Ellappan Babu, Ph. D, Sabarish Ramachandran, Ph. D, Veena Coothan-Kandaswamy, Ph. D., Selvakumar Elangovan, Ph. D, Puttur D. Prasad, Ph. D, Vadivel Ganapathy, Ph. D, et Muthusamy Thangaraju, Ph. D.
Département de biochimie et de biologie moléculaire, Medical College of Georgia, Georgia Health Sciences University, Augusta, Géorgie, États-Unis
Les utilisateurs peuvent visualiser, imprimer, copier, télécharger et exploiter le contenu de ces documents à des fins de recherche universitaire, sous réserve du respect des conditions d’utilisation complètes :
http://www.nature.com/authors/editorial_policies/license.html#terms
Correspondance : M. Thangaraju, Ph. D., Département de biochimie et de biologie moléculaire, Medical College of Georgia, Augusta, GA 30912, USA. [email protected].
Conflit d’intérêts : Les auteurs déclarent qu’il n’y a pas d’intérêt financier concurrent en relation avec le travail décrit dans ce manuscrit.
Publié dans sa forme finale sous le titre : Oncogene. 22 septembre 2011 ; 30(38) : 4026-4037. doi:10.1038/onc.2011.113.
Mots-clés : SLC5A8 ; dichloroacétate ; médicament anticancéreux ; effet Warburg ; pyruvate déshydrogénase kinase ; oxydation mitochondriale dans le cancer
Résumé
Le public et les scientifiques s’intéressent de plus en plus au dichloroacétate en tant que médicament anticancéreux potentiel. Il existe des preuves crédibles de l’activité antitumorale de ce composé, mais de fortes concentrations sont nécessaires pour obtenir un effet thérapeutique significatif. Malheureusement, ces concentrations élevées produisent des effets secondaires néfastes sur le système nerveux, ce qui exclut son utilisation dans le traitement du cancer. La base mécanistique de l’activité antitumorale du composé est sa capacité à activer le complexe pyruvate déshydrogénase par l’inhibition de la kinase pyruvate déshydrogénase. Comme le composé inhibe la kinase à des concentrations micromolaires, on ne sait pas pourquoi des doses élevées, prohibitives sur le plan thérapeutique, sont nécessaires pour supprimer la croissance tumorale. Nous avons émis l’hypothèse que l’absence de mécanismes efficaces pour l’entrée du dichloroacétate dans les cellules tumorales pouvait sous-tendre ce phénomène. Nous montrons ici que le SLC5A8 transporte très efficacement le dichloroacétate avec une haute affinité. Ce transporteur est exprimé dans les cellules normales, mais son expression est réduite au silence dans les cellules tumorales par des mécanismes épigénétiques. L’absence de ce transporteur rend les cellules tumorales résistantes à l’activité antitumorale du dichloroacétate. Cependant, si le transporteur est exprimé de manière ectopique dans les cellules tumorales, ces dernières deviennent sensibles au médicament à de faibles concentrations. Ce phénomène est évident dans les cellules du cancer du sein, du cancer du côlon et du cancer de la prostate. Les cellules normales, qui expriment le transporteur de manière constitutive, ne sont toutefois pas affectées par le composé, ce qui indique une activité thérapeutique sélective pour les cellules tumorales. Le mécanisme de l’activité antitumorale du composé est toujours sa capacité à inhiber la pyruvate déshydrogénase kinase et à forcer l’oxydation mitochondriale du pyruvate. Étant donné que la mise sous silence de SLC5A8 dans les tumeurs implique la méthylation de l’ADN et que son expression peut être induite par le traitement avec des inhibiteurs de la méthylation de l’ADN, nos résultats suggèrent que la combinaison du dichloroacétate avec un inhibiteur de la méthylation de l’ADN offrirait un moyen de réduire les doses de dichloroacétate pour éviter les effets néfastes associés aux doses élevées, mais sans compromettre l’activité antitumorale.
INTRODUCTION
Le dichloroacétate est actuellement utilisé pour le traitement de l’acidose lactique congénitale (Stacpoole et al., 2003, 2008). L’efficacité thérapeutique de ce médicament est due à sa capacité à activer le complexe pyruvate déshydrogénase (PDC) dans la matrice mitochondriale. Cependant, le complexe enzymatique n’est pas la cible directe du médicament. Le dichloroacétate est un inhibiteur de la pyruvate déshydrogénase kinase (PDK), qui phosphoryle la sous-unité E1α de la PDC et inactive le complexe (Stacpoole et al. 2003, 2008). En inhibant la PDK, le dichloroacétate empêche la phosphorylation de E1α et maintient ainsi la PDC sous forme active. L’activation de la PDC induite par le médicament facilite le métabolisme mitochondrial du pyruvate. Comme le pyruvate cytosolique peut soit être transporté dans les mitochondries et métabolisé, soit être converti en lactate dans le cytoplasme par la lactate déshydrogénase, l’activation de la PDC par le dichloroacétate et le métabolisme du pyruvate qui en résulte dans les mitochondries déplacent l’équilibre entre le pyruvate et le lactate vers le pyruvate. Cela favorise la conversion du lactate en pyruvate, réduisant ainsi les niveaux de lactate.
Ces dernières années, le dichloroacétate a suscité un intérêt considérable en tant que médicament anticancéreux potentiel (Michelakis et al., 2008). L’idée que ce médicament puisse avoir la capacité de tuer les cellules tumorales a une base rationnelle. Les cellules tumorales tirent la majeure partie de leur énergie de la glycolyse aérobie plutôt que de l’oxydation mitochondriale. Ce changement métabolique dans les cellules tumorales a été reconnu pour la première fois par Warburg (Warburg, 1956) et est donc connu sous le nom d’effet Warburg (Gatenby et Gillies, 2004 ; Kim et Dang, 2006 ; Chen et al., 2007 ; Brahimi-Horn et al., 2007 ; Vander Heiden et al., 2009 ; Ganapathy et al., 2009 ; Mathupala et al., 2010). Étant donné que la glycolyse aérobie, qui convertit le glucose en lactate, ne génère que 2 ATP alors que l’oxydation mitochondriale du pyruvate issu de la glycolyse génère 30 ATP, il semble curieux que les cellules tumorales préfèrent la voie métabolique la moins efficace pour obtenir de l’énergie. Les cellules tumorales ne souffrent cependant pas d’une carence en ATP ; en fait, elles génèrent plus d’énergie que les cellules normales pour soutenir leur croissance et leur prolifération accrues. Elles y parviennent en activant plusieurs fois la glycolyse. Il est vrai que l’oxydation mitochondriale génère plus d’ATP que la glycolyse cytoplasmique, mais dans le même temps, l’oxydation mitochondriale produit des espèces réactives de l’oxygène, qui peuvent s’avérer néfastes pour les cellules. Apparemment, les cellules tumorales reconnaissent cet aspect négatif de l’oxydation mitochondriale et optent donc pour la glycolyse comme principale source d’ATP. La glycolyse ne nécessite pas d’oxygène et ne génère pas d’espèces réactives de l’oxygène. De plus, comme la glycolyse est la principale source d’énergie, les cellules tumorales peuvent proliférer dans des conditions anaérobies, ce qui est souvent le cas des tumeurs solides. Cependant, la suppression de la fonction mitochondriale dans les tumeurs est réversible. Il est intéressant de noter que l’un des mécanismes par lesquels les cellules tumorales suppriment l’oxydation mitochondriale est l’induction de la PDK, ce qui inactive la PDC (Semenza, 2010). Par conséquent, l’oxydation du pyruvate dans les mitichondries peut être induite dans les cellules tumorales en inversant la suppression de l’activité de la PDC associée au cancer. C’est exactement ce que fait le dichloroacétate, en inhibant la PDK. Malgré cette base rationnelle pour le dichloroacétate en tant que médicament anticancéreux potentiel, il existe une controverse considérable dans la littérature concernant l’utilité clinique de ce composé pour le traitement du cancer chez l’homme. Même si l’étude de Bonnet et al (2007) a démontré l’efficacité antitumorale du dichloroacétate in vitro et in vivo chez l’animal, une étude récente de Stockwin et al (2010) a montré que de très fortes concentrations de dichloroacétate sont nécessaires pour induire la mort cellulaire dans les cellules tumorales et qu’à ces concentrations, le composé n’a aucune sélectivité pour les cellules tumorales.
