Ellappan Babu, doctor, Sabarish Ramachandran, doctor, Veena CoothanKandaswamy, doctor, Selvakumar Elangovan, doctor, Puttur D. Prasad, doctor, Vadivel Ganapathy, doctor , y Muthusamy Thangaraju, doctor.
Departamento de Bioquímica y Biología Molecular, Facultad de Medicina de Georgia, Universidad de Ciencias de la Salud de Georgia, Augusta, Georgia, EE.UU
Los usuarios pueden ver, imprimir, copiar, descargar y extraer textos y datos del contenido de dichos documentos, con fines de investigación académica, siempre que se respeten las condiciones de uso:
http://www.nature.com/authors/editorial_policies/license.html#terms
Correspondencia: M. Thangaraju, Ph. D., Department of Biochemistry and Molecular Biology, Medical College of Georgia, Augusta, GA 30912, EE.UU., [email protected].
Conflicto de intereses: Los autores declaran no tener ningún interés financiero en relación con el trabajo descrito en este manuscrito.
Publicado en su forma final editada como: Oncogene. 2011 Septiembre 22; 30(38): 4026-4037. doi:10.1038/onc.2011.113.
Palabras clave: SLC5A8; dicloroacetato; fármaco anticancerígeno; efecto Warburg; piruvato deshidrogenasa cinasa; oxidación mitocondrial en cáncer
Resumen
El dicloroacetato ha despertado un interés creciente entre el público y los científicos como posible fármaco contra el cáncer. Existen pruebas creíbles de la actividad antitumoral de este compuesto, pero se necesitan concentraciones elevadas para obtener un efecto terapéutico significativo. Desgraciadamente, estas altas concentraciones producen efectos secundarios perjudiciales que afectan al sistema nervioso, lo que impide su uso para el tratamiento del cáncer. La base mecánica de la actividad antitumoral del compuesto es su capacidad para activar el complejo piruvato deshidrogenasa mediante la inhibición de la piruvato deshidrogenasa quinasa. Dado que el compuesto inhibe la quinasa a concentraciones micromolares, se desconoce por qué son necesarias dosis terapéuticamente prohibitivas para suprimir el crecimiento tumoral. Nuestra hipótesis es que la falta de mecanismos eficaces para la entrada del dicloroacetato en las células tumorales puede ser la causa de este fenómeno. Aquí demostramos que SLC5A8 transporta dicloroacetato de forma muy eficaz y con gran afinidad. Este transportador se expresa en células normales, pero su expresión se silencia en células tumorales mediante mecanismos epigenéticos. La falta del transportador hace que las células tumorales sean resistentes a la actividad antitumoral del dicloroacetato. Sin embargo, si el transportador se expresa ectópicamente en las células tumorales, éstas se vuelven sensibles al fármaco a bajas concentraciones. Esto es evidente en células de cáncer de mama, de colon y de próstata. Sin embargo, las células normales, que expresan constitutivamente el transportador, no se ven afectadas por el compuesto, lo que indica la actividad terapéutica selectiva de las células tumorales. El mecanismo de la actividad antitumoral del compuesto sigue siendo su capacidad para inhibir la piruvato deshidrogenasa cinasa y forzar la oxidación mitocondrial del piruvato. Dado que el silenciamiento de SLC5A8 en los tumores implica la metilación del ADN y que su expresión puede ser inducida por el tratamiento con inhibidores de la metilación del ADN, nuestros hallazgos sugieren que la combinación de dicloroacetato con un inhibidor de la metilación del ADN ofrecería un medio para reducir las dosis de dicloroacetato y evitar los efectos perjudiciales asociados a dosis elevadas, pero sin comprometer la actividad antitumoral.
INTRODUCCIÓN
El dicloroacetato se utiliza actualmente para el tratamiento de la acidosis láctica congénita (Stacpoole et al., 2003, 2008). La eficacia terapéutica de este fármaco se debe a su capacidad para activar el complejo piruvato deshidrogenasa (PDC) en la matriz mitocondrial. Sin embargo, el complejo enzimático no es la diana directa del fármaco. El dicloroacetato es un inhibidor de la piruvato deshidrogenasa cinasa (PDK), que fosforila la subunidad E1α de la PDC e inactiva el complejo (Stacpoole et al. 2003, 2008). Al inhibir la PDK, el dicloroacetato impide la fosforilación de E1α y mantiene así la PDC en forma activa. La activación de la PDC inducida por el fármaco facilita el metabolismo mitocondrial del piruvato. Dado que el piruvato citosólico puede ser transportado a las mitocondrias y metabolizado o ser convertido en lactato en el citoplasma por la lactato deshidrogenasa, la activación de la PDC por el dicloroacetato y el metabolismo resultante del piruvato dentro de las mitocondrias desplaza el equilibrio entre piruvato y lactato hacia el piruvato. Esto promueve la conversión de lactato en piruvato, reduciendo así los niveles de lactato.
En los últimos años, el dicloroacetato ha despertado un gran interés como posible fármaco contra el cáncer (Michelakis et al., 2008). La idea de que este fármaco pueda tener capacidad para destruir células tumorales tiene una base racional. Las células tumorales obtienen la mayor parte de su energía de la glucólisis aeróbica en lugar de la oxidación mitocondrial. Este cambio metabólico en las células tumorales fue reconocido por primera vez por Warburg (Warburg, 1956) y se conoce como efecto Warburg (Gatenby y Gillies, 2004; Kim y Dang, 2006; Chen et al., 2007; Brahimi-Horn et al., 2007; Vander Heiden et al., 2009; Ganapathy et al., 2009; Mathupala et al., 2010). Dado que la glucólisis aeróbica, que convierte la glucosa en lactato, genera sólo 2 ATP, mientras que la oxidación mitocondrial del piruvato derivado de la glucólisis genera 30 ATP, parece desconcertante que las células tumorales prefieran la vía metabólica menos eficiente para obtener energía. Sin embargo, las células tumorales no sufren una deficiencia de ATP; de hecho, generan más energía que las células normales para apoyar su mayor crecimiento y proliferación. Esto se consigue activando la glucólisis varias veces. Es cierto que la oxidación mitocondrial genera más ATP que la glucólisis citoplasmática, pero al mismo tiempo la oxidación mitocondrial produce especies reactivas de oxígeno, que podrían resultar perjudiciales para las células. Al parecer, las células tumorales reconocen este aspecto negativo de la oxidación mitocondrial y, por tanto, optan por la glucólisis como fuente primaria de ATP. La glucólisis no requiere oxígeno y no genera especies reactivas de oxígeno. Además, con la generación de energía predominantemente a través de la glucólisis, las células tumorales pueden proliferar en condiciones aneróbicas, que a menudo existen en los tumores sólidos. Sin embargo, la supresión de la función mitocondrial en los tumores es reversible. Curiosamente, uno de los mecanismos por los que las células tumorales suprimen la oxidación mitocondrial es mediante la inducción de PDK, inactivando así PDC (Semenza, 2010). Por lo tanto, la oxidación del piruvato en las mitocondrias puede inducirse en las células tumorales revirtiendo la supresión de la actividad PDC asociada al cáncer. El dicloroacetato hace exactamente eso, inhibiendo la PDK. A pesar de esta base racional para el dicloroacetato como fármaco potencial contra el cáncer, existe una considerable controversia en la literatura sobre la utilidad clínica de este compuesto para el tratamiento del cáncer en humanos. Aunque el estudio de Bonnet et al (2007) demostró la eficacia antitumoral del dicloroacetato in vitro e in vivo en animales, un estudio reciente de Stockwin et al (2010) ha demostrado que se necesitan concentraciones muy altas de dicloroacetato para inducir la muerte celular en células tumorales y que a estas concentraciones el compuesto no tiene selectividad de células tumorales.
