er controverse over de vraag of HIF-1α-activatie bij kanker gepaard gaat met een algemene toename of afname van ROS [besproken in (28)]. Met een andere techniek hebben wij ook H2O2 in de hele cel gemeten. DCA verhoogde GBM-cel H2O2 op dosis-afhankelijke wijze (fig. 6). Bovendien verhoogde DCA ook de intracellulaire α-ketoglutaraatconcentratie op dosisafhankelijke wijze (fig. 6). Dit is verenigbaar met de toename van glucose-oxidatie die volgt op PDH-activering (3) , omdat α-ketoglutaraat een product is van de Krebs-cyclus. De Krebs-cyclus produceert de elektronendonoren die de elektronentransportketen tijdens de ademhaling voeden. Dienovereenkomstig verhoogde DCA de ademhaling in GBM-cellen met 44 ± 4% (gemiddelde ± SEM; n = 3; P < 0,05), hetgeen een algemene toename van de mitochondriale activiteit ondersteunt. De toename van H2O2 en α-ketoglutaraat kan de afname van de HIF-1α-activiteit verklaren (fig. 5B), hetgeen ook wordt bevestigd door de dosisafhankelijke afname van de VEGF-productie door GBM-cellen (fig. 6). Deze gegevens zijn consistent met de afname van de angiogenese in vivo (Fig. 4 en 5) en suggereren dat GBM-cellen op paracriene wijze signalen afgeven aan endotheelcellen. Om te bestuderen of DCA rechtstreeks angiogenese kan onderdrukken, gebruikten wij de standaardtechniek van menselijke endotheelcelbuisvorming in Matrigel. Onder fysiologische matige hypoxie veroorzaakte DCA een dosisafhankelijke directe remming van angiogenese in vitro (fig. S10). De tumoren van patiënten 2 tot 4 na behandeling met DCA vertoonden een verhoogde activiteit van het mROS-gevoelige p53 (nucleaire translocatie), hetgeen ook werd bevestigd door de verhoogde activiteit en overvloed van zijn downstream-doelwit p21 (fig. S11). Deze effecten op p53 of p21 kunnen ook de afname van HIF-gestuurde transcriptie verklaren en zijn consistent met de antiproliferatieve, naast de pro-apoptotische, effecten van DCA in GBM (fig. S12).
DISCUSSIE
De metabole modulator DCA oefent antikankereffecten uit in gekweekte cellen en knaagdieren (3, 16-20). Wij hebben nu aangetoond dat DCA kan worden gebruikt bij patiënten die lijden aan GBM. De behandeling met DCA ging bij sommige GBM-patiënten gepaard met langdurige radiologische stabilisatie of tumorregressie en vertoonde over het algemeen een goed veiligheidsprofiel. Dit vroege, first-in-human rapport vormt een argument voor uitgebreide studies met deze generieke kleine molecule bij patiënten met GBM. Onze resultaten wijzen erop dat GBM een goede kandidaat is voor metabole interventie. Het doelwit van DCA, PDKII, komt sterk tot expressie in GBM-tumoren en -cellijnen, en DCA kan de activiteit ervan in vivo remmen. GBM wordt gekenmerkt door mitochondriale hyperpolarisatie, in overeenstemming met de metabole remodeling (Warburg-effect) en de daarmee samenhangende apoptose-resistentie die kenmerkend zijn voor GBM en de meeste vaste tumoren (11). Mutaties in de genen voor cytoplasmatische en mitochondriale isocitraatdehydrogenasen zijn beschreven in GBM’s die voortkomen uit laaggradige gliomen (secundaire GBM’s) (48), maar het mechanisme waardoor deze mutaties verband houden met carcinogenese blijft onduidelijk (49, 50). Onze patiënten hadden primaire GBM’s en de mitochondriale remodeling was ten minste gedeeltelijk omkeerbaar met DCA, hetgeen suggereert dat deze niet het gevolg was van onomkeerbare disfunctie. Voorts tonen wij aan dat vermeende GBM-SC dezelfde metabole en mitochondriale remodellering kunnen ondergaan, maar in versterkte mate, omdat GBM-SC zowel in vivo als in vitro de meest gehyperpolariseerde mitochondriën hadden. Omkering van deze mitochondriale remodellering door DCA induceerde apoptose in GBM-SC, zowel in vitro als in vivo. Hoewel de omvang van de apoptose-inductie