Le dichloroacétate est ionisé et ne peut pas traverser la membrane plasmique par diffusion. On peut donc se demander comment ce composé pénètre dans les cellules et accède au PDK dans la matrice mitochondriale. À notre connaissance, il n’existe qu’un seul rapport sur le transport du dichloroacétate dans les cellules de mammifères, qui a montré que les transporteurs de monocarboxylate dans les hépatocytes et les cellules tumorales de la lettre d’Ehrlich servent de médiateurs pour l’entrée cellulaire de ce composé (Jackson et Halestrap, 1996). Comme les transporteurs de monocarboxylate sont électroneutres, la plupart des cellules, y compris les cellules tumorales qui expriment ces transporteurs, peuvent ne pas avoir la capacité de concentrer ce médicament. Récemment, nous avons, avec d’autres, identifié un nouveau transporteur pour les monocarboxylates, qui a une sélectivité de substrat similaire à celle des transporteurs de monocarboxylates, mais qui est couplé au Na+ et électrogène (Coady et al., 2004 ; Miyauchi et al., 2004). Ce transporteur, connu sous le nom de sodium-coupled monocarboxylate transporter (SMCT1) ou SLC5A8 selon la nomenclature de la Human Genome Organization, a la capacité de concentrer ses substrats contre un gradient de concentration en raison de l’implication du gradient Na+ transmembranaire et du potentiel membranaire comme forces motrices. SLC5A8 transporte l’acétate, le propionate, le butyrate, le lactate, le pyruvate, le 3-bromopyruvate, le nicotinate, le β-hydroxybutyrate et le pyroglutamate (Miyauchi et al., 2004, 2010 ; Gopal et al., 2004, 2005 ; Martin et al., 2006 ; Thangaraju et al., 2006, 2008, 2009a). Nous nous sommes demandés si ce transporteur hautement couplé à l’énergie accepterait le dichloroacétate comme substrat. Cette question est directement liée à l’activité antitumorale de ce médicament car les cellules tumorales réduisent ce transporteur au silence par des mécanismes épigénétiques (Ganapathy et al., 2005, 2008, 2009 ; Gupta et al., 2006). Par conséquent, nous avons entrepris la présente étude pour répondre à deux questions : (a) Le SLC5A8 transporte-t-il le dichloroacétate ? (b) L’activité antitumorale du médicament dépend-elle de l’expression du transporteur dans les cellules tumorales ? Les résultats de l’étude montrent que SLC5A8 est obligatoire pour l’activité antitumorale du dichloroacétate.
Résultats
SLC5A8 transporte le dichloroacétate de manière couplée au Na+
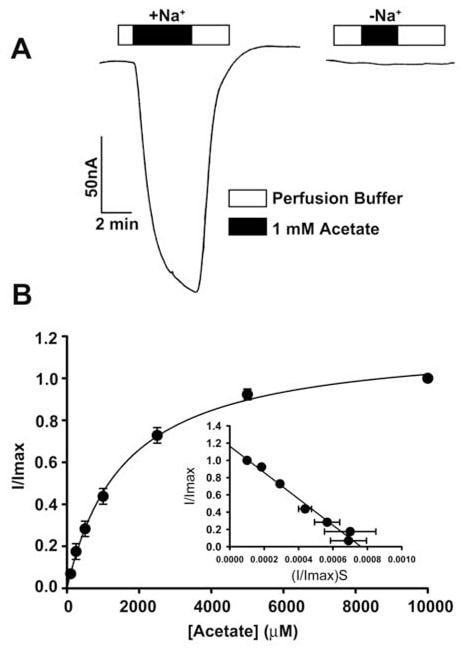
Le transport de l’acétate et de ses dérivés chlorés par le SLC5A8 humain a été étudié en utilisant le système d’expression des oocytes de X. laevis. Le transporteur humain a été exprimé dans les ovocytes de manière hétérologue par injection d’ARNc SLC5A8. La fonction de transport a été contrôlée électrophysiologiquement par la technique de voltage-clamp à deux microélectrodes. SLC5A8 fonctionne comme un transporteur couplé au Na+ pour les monocarboxylates avec une stoechiométrie Na+: monocarboxylate de 2:1. Le processus de transport est donc électrogène, associé au transfert d’une charge positive nette dans les cellules par cycle de transport. La dépolarisation résultante de la membrane peut être surveillée comme un courant entrant dans des conditions de voltage-clamp. Comme on peut le voir sur la figure 1A, l’exposition des ovocytes exprimant SLC5A8 à l’acétate (1 mM) a induit des courants entrants lorsqu’ils ont été contrôlés en présence de Na+ dans le milieu de perfusion (129 ± 9 nA ; n = 3 ovocytes). Cependant, ces courants n’ont pas été observés en l’absence de Na+. Ces données montrent que la SLC5A8 médiatise le transport de l’acétate d’une manière couplée au Na+. La cinétique de saturation a révélé que la constante de Michaelis (Kt) pour le processus de transport était de 1,6 ± 0,1 mM. Après avoir établi l’activité fonctionnelle de la SLC5A8 humaine clonée en utilisant l’acétate comme contrôle positif, nous avons examiné le transport du dichloroacétate. L’exposition d’ovocytes exprimant SLC5A8 au dichloroacétate (0,25 mM) a induit des courants entrants marqués en présence de Na+ (153 ± 28 nA ; n = 5 ovocytes) (Fig. 2A). Ces courants n’ont pas été observés en l’absence de Na+. Le processus de transport était saturable avec un Kt de 36 ± 7 μM (Fig. 2B). Ainsi, l’affinité du dichloroacétate pour le transporteur est ~45 fois plus élevée que celle de l’acétate. Les courants induits par le dichloroacétate (0,25 mM) ont augmenté avec la concentration de Na+ dans le milieu de perfusion (Fig. 2C). La relation était sigmoïdale, indiquant l’implication de plus d’un Na+ dans le processus d’activation. L’analyse des données selon l’équation de Hill a donné une valeur de 2,1 ± 0,2 pour le coefficient de Hill. Ces données montrent que la stœchiométrie Na+: dichloroacétate pour le processus de transport est de 2:1.
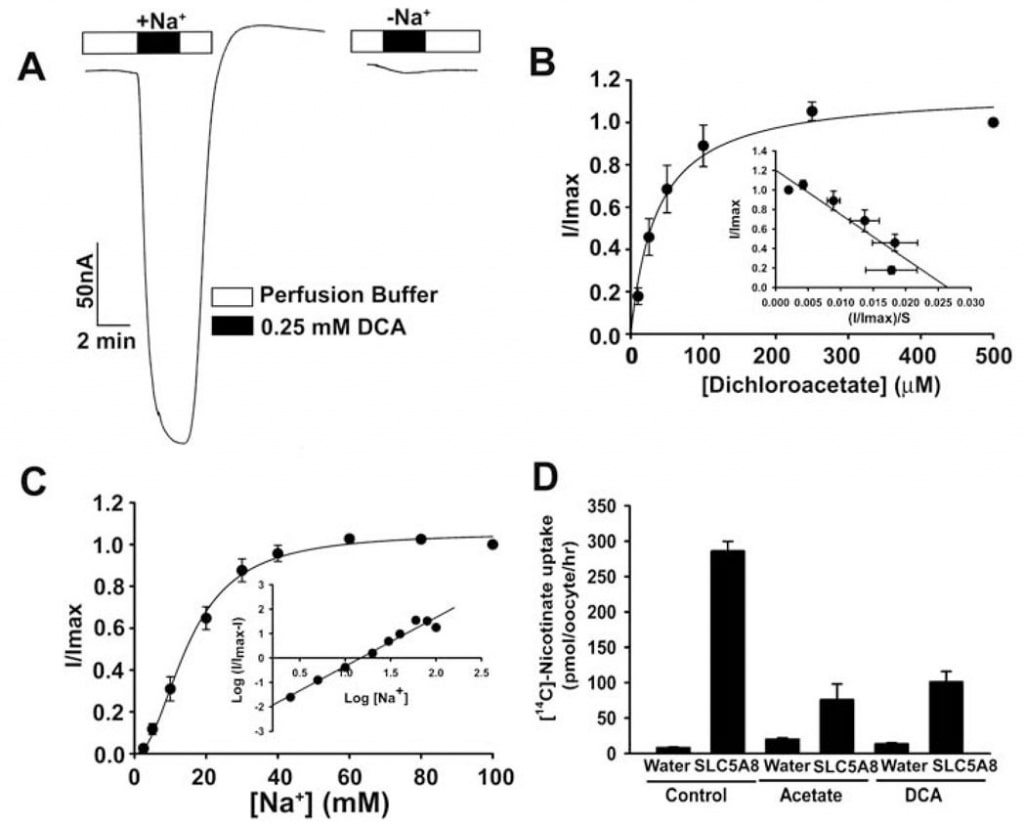
Nous avons également utilisé une méthode alternative pour évaluer le transport du dichloroacétate via SLC5A8. Cette méthode utilise le [14C]-nicotinate comme substrat pour SLC5A8. Les ovocytes injectés dans l’eau n’exprimaient pas le transporteur et ont donc été utilisés comme témoins. L’absorption du [14C]-nicotinate (50 μM) était 60 fois plus élevée dans les ovocytes exprimant SLC5A8 que dans les ovocytes injectés dans l’eau (Fig. 2D). Le captage du nicotinate spécifique à SLC5A8 était inhibé à plus de 80 % en présence d’acétate (0,25 mM) ou de dichloroacétate (0,25 mM), ce qui indique que l’acétate et le dichloroacétate entrent en compétition avec le nicotinate pour le captage via SLC5A8.
