E. D. Michelakis,1 * G. Sutendra,1 P . Dromparis,1 L. Webster,1 A. Haromy,1 E. Niven,2 C. Maguire,2 T.-L. Gammer,1 J. R. Mackey,3 D. Fulton,3 B. Abdulkarim,3 M. S. McMurtry,1 K. C. Petruk4
1Abteilungfür Medizin, Universität von Alberta, Edmonton, Alberta, Kanada T6G 2B7.
2Abteilungfür Biomedizinische Technik und diagnostische Bildgebung, Universität von Alberta, Edmonton, Alberta, Kanada T6G 2B7.
3Abteilungfür Onkologie, Universität von Alberta, Edmonton, Alberta, Kanada T6G 2B7.
4Abteilungfür Neurochirurgie, Universität von Alberta, Edmonton, Alberta, Kanada T6G 2B7.
*An wen die Korrespondenz zu richten ist. E-Mail: [email protected]
Band 2 Ausgabe 31 31ra34
Eingereicht: 11. November 2009Angenommen
: 23. April 2010Veröffentlicht
: 12. Mai 2010
Solide Tumore, einschließlich des aggressiven primären Hirntumors Glioblastoma multiforme, entwickeln eine Resistenz gegenüber dem Zelltod, die zum Teil auf eine Umstellung von mitochondrialer oxidativer Phosphorylierung auf zytoplasmatische Glykolyse zurückzuführen ist. Diese Umstellung des Stoffwechsels wird von einer mitochondrialen Hyperpolarisation begleitet. Wir haben getestet, ob das niedermolekulare Orphan-Medikament Dichloracetat (DCA) diese krebsspezifische metabolische und mitochondriale Umstrukturierung in Glioblastomen rückgängig machen kann. Frisch isolierte Glioblastome von 49 Patienten wiesen eine mitochondriale Hyperpolarisierung auf, die durch DCA rasch rückgängig gemacht wurde. In einem separaten Experiment mit fünf Glioblastom-Patienten sicherten wir prospektiv das Ausgangsmaterial und serielles Tumorgewebe, entwickelten patientenspezifische Zelllinien von Glioblastomen und mutmaßlichen Glioblastom-Stammzellen (CD133+ , nestin+ Zellen) und behandelten jeden Patienten bis zu 15 Monate lang mit oralem DCA. DCA depolarisierte die Mitochondrien, erhöhte die reaktiven Sauerstoffspezies in den Mitochondrien und induzierte Apoptose in GBM-Zellen sowie in mutmaßlichen GBM-Stammzellen, sowohl in vitro als auch in vivo. Die DCA-Therapie hemmte auch den Hypoxie-induzierbaren Faktor-1α, förderte die Aktivierung von p53 und unterdrückte die Angiogenese sowohl in vivo als auch in vitro. Die dosislimitierende Toxizität war eine dosisabhängige, reversible periphere Neuropathie, und es gab keine hämatologische, hepatische, renale oder kardiale Toxizität. Hinweise auf eine klinische Wirksamkeit lagen bei einer Dosis vor, die keine periphere Neuropathie verursachte, und bei DCA-Serumkonzentrationen, die ausreichten, um das Zielenzym von DCA, die Pyruvat-Dehydrogenase-Kinase II, zu hemmen, die in allen Glioblastomen stark exprimiert war. Die Modulation des Stoffwechsels könnte ein praktikabler therapeutischer Ansatz für die Behandlung von Glioblastomen sein.
EINLEITUNG
Das Glioblastoma multiforme (GBM) ist ein aggressiver primärer Hirntumor, der auf zugelassene Therapien extrem schlecht anspricht (1). Eine Chemotherapie mit Temozolomid (TMZ) plus Strahlentherapie (RT), die nach einer chirurgischen Entfernung des Tumors verabreicht wird, verlängert die mediane Überlebenszeit von 12,1 Monaten mit RT allein auf 14,6 Monate (1). Die mediane Zeit bis zum Fortschreiten des Tumors nach RT und TMZ beträgt nur 6,9 Monate (1). Bei rezidivierenden Gliomen sind das progressionsfreie Überleben und das Ansprechen auf TMZ deutlich schlechter (2). GBMs sind sehr vaskuläre Tumoren mit bemerkenswerter molekularer und genetischer Heterogenität (1). Eine ideale Therapie sollte die GBM-Apoptose erhöhen, die molekulare Heterogenität überwinden, die Angiogenese hemmen und die Blut-Hirn-Schranke bei minimaler systemischer Toxizität überwinden. Auf der Grundlage unserer jüngsten Erkenntnisse in Tiermodellen (3, 4)haben wir die Hypothese aufgestellt, dass das kleine Orphan-Molekül Dichloracetat (DCA) diese Kriterien erfüllt und bei der Behandlung von GBM beim Menschen wirksam sein könnte.
DCA hemmt das mitochondriale Enzym Pyruvat-Dehydrogenase-Kinase (PDK) (5). Durch die Hemmung der PDK aktiviert DCA die Pyruvat-Dehydrogenase (PDH), ein Gatekeeper-Enzym, das den Fluss von Kohlenhydraten (Pyruvat) in die Mitochondrien reguliert und das Verhältnis von Glukoseoxidation zu Glykolyse erhöht (3-5). Wenn Pyruvat im Zytoplasma verbleibt, kann es die Glykolyse abschließen, wobei Milchsäure entsteht und 2 Mol ATP pro Glukosemolekül erzeugt werden. Alternativ kann Pyruvat in verschiedene anaplerotische und Aminosäure-Biosynthesewege gelangen. Bei Aktivierung der PDH kann Pyruvat jedoch zu Acetyl-Coenzym A decarboxyliert werden, in den Krebs-Zyklus eintreten und die Glukoseoxidation in der mitochondrialen Matrix abschließen, wobei in Gegenwart von Sauerstoff bis zu 36 Mol ATP pro Glukosemolekül erzeugt werden. Die Glukoseoxidation findet nicht statt, wenn kein Pyruvat in die Mitochondrien gelangt (z. B. in kranken Mitochondrien oder wenn die PDH gehemmt ist) oder wenn kein Sauerstoff vorhanden ist.
Warburg (6) zeigte erstmals, dass der Stoffwechsel von Krebszellen auch unter Normoxie durch ein erhöhtes Verhältnis von zytoplasmatischer Glykolyse zu mitochondrialer Glukoseoxidation gekennzeichnet ist. Obwohl der Mechanismus dieses „Warburg-Effekts“ unbekannt ist und ein ätiologischer Zusammenhang mit der Krebsentstehung nicht bewiesen ist (7), wächst das Interesse am Stoffwechsel als Ziel für Krebstherapien (8-11). Die energetische Umstellung von mitochondrialer Glukoseoxidation auf zytoplasmatische Glykolyse könnte Krebszellen einen Proliferationsvorteil verschaffen (11). So haben die meisten glykolytischen Enzyme auch direkte antiapoptotische Wirkungen (12); Milchsäure fördert die Angiogenese und den Abbau der interstitiellen Matrix und erleichtert so die Metastasierung (11); und eine verminderte Mitochondrienfunktion wird mit einer Hemmung der mitochondrienabhängigen Apoptose in Verbindung gebracht (3). GBM weist einen ausgeprägten glykolytischen Phänotyp auf, und es ist bekannt, dass eine Reihe von molekularen Anomalien, die bei GBM auftreten, die mitochondriale Glukoseoxidation unterdrücken und die zytoplasmatische Glykolyse fördern (1), einschließlich der Aktivierung des Phosphatidylinositol-3-Kinase-AKT- oder Myc-Wegs oder der Unterdrückung des p53-Wegs (9, 10).