door DCA niet groot is (in vergelijking met cytotoxische middelen), is zij relatief selectief, waarbij niet-kankercellen worden ontzien (3), en kan zij, omdat het GBM-SC betreft, resulteren in een duurzamer klinisch effect. Patiënt 3 overleed en patiënt 4 had een recidief binnen de eerste 3 maanden toen DCA geen duurzame therapeutische waarden had bereikt (zoals blijkt uit het feit dat de dalspiegelconcentraties niet detecteerbaar waren). Patiënten kunnen dus onderbehandeld zijn bij de initiële toediening van DCA en het risico lopen op initiële ziekteprogressie. Door de weerstand tegen mitochondriale apoptose te verminderen, kan DCA mogelijk de effecten van standaardtherapieën versterken. De effecten van DCA plus TMZ op de apoptose van GBMSC zouden inderdaad ten grondslag kunnen liggen aan de voordelen op lange termijn van patiënt 5. De effecten van DCA op kankercellen worden nagebootst door PDKII small interfering RNA, en DCA heeft geen andere effecten dan die welke worden waargenomen na uitschakeling van PDKII (3). Dit suggereert dat het mechanisme van de antikankereffecten van DCA de remming is van PDKII, een enzym dat in verhoogde concentraties voorkomt in GBM. Door remming van PDKII normaliseert DCA de verhoogde verhouding tussen glycolyse en glucose-oxidatie in kankercellen, waardoor de mitochondriale functie toeneemt (3). Dit mechanisme wordt ondersteund door onze huidige bevindingen dat DCA zowel de GBM-celademhaling als de a-ketoglutaraatconcentratie verhoogt. In tegenstelling tot de gecontroleerde omstandigheden van de celkweek, kunnen echter nog andere mechanismen bijdragen tot de effecten van DCA op het metabolisme en de apoptose van kankercellen in vivo. Naast de eerder beschreven effecten van DCA op kankercellen (3) zijn onze gegevens over HXKII, mROS, HIF-1α, p53 en p21 consistent met de door DCA geïnduceerde remming van angiogenese, inductie van apoptose en onderdrukking van proliferatie in zowel GBM als GBM-SC, zoals samengevat in het aanvullend materiaal en fig. S12. DCA had geen andere duidelijke toxiciteit dan een eerder beschreven dosisafhankelijke, omkeerbare nietemyeliniserende neurotoxiciteit (32, 34) die minimaal of afwezig was bij de orale dosis van 6,25 mg/kg, tweemaal daags. Deze dosis vertoonde biologische en klinische werkzaamheid en bereikte plasmaconcentraties op waarden die vereist zijn voor PDK-remming (31). Met het kleine aantal behandelde patiënten in onze studie kunnen geen harde conclusies worden getrokken over DCA als therapie voor GBM. Ons werk ondersteunt de noodzaak van verdere studies met DCA in GBM, met de nadruk op protocollen voor combinatietherapie. GBM kan ook kwetsbaar zijn voor andere geneesmiddelen in de opkomende familie van metabole modulatoren, wat wijst op een nieuwe aanpak in de behandeling van deze ongeneeslijke kanker.
AANVULLEND MATERIAAL
www.sciencetranslationalmedicine.org/cgi/content/full/2/31/31ra34/DC1
Materialen en methoden
Resultaten
Discussie
Fig. S1. Moleculaire karakterisering van GBM-tumoren.
Fig. S2. Evolutie van de tumorrespons bij patiënt 1.
Fig. S3. Evolutie van de tumorrespons bij patiënt 4.
Fig. S4. Evolutie van de tumorrespons bij patiënt 5.
Fig. S5. GBM MRI van patiënt 3.
Fig. S6. Mitochondriaal membraan potentieel in GBM-SC van vers uitgesneden GBM weefsel.
Fig. S7. Karakterisering van primaire GBM-cellen en GBM-SC.
Fig. S8. HXKII in GBM-cellen afkomstig van patiënten voor en na chronische DCA-behandeling.
Fig. S9 Effecten van DCA-therapie op GBM-SC en vasculaire apoptose.
Fig. S10. Effecten van DCA op angiogenese in vitro.
Fig. S11. Effecten van DCA behandeling op p53 en p21 activiteit in vivo.
Fig. S12. Een voorgesteld uitgebreid mechanisme voor de antikankereffecten van DCA in GBM (zie
Aanvullende discussie).