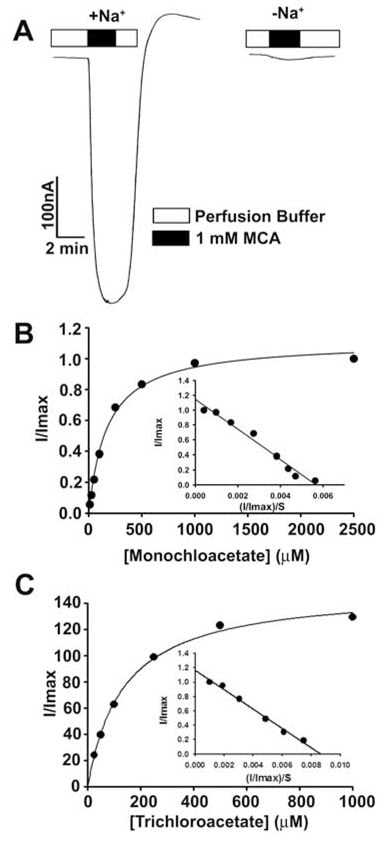
Ces résultats montrent que le dichloroacétate est un substrat de haute affinité pour le SLC5A8 humain. Nous avons ensuite étudié le transport du monochloroacétate et du trichloroacétate à titre de comparaison (Fig. 3). Les deux composés ont été transportés via SLC5A8 de manière couplée au Na+ et saturable. Le Kt était de 177 ± 16μM pour le monochloroacétate et de 134 ± 11 μM pour le trichloroacétate.
L’apoptose induite par le dichloroacétate dans les cellules cancéreuses nécessite SLC5A8
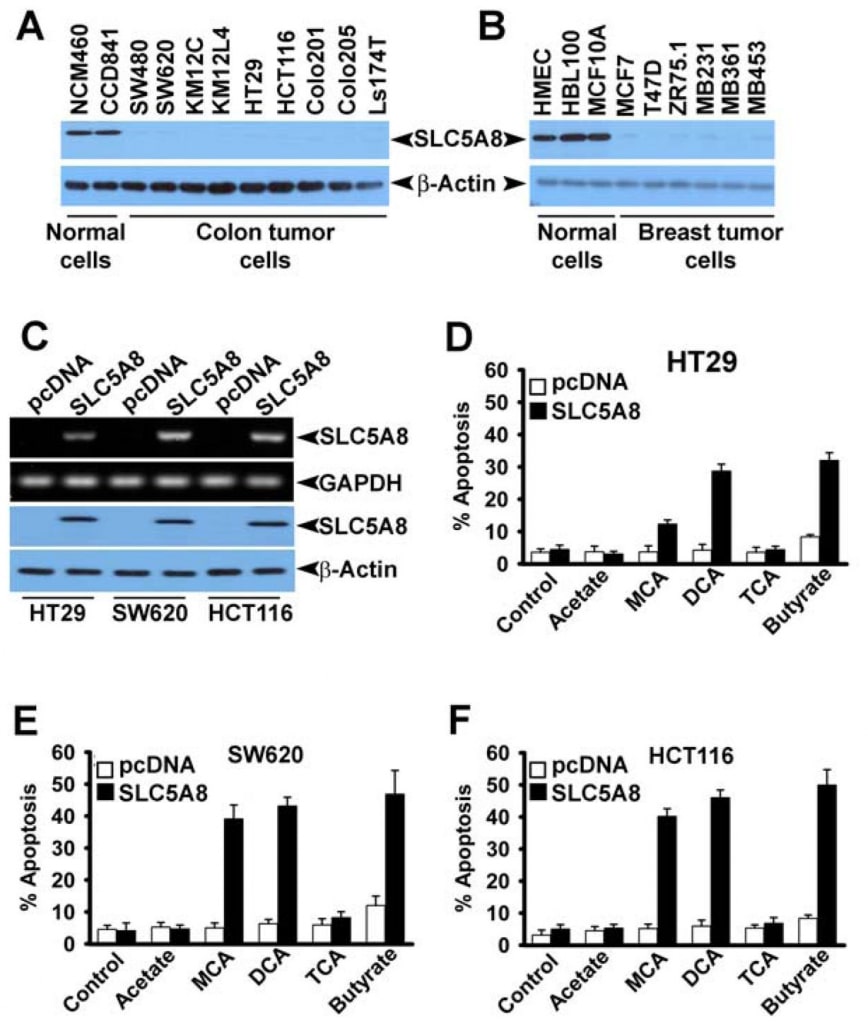
Bien que plusieurs études aient montré que le dichloroacétate induit l’apoptose dans une variété de lignées cellulaires cancéreuses (Bonnet et al., 2007 ; Wong et al., 2008 ; Cao et al., 2008), une enquête récente n’a pas été en mesure de confirmer ces résultats (Stockwin et al., 2010). Les études de Bonnet et al (2007) ont montré que le dichloroacétate à une concentration de 0,5 mM était capable d’induire des changements métaboliques spécifiquement dans les cellules cancéreuses et non dans les cellules normales. Ces modifications comprennent la dépolarisation de la membrane mitochondriale, la suppression de la glycolyse, l’augmentation de l’oxydation mitochondriale, la production d’espèces réactives de l’oxygène, l’induction du canal potassique Kv1.5 de la membrane plasmique et la libération de facteurs pro-apoptotiques par les mitochondries. Wong et al (2008) ont ensuite montré que le dichloroacétate provoquait l’apoptose des cellules cancéreuses de l’endomètre, et Cao et al (2008) ont montré que ce composé sensibilisait les cellules cancéreuses de la prostate aux radiations. En revanche, les études de Stockwin et al (2010) ont démontré que même si le dichloroacétate était capable d’induire une dépolarisation mitochondriale et la génération d’espèces réactives de l’oxygène, ces changements se produisaient aussi bien dans les cellules cancéreuses que dans les cellules normales. En outre, une très forte concentration du composé (≥25 mM) était nécessaire pour induire l’apoptose. Sur la base des résultats de notre étude actuelle, selon lesquels SLC5A8 est le médiateur de l’entrée active, couplée à l’énergie, du dichloroacétate dans les cellules, et du fait que les cellules cancéreuses réduisent le transporteur au silence, nous nous sommes demandé si l’absence du transporteur dans les cellules cancéreuses était la raison de l’absence d’apoptose détectable à de faibles concentrations du composé observée par Stockwin et al (2010). Nous avons abordé cette question en utilisant trois différentes lignées cellulaires humaines de cancer du côlon (HCT116, SW620, et HT29). Ces trois lignées cellulaires n’expriment pas SLC5A8 (Thangaraju et al., 2008).