El dicloroacetato está ionizado y no puede atravesar la membrana plasmática por difusión. Esto plantea la cuestión de cómo este compuesto entra en las células y accede a la PDK dentro de la matriz mitocondrial. Hasta donde sabemos, sólo existe un único informe sobre el transporte de dicloroacetato en células de mamíferos, que ha demostrado que los transportadores de monocarboxilato en hepatocitos y células tumorales de Ehrlich Lettre median la entrada celular de este compuesto (Jackson y Halestrap, 1996). Dado que los transportadores de monocarboxilato son electroneutrales, es posible que la mayoría de las células, incluidas las tumorales, que expresan estos transportadores no tengan la capacidad de concentrar este fármaco. Recientemente, nosotros y otros hemos identificado un nuevo transportador de monocarboxilatos, que tiene una selectividad de sustrato similar a la de los transportadores de monocarboxilatos, pero está acoplado al Na+ y es electrogénico (Coady et al., 2004; Miyauchi et al., 2004). Este transportador, conocido como transportador de monocarboxilato acoplado a sodio (SMCT1) o SLC5A8 según la nomenclatura de la Organización del Genoma Humano, tiene la capacidad de concentrar sus sustratos contra un gradiente de concentración debido a la implicación del gradiente transmembrana de Na+ y el potencial de membrana como fuerzas impulsoras. SLC5A8 transporta acetato, propionato, butirato, lactato, piruvato, 3-bromopiruvato, nicotinato, β-hidroxibutirato y piroglutamato (Miyauchi et al., 2004, 2010; Gopal et al., 2004, 2005; Martin et al., 2006; Thangaraju et al., 2006, 2008, 2009a). Nos preguntamos si este transportador altamente acoplado energéticamente aceptaría el dicloroacetato como sustrato. Esta cuestión es directamente relevante para la actividad antitumoral de este fármaco porque las células tumorales silencian este transportador mediante mecanismos epigenéticos (Ganapathy et al., 2005, 2008, 2009; Gupta et al., 2006). Por lo tanto, emprendimos el presente estudio para abordar dos cuestiones: (a) ¿SLC5A8 transporta dicloroacetato? (b) ¿Depende la actividad antitumoral del fármaco de la expresión del transportador en las células tumorales? Los resultados del estudio muestran que SLC5A8 es obligatorio para la actividad antitumoral del dicloroacetato.
Resultados
SLC5A8transporta dicloroacetato de forma acoplada al Na+
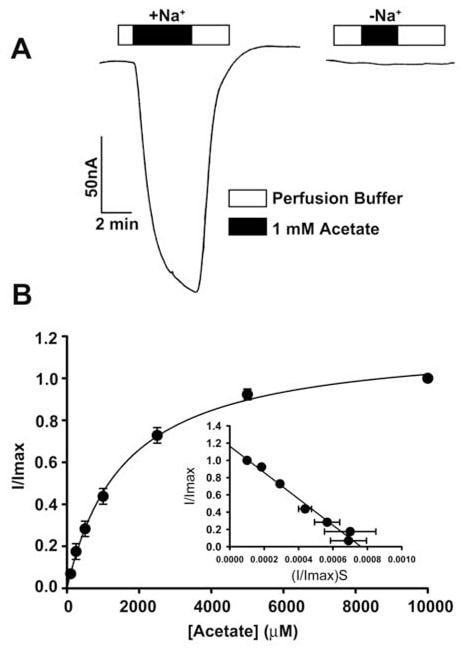
Se estudió el transporte de acetato y sus derivados clorados por SLC5A8 humano utilizando el sistema de expresión de oocitos de X. laevis. El transportador humano se expresó en oocitos de forma heteróloga mediante la inyección de ARNc SLC5A8. La función de transporte se monitorizó electrofisiológicamente mediante la técnica de pinzamiento de voltaje con dos microelectrodos. SLC5A8 funciona como un transportador acoplado a Na+ para monocarboxilatos con una estequiometría Na+: monocarboxilato de 2:1. El proceso de transporte es, por tanto, electrogénico. El proceso de transporte es, por tanto, electrogénico, asociado a la transferencia de una carga positiva neta a las células por ciclo de transporte. La despolarización resultante de la membrana puede ser monitorizada como una corriente entrante bajo condiciones de pinza de voltaje. Como puede observarse en la Fig. 1A, la exposición de oocitos que expresan SLC5A8 a acetato (1 mM) indujo corrientes entrantes cuando se monitorizaron en presencia de Na+ en el medio de perfusión (129 ± 9 nA; n = 3 oocitos). Sin embargo, estas corrientes no se observaron en ausencia de Na+. Estos datos demuestran que SLC5A8 media el transporte de acetato de una manera acoplada al Na+. La cinética de saturación reveló que la constante de Michaelis (Kt) para el proceso de transporte era de 1,6 ± 0,1 mM. Tras establecer la actividad funcional del SLC5A8 humano clonado utilizando acetato como control positivo, examinamos el transporte de dicloroacetato. La exposición de oocitos que expresaban SLC5A8 a dicloroacetato (0,25 mM) indujo marcadas corrientes entrantes en presencia de Na+ (153 ± 28 nA; n = 5 oocitos) (Fig. 2A). Estas corrientes no se observaron en ausencia de Na+. El proceso de transporte fue saturable con un Kt de 36 ± 7 μM (Fig. 2B). Así, la afinidad del dicloroacetato por el transportador es ~45 veces mayor que la del acetato. Las corrientes inducidas por dicloroacetato (0,25 mM) aumentaron al aumentar la concentración de Na+ en el medio de perfusión (Fig. 2C). La relación fue sigmoidal, indicando la participación de más de un Na+ en el proceso de activación. El análisis de los datos según la ecuación de Hill dio un valor de 2,1 ± 0,2 para el coeficiente de Hill. Estos datos muestran que la estequiometría Na+: dicloroaceato para el proceso de transporte es 2:1.
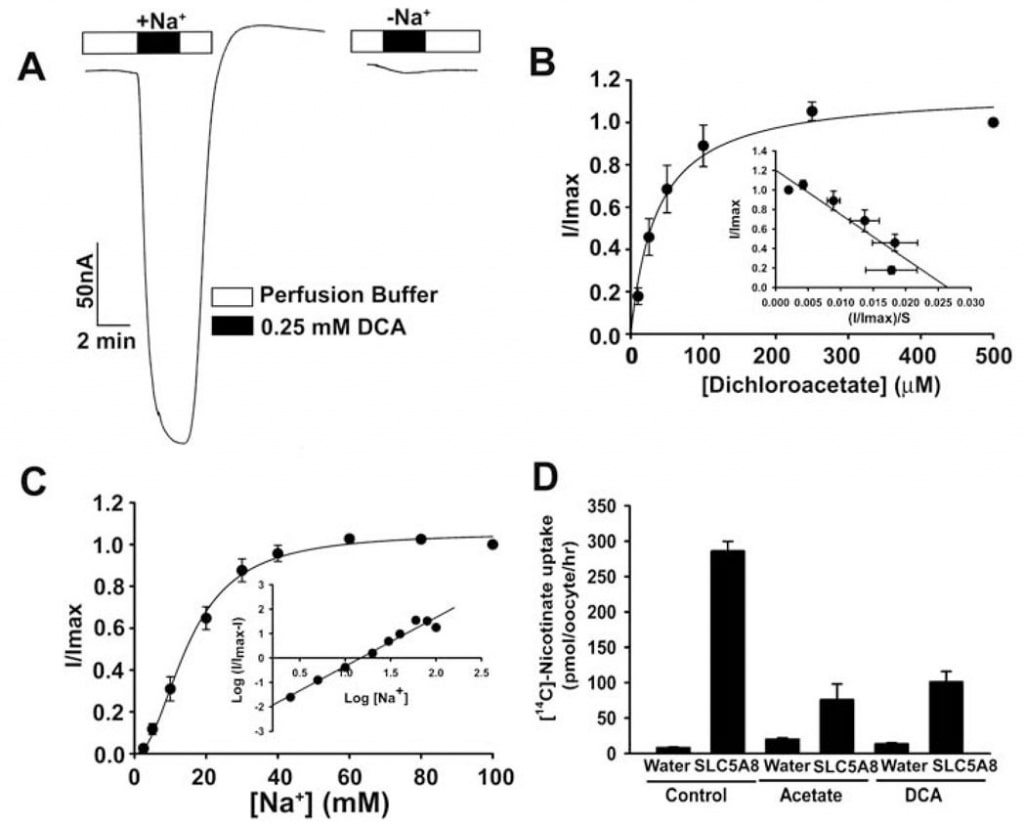
También utilizamos un método alternativo para evaluar el transporte de dicloroacetato a través de SLC5A8. Este método utilizó [14C]-nicotinato como sustrato para SLC5A8. Los ovocitos inyectados en agua no expresaban el transportador, por lo que se utilizaron como control. La captación de [14C]-nicotinato (50 μM) fue 60 veces mayor en los ovocitos que expresaban SLC5A8 que en los ovocitos inyectados con agua (Fig. 2D). La captación de nicotinato específica de SLC5A8 se inhibió en más de un 80% en presencia de acetato (0,25 mM) o dicloroacetato (0,25 mM), lo que indica que el acetato y el dicloroacetato compiten con el nicotinato en la captación a través de SLC5A8.