Tabel S1. Laboratorium- en klinische parameters van vijf GBM-patiënten voor en na DCA-behandeling.
Referenties
REFERENTIES
1 1. P. Y. Wen, S. Kesari, Kwaadaardige gliomen bij volwassenen. N. Engl. J. Med. 359, 492-507 (2008).
2 W. K. Yung, R. E. Albright, J. Olson, R. Fredericks, K. Fink, M. D. Prados, M. Brada, A. Spence, R. J. Hohl, W. Shapiro, M. Glantz, H. Greenberg, R. G. Selker, N. A. Vick, R. Rampling, H. Friedman, P. Phillips, J. Bruner, N. Yue, D. Osoba, S. Zaknoen, V. A. Levin, A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br. J. Cancer 83, 588-593 (2000).
3 S. Bonnet, S. L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C. T. Lee, G. D. Lopaschuk, L. Puttagunta, S. Bonnet, G. Harry, K. Hashimoto, C. J. Porter, M. A. Andrade, B. Thebaud, E. D. Michelakis, A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 11, 37-51 (2007).
4 E. D. Michelakis, L. Webster, J. R. Mackey, Dichloroacetate (DCA) as a potential metabolictargeting therapy for cancer. Br. J. Cancer 99, 989-994 (2008).
5 P. W. Stacpoole, De farmacologie van dichlooracetaat. Metabolisme 38, 1124-1144 (1989).
6 O. Warburg, Ueber den Stoffwechsel der Tumoren (Constable, Londen, 1930).
7 S. Weinhouse, De Warburg-hypothese vijftig jaar later. Z. Krebsforsch. Klin. Onkol. Cancer Res. Clin. Oncol. 87, 115-126 (1976).
8 M. G. Vander Heiden, L. C. Cantley, C. B. Thompson, Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 324, 1029-1033 (2009).
9 J. G. Pan, T. W. Mak, Metabolic targeting as an anticancer strategy: Dawn of a new era? Sci. STKE 2007, pe14 (2007).
10 J. W. Kim, C. V. Dang, Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 66, 8927-8930 (2006).
11 R. A. Gatenby, R. J. Gillies, Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891-899 (2004).
12 J. W. Kim, C. V. Dang, Veelzijdige rollen van glycolytische enzymen. Trends Biochem. Sci. 30, 142-150 (2005).
13 L. B. Chen, Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 4, 155-181 (1988).
14 G. Kroemer, L. Galluzzi, C. Brenner, Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99-163 (2007).
15. N. Zamzami, G. Kroemer, The mitochondrion in apoptosis: How Pandora’s box opens. Nat. Rev. Mol. Cell Biol. 2, 67-71 (2001).
16 R. A. Cairns, I. Papandreou, P. D. Sutphin, N. C. Denko, Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc. Natl. Acad. Sci. U.S.A. 104, 9445-9450 (2007).
17 W. Cao, S. Yacoub, K. T. Shiverick, K. Namiki, Y. Sakai, S. Porvasnik, C. Urbanek, C. J. Rosser, Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostaat 68, 1223-1231 (2008).
18 S. Dhar, S. J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. U.S.A. 106, 22199-22204 (2009).
19 R. C. Sun, M. Fadia, J. E. Dahlstrom, C. R. Parish, P. G. Board, A. C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 120, 253-260 (2010).
20 J. Y. Wong, G. S. Huggins, M. Debidda, N. C. Munshi, I. De Vivo, Dichloroacetate induceert apoptose in endometriumkankercellen. Gynecol. Oncol. 109, 394-402 (2008).
21 S. Wang, S. S. Leonard, J. Ye, M. Ding, X. Shi, The role of hydroxyl radical as a messenger in Cr(VI)-induced p53 activation. Am. J. Physiol. Cell Physiol. 279, C868-C875 (2000).
22 C. Huang, Z. Zhang, M. Ding, J. Li, J. Ye, S. S. Leonard, H. M. Shen, L. Butterworth, Y. Lu, M. Costa, Y. Rojanasakul, V. Castranova, V. Vallyathan, X. Shi, Vanadate induceert p53-transactivatie via waterstofperoxide en veroorzaakt apoptose. J. Biol. Chem. 275, 32516-32522 (2000).
23 T. Schmid, J. Zhou, R. Köhl, B. Brüne, p300 verlicht p53-geïnduceerde transcriptionele repressie van hypoxia-induceerbare factor-1 (HIF-1). Biochem. J. 380, 289-295 (2004).