Nous avons également confirmé l’absence d’expression de SLC5A8 dans les lignées cellulaires humaines de cancer du colon et du sein au niveau des protéines (Fig. 4A et B). Nous avons exprimé SLC5A8 dans les lignées cellulaires HCT116, SW620 et HT29 par transfection transitoire d’une construction d’expression mammalienne et confirmé l’expression par RT-PCR et analyse Western blot (Fig. 4C). Les cellules transfectées avec un vecteur vide ont servi de témoins. Nous avons ensuite exposé les cellules témoins et les cellules exprimant SLC5A8 au dichloroacétate (1 mM) pendant 48 heures et avons surveillé l’apoptose (Fig. 4D-F). Les résultats ont été intéressants. Dans les cellules témoins, qui n’expriment pas le transporteur, le dichloroacétate n’a pas eu d’effet significatif. Cependant, dans des conditions identiques, les cellules exprimant SLC5A8 ont subi une apoptose marquée. Ce phénomène a été observé dans les trois lignées cellulaires de cancer du côlon. Le monochloroacétate a également eu un comportement similaire à celui du dichloroacétate dans les cellules HCT116 et SW620, mais pas dans les cellules HT29, où la capacité du monochloroacétate à induire l’apoptose était nettement inférieure à celle du dichloroacétate. L’acétate et le trichloroacétate n’ont pas eu d’effet notable. Nous avons utilisé le butyrate comme contrôle positif dans ces expériences en nous basant sur notre rapport précédent selon lequel l’induction de l’apoptose par le butyrate dans les lignées cellulaires du cancer du côlon dépend obligatoirement de l’expression de SLC5A8 (Thangaraju et al., 2008).
Nous avons ensuite voulu déterminer si l’apoptose induite par le dichloroacétate présente une sélectivité des cellules tumorales et également si l’effet est observé dans des lignées cellulaires cancéreuses d’origine tissulaire autre que le côlon. Pour cela, nous avons choisi CCD841 et MCF10A comme représentantes des lignées cellulaires normales (CCD841, côlon ; MCF10A, épithélium mammaire) et quatre lignées cellulaires cancéreuses humaines : MCF7 (une lignée cellulaire de cancer du sein à récepteurs d’œstrogènes positifs), MB231 (une lignée cellulaire de cancer du sein à récepteurs d’œstrogènes négatifs), DU145 (une lignée cellulaire de cancer de la prostate insensible aux androgènes) et LNCaP (une lignée cellulaire de cancer de la prostate sensible aux androgènes). Comme indiqué précédemment (Thangaraju et al., 2006, 2008), les lignées cellulaires normales CCD841 et MCF10A ont exprimé des niveaux détectables de SLC5A8, tant au niveau de l’ARNm que des protéines (Fig. 5A). En revanche, aucune des quatre lignées cellulaires cancéreuses examinées ici n’exprimait le transporteur. L’expression est devenue évidente dans les lignées de cellules cancéreuses après transfection transitoire d’une construction d’expression mammalienne. Dans les cellules normales, les niveaux d’expression ont augmenté après transfection. En utilisant ces lignées cellulaires, nous avons comparé la capacité du dichloroacétate à induire l’apoptose entre les lignées cellulaires normales et cancéreuses (Fig. 5B). Nous n’avons trouvé aucune différence significative dans l’apoptose des lignées cellulaires normales avec et sans traitement au dichloroacétate (1 mM). L’augmentation des niveaux d’expression de SLC5A8 n’a pas non plus eu d’effet sur l’ampleur de l’apoptose. En revanche, le dichloroacétate a pu induire une apoptose marquée dans les quatre lignées cellulaires cancéreuses, mais uniquement si les cellules exprimaient le transporteur. Sans l’expression du transporteur, les lignées cellulaires cancéreuses n’ont pas subi d’apoptose lors du traitement au dichloroacétate. Ces résultats montrent que le dichloroacétate est capable de provoquer l’apoptose, même à des concentrations aussi faibles que 1 mM, de manière sélective pour les cellules cancéreuses, mais seulement si SLC5A8 est exprimé. Ces observations ont également été confirmées par l’expression stable de SLC5A8 (inductible par la doxycyline) médiée par le virus de la lentille dans deux lignées cellulaires de cancer du sein, MCF7 et MB231 (Fig. 5C, D). Il a été bien établi que l’extinction de SLC5A8 est associée à la méthylation de l’ADN et que le traitement des cellules cancéreuses avec des agents de déméthylation de l’ADN réactive l’expression de SLC5A8. Nous avons donc traité des lignées cellulaires normales et cancéreuses avec de la 5′-aza-2-désoxycytidine (5-Azadc), un agent de déméthylation de l’ADN, et la réactivation de SLC5A8 a été confirmée par une analyse par immunoblotting (Fig. 5E). le traitement au 5-Azadc n’a pas modifié l’expression de la protéine SLC5A8 dans les cellules épithéliales normales du côlon et du sein, alors qu’il a réactivé l’expression de SLC5A8 dans toutes les lignées cellulaires cancéreuses. En outre, le traitement de ces cellules par le 5-Azadc lui-même a induit une apoptose significative ; cependant, le traitement de ces cellules par le dichloroacétate (1 mM) a considérablement renforcé l’apoptose induite par le 5-Azadc (Fig. 5F).