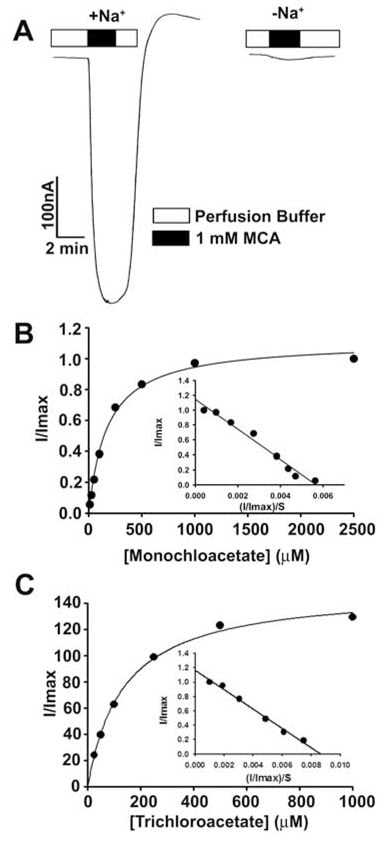
Estos resultados demostraron que el dicloroacetato es un sustrato de alta afinidad para el SLC5A8 humano. A continuación, estudiamos el transporte de monocloroacetato y tricloroacetato con fines comparativos (Fig. 3). Ambos compuestos fueron transportados a través de SLC5A8 de una manera acoplada al Na+ y saturable. El Kt fue de 177 ± 16μM para el monocloroacetato y de 134 ± 11 μM para el tricloroacetato.
Laapoptosis inducida por dicloroacetatoen células cancerosas requiere SLC5A8
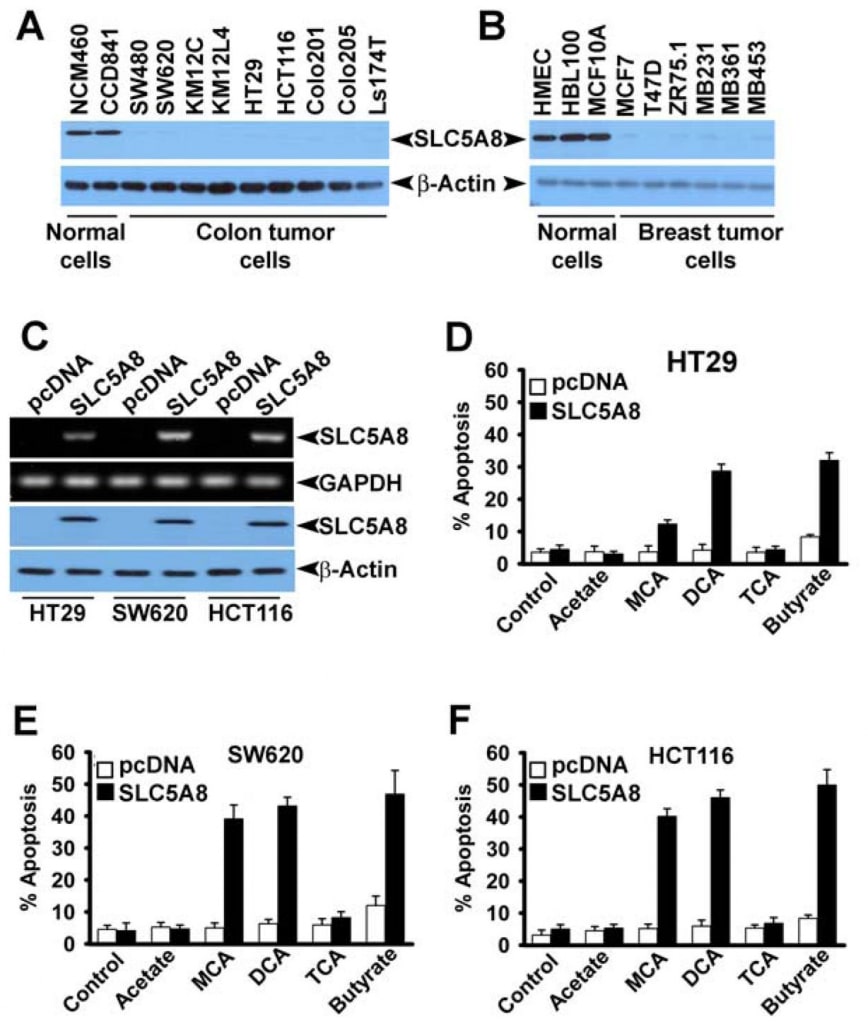
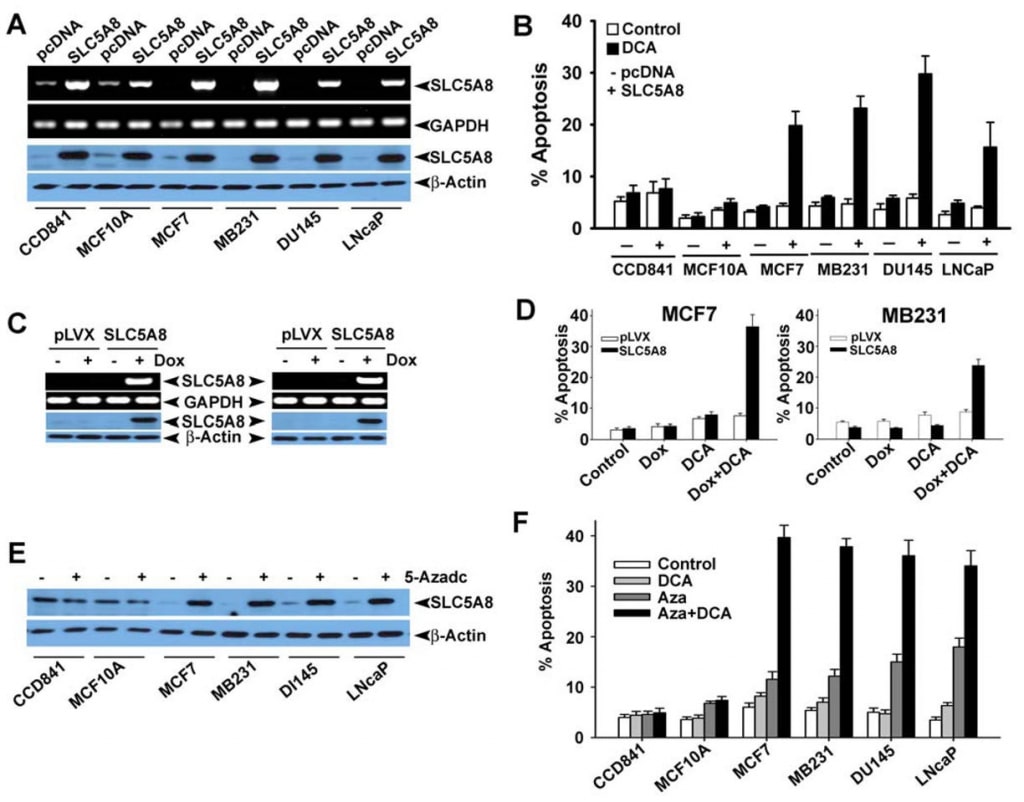
Aunque varios estudios han demostrado que el dicloroacetato induce la apoptosis en diversas líneas celulares cancerosas (Bonnet et al., 2007; Wong et al., 2008; Cao et al., 2008), una investigación reciente no pudo confirmar estos hallazgos (Stockwin et al., 2010). Los estudios de Bonnet et al. (2007) demostraron que el dicloroacetato a una concentración de 0,5 mM era capaz de inducir cambios metabólicos específicamente en células cancerosas y no en células normales. Estos cambios incluían la despolarización de la membrana mitocondrial, la supresión de la glucólisis, el aumento de la oxidación mitocondrial, la producción de especies reactivas de oxígeno, la inducción del canal de potasio de la membrana plasmática Kv1.5 y la liberación de factores pro-apoptóticos de las mitocondrias. Wong et al (2008) demostraron posteriormente que el dicloroacetato causaba apoptosis en células de cáncer de endometrio, y Cao et al (2008) demostraron que el compuesto sensibilizaba las células de cáncer de próstata a la radiación. En cambio, los estudios de Stockwin et al (2010) demostraron que, aunque el dicloroacetato era capaz de inducir la despolarización mitocondrial y la generación de especies reactivas del oxígeno, estos cambios se producían tanto en las células cancerosas como en las normales. Además, se requería una concentración muy alta del compuesto (≥25 mM) para inducir la apoptosis. Basándonos en los hallazgos de nuestro presente estudio de que SLC5A8 media la entrada activa acoplada a energía de dicloroacetato en las células y en el hecho de que las células cancerosas silencian el transportador, nos preguntamos si la ausencia del transportador en las células cancerosas era la razón de la falta de apoptosis detectable a bajas concentraciones del compuesto observada por Stockwin et al (2010). Abordamos esta cuestión utilizando tres líneas celulares diferentes de cáncer de colon humano (HCT116, SW620 y HT29). Estas tres líneas celulares no expresan SLC5A8 (Thangaraju et al., 2008).