Die Mitochondrien von Krebszellen sind im Vergleich zu denen von Nicht-Krebszellen hyperpolarisiert (3, 13), ein Zustand, der mit einer unterdrückten Mitochondrienfunktion einhergeht. Obwohl dies umstritten ist [siehe (14)], hängt der Ausfluss proapoptotischer Mediatoren durch die mitochondriale Übergangspore (MTP) zum Teil vom mitochondrialen Membranpotenzial (ΔΨm) ab, so dass die mitochondriale Hyperpolarisation einen Zustand der Apoptoseresistenz kennzeichnen kann (3, 15). Wir haben gezeigt, dass dieser Zustand in Krebszellen durch DCA umgekehrt werden kann, das durch Hemmung der PDK den Eintritt von Pyruvat in die Mitochondrien fördert, wodurch der Anstieg des Verhältnisses zwischen Glykolyse und Glukoseoxidation umgekehrt wird, die Funktion der Mitochondrien verbessert und die mitochondriale Hyperpolarisation umgekehrt wird (3). DCA verringert daher das Tumorwachstum in vitro und in vivo, ohne die Mitochondrien und das Gewebe von Nicht-Krebszellen zu beeinträchtigen (3, 16-20). Der Anstieg der mitochondrialen Atmung geht mit einer erhöhten Produktion von reaktiven Sauerstoffspezies (mROS) in den Mitochondrien, vor allem Superoxid, einher. Superoxid kann zuH2O2dismutiert werden, einer relativ stabilen mROS, die über die Mitochondrien hinaus auch andere zelluläre Strukturen erreichen kann. So kannH2O2beispielsweise redoxempfindliche spannungsabhängige Kaliumkanäle in der Plasmamembran aktivieren und, zumindest in einigen Geweben, einen Rückgang des intrazellulären Kalziums bewirken (3, 4). Zu den weiteren redoxsensiblen Zielen kann p53 gehören, das bei Oxidation aktiviert wird (21, 22). Die p53-Achse ist bei GBM gehemmt, was zur erhöhten Proliferation von GBM-Zellen beiträgt (1). p53 unterdrückt auch die Hypoxie-induzierbare Faktor-1α (HIF-1α)-stimulierte Transkription, da p53 und HIF-1α um denselben Kotranskriptionsfaktor konkurrieren (23, 24). HIF-1α erhöht die Expression von Glukosetransportern und mehreren glykolytischen Enzymen sowie von PDK, wodurch der glykolytische Phänotyp aufrechterhalten wird (25, 26). Darüber hinaus erhöht HIF-1α die Expression des vaskulären endothelialen Wachstumsfaktors (VEGF) und fördert so die Angiogenese. Die Angiogenese kann auch durch normoxische HIF-1α-Aktivierung gefördert werden. Da Mitochondrien wichtige Sauerstoffsensoren sind (27), können gehemmte Mitochondrien pseudohypoxische Redoxsignale übermitteln und HIF-1α auch bei Normoxie aktivieren (28-30). Darüber hinaus kann eine Abnahme von α-Ketoglutarat, einem direkten Produkt des Krebs-Zyklus, ebenfalls die HIF-Aktivierung fördern, da es ein Kofaktor für die Prolylhydroxylierungsreaktion ist, die HIF-1α abbaut (30).
Wir stellten die Hypothese auf, dass oral verabreichtes DCA, das die Blut-Hirn-Schranke überwindet, das GBM-Wachstum in vivo verringern würde. Wir vermuteten ferner, dass dies durch (i) die Umkehrung des glykolytischen Phänotyps und die Normalisierung von ΔΨm, was die mitochondrienabhängige Apoptose fördern würde, (ii) die Erhöhung von mROS und die Förderung der p53-Aktivierung und (iii) die Erhöhung der α-Ketoglutarat-Konzentrationen geschehen könnte. Die letzten beiden Wirkungen würden zu einer Hemmung von HIF-1α, einem Rückgang von VEGF und einer Hemmung der Angiogenese führen.
Ergebnisse
Auswirkungen von DCA auf Mitochondrien aus 49 frisch isolierten GBM-Tumoren
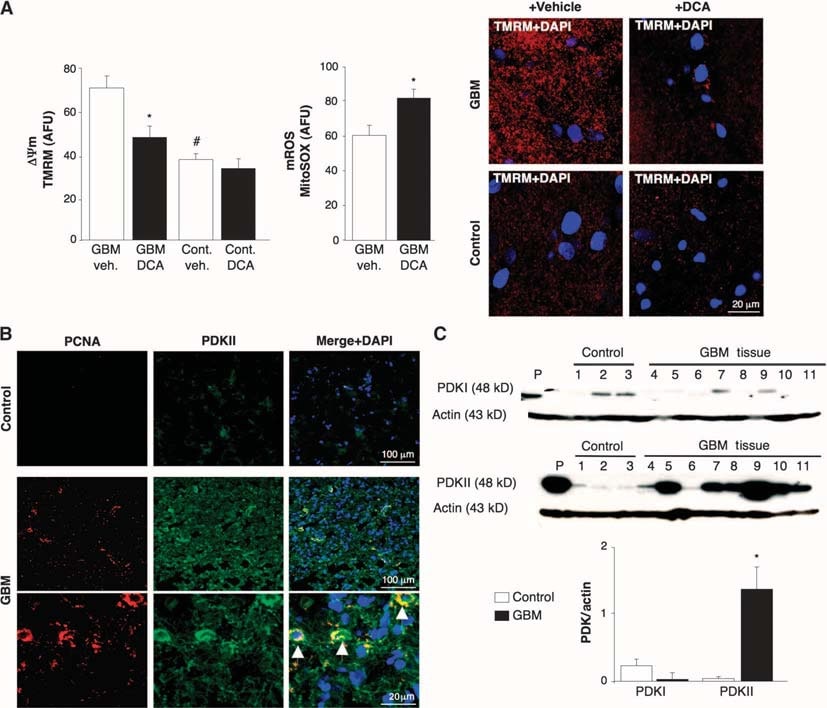
Um festzustellen, ob das humane GBM ein Ziel für eine Stoffwechseltherapie mit DCA sein könnte, untersuchten wir 49 frisch exzidierte konsekutive primäre GBMs (60% männlich, 48 ± 11 Jahre). Zusätzlich zu den klinischen und neuropathologischen Berichten bestätigten wir die GBM-Identität durch Immunhistochemie, die eine Expression von glial fibrillärem saurem Protein (GFAP), aber kein bIII-Tubulin oder Oligodendrozytenmarker zeigte (Abb. S1). ΔΨm war in den frisch isolierten GBMs im Vergleich zu nicht krebsbedingtem Hirngewebe, das bei einer Epilepsieoperation gewonnen wurde, erhöht (n = 3) (Abb. 1A). DCA, aber nicht das Vehikel (normale Kochsalzlösung), verursachte eine mitochondriale Depolarisation im GBM, aber nicht im normalen Hirngewebe. DCA erhöhte auch die mROS des GBM (Abb. 1A). Dies deutet darauf hin, dass der metabolische und mitochondriale Umbau in GBM teilweise reversibel ist und dass dieser Umbau zumindest teilweise durch PDK reguliert wird. Die Reaktion auf DCA steht im Einklang mit einer höheren Konzentration von PDKII [die am häufigsten ubiquitär exprimierte Isoform und diejenige mit dem niedrigstenKi für DCA (31)] in GBM als in nicht krebsbedingtem Hirngewebe, wie mit Immunhistochemie und Immunoblots festgestellt wurde (Abb. 1, B und C). Die Zellen mit den höchsten PDKII-Konzentrationen enthielten auch proliferierendes Zellkern-Antigen (PCNA), was darauf hindeutet, dass diese Zellen proliferierten (Abb. 1B). Diese Daten, die über einen Zeitraum von zwei Jahren gesammelt wurden, untermauerten die Gründe für die anschließende Verabreichung von DCA an Patienten mit GBM (4).