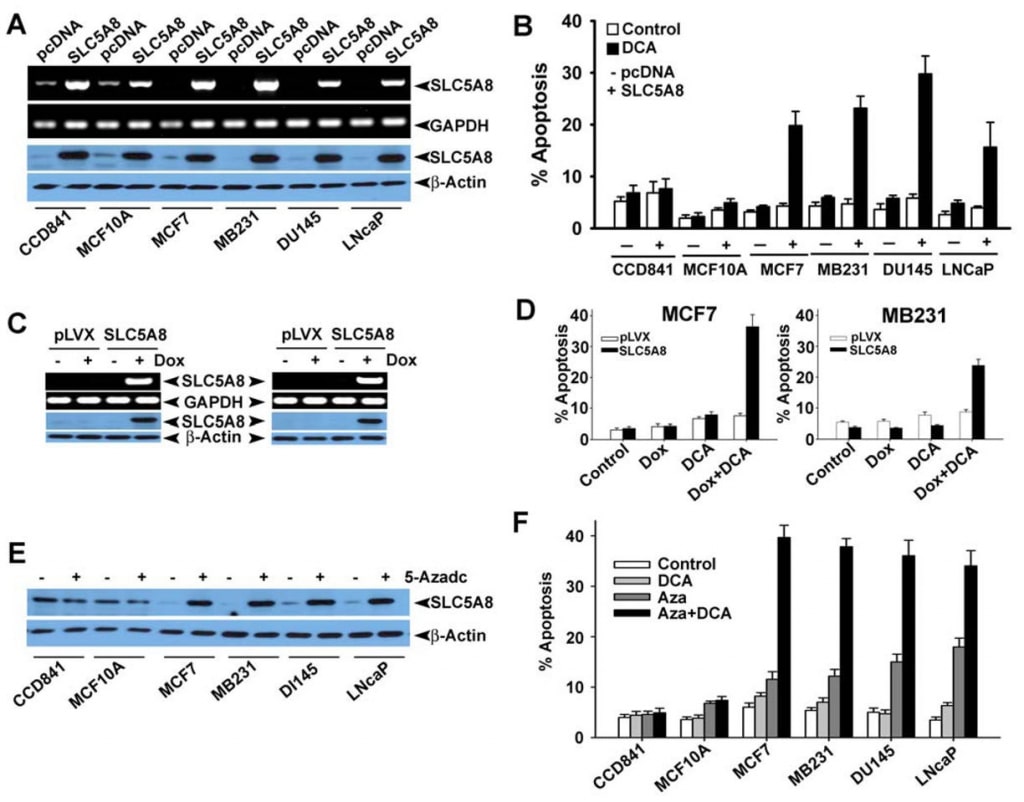
Effet du dichloroacétate sur les niveaux intracellulaires de pyruvate
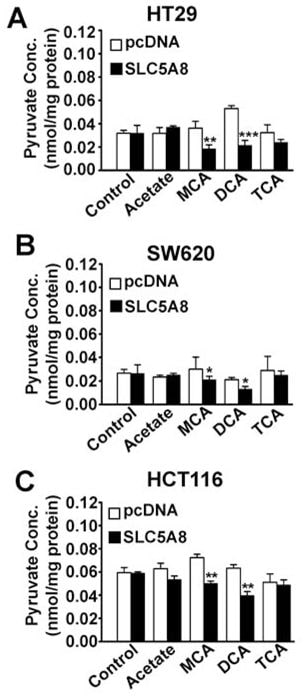
Quel est le mécanisme responsable de l’induction de l’apoptose par SLC5A8/ dichloroacétate dans les cellules cancéreuses ? SLC5A8 est un transporteur actif pour le dichloroacétate avec un Kt de 36 ± 7μM. Par conséquent, le composé sera concentré dans les cellules cancéreuses si le transporteur est exprimé. Cela expliquerait la nécessité obligatoire de SLC5A8 pour de faibles concentrations de dichloroacétate pour induire l’apoptose dans ces cellules. Si le transporteur n’est pas exprimé, les cellules peuvent ne pas accumuler le composé à des niveaux suffisants pour provoquer l’apoptose. Une fois accumulé dans les cellules cancéreuses, comment le dichloroacétate agit-il pour induire l’apoptose ? Le seul mécanisme d’action connu du dichloroacétate est sa capacité à activer la PDC par inhibition de la PDK (Stacpoole et al., 1998, 2003, 2008). Il en résulterait une stimulation de l’oxydation mitochondriale, la génération d’espèces réactives de l’oxygène, la dépolarisation du potentiel de la membrane mitochondriale et l’induction de l’apoptose. L’activation de la PDC par le dichloroacétate dans des cellules intactes diminuerait les niveaux intracellulaires de pyruvate. Sur cette base, nous avons émis l’hypothèse que le dichloroacétate diminuerait les niveaux de pyruvate dans les cellules cancéreuses et que cet effet dépendrait obligatoirement de l’expression de SLC5A8. Nous avons testé cette hypothèse dans trois lignées cellulaires cancéreuses différentes (HT29, SW620 et HCT116) avec et sans expression exogène de SLC5A8 (Fig. 6). L’expression du transporteur par lui-même n’a pas eu d’effet sur les niveaux intracellulaires de pyruvate. Le dichloroacétate a été capable de diminuer les niveaux de pyruvate de manière significative dans les trois lignées cellulaires cancéreuses, mais seulement lorsque le transporteur était exprimé. Il est intéressant de noter que le monochloroacétate a également eu un effet similaire, là encore uniquement en présence de SLC5A8 ; en revanche, le trichloroacétate et l’acétate n’ont pas été capables d’affecter les niveaux de pyruvate.
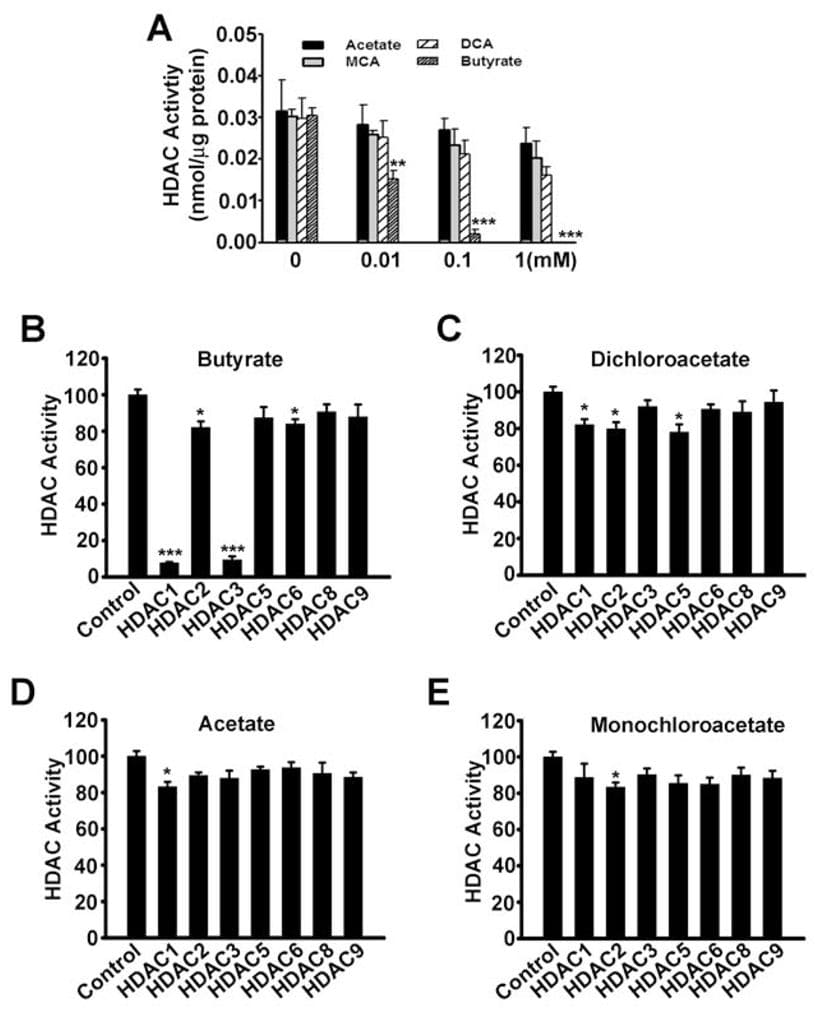
Jusqu’à présent, le seul mécanisme par lequel SLC5A8 fonctionne comme un suppresseur de tumeur était sa capacité à concentrer à l’intérieur des cellules le butyrate et le pyruvate, qui sont des inhibiteurs des histones désacétylases. Nous avons montré dans la présente étude que le dichloroacétate induit également l’apoptose dans les cellules cancéreuses et que cet effet dépend de l’expression de SLC5A8. Ces résultats sont similaires à ceux obtenus avec le butyrate et le pyruvate. Nous nous sommes donc demandé si l’inhibition des histones désacétylases pouvait jouer un rôle dans l’apoptose induite par le dichloroacétate dans les cellules cancéreuses. Cela semblait toutefois peu probable car l’acétate n’est pas un inhibiteur des histones désacétylases (Thangaraju et al., 2006). Cependant, nous ne pouvions pas exclure la possibilité que le dichloroacétate puisse fonctionner comme un inhibiteur des histones désacétylases. Nous avons donc examiné l’influence de l’acétate, du monochloroacétate, du dichloroacétate et du butyrate sur l’activité des histones désacétylases. Tout d’abord, nous avons utilisé des lysats de cellules SW620 comme source d’histone désacétylases (Fig. 7A). Le butyrate a été utilisé comme contrôle positif dans ces expériences. Comme prévu, le butyrate a inhibé l’activité des histones désacétylases. Dans des conditions identiques, le dichloroacétate était au moins 100 fois moins puissant que le butyrate pour inhiber les histones désacétylases. Nous avons ensuite utilisé des isoformes humaines recombinantes d’histone désacétylases pour déterminer si des isoformes spécifiques de l’enzyme pouvaient être inhibées par le dichloroacétate. Nos études précédentes ont montré que le butyrate est un inhibiteur spécifique des isoformes HDAC1 et HDAC3 de l’histone désacétylase (Thangaraju et al., 2009b). Dans la présente étude, nous avons constaté la même chose avec le butyrate (Fig. 7B). Le dichloroacétate a également inhibé les isoformes HDAC1, HDAC2 et HDAC5 de manière significative, mais la puissance était très faible (~20% d’inhibition à une concentration de 1 mM) (Fig. 7C). L’acétate (Fig. 7D) et le monochloroacétate (Fig. 7E) n’ont eu aucun effet sur aucune des isoformes examinées dans l’étude.