También confirmamos la ausencia de expresión de SLC5A8 en líneas celulares humanas de cáncer de colon y mama a nivel proteico (Fig. 4A y B). Expresamos SLC5A8 en las líneas celulares HCT116, SW620 y HT29 mediante transfección transitoria de un constructo de expresión de mamífero y confirmamos la expresión mediante RT-PCR y análisis Western blot (Fig. 4C). Las células transfectadas con el vector vacío sirvieron de control. A continuación, expusimos las células de control y las que expresaban SLC5A8 a dicloroacetato (1 mM) durante 48 h y monitorizamos la apoptosis (Fig. 4D-F). Los resultados fueron interesantes. En las células de control, que no expresaban el transportador, el dicloroacetato no tuvo ningún efecto significativo. Sin embargo, en condiciones idénticas, las células que expresaban SLC5A8 sufrieron apoptosis en gran medida. Este fenómeno se observó en las tres líneas celulares de cáncer de colon. El monocloroacetato también se comportó de forma similar al dicloroacetato en las células HCT116 y SW620, aunque no en las células HT29, en las que la capacidad del monocloroacetato para inducir la apoptosis fue significativamente inferior a la del dicloroacetato. El acetato y el tricloroacetato no tuvieron ningún efecto apreciable. Utilizamos butirato como control positivo en estos experimentos basándonos en nuestro informe anterior de que la inducción de la apoptosis por butirato en líneas celulares de cáncer de colon depende obligatoriamente de la expresión de SLC5A8 (Thangaraju et al., 2008).
A continuación quisimos determinar si la apoptosis inducida por dicloroacetato presenta selectividad de células tumorales y también si el efecto se observa en líneas celulares cancerosas de origen tisular distinto del colon. Para ello, seleccionamos CCD841 y MCF10A como representantes de líneas celulares normales (CCD841, colon; MCF10A, epitelio mamario) y cuatro líneas celulares de cáncer humano: MCF7 (línea celular de cáncer de mama con receptor de estrógenos positivo), MB231 (línea celular de cáncer de mama con receptor de estrógenos negativo), DU145 (línea celular de cáncer de próstata insensible a los andrógenos) y LNCaP (línea celular de cáncer de próstata sensible a los andrógenos). Como ya se informó anteriormente (Thangaraju et al., 2006, 2008), las líneas celulares normales CCD841 y MCF10A expresaron niveles detectables de SLC5A8, tanto a nivel de ARNm como de proteína (Fig. 5A). En cambio, ninguna de las cuatro líneas celulares cancerosas aquí examinadas expresaba el transportador. La expresión se hizo evidente en las líneas celulares cancerosas tras la transfección transitoria de un constructo de expresión de mamífero. En las células normales, los niveles de expresión aumentaron tras la transfección. Utilizando estas líneas celulares, comparamos la capacidad del dicloroacetato para inducir la apoptosis entre líneas celulares normales y cancerosas (Fig. 5B). No encontramos diferencias significativas en la apoptosis en las líneas celulares normales con y sin tratamiento con dicloroacetato (1 mM). El aumento de los niveles de expresión de SLC5A8 tampoco tuvo ningún efecto sobre el grado de apoptosis. En cambio, el dicloroacetato fue capaz de inducir una apoptosis marcada en las cuatro líneas celulares cancerosas, pero sólo si las células expresaban el transportador. Sin la expresión del transportador, las líneas celulares cancerosas no sufrieron apoptosis tras el tratamiento con dicloroacetato. Estos resultados demuestran que el dicloroacetato es capaz de causar apoptosis incluso a concentraciones tan bajas como 1 mM de forma selectiva para las células cancerosas, pero sólo si se expresa SLC5A8. Estas observaciones también se confirmaron con la expresión estable de SLC5A8 mediada por virus lenti (inducible por doxicilina) en dos líneas celulares de cáncer de mama, MCF7 y MB231 (Fig. 5C, D). Está bien establecido que el silenciamiento de SLC5A8 está asociado a la metilación del ADN y que el tratamiento de células cancerosas con agentes desmetilantes del ADN reactiva la expresión de SLC5A8. Por lo tanto, tratamos líneas celulares normales y cancerosas con 5′-aza-2-deoxicitidina (5-Azadc), un agente desmetilante del ADN, y la reactivación de SLC5A8 se confirmó mediante análisis de inmunotransferencia (Fig. 5E). el tratamiento con 5-Azadc no alteró la expresión proteica de SLC5A8 en las células epiteliales normales de colon y mamarias, mientras que reactivó la expresión de SLC5A8 en todas las líneas celulares cancerosas. Además, el tratamiento de estas células con 5-Azadc indujo por sí mismo una apoptosis significativa; sin embargo, el tratamiento de estas células con dicloroacetato (1 mM) potenció drásticamente la apoptosis inducida por 5-Azadc (Fig. 5F).
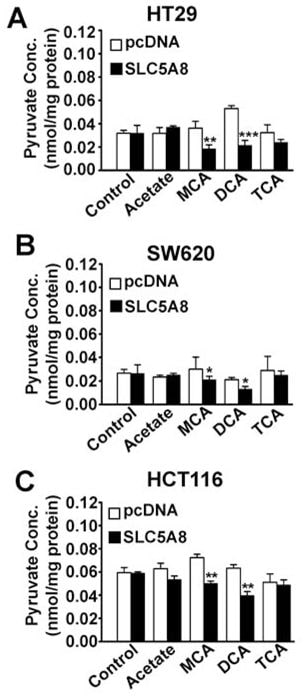
Efecto del dicloroacetato en los niveles intracelulares de piruvato
¿Cuál es el mecanismo responsable de la inducción de la apoptosis por SLC5A8/dicloroacetato en células cancerosas? SLC5A8 es un transportador activo para el dicloroacetato con un Kt de 36 ± 7μM. Por lo tanto, el compuesto se concentrará en las células cancerosas si se expresa el transportador. Esto explicaría el requisito obligatorio de SLC5A8 para que concentraciones bajas de dicloroacetato induzcan la apoptosis en estas células. Si el transportador no se expresa, es posible que las células no acumulen el compuesto a niveles suficientes para provocar la apoptosis. Una vez acumulado en el interior de las células cancerosas, ¿cómo actúa el dicloroacetato para inducir la apoptosis? El único mecanismo de acción conocido del dicloroacetato es su capacidad para activar la PDC mediante la inhibición de la PDK (Stacpoole et al., 1998, 2003, 2008). Esto daría lugar a la estimulación de la oxidación mitocondrial, la generación de especies reactivas de oxígeno, la despolarización del potencial de membrana mitocondrial y la inducción de la apoptosis. La activación de la PDC por dicloroacetato en células intactas disminuiría los niveles intracelulares de piruvato. Basándonos en esto, planteamos la hipótesis de que el dicloroacetato disminuiría los niveles de piruvato en las células cancerosas y que el efecto dependería obligatoriamente de la expresión de SLC5A8. Pusimos a prueba esta hipótesis en tres líneas celulares de cáncer diferentes (HT29, SW620 y HCT116) con y sin expresión exógena de SLC5A8 (Fig. 6). La expresión del transportador por sí misma no tuvo ningún efecto sobre los niveles intracelulares de piruvato. El dicloroacetato fue capaz de disminuir los niveles de piruvato de forma significativa en las tres líneas celulares de cáncer, pero sólo cuando se expresaba el transportador. Curiosamente, el monocloroacetato también tuvo un efecto similar, de nuevo sólo en presencia de SLC5A8; por el contrario, el tricloroacetato y el acetato no fueron capaces de afectar a los niveles de piruvato.