Klinische Wirkungen von DCA bei fünf Patienten mit GBM
Anschließend behandelten wir mit DCA fünf aufeinander folgende Patienten mit primärem GBM, die von unserem Hirntumorprogramm überwiesen wurden und von denen Gewebe aus der letzten Debulking-Operation verfügbar war. Drei Patienten (Patienten 1 bis 3) hatten ein GBM-Rezidiv, bei dem die Krankheit nach mehreren Chemotherapien (zusätzlich zur Standardbehandlung mit Operation, RT und TMZ) fortgeschritten war und das für eine palliative Therapie in Frage kam. Zwei weitere Patienten (Patienten 4 und 5) wurden neu diagnostiziert, und nach der ersten Debulking-Operation wurde DCA zusätzlich zur Standardbehandlung mit RT und TMZ verabreicht. Bei Patient 4 folgte auf eine dreimonatige Vorbehandlung mit DCA eine zusätzliche Behandlung mit RT und TMZ, während bei Patient 5 DCA gleichzeitig mit RT und TMZ nach der Debulking-Operation begonnen wurde. Wenn bei den Patienten eine erneute Operation oder Autopsie erforderlich war, wurde das Gewebe der letzten Debulking-Operation (vor der DCA-Verabreichung) mit dem Gewebe nach der DCA-Behandlung verglichen. Die klinischen Daten der Patienten sind in Tabelle S1 zusammengefasst. DCA wird seit mehr als 30 Jahren an Patienten verabreicht, hauptsächlich zur Behandlung von angeborenen Fehlern des mitochondrialen Stoffwechsels, und es liegen pharmakokinetische und pharmakodynamische Daten vor (5, 32-34). Wir behandelten die Patienten einen Monat lang mit einer Anfangsdosis von 12,5 mg/kg zweimal täglich oral, danach wurde die Dosis auf 25 mg/kg zweimal täglich oral erhöht. Anschließend folgte ein Protokoll zur Deeskalation der Dosis, wobei die Dosis um 50 % verringert wurde, wenn dosislimitierende Toxizität auftrat. Die Patienten wurden bis zu 15 Monate lang klinisch beobachtet. Bei keinem der Patienten traten hämatologische, hepatische, renale oder kardiale Toxizitäten auf (Tabelle S1). Periphere Neuropathie war die einzige offensichtliche Toxizität. Bei den Patienten traten in unterschiedlichem Maße dosisabhängige periphere Neuropathien auf, die reversibel waren, was frühere Studien bestätigte (35-37). Als die Dosis auf 6,25 mg/kg zweimal täglich oral reduziert wurde, hatte keiner der Patienten eine klinisch signifikante periphere Neuropathie (Tabelle S1). Die anfängliche Halbwertszeit von DCA beträgt <1 Stunde. DCA hemmt seinen eigenen Metabolismus, und die Serumkonzentrationen steigen an und erreichen schließlich ein Plateau (34). Bei unseren Patienten blieben die DCA-Plasmakonzentrationen in den ersten 2 bis 3 Monaten nicht nachweisbar, erreichten danach aber therapeutische Konzentrationen. Bei einer Dosis von 6,25 mg/kg oral zweimal täglich über mindestens 3 Monate betrugen die DCA-Trogkonzentrationen 0,44 ± 0,16 mM (Mittelwert ± SD; n = 4) (Tabelle S1). Diese Werte sind vergleichbar mit denen, die bei der chronischen DCA-Behandlung von Erwachsenen mit mitochondrialen Defekten (34) und liegen im gleichen Bereich wie derKi-Wert von DCA für PDKII (0,2 mM) (31). Die Patienten 1, 4 und 5 zeigten einige Anzeichen einer radiologischen Rückbildung in der Magnetresonanztomographie (MRT) (Abb. 2A und Abb. S2 bis S4). Patient 3 hatte zu Beginn der Behandlung einen sehr großen Tumor mit Hirnödem (Abb. S5), obwohl er hohe Steroiddosen erhielt, und einen niedrigen Karnofsky-Score, der sich weiter verschlechterte. Er starb 3 Monate nach Beginn der DCA-Therapie an den Komplikationen des Hirnödems. Bei Patient 2 war im 11. Monat der DCA-Therapie die Drainage einer Zyste und eine Entstauung erforderlich. Bei Patient 4 kam es im dritten Monat der DCA-Therapie zu einer radiologischen Progression, woraufhin eine weitere Entstippung durchgeführt und zusätzlich zu DCA eine RT plus TMZ gegeben wurde. Mit Ausnahme von Patient 3 waren alle Patienten im Monat 15 der DCA-Therapie klinisch stabil und lebten im Monat 18 (telefonische Nachuntersuchung). Weitere klinische Details sind im ergänzenden Material beschrieben.