Discussion
Depuis la publication dans Cancer Cell de l’article de Bonnet et al (2007) qui a montré que le dichloroacétate favorise l’apoptose dans les lignées de cellules cancéreuses in vitro et inhibe la croissance du cancer dans les xénogreffes de souris in vivo, l’intérêt du public pour l’utilité potentielle de ce composé en tant que médicament anticancéreux n’a cessé de croître. Le dichloroaetate a été utilisé comme médicament pour le traitement de l’acidose lactique congénitale et, par conséquent, les détails de la pharmacocinétique et du profil de toxicité du médicament ont été élaborés (Stacpoole et al., 1998). Les récentes découvertes selon lesquelles le médicament pourrait également être utile dans le traitement du cancer ont incité de nombreux patients cancéreux à utiliser ce médicament non approuvé sans la recommandation de leur médecin (Pearson, 2007). Il est intéressant de noter qu’il n’y a pas eu d’essais cliniques contrôlés pour documenter l’efficacité thérapeutique du dichloroacétate comme médicament anticancéreux. Comme la structure du dichloroacétate ne peut pas être brevetée, les sociétés pharmaceutiques n’étaient apparemment pas intéressées à développer le médicament pour le traitement du cancer par le biais d’essais cliniques, ce qui a incité le chercheur principal de la publication originale dans Cancer Cell à réunir des fonds pour ses propres petits essais cliniques (Pearson, 2007). Les résultats de l’essai clinique ont été publiés récemment (Machelakis et al., 2010), ce qui a montré que le médicament est efficace chez les patients atteints de gliobastome. Malheureusement, même si le médicament était efficace in vivo dans le traitement du glioblastome, de fortes concentrations du médicament ont dû être utilisées pour obtenir une efficacité thérapeutique, ce qui a entraîné une neuropathie périphérique. C’est le seul effet secondaire indésirable qui a limité l’utilisation de doses plus élevées du médicament pour une plus grande efficacité thérapeutique. Il n’y a eu aucun signe de toxicité hématologique, hépatique, rénale ou cardiaque. Plusieurs études ont documenté des complications neurologiques avec des doses élevées de dichloroacétate chez l’animal et chez l’homme (Calcutt et al., 2009 ; Wiemer et Sachdev, 2009 ; Brandsma et al., 2010). Les complications neurologiques ne surviennent qu’à des doses très élevées de dichloroacétate : 12.5 mg/kg/jour pendant 3 mois (Bonnet et al., 2007) ou 15 mg/kg/jour pendant 4 semaines (Brandsma et al., 2010). La nécessité de fortes doses de dichloroacétate pour être thérapeutiquement efficace in vivo concorde avec la plupart des données in vitro récemment publiées qui montrent que des concentrations de 10 mM ou plus sont nécessaires pour provoquer l’apoptose dans les lignées cellulaires cancéreuses (Stockwin et al., 2010 ; Madhok et al., 2010 ; Xiao et al., 2010 ; Heshe et al., 2010 ; Sun et al., 2010).
Il existe des preuves convaincantes que le dichloroacétate a la capacité d’inverser le phénotype métabolique des cellules cancéreuses et de favoriser leur mort, mais l’effet antitumoral n’est observé qu’à des concentrations très élevées, tant in vitro qu’in vivo. Étant donné que la toxicité neurologique se produit à ces concentrations thérapeutiquement efficaces, l’utilité du composé comme médicament anticancéreux est limitée chez l’homme. Il est intéressant de noter que la seule cible moléculaire de l’activité antitumorale du dichloroacétate est la PDK, qui est inhibée par le composé à des concentrations micromolaires (constante d’inhibition, 50-100 μM) (Whitehouse et al., 1974 ; Cooper et al., 1974). Pourquoi alors des concentrations millimolaires élevées de dichloroacétate sont-elles nécessaires in vitro pour tuer les cellules cancéreuses ? Il faut répondre à cette question afin d’exploiter le potentiel de ce composé comme médicament anticancéreux. Nos études actuelles apportent une réponse satisfaisante à cette importante question. Les cellules cancéreuses n’expriment aucun système de transport capable de transférer efficacement le composé du milieu extracellulaire à sa cible intracellulaire. Les transporteurs monocarboxylates qui sont exprimés dans les cellules cancéreuses transportent le dichloroacétate avec un Kt de 0,6 mM (Jackson et Halestrap, 1996). Il s’agit d’un processus de transport à faible affinité. De plus, les transporteurs de monocarboxylate ne sont pas très concentrés. De plus, les cellules tumorales génèrent de grandes quantités de lactate, qui est également un substrat pour les transporteurs de monocarboxylate avec une affinité comparable à celle du dichloroacétate, provoquant ainsi une compétition pour le processus de transport. Les présentes études montrent que SLC5A8 transporte le dichloroacétate avec une affinité nettement supérieure à celle des transporteurs monocarboxylates (Kt: 36 μM contre 600 μM). Plus important encore est le fait que SLC5A8 est beaucoup plus efficace que les transporteurs monocarboxylates pour transporter le dichloroacétate en raison de l’énergisation du premier par un gradient électrochimique transmembranaire de Na+. Il est intéressant de noter que l’affinité pour le transporteur change drastiquement par la chloration du second carbone dans l’acétate (Kt: 1572 ± 101 μM pour l’acétate ; 177 ± 16 μM pour le monochloroacétate ; 36 ± 7 μM pour le dichloroacétate ; 134 ± 11 μM pour le trichloroacétate). Nos résultats nous amènent à conclure que les cellules tumorales sont résistantes à l’apoptose induite par le dichloroacétate parce que ces cellules ne possèdent pas de systèmes de transport efficaces pour le médicament. C’est la raison la plus probable pour laquelle de très fortes concentrations du composé sont nécessaires pour induire la mort cellulaire dans les cellules tumorales. Il est également possible que la mort cellulaire observée dans les cellules tumorales à ces concentrations élevées ne soit pas due à l’activation de la PDC car des effets similaires sont également observés dans les cellules normales (Stockwin et al., 2010).
Dans la présente étude, nous avons montré que, si SLC5A8 est exprimé dans les cellules tumorales, le dichloroacétate induit la mort cellulaire à une concentration relativement faible (1 mM) de manière sélective pour les cellules tumorales. Dans des conditions identiques, les cellules normales (CCD841 et MCF10A) ne sont pas affectées. La résistance des cellules normales au dichloroacétate n’est pas due à l’absence d’un système de transport pour le médicament. Les cellules normales expriment SLC5A8 de manière constitutive, et nous avons montré que les cellules ne subissent pas d’apoptose avec ou sans expression supplémentaire du transporteur. Cela confirme l’effet spécifique du dichloroacétate sur les cellules tumorales. Nos études ont également montré que l’efficacité du dichloroacétate en tant qu’inducteur d’apoptose est observée non seulement avec les lignées cellulaires du cancer du côlon, mais aussi avec les lignées cellulaires du cancer du sein et de la prostate. Le mécanisme de la mort cellulaire induite par le dichloroacétate dans les cellules cancéreuses est principalement dû à l’activation de l’oxydation mitochondriale du pyruvate. Cette hypothèse est étayée par le fait que les niveaux intracellulaires de pyruvate dans les cellules cancéreuses diminuent en réponse au traitement par le dichloroacétate d’une manière SLC5A8-dépendante, ce qui suggère que l’entrée concentrée du médicament dans les cellules cancéreuses, médiée par le SLC5A8 exprimé de manière exogène, est responsable de cet effet. Le dichloroacétate n’inhibe pas les histones désacétylases, ce qui exclut l’inhibition des histones désacétylases comme mécanisme potentiel d’apoptose des cellules tumorales induite par le composé.