24 M. V. Blagosklonny, W. G. An, L. Y. Romanova, J. Trepel, T. Fojo, L. Neckers, p53 remt hypoxia-inducible factor-gestimuleerde transcriptie. J. Biol. Chem. 273, 11995-11998 (1998).
25 J. W. Kim, I. Tchernyshyov, G. L. Semenza, C. V. Dang, HIF-1-gemedieerde expressie van pyruvaat dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177-185 (2006).
26 G. L. Semenza, D. Artemov, A. Bedi, Z. Bhujwalla, K. Chiles, D. Feldser, E. Laughner, R. Ravi, J. Simons, P. Taghavi, H. Zhong, ‘Het metabolisme van tumoren’: 70 jaar later. Novartis Found. Symp. 240, 251-260 (2001).
27 E. K. Weir, J. López-Barneo, K. J. Buckler, S. L. Archer, Acute oxygen-sensing mechanisms. N. Engl. J. Med. 353, 2042-2055 (2005).
28 N. C. Denko, Hypoxia, HIF1 and glucose metabolism in the solid tumor. Nat. Rev. Cancer 8, 705-713 (2008).
29 G. L. Semenza, Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 3, 721-732 (2003).
30 E. D. MacKenzie, M. A. Selak, D. A. Tennant, L. J. Payne, S. Crosby, C. M. Frederiksen, D. G. Watson, E. Gottlieb, Cell-permeating a-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell. Biol. 27, 3282-3289 (2007).
31 M. M. Bowker-Kinley, W. I. Davis, P. Wu, R. A. Harris, K. M. Popov, Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329 (Pt 1), 191-196 (1998).
32 P. W. Stacpoole, D. S. Kerr, C. Barnes, S. T. Bunch, P. R. Carney, E. M. Fennell, N. M. Felitsyn, R. L. Gilmore, M. Greer, G. N. Henderson, A. D. Hutson, R. E. Neiberger, R. G. O’Brien, L. A. Perkins, R. G. Quisling, A. L. Shroads, J. J. Shuster, J. H. Silverstein, D. W. Theriaque, E. Valenstein, Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 117, 1519-1531 (2006).
33 P. W. Stacpoole, A. C. Lorenz, R. G. Thomas, E. M. Harman, Dichloroacetate in the treatment of lactic acidosis. Ann. Intern. Med. 108, 58-63 (1988).
34 P. W. Stacpoole, T. L. Kurtz, Z. Han, T. Langaee, Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv. Drug Deliv. Rev. 60, 1478-1487 (2008).
35 P. Kaufmann, K. Engelstad, Y. Wei, S. Jhung, M. C. Sano, D. C. Shungu, W. S. Millar, X. Hong, C. L. Gooch, X. Mao, J. M. Pascual, M. Hirano, P. W. Stacpoole, S. DiMauro, D. C. De Vivo, Dichloroacetate causes toxic neuropathy in MELAS: A randomized, controlled clinical trial. Neurologie 66, 324-330 (2006).
36 P. W. Stacpoole, L. R. Gilbert, R. E. Neiberger, P. R. Carney, E. Valenstein, D. W. Theriaque, J. J. Shuster, Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121, e1223-e1228 (2008).
37 P. W. Stacpoole, G. N. Henderson, Z. Yan, M. O. James, Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 106 (Suppl. 4), 989-994 (1998).
38 N. Sanai, A. Alvarez-Buylla, M. S. Berger, Neural stem cells and the origin of gliomas. N. Engl. J. Med. 353, 811-822 (2005).
39 F. Zindy, T. Uziel, O. Ayrault, C. Calabrese, M. Valentine, J. E. Rehg, R. J. Gilbertson, C. J. Sherr, M. F. Roussel, Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 67, 2676-2684 (2007).
40 T. Hide, T. Takezaki, H. Nakamura, J. Kuratsu, T. Kondo, Brain tumor stem cells as research and treatment targets. Brain Tumor Pathol. 25, 67-72 (2008).
41 C. Calabrese, H. Poppleton, M. Kocak, T. L. Hogg, C. Fuller, B. Hamner, E. Y. Oh, M. W. Gaber, D. Finklestein, M. Allen, A. Frank, I. T. Bayazitov, S. S. Zakharenko, A. Gajjar, A. Davidoff, R. J. Gilbertson, A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69-82 (2007).