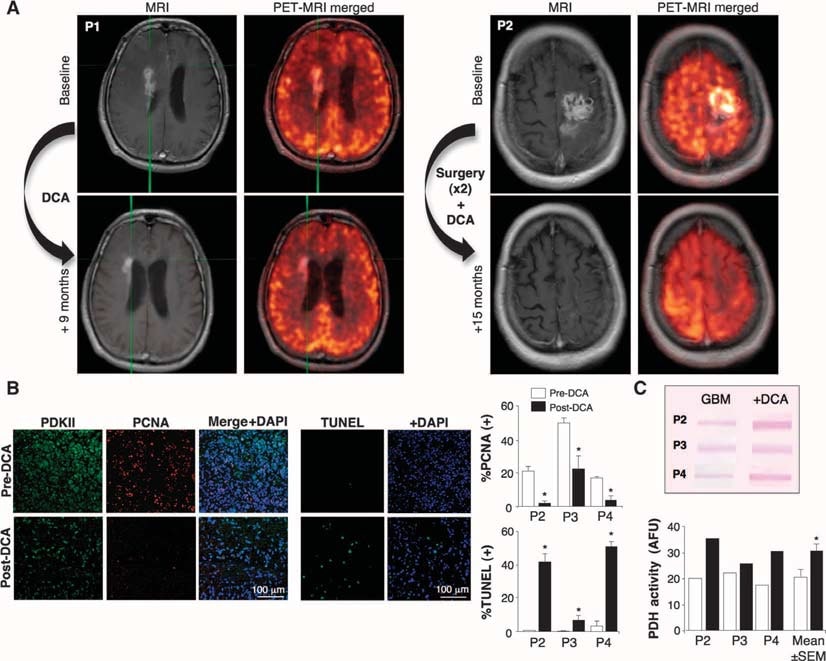
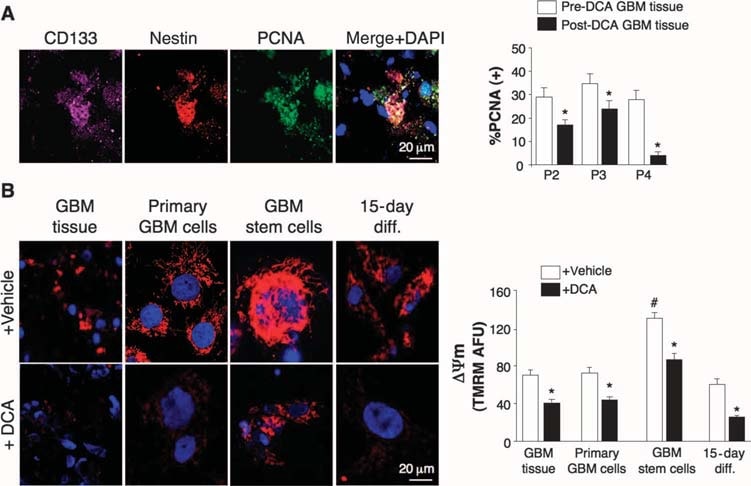
Auswirkungen von DCA auf GBM-Tumore in vivo, primäre GBM-Zelllinien und mutmaßliche GBM-SC, die von den DCA-behandelten Patienten stammen
Wir führten Experimente an Geweben dieser fünf Patienten durch und konnten Vergleiche an Geweben vor und nach der DCA-Behandlung bei den Patienten 2 bis 4 anstellen; bei den Patienten 1 und 5 hatten wir nur „Vorher“-Gewebe. Im Vergleich zum Gewebe vor der DCA-Behandlung wies das GBM-Gewebe aller drei Patienten nach der DCA-Behandlung eine verringerte Anzahl von Zellen pro Volumeneinheit, eine verringerte Proliferation und eine erhöhte Apoptose auf (Abb. 2B) sowie eine erhöhte enzymatische Aktivität der PDH im Gewebe, was auf eine wirksame Hemmung der PDK in vivo schließen lässt (Abb. 2C). Mutmaßliche GBM-Krebsstammzellen (GBM-SCs) könnten für die Resistenz und das Wiederauftreten von GBM nach der Behandlung verantwortlich sein (38-43). Diese Zellen sind als CD133+ / Nestin+ GBM-SC charakterisiert und bilden Nischen um Kapillaren (41). In solchen vaskulären GBM-SC-Einheiten können GBM-SC die Angiogenese induzieren, während ihr molekularer Stammzellphänotyp durch ihren Zugang zu zirkulierenden Wachstumsfaktoren aufrechterhalten wird (44). Die GBM-SC-Proliferation ist mit einem besonders schlechten klinischen Ergebnis verbunden (42). CD133+ /nestin+ GBM-SC exprimierten PCNA in vivo in allen Tumoren vor der DCA-Therapie, was darauf hindeutet, dass sie sich teilen, aber der Prozentsatz der CD133+ / nestin+ Zellen, die PCNA exprimierten, war nach der DCA-Therapie bei den Patienten 2 bis 4 signifikant verringert (Abb. 3A). Die gleichzeitige Färbung mit einem CD133-Antikörper und Tetramethyl-Rhodamin-Methylester (TMRM) zeigte, dass CD133+-Zellen im Vergleich zu benachbarten Nicht-GBM-SC in vivo die höchste ΔΨm aufweisen (Abb. S6). In den aus dem Tumor stammenden primären Zelllinien exprimierten ~10 % der Zellen sowohl CD133 als auch Nestin, während >90 % der Zellen den reifen Marker GFAP (aber nicht bIII-Tubulin oder Oligodendrozyten) exprimierten (Abb. S7), ähnlich wie bei der Histopathologie von GBM (Abb. S1). Wir isolierten mutmaßliche GBM-SC aus GBM-Tumoren und kultivierten sie mit den entsprechenden Wachstumsfaktoren (humaner Fibroblasten-Wachstumsfaktor, 20 ng/ml; humaner epidermaler Wachstumsfaktor, 20 ng/ml). Diese Zellen wiesen eine sehr hohe Expression von CD133 und Nestin auf, hatten eine sehr geringe Expression von reifen Glia-Markern (Abb. S7) und bildeten charakteristische Neurosphären (Abb. 4 und Abb. S7), ein unabhängiger Prädiktor für einen schlechten klinischen Ausgang (43). Wir haben DYm in frisch exzidierten Tumoren, in primären Zelllinien und in aus diesen Tumoren isolierten GBM-SC sowie in differenzierten Zellen aus GBM-SC gemessen (Abb. 3B). Das höchste Potenzial wurde in den mutmaßlichen GBM-SC gefunden. Sowohl die primären als auch die von GBM-SC abgeleiteten sekundären GBM-Zellen (15-tägige Differenzierung) hatten ein ähnliches mitochondriales Potenzial wie die Elterntumoren. DCA (0,5 mM für 24 Stunden) verringerte das Potenzial in allen Zellgruppen. Obwohl die Ursache für das erhöhte ΔΨm bei Krebs (3, 13) noch nicht vollständig geklärt ist, wurde vorgeschlagen, dass sie zum Teil durch eine Translokation von Hexokinase II (HXKII), einem Schlüsselenzym der Glykolyse, vom Zytoplasma zur äußeren Mitochondrienmembran verursacht wird (45, 46). Dort kann HXKII an den spannungsabhängigen Anionenkanal (eine Komponente des MTP) binden und diesen hemmen, wodurch der ΔΨm und die apoptotische Schwelle erhöht werden. Die Hemmung dieser Translokation verringert das ΔΨm des Krebses und hebt die Apoptoseresistenz auf (45, 46). Unsere primären Zelllinien, die aus prä-DCA-Tumoren gewonnen wurden, zeigten eine anhaltende mitochondriale Translokation von HXKII, was den Anstieg von ΔΨm erklären könnte. Die HXKII-Translokation war in primären Zelllinien aus Tumoren nach DCA-Behandlung nicht vorhanden (Abb. S8), was mit der Vorstellung vereinbar ist, dass DCA eine Unterdrückung der Glykolyse und eine Verringerung von ΔΨm induziert. Wie in den Tumoren war auch in den GBM-Zelllinien der Patienten 2 bis 4 PDKII in hohen Konzentrationen vorhanden, obwohl auch die anderen bekannten Isoenzyme exprimiert wurden (Abb. S9A). Wenn man die GBM-SCs in sekundäre GBM-Zelllinien differenzieren ließ, sank der Anteil der Zellen mit GBM-SC-Markern auf einen ähnlichen Wert wie bei den primären Zelllinien (~10 %). Wenn man sie jedoch in Gegenwart von DCA (0,5 mM) differenzieren ließ, sank der Anteil der Zellen mit GBM-SC-Markern noch weiter auf ~5% (Abb. S7). DCA induzierte in der Tat Apoptose in GBM-SC in vitro (Abb. 4 und Abb. S9B) sowie in primären GBM-Zelllinien (Abb. S9C). Die Apoptose wurde in GBM-SCs durch die Kombination von DCA und TMZ weiter erhöht (Abb. 4 und Abb. S9B), was eine Begründung für die Kombinationstherapie liefert. Die GBM-SC-Apoptose fand auch in vivo in den Tumoren nach der DCA-Behandlung statt, was durch die Kolokalisierung von Nestin, CD133 und die durch terminale Desoxynukleotidyltransferase vermittelte Desoxyuridintriphosphat-Nick-End-Labeling (TUNEL)-Färbung gezeigt wurde (Abb. 4 und Abb. S9D).