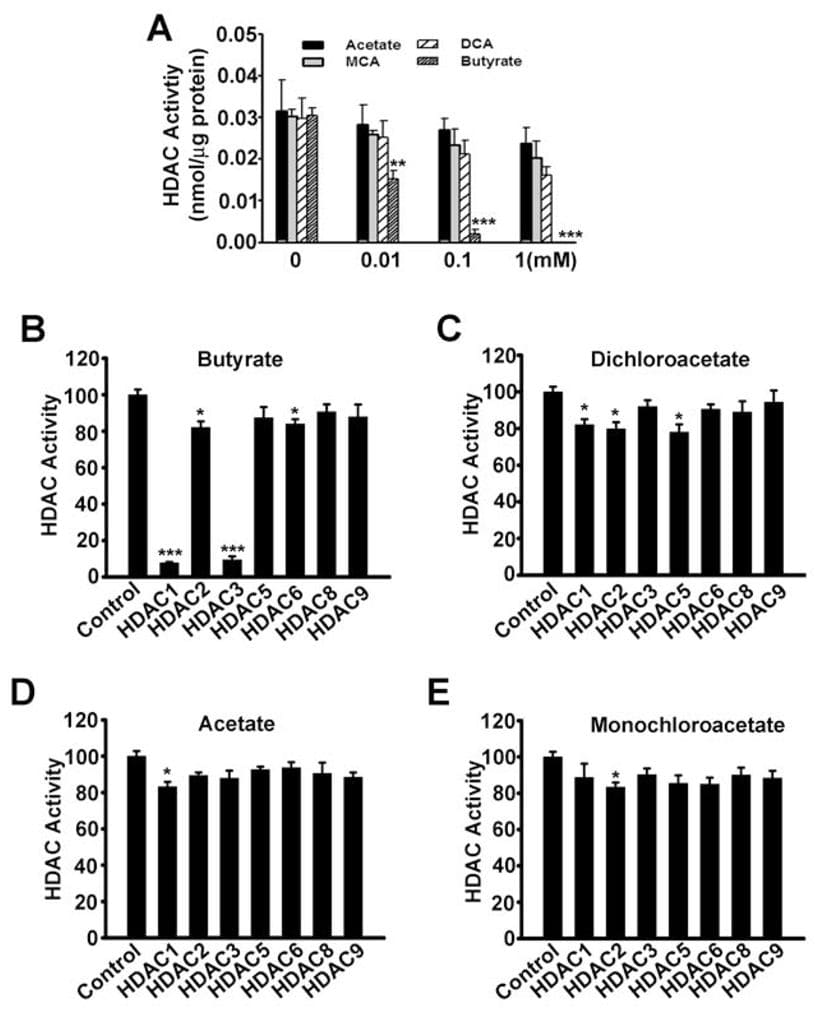
Hasta ahora, el único mecanismo por el que SLC5A8 funciona como supresor tumoral era a través de su capacidad para concentrar en el interior de las células butirato y piruvato, que son inhibidores de las histonas desacetilasas. En el presente estudio hemos demostrado que el dicloroacetato también induce la apoptosis en células cancerosas y que el efecto depende de la expresión de SLC5A8. Estos resultados son similares a los obtenidos con el butirato y el piruvato. Por lo tanto, nos preguntamos si existe alguna implicación de la inhibición de las histonas deacetilasas en la apoptosis inducida por dicloroacetato en células cancerosas. Sin embargo, esto parecía poco probable porque el acetato no es un inhibidor de las histonas deacetilasas (Thangaraju et al., 2006). Sin embargo, no podíamos descartar la posibilidad de que el dicloroacetato funcionara como inhibidor de las histonas desacetilasas. Por lo tanto, examinamos la influencia del acetato, monocloroacetato, dicloroacetato y butirato en la actividad de las histonas desacetilasas. En primer lugar, utilizamos lisados de células SW620 como fuente de histona desacetilasas (Fig. 7A). El butirato se utilizó como control positivo en estos experimentos. Como era de esperar, el butirato inhibió la actividad de las histonas desacetilasas. En condiciones idénticas, el dicloroacetato fue al menos 100 veces menos potente que el butirato en la inhibición de las histonas desacetilasas. A continuación, utilizamos isoformas recombinantes de histona deacetilasa humana para determinar si el dicloroacetato podía inhibir isoformas específicas de la enzima. Nuestros estudios anteriores han demostrado que el butirato es un inhibidor específico de las isoformas de histona deacetilasa HDAC1 y HDAC3 (Thangaraju et al., 2009b). En el presente estudio, encontramos lo mismo con butirato (Fig. 7B). El dicloroacetato también inhibió las isoformas HDAC1, HDAC2 y HDAC5 de forma significativa, pero la potencia fue muy baja (~20% de inhibición a una concentración de 1 mM) (Fig. 7C). El acetato (Fig. 7D) y el monocloroacetato (Fig. 7E) no tuvieron ningún efecto sobre ninguna de las isoformas examinadas en el estudio.
Discusión
Desde la publicación en Cancer Cell del artículo de Bonnet et al (2007) que demostraba que el dicloroacetato promueve la apoptosis en líneas celulares cancerosas in vitro e inhibe el crecimiento del cáncer en xenoinjertos de ratón in vivo, ha aumentado constantemente el interés público por la posible utilidad de este compuesto como fármaco anticanceroso. El dicloroaetato se ha venido utilizando como fármaco para el tratamiento de la acidosis láctica congénita y, por tanto, se han elaborado detalles sobre la farmacocinética y el perfil de toxicidad del fármaco (Stacpoole at al., 1998). Los recientes descubrimientos de que el fármaco también puede ser útil en el tratamiento del cáncer han llevado a muchos pacientes oncológicos a utilizar este fármaco no aprobado sin la recomendación de sus médicos (Pearson, 2007). Curiosamente, no se han realizado ensayos clínicos controlados que documenten la eficacia terapéutica del dicloroacetato como medicamento contra el cáncer. Dado que la estructura del dicloroacetato no se puede patentar, al parecer las empresas farmacéuticas no estaban interesadas en desarrollar el fármaco para la terapia del cáncer mediante ensayos clínicos, lo que impulsó al investigador principal de la publicación original en Cancer Cell a recaudar fondos para realizar sus propios ensayos clínicos a pequeña escala (Pearson, 2007). Los resultados del ensayo clínico se han publicado recientemente (Machelakis et al., 2010), lo que ha demostrado que el fármaco es eficaz en pacientes con gliobastoma. Desgraciadamente, aunque el fármaco fue eficaz in vivo en el tratamiento del glioblastoma, fue necesario utilizar altas concentraciones del fármaco para obtener eficacia terapéutica, lo que provocó neuropatía periférica. Éste fue el único efecto secundario indeseable que limitó el uso de dosis más elevadas del fármaco para lograr una mayor eficacia terapéutica. No hubo indicios de toxicidad hematológica, hepática, renal o cardiaca. Varios estudios han documentado complicaciones neurológicas con dosis altas de dicloroacetato en animales y en humanos (Calcutt et al., 2009; Wiemer y Sachdev, 2009; Brandsma et al., 2010). Las complicaciones neurológicas sólo se producen con dosis muy elevadas de dicloroacetato: 12.5 mg/kg/día durante 3 meses (Bonnet et al., 2007) o 15 mg/kg/día durante 4 semanas (Brandsma et al., 2010). La necesidad de altas dosis de dicloroacetato para ser terapéuticamente eficaz in vivo concuerda con la mayoría de los datos in vitro publicados recientemente que muestran que son necesarias concentraciones de 10 mM o superiores para causar apoptosis en líneas celulares de cáncer (Stockwin et al., 2010; Madhok et al., 2010; Xiao et al., 2010; Heshe et al., 2010; Sun et al., 2010).