42 R. Pallini, L. Ricci-Vitiani, G. L. Banna, M. Signore, D. Lombardi, M. Todaro, G. Stassi, M. Martini, G. Maira, L. M. Larocca, R. De Maria, Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin. Cancer Res. 14, 8205-8212 (2008).
43 D. R. Laks, M. Masterman-Smith, K. Visnyei, B. Angenieux, N. M. Orozco, I. Foran, W. H. Yong, H. V. Vinters, L. M. Liau, J. A. Lazareff, P. S. Mischel, T. F. Cloughesy, S. Horvath, H. I. Kornblum, Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 27, 980-987 (2009).
44 R. J. Gilbertson, J. N. Rich, Making a tumor’s bed: Glioblastoma stamcellen en de vasculaire niche. Nat. Rev. Cancer 7, 733-736 (2007).
45 J. G. Pastorino, J. B. Hoek, Hexokinase II: The integration of energy metabolism and control of apoptosis. Curr. Med. Chem. 10, 1535-1551 (2003).
46 J. G. Pastorino, J. B. Hoek, N. Shulga, Activation of glycogen synthase kinase 3b disrupts the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545-10554 (2005).
47M. Diehn, R. W. Cho, N. A. Lobo, T. Kalisky, M. J. Dorie, A. N. Kulp, D. Qian, J. S. Lam, L. E. Ailles, M. Wong, B. Joshua, M. J. Kaplan, I. Wapnir, F. M. Dirbas, G. Somlo, C. Garberoglio, B. Paz, J. Shen, S. K. Lau, S. R. Quake, J. M. Brown, I. L. Weissman, M. F. Clarke, Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780-783 (2009).
48 H. Yan, D. W. Parsons, G. Jin, R. McLendon, B. A. Rasheed, W. Yuan, I. Kos, I. Batinic-Haberle, S. Jones, G. J. Riggins, H. Friedman, A. Friedman, D. Reardon, J. Herndon, K. W. Kinzler, V. E. Velculescu, B. Vogelstein, D. D. Bigner, IDH1- en IDH2-mutaties in gliomen. N. Engl. J. Med. 360, 765-773 (2009).
49 C. B. Thompson, Metabole enzymen als oncogenen of tumorsuppressoren. N. Engl. J. Med. 360, 813-815 (2009).
50 L. Dang, D. W. White, S. Gross, B. D. Bennett, M. A. Bittinger, E. M. Driggers, V. R. Fantin, H. G. Jang, S. Jin, M. C. Keenan, K. M. Marks, R. M. Prins, P. S. Ward, K. E. Yen, L. M. Liau, J. D. Rabinowitz, L. C. Cantley, C. B. Thompson, M. G. Vander Heiden, S. M. Su, Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739-744 (2009).
51 Financiering: Deze studie werd gefinancierd door de Hecht Foundation (Vancouver, British Columbia, Canada; E.D.M.), de Canada Institutes for Health Research en het Canada Research Chairs Program (E.D.M.), en door publieke donaties aan het DCA-programma (ontvangen en beheerd door de Regents van de Universiteit van Alberta en de Faculteit Geneeskunde). De auteurs erkennen de steun van de Alberta Health Services (D. Gordon, Senior VicePresident, Major Tertiary Hospitals). Bijdragen van de auteurs: E.D.M. ontwierp de studies, hield toezicht op de mechanistische studies, zorgde voor de financiering, analyseerde de gegevens en schreef het manuscript. K.C.P. ontwierp de studies mee, hield toezicht op alle klinische studies en schreef mee aan het manuscript. G.S. en P.D. voerden alle mechanistische studies uit en redigeerden het manuscript. L.W. coördineerde alle studies, droeg bij aan het verzamelen van gegevens, analyseerde de klinische gegevens en redigeerde het manuscript. A.H., E.N., C.M., T.-L.G., en M.S.M. droegen bij aan de gegevensverzameling en -analyse en redigeerden het manuscript. J.R.M., D.F., en B.A. ontwierpen mee de klinische studies, droegen bij tot het verzamelen van gegevens en redigeerden het manuscript. Concurrerende belangen: E.D.M. is mede-eigenaar van een hangend gebruiksoctrooi op het gebruik van DCA als kankertherapie. Er is geen actieve of geplande commercialisering van dit octrooi.