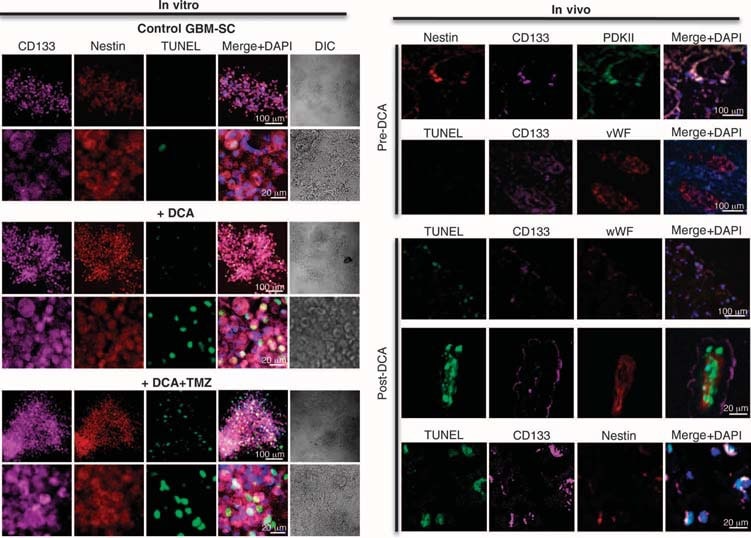
Auswirkungen von DCA auf die GBM-SC-Mikrogefäßeinheit und die Angiogenese in vivo und in vitro
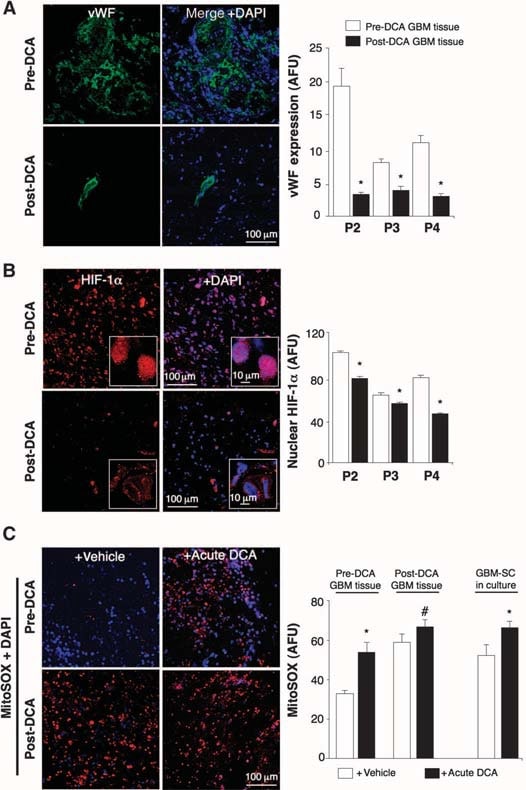
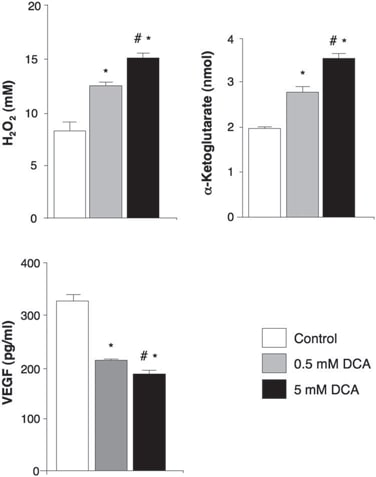
In den unbehandelten Geweben vor der DCA-Behandlung wurden Nischen von GBM-SC um Mikrogefäßbetten herum gefunden [von Willebrand-Faktor (vWF)-Färbung], wie berichtet (41). Diese GBM-SC-Mikrogefäßeinheit (44) wurde durch die DCA-Behandlung zerstört, da neben GBM-SC auch die Apoptose in mikrovaskulären Endothelzellen erhöht war (Abb. 4 und Abb. S9D), was auf eine mögliche Hemmung der Angiogenese schließen lässt. In der Tat deutet die verringerte vWF-Färbung in den Tumoren nach der DCA-Behandlung auf eine verringerte Vaskularität hin (Abb. 5A). HIF-1α wurde in den Tumoren vor der DCA-Behandlung stark exprimiert oder aktiviert (Kernlokalisierung) und in den Tumoren nach der DCA-Behandlung gehemmt (Abb. 5B). Nach der DCA-Behandlung wiesen die Tumore der Patienten 2 bis 4 einen signifikanten Anstieg von mROS in vivo auf (Superoxid gemessen durch mitoSOX), verglichen mit den prä-DCA-Tumoren derselben Patienten (Abb. 5C). In den prä-DCA-Tumoren erhöhte akutes DCA die mROS auf Werte, die in den Tumoren nach der DCA-Behandlung beobachtet wurden. Im Gegensatz dazu erhöhte akutes DCA in den post-DCA-Tumoren die mROS nur minimal, was auf eine fast maximale Wirkung in vivo schließen lässt. DCA erhöhte auch die mROS in GBM-SC (Abb. 5C). Niedrige ROS-Konzentrationen in Krebsstammzellen können eine Resistenz gegen Apoptose widerspiegeln, und es wird angenommen, dass Therapien, die die ROS-Konzentration in Krebsstammzellen erhöhen, wirksamer sind (47). Obwohl wir mROS (mitochondriales Superoxid) untersucht haben, ist umstritten, ob die HIF-1α-Aktivierung bei Krebs mit einer allgemeinen Zunahme oder Abnahme von ROS einhergeht [nachzulesen in (28)]. Mit einer anderen Technik haben wir auch H2O2 in der gesamten Zelle gemessen. DCA erhöhte dieH2O2-Konzentrationin GBM-Zellen in einer dosisabhängigen Weise (Abb. 6). Darüber hinaus erhöhte DCA auch die intrazellulären α-Ketoglutarat-Konzentrationen in einer dosisabhängigen Weise (Abb. 6). Dies ist mit dem Anstieg der Glukoseoxidation vereinbar, der auf die PDH-Aktivierung folgt (3) denn α-Ketoglutarat ist ein Produkt des Krebszyklus. Der Krebs-Zyklus produziert die Elektronendonatoren, die während der Atmung in die Elektronentransportkette eingespeist werden. Dementsprechend erhöhte DCA die Atmungsrate in GBM-Zellen um 44 ± 4 % (Mittelwert ± SEM; n = 3; P < 0,05), was für eine allgemeine Zunahme der Mitochondrienaktivität spricht. Der Anstieg vonH2O2und α-Ketoglutarat kann den Rückgang der HIF-1α-Aktivität erklären (Abb. 5B), was auch durch den dosisabhängigen Rückgang der VEGF-Produktion durch GBM-Zellen bestätigt wird (Abb. 6). Diese Daten stimmen mit dem Rückgang der Angiogenese in vivo überein (Abb. 4 und 5) und legen nahe, dass GBM-Zellen auf parakrine Weise Signale an Endothelzellen senden. Um zu untersuchen, ob DCA die Angiogenese direkt unterdrücken kann, verwendeten wir die Standardtechnik der Bildung von Röhren aus menschlichen Endothelzellen in Matrigel. Unter physiologischer moderater Hypoxie verursachte DCA eine dosisabhängige direkte Hemmung der Angiogenese in vitro (Abb. S10). Die Tumore der Patienten 2 bis 4 zeigten nach der DCA-Behandlung eine erhöhte Aktivität des mROS-sensitiven p53 (Kerntranslokation), was auch durch die erhöhte Aktivität und Häufigkeit seines nachgeschalteten Ziels p21 bestätigt wurde (Abb. S11). Diese Wirkungen auf p53 oder p21 können auch den Rückgang der HIF-gesteuerten Transkription erklären und stehen im Einklang mit den antiproliferativen, zusätzlich zu den proapoptotischen Wirkungen von DCA bei GBM (Abb. S12).