Existen pruebas convincentes de que el dicloroacetato tiene la capacidad de invertir el fenotipo metabólico de las células cancerosas y promover su muerte, pero el efecto antitumoral sólo se observa a concentraciones muy altas tanto in vitro como in vivo. Dado que a estas concentraciones terapéuticamente eficaces se produce toxicidad neurológica, la utilidad del compuesto como fármaco anticanceroso es limitada en humanos. Curiosamente, la única diana molecular de la actividad antitumoral del dicloroacetato es la PDK, que es inhibida por el compuesto a concentraciones micromolares (constante de inhibición, 50-100 μM) (Whitehouse et al., 1974; Cooper et al., 1974). Entonces, ¿por qué se necesitan altas concentraciones milimolares de dicloroacetato in vitro para destruir las células cancerosas? Hay que responder a esta pregunta para poder explotar el potencial de este compuesto como fármaco anticanceroso. Nuestros estudios actuales proporcionan una respuesta satisfactoria a esta importante cuestión. Las células cancerosas no expresan ningún sistema de transporte que pueda transferir eficazmente el compuesto desde el medio extracelular a su diana intracelular. Los transportadores de monocarboxilato que se expresan en las células cancerosas transportan dicloroacetato con un Kt de 0,6 mM (Jackson y Halestrap, 1996). Se trata de un proceso de transporte de baja afinidad. Además, los transportadores de monocarboxilato no son altamente concentrativos. Además, las células tumorales generan grandes cantidades de lactato, que también es un sustrato para los transportadores de monocarboxilato con una afinidad comparable a la del dicloroacetato, lo que provoca competencia en el proceso de transporte. Los presentes estudios demuestran que SLC5A8 transporta dicloroacetato con una afinidad considerablemente mayor que la de los transportadores de monocarboxilato (Kt: 36 μM frente a 600 μM). Aún más importante es el hecho de que SLC5A8 es mucho más eficaz que los transportadores de monocarboxilato en el transporte de dicloroacetato debido a la energización del primero por un gradiente electroquímico transmembrana de Na+. Es interesante observar que la afinidad por el transportador cambia drásticamente por la cloración del segundo carbono en el acetato (Kt: 1572 ± 101 μM para el acetato; 177 ± 16 μM para el monocloroacetato; 36 ± 7 μM para el dicloroacetato; 134 ± 11 μM para el tricloroacetato). Nuestros resultados nos llevan a concluir que las células tumorales son resistentes a la apoptosis inducida por dicloroacetato porque estas células no poseen sistemas de transporte eficaces para el fármaco. Esta es la razón más probable por la que se necesitan concentraciones muy elevadas del compuesto para inducir la muerte celular en las células tumorales. También es posible que la muerte celular observada en células tumorales a estas altas concentraciones no se deba a la activación de la PDC porque también se observan efectos similares en células normales (Stockwin et al., 2010).
En el presente estudio, hemos demostrado que, si SLC5A8 se expresa en células tumorales, el dicloroacetato induce la muerte celular a una concentración relativamente más baja (1 mM) de forma selectiva para células tumorales. En condiciones idénticas, las células normales (CCD841 y MCF10A) no se ven afectadas. La resistencia de las células normales al dicloroacetato no se debe a la ausencia de un sistema de transporte para el fármaco. Las células normales expresan SLC5A8 de forma constitutiva, y hemos demostrado que las células no sufren apoptosis con o sin expresión adicional del transportador. Esto confirma el efecto específico del dicloroacetato sobre las células tumorales. Nuestros estudios también han demostrado que la eficacia del dicloroacetato como inductor de la apoptosis se observa no sólo con líneas celulares de cáncer de colon, sino también con líneas celulares de cáncer de mama y líneas celulares de cáncer de próstata. El mecanismo de la muerte celular inducida por el dicloroacetato en las células cancerosas se debe principalmente a la activación de la oxidación mitocondrial del piruvato. Esto se ve apoyado por los hallazgos de que los niveles intracelulares de piruvato en las células cancerosas disminuyen en respuesta al tratamiento con dicloroacetato de una manera dependiente de SLC5A8, lo que sugiere que la entrada concentrativa del fármaco en las células cancerosas mediada por el SLC5A8 expresado exógenamente es responsable de este efecto. El dicloroacetato no inhibe las histonas deacetilasas, lo que excluye la inhibición de las histonas deacetilasas como mecanismo potencial de apoptosis en células tumorales inducida por el compuesto.
Los hallazgos de que el dicloroacetato induce selectivamente la muerte celular en células tumorales a bajas concentraciones, pero sólo si se expresa SLC5A8, tienen importancia clínica y terapéutica. La capacidad del dicloroacetato para activar la PDC a través de la inhibición de la PDK en las células cancerosas proporciona una base mecánicamente racional para la actividad antitumoral del compuesto. Sin embargo, las células cancerosas son resistentes al fármaco debido a la ausencia de un transportador eficaz para el fármaco, lo que obliga a utilizar altas concentraciones del compuesto para inducir la muerte celular, lo que desgraciadamente provoca efectos secundarios perjudiciales como la neuropatía. En el presente estudio hemos demostrado que SLC5A8 sirve como transportador activo para el dicloroacetato. Sin embargo, dado que la expresión del transportador está silenciada en las células tumorales, ¿cómo pueden ser relevantes los presentes hallazgos para el potencial uso terapéutico del fármaco? El silenciamiento de SLC5A8 en las células cancerosas se produce a través de mecanismos epigenéticos que implican la metilación del ADN; el tratamiento de las células cancerosas con 5′-azacitidina, un inhibidor de la metilación del ADN, reactiva la expresión del gen (Li et al., 2003; Ueno et al., 2004; Hong et al., 2005; Porra et al., 2005; Thangaraju et al., 2006; Park et al., 2007, 2008). La metilación del ADN desempeña un papel fundamental en el silenciamiento de genes supresores de tumores en diversos tipos de cáncer, y los inhibidores de la metilación del ADN son prometedores como fármacos contra el cáncer (Baylin, 2005). Dos compuestos con actividad inhibidora de la metilación del ADN están en uso clínico para el tratamiento de neoplasias hematológicas. Se trata de la 5′-aza-2′-deoxicitidina, también conocida como decitabina (nombre comercial, Dacogen) y la 5′-azacitidina (nombre comercial, Vidaza). Estudios in vitro han demostrado que el tratamiento de una variedad de líneas celulares de cáncer con estos compuestos reactiva la expresión de SLC5A8. Especulamos que el mismo fenómeno se produciría también in vivo. Por lo tanto, es probable que una combinación de un inhibidor de la metilación del ADN y dicloroacetato sea eficaz para el tratamiento del cáncer porque el inhibidor de la metilación del ADN induciría la expresión de SLC5A8 en los tumores, que entonces transportaría eficazmente el dicloroacetato a las células tumorales para provocar su actividad antitumoral. Este modo de tratamiento reduciría considerablemente la concentración de dicloroacetato necesaria para la eficacia in vivo como agente anticanceroso, proporcionando así potencialmente selectividad tumoral y evitando también los efectos secundarios perjudiciales como la neuropatía. Los resultados del presente estudio proporcionan una base racional para este tipo de terapia combinada.
Materiales y métodos
Materiales
[ 14C]-Nicotinate se adquirió de American Radiolabeled Chemicals (St. Louis, MO). El acetato y sus derivados clorados se obtuvieron de Sigma (St. Louis, MO). SLC5A8 se clonó originalmente a partir de intestino humano (Miyauchi et al., 2004).
Sistema de expresión de ovocitos de X. laevis
Se sintetizó ARNc capsulado a partir de ADNc de SLC5A8 humano (clonado en pGH19, un vector de expresión de ovocitos de X. laevis) utilizando el kit mMESSAGE-mMACHINE (Ambion, Austin, TX). Los ovocitos maduros de X. laevis se aislaron mediante tratamiento con colagenasa A (1,6 mg/ml), se defoliaron manualmente y se mantuvieron a 18 °C en medio de Barth modificado, suplementado con 25 μg/ml de gentamicina. Se inyectaron 50 ng de ARNc en los ovocitos. Los ovocitos inyectados en agua sirvieron de control. Los estudios electrofisiológicos se realizaron mediante el método de pinza de voltaje de dos microelectrodos. Los oocitos se perifundieron con un tampón que contenía NaCl (100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, pH 7,5), seguido del mismo tampón que contenía acetato o sus derivados clorados. El potencial de membrana se fijó a -50 mV. Las diferencias entre las corrientes en estado estacionario medidas en presencia y en ausencia de sustratos se consideraron corrientes inducidas por sustratos. En el análisis de la cinética de saturación de las corrientes inducidas por sustrato, el parámetro cinético Kt (es decir, la concentración de sustrato necesaria para la inducción de la corriente semimáxima) se calculó ajustando la ecuación de Michaelis-Menten a los valores de las corrientes inducidas por sustrato. La cinética de activación por Na+ de las corrientes inducidas por sustrato se analizó midiendo las corrientes específicas de sustrato en presencia de concentraciones crecientes de Na+. Los datos de las corrientes dependientes del Na+ se analizaron según la ecuación de Hill para determinar el coeficiente de Hill (el número de iones Na+ implicados en el proceso de activación). Dado que los niveles de expresión variaban significativamente de un ovocito a otro, los análisis cinéticos se realizaron normalizando los niveles de expresión. Esto se hizo tomando como 1 la corriente específica de SLC5A8 inducida al máximo en cada experimento cinético en oocitos individuales. Cada experimento se repitió con 3 o 4 oocitos diferentes. La actividad del SLC5A8 expresado heterológicamente en oocitos también se monitorizó mediante la captación de [14C]-nicotinato. La concentración de nicotinato en estos experimentos fue de 50 μM y el tiempo de incubación fue de 1 h.