Les résultats selon lesquels le dichloroacétate induit la mort cellulaire de manière sélective dans les cellules tumorales à de faibles concentrations, mais uniquement si SLC5A8 est exprimé, ont une signification clinique et thérapeutique. La capacité du dichloroacétate à activer la PDC via l’inhibition de la PDK dans les cellules cancéreuses fournit une base mécaniquement rationnelle pour l’activité antitumorale du composé. Mais les cellules cancéreuses sont résistantes au médicament en raison de l’absence d’un transporteur efficace pour le médicament, ce qui nécessite l’utilisation de fortes concentrations du composé pour induire la mort cellulaire, ce qui entraîne malheureusement des effets secondaires néfastes tels que la neuropathie. Nous avons démontré dans la présente étude que SLC5A8 sert de transporteur actif pour le dichloroacétate. Cependant, étant donné que l’expression du transporteur est réduite au silence dans les cellules tumorales, comment les présentes conclusions peuvent-elles être pertinentes pour l’utilisation thérapeutique potentielle du médicament ? L’extinction de SLC5A8 dans les cellules cancéreuses se produit par des mécanismes épigénétiques impliquant la méthylation de l’ADN ; le traitement des cellules cancéreuses avec la 5′-azacytidine, un inhibiteur de la méthylation de l’ADN, réactive l’expression du gène (Li et al., 2003 ; Ueno et al., 2004 ; Hong et al., 2005 ; Porra et al., 2005 ; Thangaraju et al., 2006 ; Park et al., 2007, 2008). La méthylation de l’ADN joue un rôle essentiel dans l’extinction des gènes suppresseurs de tumeurs dans une variété de cancers, et les inhibiteurs de la méthylation de l’ADN sont prometteurs en tant que médicaments anticancéreux (Baylin, 2005). Deux composés ayant une activité d’inhibition de la méthylation de l’ADN sont utilisés en clinique pour le traitement des hémopathies malignes. Il s’agit de la 5′-aza-2′-déoxycytidine, également connue sous le nom de décitabine (nom commercial, Dacogen) et de la 5′-azacytidine (nom commercial, Vidaza). Des études in vitro ont montré que le traitement d’une variété de lignées cellulaires cancéreuses avec ces composés réactive l’expression de SLC5A8. Nous pensons que le même phénomène pourrait se produire in vivo. Par conséquent, une combinaison d’un inhibiteur de la méthylation de l’ADN et de dichloroacétate est susceptible d’être efficace pour le traitement du cancer, car l’inhibiteur de la méthylation de l’ADN induirait l’expression de SLC5A8 dans les tumeurs, qui transporterait ensuite efficacement le dichloroacétate dans les cellules tumorales pour déclencher son activité antitumorale. Ce mode de traitement réduirait considérablement la concentration de dichloroacétate nécessaire à l’efficacité in vivo en tant qu’agent anticancéreux, ce qui permettrait d’obtenir une sélectivité tumorale et d’éviter les effets secondaires néfastes tels que la neuropathie. Les résultats de la présente étude fournissent une base rationnelle pour une telle thérapie combinée.
Matériel et méthodes
Matériaux
[ 14C]-Nicotinate a été acheté chez American Radiolabeled Chemicals (St. Louis, MO). L’acétate et ses dérivés chlorés ont été obtenus auprès de Sigma (St. Louis, MO). SLC5A8 a été initialement cloné à partir de l’intestin humain (Miyauchi et al., 2004).
Système d’expression des ovocytes de X. laevis
L’ARNc coiffé de l’ADNc de SLC5A8 humain (cloné dans pGH19, un vecteur d’expression des ovocytes de X. laevis) a été synthétisé à l’aide du kit mMESSAGE-mMACHINE (Ambion, Austin, TX). Les ovocytes matures de X. laevis ont été isolés par traitement à la collagénase A (1,6 mg/ml), défolliculés manuellement et maintenus à 18 °C dans un milieu de Barth modifié, complété par 25 μg/ml de gentamicine. Les ovocytes ont été injectés avec 50 ng d’ARNc. Les ovocytes injectés dans l’eau ont servi de témoins. Les études électrophysiologiques ont été réalisées par la méthode de voltage-clamp à deux microélectrodes. Les ovocytes ont été périfusés avec un tampon contenant du NaCl (100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, pH 7,5), suivi du même tampon contenant de l’acétate ou ses dérivés chlorés. Le potentiel de la membrane a été fixé à -50 mV. Les différences entre les courants à l’état d’équilibre mesurés en présence et en l’absence de substrats ont été considérées comme les courants induits par les substrats. Dans l’analyse de la cinétique de saturation des courants induits par le substrat, le paramètre cinétique Kt (c’est-à-dire la concentration de substrat nécessaire à l’induction d’un courant semi-maximal) a été calculé en ajustant l’équation de Michaelis-Menten aux valeurs des courants induits par le substrat. La cinétique d’activation par le Na+ des courants induits par le substrat a été analysée en mesurant les courants spécifiques au substrat en présence de concentrations croissantes de Na+. Les données relatives aux courants dépendants du Na+ ont été analysées selon l’équation de Hill afin de déterminer le coefficient de Hill (le nombre d’ions Na+ impliqués dans le processus d’activation). Comme les niveaux d’expression varient considérablement d’un ovocyte à l’autre, les analyses cinétiques ont été effectuées en normalisant les niveaux d’expression. Pour ce faire, on a pris comme valeur 1 le courant spécifique de SLC5A8 maximalement induit dans chaque expérience cinétique dans des ovocytes individuels. Chaque expérience a été répétée avec 3 ou 4 ovocytes différents. L’activité de la SLC5A8 exprimée de façon hétérologue dans les ovocytes a également été contrôlée par l’absorption de [14C]-nicotinate. La concentration de nicotinate dans ces expériences était de 50 μM et le temps d’incubation était de 1 h.
Le protocole d’utilisation des grenouilles dans ces expériences a été approuvé par le comité institutionnel de soins et d’utilisation des animaux.
Expression ectopique de SLC5A8 dans des lignées cellulaires en culture
Les cellules ont été ensemencées dans des boîtes de culture de 35 mm et cultivées en l’absence de pyruvate. Les cellules ont été transfectées avec de l’ADNpc ou de l’ADNc humain SLC5A8 en utilisant Fugene 6 et Opti-MEM. La pEGFP-N1 a été utilisée pour la co-transfection afin de déterminer l’efficacité de la transfection. Après 24 h, les cellules ont été traitées avec ou sans acétate ou ses dérivés chlorés (1 mM) pendant 24 h. Pour l’analyse FACS, les cellules ont été fixées dans de l’éthanol à 50 %, traitées avec 0,1 % de citrate de sodium, 1 mg/ml de RNase A et 50 μg/ml d’iodure de propidium, puis soumises à une analyse de tri cellulaire activé par fluorescence (FACS ; FACS Caliber, Becton Dickinson).
Génération de lignées cellulaires stables exprimant SLC5A8
Nous avons généré des clones stables exprimant SLC5A8 dans deux lignées cellulaires, MCF7et MDA-MB231, pour exprimer conditionnellement SLC5A8 en plaçant l’ADNc de SLC5A8 sous le contrôle du promoteur dépendant de Tet (Tet-On Advanced, Clontech). Le système comprend deux éléments, le vecteur régulateur (pLVX-Tet-On Advanced) et le vecteur de réponse (pLVX-Tight-Puro). En bref, le virus lentique recombinant a été produit par co-transfection dans des cellules 293FT du vecteur régulateur (pLVX-Tet-On Advanced) et de trois autres vecteurs auxiliaires, pLP-1, pLP-2 et pVSVG (Invitrogen) en utilisant le réactif de transfection Lipofectamine 2000. Le surnageant viral lenti a été récolté 72 h après la transfection et filtré à travers une membrane de 0,45 μm. Les cellules MCF7 et MDA-MB231 ont été infectées pendant 24 h avec le virus lentille dans un milieu contenant 8 μg/ml de polybrène et cultivées pendant 48 h supplémentaires. Les cellules ont été sélectionnées avec G418 (2 mg/ml) et l’expression de la protéine rTet R a été vérifiée par Western blot. SLC5A8 a été sous-cloné dans pLVX-Tight-Puro et SLC5A8 pLVX-Tight-Puro a été transduit dans des cellules MCF7 et MDA-MB231 exprimant la protéine rTetR avec la même procédure décrite ci-dessus. Les cellules ont été sélectionnées avec 2 μg/ml de puromycine et maintenues dans un milieu contenant du FBS exempt de Tet, 0,25 μg/ml de puromycine et 250 μg/ml de G418. L’induction de l’ARNm de SLC5A8 lors de l’ajout de doxycycline a été confirmée par RT-PCR.