DISKUSSION
Der Stoffwechselmodulator DCA wirkt in kultivierten Zellen und bei Nagetieren krebshemmend (3, 16-20). Wir haben nun gezeigt, dass DCA bei Patienten mit GBM eingesetzt werden kann. Die Behandlung mit DCA führte bei einigen GBM-Patienten zu einer verlängerten radiologischen Stabilisierung oder Rückbildung des Tumors und wies im Allgemeinen ein gutes Sicherheitsprofil auf. Dieser frühe Bericht über die erste Anwendung beim Menschen liefert eine Begründung für erweiterte Studien mit diesem generischen kleinen Molekül bei Patienten mit GBM. Unsere Ergebnisse deuten darauf hin, dass GBM ein guter Kandidat für Stoffwechselinterventionen ist. Das Zielmolekül von DCA, PDKII, wird in GBM-Tumoren und Zelllinien stark exprimiert, und DCA kann seine Aktivität in vivo hemmen. GBM zeichnet sich durch eine mitochondriale Hyperpolarisation aus, die mit dem metabolischen Umbau (Warburg-Effekt) und der damit verbundenen Apoptoseresistenz übereinstimmt, die GBM und die meisten soliden Tumoren kennzeichnen (11). Mutationen in den Genen für zytoplasmatische und mitochondriale Isocitrat-Dehydrogenasen wurden in GBMs beschrieben, die aus Gliomen niedrigeren Grades (sekundären GBMs) entstanden sind (48)aber der Mechanismus, durch den diese Mutationen mit der Krebsentstehung zusammenhängen, bleibt unklar (49, 50). Bei unseren Patienten handelte es sich um primäre GBMs, und der mitochondriale Umbau war zumindest teilweise mit DCA reversibel, was darauf hindeutet, dass es sich nicht um eine irreversible Funktionsstörung handelte. Darüber hinaus zeigen wir, dass mutmaßliche GBM-SC möglicherweise denselben metabolischen und mitochondrialen Umbau durchlaufen, allerdings in einem höheren Maße, da GBM-SC sowohl in vivo als auch in vitro die am stärksten hyperpolarisierten Mitochondrien aufwiesen. Die Umkehrung dieses mitochondrialen Umbaus durch DCA löste bei GBM-SC sowohl in vitro als auch in vivo Apoptose aus. Obwohl das Ausmaß der Apoptoseinduktion durch DCA nicht hoch ist (im Vergleich zu zytotoxischen Mitteln), ist sie relativ selektiv und verschont Nicht-Krebszellen (3)und kann, da es sich um GBM-SC handelt, zu einer nachhaltigeren klinischen Wirkung führen. Patient 3 starb, und bei Patient 4 kam es innerhalb der ersten drei Monate zu einem Rezidiv, als DCA noch keine anhaltenden therapeutischen Werte erreicht hatte (wie die Tatsache zeigt, dass die Talspiegelkonzentrationen im Plasma nicht nachweisbar waren). Somit können Patienten bei der ersten DCA-Verabreichung unterbehandelt werden und sind dem Risiko einer ersten Krankheitsprogression ausgesetzt. Durch die Verringerung der Resistenz gegen die mitochondrienabhängige Apoptose kann DCA möglicherweise die Wirkung von Standardtherapien verstärken. In der Tat könnten die Auswirkungen von DCA plus TMZ auf die GBMSC-Apoptose die Ursache für die langfristigen Vorteile bei Patient 5 sein. Die Wirkungen von DCA auf Krebszellen werden durch PDKII small interfering RNA nachgeahmt, und DCA hat keine Wirkungen, die über die nach PDKII-Knockdown beobachteten hinausgehen (3). Dies deutet darauf hin, dass der Mechanismus der krebshemmenden Wirkung von DCA in der Hemmung von PDKII besteht, einem Enzym, das in GBM in erhöhten Konzentrationen vorkommt. Durch die Hemmung von PDKII normalisiert DCA das erhöhte Verhältnis von Glykolyse zu Glukoseoxidation in Krebszellen und erhöht die mitochondriale Funktion (3). Dieser Mechanismus wird durch unsere aktuellen Ergebnisse gestützt, dass DCA sowohl die Atmung der GBM-Zellen als auch die a-Ketoglutarat-Konzentration erhöht. Im Gegensatz zu den kontrollierten Bedingungen in der Zellkultur könnten jedoch zusätzliche Mechanismen zu den Auswirkungen von DCA auf den Stoffwechsel der Krebszellen und die Apoptose in vivo beitragen. Zusätzlich zu den bereits beschriebenen Wirkungen von DCA auf Krebszellen (3)sind unsere Daten zu HXKII, mROS, HIF-1α, p53 und p21 konsistent mit der DCA-induzierten Hemmung der Angiogenese, der Induktion von Apoptose und der Unterdrückung der Proliferation sowohl bei GBM als auch bei GBM-SC, wie im ergänzenden Material und in Abb. S12 zusammengefasst. S12. DCA hatte keine offensichtliche Toxizität, abgesehen von einer zuvor beschriebenen dosisabhängigen, reversiblen, nicht-myelinisierenden Neurotoxizität (32, 34) die bei der zweimal täglich oral verabreichten Dosis von 6,25 mg/kg minimal oder gar nicht vorhanden war. Diese Dosis zeigte biologische und klinische Wirksamkeit und erreichte Plasmakonzentrationen, die für eine PDK-Hemmung erforderlich sind (31). Angesichts der geringen Zahl der in unserer Studie behandelten Patienten können keine eindeutigen Schlussfolgerungen hinsichtlich DCA als Therapie für GBM gezogen werden. Unsere Arbeit unterstreicht die Notwendigkeit weiterer Studien mit DCA bei GBM, wobei der Schwerpunkt auf Kombinationstherapieprotokollen liegen sollte. GBM könnte auch für andere Medikamente aus der neuen Familie der Stoffwechselmodulatoren anfällig sein, was auf einen neuen Ansatz bei der Behandlung dieses unheilbaren Krebses hindeutet.
ERGÄNZENDES MATERIAL
www.sciencetranslationalmedicine.org/cgi/content/full/2/31/31ra34/DC1Materials und MethodenErgebnisseDiskussionAbb
. S1. Molekulare Charakterisierung von GBM-Tumoren.
Abb. S2. Entwicklung des Tumoransprechens bei Patient 1.
Abb. S3. Entwicklung des Tumoransprechens bei Patient 4.
Abb. S4. Entwicklung des Tumoransprechens bei Patient 5.
Abb. S5. GBM-MRT von Patient 3.
Abb. S6. Mitochondriales Membranpotenzial in GBM-SC aus frisch entnommenem GBM-Gewebe.
Abb. S7. Charakterisierung von primären GBM-Zellen und GBM-SC.