El protocolo para el uso de ranas en estos experimentos fue aprobado por el Comité Institucional para el Cuidado y Uso de Animales.
Expresión ectópica de SLC5A8 en líneas celulares cultivadas
Se sembraron células en placas de cultivo de 35 mm y se cultivaron en ausencia de piruvato. Las células se transfectaron con pcADN o ADNc de SLC5A8 humano utilizando Fugene 6 y Opti-MEM. Se utilizó pEGFP-N1 para la cotransfección con el fin de determinar la eficacia de la transfección. Tras 24 h, las células se trataron con o sin acetato o sus derivados clorados (1 mM) durante 24 h. Para el análisis FACS, las células se fijaron en etanol al 50%, se trataron con citrato sódico al 0,1%, 1 mg/ml de RNasa A y 50 μg/ml de yoduro de propidio, y a continuación se sometieron a un análisis de clasificación celular activada por fluorescencia (FACS; FACS Caliber, Becton Dickinson).
Generación de líneas celulares estables que expresan SLC5A8
Generamos clones estables que expresan SLC5A8 en dos líneas celulares, MCF7y MDA-MB231, para expresar condicionalmente SLC5A8 colocando el ADNc de SLC5A8 bajo el control del promotor dependiente de Tet (Tet-On Advanced, Clontech). El sistema viene con dos elementos, el vector regulador (pLVX-Tet-On Advanced) y el vector de respuesta (pLVX-Tight-Puro). Brevemente, el virus lenti recombinante se produjo por co-transfección en células 293FT del vector regulador (pLVX-Tet-On Advanced) y otros tres vectores ayudantes, pLP-1, pLP-2 y pVSVG (Invitrogen) utilizando el reactivo de transfección Lipofectamine 2000. El sobrenadante viral Lenti se recogió 72 h después de la transfección y se filtró a través de una membrana de 0,45 μm. Las células MCF7 y MDA-MB231 se infectaron durante 24 h con el virus lenti en medio que contenía 8 μg/ml de polibreno y se cultivaron durante 48 h adicionales. Las células se seleccionaron con G418 (2 mg/ml) y la expresión de la proteína rTet R se comprobó con Western blot. SLC5A8 se subclonó en pLVX-Tight-Puro y SLC5A8 pLVX-Tight-Puro se transdujo en células MCF7 y MDA-MB231 expresando la proteína rTetR con el mismo procedimiento descrito anteriormente. Las células se seleccionaron con 2 μg/ml de puromicina y se mantuvieron en medio que contenía FBS libre de Tet, 0,25 μg/ml de puromicina y 250 μg/ml de G418. La inducción del ARNm de SLC5A8 tras la adición de doxiciclina se confirmó mediante RT-PCR.
Reactivación de la expresión de SLC5A8 mediante el agente desmetilante del ADN
Lascélulas se sembraron en placas de 10 cm a muy baja densidad (0,5 ×106 células/placa) y se cultivaron en el medio respectivo sin piruvato sódico. Tras 24 h, las células se trataron con 5′-Azadc (2 μg/ml) durante 72 h. El medio se sustituyó con medio fresco que contenía 5′-Azadc cada 24 h. Tras el tratamiento con 5′-Azadc, las células se trataron con DCA (1 mM) durante 48 h. Tras el tratamiento, las células se cosecharon y se procesaron para la extracción de proteínas, así como para el análisis FACS.
Transcripción inversa-PCR
El ARN preparado a partir de líneas celulares normales y cancerosas se utilizó para la RT PCR semicuantitativa. El ARN (2 μg) se transcribió inversamente en ADNc utilizando el sistema GeneAmp PCR (Roche). Los cebadores de PCR para genes específicos se diseñaron basándose en las secuencias de nucleótidos disponibles en GenBank. La transcripción inversa-PCR (RT-PCR) se repitió dos veces con cada muestra de ARN. Se utilizó GAPDH (gliceraldehído-3-fosfato deshidrogenasa) como control interno.
Análisis Western blot
Se fraccionaron 50 microgramos de proteína mediante SDS-PAGE, y las proteínas fraccionadas se transfirieron a una membrana de nitrocelulosa (Whatman GmbH). Las membranas se bloquearon con leche en polvo desgrasada al 5% y se expusieron al anticuerpo primario anti-SLC5A8 (Cat. # ARP44110, Aviva System Biology) a 4 °C durante la noche, seguido de tratamiento con anticuerpos secundarios apropiados. Las proteínas se visualizaron mediante ECL SuperSignal Western System (GE Healthcare).
Análisis de apoptosis
Las células se fijaron en etanol al 50%, se trataron con citrato sódico al 0,1%, 1 mg/mL de RNasa A y 50 μg/mL de yoduro de propidio, y se sometieron a análisis de clasificación celular activada por fluorescencia (FACS, Becton Dickinson).
Medición de la actividad HDAC
Para determinar la actividad HDAC se utilizó un kit comercial (Biovision, Mountain View, CA). Cuando se utilizó el lisado celular de la línea celular de cáncer de colon SW620 como fuente de actividad HDAC, se añadieron 100 μg de proteína en el lisado al ensayo enzimático en presencia o ausencia de acetato o sus derivados clorados (1 mM). La actividad de las isoformas humanas recombinantes de HDAC también se midió utilizando el mismo kit. Las isoformas HDAC recombinantes se adquirieron a Cayman Chemical Company.
Medición de los niveles intracelulares de piruvato
Las células se sembraron en placas de cultivo de 35 mm y se cultivaron en ausencia de piruvato. Las células se transfectaron con pcDNA o cDNA SLC5A8 humano utilizando Fugene 6 y Opti-MEM. Tras 24 h, las células se trataron con o sin acetato o sus derivados clorados (1 mM) durante 24 h. A continuación, se utilizaron lisados celulares para medir el piruvato mediante un kit comercial (Biovision, Mountain View, CA).
Agradecimientos
El trabajo aquí descrito está financiado en parte por la beca CA131402 de los Institutos Nacionales de Salud.
REFERENCIAS
1 Baylin SB. Metilación del ADN y silenciamiento génico en el cáncer. Nat Clin Pract Oncol. 2005; 2:S4-S11. [PubMed: 16341240]
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondriaK + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Célula del Cáncer. 2007; 11:37-51. [PubMed: 17222789]
2Brahimi-HornMC, Chiche J, Pouyssegur J. Hypoxia signaling controls metabolic demand. Curr Opin Cell Biol. 2007; 19:223-229. [PubMed: 17303407]
2BrandsmaD, Dorlo TP, Haanen JH, Beijnen JH, Boogerd W. Severe encephalopathy and polyneuropathy induced by dichloroacetate. J Neurol. 2010 En prensa.