Abb. S8. HXKII in GBM-Zellen von Patienten vor und nach chronischer DCA-Behandlung.
Abb. S9 Auswirkungen der DCA-Therapie auf GBM-SC und vaskuläre Apoptose.
Abb. S10. Auswirkungen von DCA auf die Angiogenese in vitro.
Abb. S11. Auswirkungen der DCA-Behandlung auf die Aktivität von p53 und p21 in vivo.
Abb. S12. Ein vorgeschlagener umfassender Mechanismus für die krebshemmenden Wirkungen von DCA bei GBM (siehe Ergänzende
Diskussion).
Tabelle S1. Labor- und klinische Parameter von fünf GBM-Patienten vor und nach der DCA-Behandlung.
Referenzen
REFERENZEN
1 1. P. Y. Wen, S. Kesari, Maligne Gliome bei Erwachsenen. N. Engl. J. Med. 359, 492-507 (2008).
2 W. K. Yung, R. E. Albright, J. Olson, R. Fredericks, K. Fink, M. D. Prados, M. Brada, A. Spence, R. J. Hohl, W. Shapiro, M. Glantz, H. Greenberg, R. G. Selker, N. A. Vick, R. Rampling, H. Friedman, P. Phillips, J. Bruner, N. Yue, D. Osoba, S. Zaknoen, V. A. Levin, A phase II study of temozolomide vs. procarbazine in patients with glioblastoma multiforme at first relapse. Br. J. Cancer 83, 588-593 (2000).
3 S. Bonnet, S. L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, C. T. Lee, G. D. Lopaschuk, L. Puttagunta, S. Bonnet, G. Harry, K. Hashimoto, C. J. Porter, M. A. Andrade, B. Thebaud, E. D. Michelakis, A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle 11, 37-51 (2007).
4 E. D. Michelakis, L. Webster, J. R. Mackey, Dichloracetat (DCA) als potenzielle metabolisierende Therapie für Krebs. Br. J. Cancer 99, 989-994 (2008).
5 P. W. Stacpoole, The pharmacology of dichloroacetate. Stoffwechsel 38, 1124-1144 (1989).
6 O. Warburg, Ueber den Stoffwechsel der Tumoren (Constable, London, 1930).
7 S. Weinhouse, Die Warburg-Hypothese fünfzig Jahre später. Z. Krebsforsch. Klin. Onkol. Cancer Res. Clin. Oncol. 87, 115-126 (1976).
8 M. G. Vander Heiden, L. C. Cantley, C. B. Thompson, Understanding the Warburg effect: Die metabolischen Anforderungen der Zellproliferation. Wissenschaft 324, 1029-1033 (2009).
9 J. G. Pan, T. W. Mak, Metabolic targeting as an anticancer strategy: Dawn of a new era? Sci. STKE 2007, pe14 (2007).
10 J. W. Kim, C. V. Dang, Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 66, 8927-8930 (2006).
11 R. A. Gatenby, R. J. Gillies, Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 4, 891-899 (2004).
12 J. W. Kim, C. V. Dang, Multifaceted roles of glycolytic enzymes. Trends Biochem. Sci. 30, 142-150 (2005).
13 L. B. Chen, Mitochondrial membrane potential in living cells. Annu. Rev. Cell Biol. 4, 155-181 (1988).
14 G. Kroemer, L. Galluzzi, C. Brenner, Mitochondrial membrane permeabilization in cell death. Physiol. Rev. 87, 99-163 (2007).
15. N. Zamzami, G. Kroemer, Das Mitochondrium in der Apoptose: Wie sich die Büchse der Pandora öffnet. Nat. Rev. Mol. Cell Biol. 2, 67-71 (2001).
16 R. A. Cairns, I. Papandreou, P. D. Sutphin, N. C. Denko, Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc. Natl. Acad. Sci. U.S.A. 104, 9445-9450 (2007).
17 W. Cao, S. Yacoub, K. T. Shiverick, K. Namiki, Y. Sakai, S. Porvasnik, C. Urbanek, C. J. Rosser, Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 68, 1223-1231 (2008).
18 S. Dhar, S. J. Lippard, Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc. Natl. Acad. Sci. U.S.A. 106, 22199-22204 (2009).
19 R. C. Sun, M. Fadia, J. E. Dahlstrom, C. R. Parish, P. G. Board, A. C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res. Treat. 120, 253-260 (2010).
20 J. Y. Wong, G. S. Huggins, M. Debidda, N. C. Munshi, I. De Vivo, Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol. Oncol. 109, 394-402 (2008).
21 S. Wang, S. S. Leonard, J. Ye, M. Ding, X. Shi, The role of hydroxyl radical as a messenger in Cr(VI)-induced p53 activation. Am. J. Physiol. Cell Physiol. 279, C868-C875 (2000).
22 C. Huang, Z. Zhang, M. Ding, J. Li, J. Ye, S. S. Leonard, H. M. Shen, L. Butterworth, Y. Lu, M. Costa, Y. Rojanasakul, V. Castranova, V. Vallyathan, X. Shi, Vanadate induces p53 transactivation through hydrogen peroxide and causes apoptosis. J. Biol. Chem. 275, 32516-32522 (2000).
23 T. Schmid, J. Zhou, R. Köhl, B. Brüne, p300 entlastet die p53-verursachte transkriptionelle Unterdrückung des Hypoxie-induzierbaren Faktors-1 (HIF-1). Biochem. J. 380, 289-295 (2004).
24 M. V. Blagosklonny, W. G. An, L. Y. Romanova, J. Trepel, T. Fojo, L. Neckers, p53 inhibiert Hypoxie-induzierbare Faktor-stimulierte Transkription. J. Biol. Chem. 273, 11995-11998 (1998).
25 J. W. Kim, I. Tchernyshyov, G. L. Semenza, C. V. Dang, HIF-1-mediated expression of pyruvate dehydrogenase kinase: Ein metabolischer Schalter, der für die zelluläre Anpassung an Hypoxie erforderlich ist. Cell Metab. 3, 177-185 (2006).
26 G. L. Semenza, D. Artemov, A. Bedi, Z. Bhujwalla, K. Chiles, D. Feldser, E. Laughner, R. Ravi, J. Simons, P. Taghavi, H. Zhong, ‚The metabolism of tumours‘: 70 years later. Novartis Found. Symp. 240, 251-260 (2001).
27 E. K. Weir, J. López-Barneo, K. J. Buckler, S. L. Archer, Acute oxygen-sensing mechanisms. N. Engl. J. Med. 353, 2042-2055 (2005).
28 N. C. Denko, Hypoxia, HIF1 and glucose metabolism in the solid tumour. Nat. Rev. Cancer 8, 705-713 (2008).
29 G. L. Semenza, Zielgerichtetes HIF-1 für die Krebstherapie. Nat. Rev. Cancer 3, 721-732 (2003).
30 E. D. MacKenzie, M. A. Selak, D. A. Tennant, L. J. Payne, S. Crosby, C. M. Frederiksen, D. G. Watson, E. Gottlieb, Cell-permeating a-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol. Cell. Biol. 27, 3282-3289 (2007).
31 M. M. Bowker-Kinley, W. I. Davis, P. Wu, R. A. Harris, K. M. Popov, Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 329 (Pt 1), 191-196 (1998).
32 P. W. Stacpoole, D. S. Kerr, C. Barnes, S. T. Bunch, P. R. Carney, E. M. Fennell, N. M. Felitsyn, R. L. Gilmore, M. Greer, G. N. Henderson, A. D. Hutson, R. E. Neiberger, R. G. O’Brien, L. A. Perkins, R. G. Quisling, A. L. Shroads, J. J. Shuster, J. H. Silverstein, D. W. Theriaque, E. Valenstein, Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 117, 1519-1531 (2006).
33 P. W. Stacpoole, A. C. Lorenz, R. G. Thomas, E. M. Harman, Dichloracetate in the treatment of lactic acidosis. Ann. Intern. Med. 108, 58-63 (1988).
34 P. W. Stacpoole, T. L. Kurtz, Z. Han, T. Langaee, Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv. Drug Deliv. Rev. 60, 1478-1487 (2008).
35 P. Kaufmann, K. Engelstad, Y. Wei, S. Jhung, M. C. Sano, D. C. Shungu, W. S. Millar, X. Hong, C. L. Gooch, X. Mao, J. M. Pascual, M. Hirano, P. W. Stacpoole, S. DiMauro, D. C. De Vivo, Dichloroacetate causes toxic neuropathy in MELAS: A randomized, controlled clinical trial. Neurologie 66, 324-330 (2006).
36 P. W. Stacpoole, L. R. Gilbert, R. E. Neiberger, P. R. Carney, E. Valenstein, D. W. Theriaque, J. J. Shuster, Evaluation of long-term treatment of children with congenital lactic acidosis with dichloroacetate. Pediatrics 121, e1223-e1228 (2008).
37 P. W. Stacpoole, G. N. Henderson, Z. Yan, M. O. James, Clinical pharmacology and toxicology of dichloroacetate. Environ. Health Perspect. 106 (Suppl. 4), 989-994 (1998).
38 N. Sanai, A. Alvarez-Buylla, M. S. Berger, Neural stem cells and the origin of gliomas. N. Engl. J. Med. 353, 811-822 (2005).
39 F. Zindy, T. Uziel, O. Ayrault, C. Calabrese, M. Valentine, J. E. Rehg, R. J. Gilbertson, C. J. Sherr, M. F. Roussel, Genetic alterations in mouse medulloblastomas and generation of tumors de novo from primary cerebellar granule neuron precursors. Cancer Res. 67, 2676-2684 (2007).
40 T. Hide, T. Takezaki, H. Nakamura, J. Kuratsu, T. Kondo, Brain tumor stem cells as research and treatment targets. Brain Tumor Pathol. 25, 67-72 (2008).
41 C. Calabrese, H. Poppleton, M. Kocak, T. L. Hogg, C. Fuller, B. Hamner, E. Y. Oh, M. W. Gaber, D. Finklestein, M. Allen, A. Frank, I. T. Bayazitov, S. S. Zakharenko, A. Gajjar, A. Davidoff, R. J. Gilbertson, A perivascular niche for brain tumor stem cells. Cancer Cell 11, 69-82 (2007).
42 R. Pallini, L. Ricci-Vitiani, G. L. Banna, M. Signore, D. Lombardi, M. Todaro, G. Stassi, M. Martini, G. Maira, L. M. Larocca, R. De Maria, Cancer stem cell analysis and clinical outcome in patients with glioblastoma multiforme. Clin. Cancer Res. 14, 8205-8212 (2008).
43 D. R. Laks, M. Masterman-Smith, K. Visnyei, B. Angenieux, N. M. Orozco, I. Foran, W. H. Yong, H. V. Vinters, L. M. Liau, J. A. Lazareff, P. S. Mischel, T. F. Cloughesy, S. Horvath, H. I. Kornblum, Neurosphere formation is an independent predictor of clinical outcome in malignant glioma. Stem Cells 27, 980-987 (2009).
44 R. J. Gilbertson, J. N. Rich, Making a tumour’s bed: Glioblastom-Stammzellen und die vaskuläre Nische. Nat. Rev. Cancer 7, 733-736 (2007).
45 J. G. Pastorino, J. B. Hoek, Hexokinase II: Die Integration von Energiestoffwechsel und Apoptosekontrolle. Curr. Med. Chem. 10, 1535-1551 (2003).
46 J. G. Pastorino, J. B. Hoek, N. Shulga, Activation of glycogen synthase kinase 3b disrupted the binding of hexokinase II to mitochondria by phosphorylating voltage-dependent anion channel and potentiates chemotherapy-induced cytotoxicity. Cancer Res. 65, 10545-10554 (2005).
47M. Diehn, R. W. Cho, N. A. Lobo, T. Kalisky, M. J. Dorie, A. N. Kulp, D. Qian, J. S. Lam, L. E. Ailles, M. Wong, B. Joshua, M. J. Kaplan, I. Wapnir, F. M. Dirbas, G. Somlo, C. Garberoglio, B. Paz, J. Shen, S. K. Lau, S. R. Quake, J. M. Brown, I. L. Weissman, M. F. Clarke, Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780-783 (2009).
48 H. Yan, D. W. Parsons, G. Jin, R. McLendon, B. A. Rasheed, W. Yuan, I. Kos, I. Batinic-Haberle, S. Jones, G. J. Riggins, H. Friedman, A. Friedman, D. Reardon, J. Herndon, K. W. Kinzler, V. E. Velculescu, B. Vogelstein, D. D. Bigner, IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765-773 (2009).
49 C. B. Thompson, Metabolische Enzyme als Onkogene oder Tumorsuppressoren. N. Engl. J. Med. 360, 813-815 (2009).
50 L. Dang, D. W. White, S. Gross, B. D. Bennett, M. A. Bittinger, E. M. Driggers, V. R. Fantin, H. G. Jang, S. Jin, M. C. Keenan, K. M. Marks, R. M. Prins, P. S. Ward, K. E. Yen, L. M. Liau, J. D. Rabinowitz, L. C. Cantley, C. B. Thompson, M. G. Vander Heiden, S. M. Su, Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739-744 (2009).
51 Finanzierung: Diese Studie wurde von der Hecht Foundation (Vancouver, British Columbia, Kanada; E.D.M.), den Canada Institutes for Health Research und dem Canada Research Chairs Program (E.D.M.) sowie durch öffentliche Spenden für das DCA-Programm (erhalten und verwaltet von den Regenten der Universität von Alberta und der medizinischen Fakultät) finanziert. Die Autoren möchten sich für die Unterstützung durch die Alberta Health Services (D. Gordon, Senior VicePresident, Major Tertiary Hospitals) bedanken. Beiträge der Autoren: E.D.M. konzipierte die Studien, überwachte die mechanistischen Studien, sicherte die Finanzierung, analysierte die Daten und schrieb das Manuskript. K.C.P. konzipierte die Studien mit, überwachte alle klinischen Studien und schrieb das Manuskript mit. G.S. und P.D. führten alle mechanistischen Studien durch und redigierten das Manuskript. L.W. koordinierte alle Studien, trug zur Datenerfassung bei, analysierte die klinischen Daten und redigierte das Manuskript. A.H., E.N., C.M., T.-L.G. und M.S.M. trugen zur Datenerfassung und Datenanalyse bei und redigierten das Manuskript. J.R.M., D.F. und B.A. gestalteten die klinischen Studien mit, trugen zur Datenerfassung bei und redigierten das Manuskript. Konkurrierende Interessen: E.D.M. ist Miteigentümer eines anhängigen Patents für die Verwendung von DCA als Krebstherapie. Eine aktive oder geplante Vermarktung dieses Patents ist nicht erfolgt.