2CalcuttNA, Lopez VL, Bautista AD, Mizisin LM, Torres BR, Shroads AL, et al. Peripheral neuropathy in rats exposed to dichloroacetate. J Neuropathol Exp Neurol. 2009; 68:985-993. [PubMed: 19680144]
3CaoW, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Dichloroacetate (DCA) sensitizes both wild type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 2008; 68:1223-1231. [PubMed: 18465755]
4ChenZ, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007; 39:267-274. [PubMed: 17551814]
5CoadyMJ, Chang MH, Charron FM, Plata C, Wallendorff B, Sah JF, et al. The tumour suppressor gene SLC5A8 expresses a Na+-monocarboxylate cotransporter. J Physiol. 2004; 557:719-731. [PubMed: 15090606]
6CooperRH, Randle PJ, Denton RM. Regulation of heart muscle pyruvate dehydrogenase kinase. Biochem J. 1974; 143:625-641. [PubMed: 4462746]
7GanapathyV, Gopal E, Miyauchi S, Prasad PD. Biological functions of SLC5A8, a candidate tumor suppressor. Biochem Soc Trans. 2005; 33:237-240. [PubMed: 15667316]
8GanapathyV, Thangaraju M, Gopal E, Itagaki S, Miyauchi S, Prasad PD. Sodium-coupled monocarboxylate transporters in normal tissues and in cancer. AAPS J. 2008; 10:193-199. [PubMed: 18446519]
9GanapathyV, Thangaraju M, Prasad PD. Nutrient transporters in cancer: Relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009; 121:29-40. [PubMed: 18992769]
10GatenbyRA, Gillies RJ. ¿Por qué los cánceres tienen alta glucólisis aeróbica? Nat Rev Cancer. 2004; 4:891- 899. [PubMed: 15516961]
11GopalE, Fei YJ, Sugawara M, Miyauchi S, Zhuang L, Martin PM, et al. Expression of Slc5a8 in kidney and its role in Na+-coupled transport of lactate. J Biol Chem. 2004; 279:44522-44532. [PubMed: 15322102]
12GopalE, Fei YJ, Miyauchi S, Zhuang L, Prasad PD, Ganapathy V. Sodium-coupled and electrogenic transport of B-complex vitamin nicotinic acid by Slc5a8, a member of the Na+/glucose cotransporter gene family. Biochem J. 2005; 388:309-316. [PubMed: 15651982]
13GuptaN, Martin PM, Prasad PD, Ganapathy V. SLC5A8 (SMCT1)-mediated transport of butyrate forms the basis for the tumor suppressive function of the transporter. Life Sci. 2006; 78:2419- 2425. [PubMed: 16375929]
14HesheD, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol. 2010 En prensa.
15HongC, Maunakea A, Jun P, Bollen AW, Hodgson JG, Goldenberg DD, et al. Mecanismos epigenéticos compartidos en gliomas humanos y de ratón inactivan la expresión del supresor del crecimiento SLC5A8. Cancer Res. 2005; 65:3617-3623. [PubMed: 15867356]
16JacksonVN, Halestrap AP. La cinética, el sustrato y la especificidad del inhibidor del transportador de monocarboxilato (lactato) de células de hígado de rata determinado utilizando el indicador de pH intracelular fluorescente, 2′, 7′-bis (carboxietil)-5(6)-carboxifluoresceína. J Biol Chem. 1996; 271:861-868. [PubMed: 8557697]
17KimJW, Dang CV. Sweet tooth molecular del cáncer y el efecto Warburg. Cancer Res. 2006; 66:8927-8930. [PubMed: 16982728]
19LiH, Myeroff L, Smiraglia D, Romero MF, Pretlow TP, Kasturi L, et al. SLC5A8, a sodium transporter, is a tumor suppressor gene silenced by methylation in human colon aberrant crypt foci and cancers. Proc Natl Acad Sci USA. 2003; 100:8412-8417. [PubMed: 12829793]
20MadhokBM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloroacetate induces apoptosis and cellcycle arrest in colorectal cancer cells. Br J Cancer. 2010; 102:1746-1752. [PubMed: 20485289]
21MartinPM, Gopal E, Ananth S, Zhuang L, Itagaki S, Prasad BM, et al. Identity of SMCT1 (SLC5A8) as a neuron-specific Na+-coupled transporter for active uptake of L-lactate and ketone bodies in the brain. J Neurochem. 2006; 98:279-288. [PubMed: 16805814]
22MathupalaSP, KOYH, Pedersen PL. The pivotal roles of mitochondria in cancer: Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta. 2010; 1797:1225-1230. [PubMed: 20381449]
23MichelakisED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008; 99:989-994. [PubMed: 18766181]
24MichelakisED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010; 2:31-34.
25MiyauchiS, Gopal E, Fei YJ, Ganapathy V. Functional identification of SLC5A8, a tumor suppressor downregulated in colon cancer, as a Na+-coupled transporter for short-chain fatty acids. J Biol Chem. 2004; 279:13293-13296. [PubMed: 14966140]
26MiyauchiS, Gopal E, Babu E, Sonne SR, Kubo Y, Umapathy NS, et al. Sodium-coupled electrogenic transport of pyroglutamate (5-oxoproline) via SLC5A8, a monocarboxylate transporter. Biochim Biophys Acta. 2010; 1798:1164-1171. [PubMed: 20211600]
27ParkJY, Zheng W, Kim D, Cheng JQ, Kumar N, Ahmad N, et al. Candidate tumor suppressor gene SLC5A8 is frequently down-regulated by promoter hypermethylation in prostate cancer. Cancer Detect Prev. 2007; 31:359-365. [PubMed: 18037591]
28ParkJY, Helm JF, Zheng W, Ly QP, Hodul PJ, Centeno BA, et al. Silencing of the candidate tumor suppressor gene solute carrier family 5 member 8 (SLC5A8) in human pancreatic cancer. Pancreas. 2008; 36:e32-e39. [PubMed: 18437076]
29PearsonH. Cancer patients opt for unpproved drug. Nature. 2007; 446:474-475. [PubMed: 17392750]
30PorraV, Ferraro-Peyret C, Durand C, Selmi-Ruby S, Giroud H, Berger-Dutrieux N, et al. Silencing of the tumor suppressor gene SLC5A8 is associated with BRAF mutations is classical papillary thyroid carcinomas. J Clin Endocrinol Metab. 2005; 90:3028-3035. [PubMed: 15687339]
31SemenzaGL. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev. 2010; 20:51-56. [PubMed: 19942427]
32StacpoolePW, Henderson GN, Yan Z, Cornett R, James MO. Farmacocinética, metabolismo y toxicología del dicloroacetato. Drug Metab Rev. 1998; 30:499-539. [PubMed: 9710704]
33StacpoolePW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003; 43:683-691. [PubMed: 12856382]
34StacpoolePW, Kurtz TL, Han Z, Langaee T. Papel del dicloroacetato en el tratamiento de enfermedades mitocondriales genéticas. Adv Drug Deliv Rev. 2008; 60:1478-1487. [PubMed: 18647626]
35StockwinLH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Sodium dichloroacetate (DCA) selectively targets cells with defects in the mitochondrial ETC. Int J Cancer. 2010 en prensa.
36SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat. 2010; 120:253-260. [PubMed: 19543830]
37ThangarajuM, Gopal E, Martin PM, Ananth S, Smith SB, Prasad PD, et al. SLC5A8 triggers tumor cell apoptosis through pyruvate-dependent inhibition of histone deacetylases. Cancer Res. 2006; 66:11560-11564. [PubMed: 17178845]
38ThangarajuM, Cresci G, Itagaki S, Mellinger J, Browning DD, Berger FG, et al. Sodium-coupled transport of the short-chain fatty acid butyrate by SLC5A8 and its relevance to colon cancer. J Gastrointest Surg. 2008; 12:1773-1782. [PubMed: 18661192]
39ThangarajuM, Karunakaran S, Itagaki S, Gopal E, Elangovan S, Prasad PD, et al. Transport via SLC5A8 with subsequent inhibition of histone deacetylases HDAC1 and HDAC3 underlies the antitumor activity of 3-bromopyruvate. Cancer. 2009a; 115:4655-4666. [PubMed: 19637353]
40ThangarajuM, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J. 2009b; 417:379-389. [PubMed: 18789002]
41UenoM, Toyota M, Akino K, Suzuki H, Kusano M, Satoh A, et al. Aberrant methylation and histone deacetylation associated with silencing of SLC5A8 in gastric cancer. Tumour Biol. 2004; 25:134- 140. [PubMed: 15361710]
42VanderHeiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029-1033. [PubMed: 19460998]
43WarburgO. On the origin of cancer cells. Science. 1956; 123:309-314. [PubMed: 13298683]
44WeimerLH, Sachdev N. Update on medication-induced peripheral neuropathy. Curr Neurol Neurosci Rep. 2009; 9:69-75. [PubMed: 19080756]
45WhitehouseS, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974; 141:761-774. [PubMed: 4478069]
46WongJY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008; 109:394-402. [PubMed: 18423823]
47XiaoL, Li X, Niu N, Qian J, Xie G, Wang Y. Dicloroacetato (DCA) mejora la muerte de células tumorales en combinación con adenovirus oncolítico armado con MDA-7/IL-24. Mol Cell Biochem. 2010; 340:31-40. [PubMed: 20165905]