Réactivation de l’expression de SLC5A8 par l’agent de déméthylation de l’ADN
Les cellules ont été ensemencées dans des plats de 10 cm à très faible densité (0,5 ×106 cellules/plateau) et cultivées dans un milieu respectif sans pyruvate de sodium. Après 24 h, les cellules ont été traitées avec 5′-Azadc (2 μg/ml) pendant 72 h. Le milieu a été remplacé par du milieu frais contenant 5′-Azadc toutes les 24 h. Après le traitement avec 5′-Azadc, les cellules ont été traitées avec DCA (1 mM) pendant 48 h. Après le traitement, les cellules ont été récoltées et traitées pour l’extraction des protéines ainsi que pour l’analyse FACS.
Transcription inverse-PCR
L’ARN préparé à partir de lignées de cellules normales et cancéreuses a été utilisé pour la RT PCR semi-quantitative. L’ARN (2 μg) a été transcrit de manière inverse en ADNc à l’aide du système GeneAmp PCR (Roche). Les amorces PCR pour les gènes spécifiques ont été conçues sur la base des séquences nucléotidiques disponibles dans la GenBank. La transcription inverse-PCR (RT-PCR) a été répétée deux fois avec chaque échantillon d’ARN. La GAPDH (Glyceraldehyde-3-phosphate dehydrogenase) a été utilisée comme contrôle interne.
Analyse Western blot
Cinquante microgrammes de protéines ont été fractionnés par SDS-PAGE, et les protéines fractionnées ont été transférées sur une membrane de nitrocellulose (Whatman GmbH). Les membranes ont été bloquées avec du lait sec non gras à 5 %, puis exposées à l’anticorps primaire anti-SLC5A8 (Cat. # ARP44110, Aviva System Biology) à 4 °C pendant une nuit, suivi du traitement avec les anticorps secondaires appropriés. Les protéines ont été visualisées par le système Western ECL SuperSignal (GE Healthcare).
Analyse de l’apoptose
Les cellules ont été fixées dans de l’éthanol à 50 %, traitées avec du citrate de sodium à 0,1 %, de la RNase A à 1 mg/mL et de l’iodure de propidium à 50 μg/mL, puis soumises à une analyse de tri cellulaire activé par fluorescence (FACS, Becton Dickinson).
Mesure de l’activité HDAC
Un kit disponible dans le commerce (Biovision, Mountain View, CA) a été utilisé pour déterminer l’activité HDAC. Lorsque le lysat cellulaire de la lignée de cellules cancéreuses du côlon SW620 a été utilisé comme source d’activité HDAC, 100 μg de protéines du lysat ont été ajoutés au dosage enzymatique en présence ou en l’absence d’acétate ou de ses dérivés chlorés (1 mM). L’activité des isoformes humaines recombinantes d’HDAC a également été mesurée à l’aide du même kit. Les isoformes d’HDAC recombinantes ont été achetées auprès de Cayman Chemical Company.
Mesure des niveaux intracellulaires de pyruvate
Les cellules ont été ensemencées dans des boîtes de culture de 35 mm et cultivées en l’absence de pyruvate. Les cellules ont été transfectées avec l’ADNpc ou l’ADNc humain SLC5A8 en utilisant Fugene 6 et Opti-MEM. Après 24 h, les cellules ont été traitées avec ou sans acétate ou ses dérivés chlorés (1 mM) pendant 24 h. Les lysats cellulaires ont ensuite été utilisés pour mesurer le pyruvate à l’aide d’un kit disponible dans le commerce (Biovision, Mountain View, CA).
Remerciements
Le travail décrit ici est soutenu en partie par la subvention CA131402 des National Institutes of Health.
RÉFÉRENCES
1 Baylin SB. Méthylation de l’ADN et extinction des gènes dans le cancer. Nat Clin Pract Oncol. 2005 ; 2:S4-S11. [PubMed : 16341240]
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondriaK + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007 ; 11:37-51. [PubMed : 17222789]
2Brahimi-HornMC, Chiche J, Pouyssegur J. Hypoxia signaling controls metabolic demand. Curr Opin Cell Biol. 2007 ; 19:223-229. [PubMed : 17303407]
2BrandsmaD, Dorlo TP, Haanen JH, Beijnen JH, Boogerd W. Encéphalopathie et polyneuropathie sévères induites par le dichloroacétate. J Neurol. 2010 Sous presse.
2CalcuttNA, Lopez VL, Bautista AD, Mizisin LM, Torres BR, Shroads AL, et al. Peripheral neuropathy in rats exposed to dichloroacetate. J Neuropathol Exp Neurol. 2009 ; 68:985-993. [PubMed : 19680144]
3CaoW, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Le dichloroacétate (DCA) sensibilise in vitro les cellules cancéreuses de la prostate de type sauvage et celles qui sur-expriment Bcl-2 aux radiations. Prostate. 2008 ; 68:1223-1231. [PubMed : 18465755]
4ChenZ, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007 ; 39:267-274. [PubMed : 17551814]
5CoadyMJ, Chang MH, Charron FM, Plata C, Wallendorff B, Sah JF, et al. The tumour suppressor gene SLC5A8 expresses a Na+-monocarboxylate cotransporter. J Physiol. 2004 ; 557:719-731. [PubMed : 15090606]
6CooperRH, Randle PJ, Denton RM. Regulation of heart muscle pyruvate dehydrogenase kinase. Biochem J. 1974 ; 143:625-641. [PubMed : 4462746]
7GanapathyV, Gopal E, Miyauchi S, Prasad PD. Biological functions of SLC5A8, a candidate tumor suppressor. Biochem Soc Trans. 2005 ; 33:237-240. [PubMed : 15667316]
8GanapathyV, Thangaraju M, Gopal E, Itagaki S, Miyauchi S, Prasad PD. Transporteurs monocarboxylates couplés au sodium dans les tissus normaux et dans le cancer. AAPS J. 2008 ; 10:193-199. [PubMed : 18446519]
9GanapathyV, Thangaraju M, Prasad PD. Nutrient transporters in cancer : Relevance to Warburg hypothesis and beyond. Pharmacol Ther. 2009 ; 121:29-40. [PubMed : 18992769]
10GatenbyRA, Gillies RJ. Pourquoi les cancers ont-ils une glycolyse aérobie élevée ? Nat Rev Cancer. 2004 ; 4:891- 899. [PubMed : 15516961]
11GopalE, Fei YJ, Sugawara M, Miyauchi S, Zhuang L, Martin PM, et al. Expression de Slc5a8 dans le rein et son rôle dans le transport Na+-couplé du lactate. J Biol Chem. 2004 ; 279:44522-44532. [PubMed : 15322102]
12GopalE, Fei YJ, Miyauchi S, Zhuang L, Prasad PD, Ganapathy V. Sodium-coupled and electrogenic transport of B-complex vitamin nicotinic acid by Slc5a8, a member of the Na+/glucose cotransporter gene family. Biochem J. 2005 ; 388:309-316. [PubMed : 15651982]
13GuptaN, Martin PM, Prasad PD, Ganapathy V. SLC5A8 (SMCT1)-mediated transport of butyrate forms the basis for the tumor suppressive function of the transporter. Life Sci. 2006 ; 78:2419- 2425. [PubMed : 16375929]
14HesheD, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. La thérapie métabolique ciblée par le dichloroacétate vainc la cytotoxicité des médicaments anticancéreux standard. Cancer Chemother Pharmacol. 2010 Sous presse.
15HongC, Maunakea A, Jun P, Bollen AW, Hodgson JG, Goldenberg DD, et al. Shared epigenetic mechanisms in human and mouse gliomas inactivate expression of the growth suppressor SLC5A8. Cancer Res. 2005 ; 65:3617-3623. [PubMed : 15867356]
16JacksonVN, Halestrap AP. La cinétique, le substrat et la spécificité de l’inhibiteur du transporteur de monocarboxylate (lactate) des cellules hépatiques de rat déterminés en utilisant l’indicateur de pH intracellulaire fluorescent, 2′, 7′-bis (carboxyéthyl)-5(6)-carboxyfluorescéine. J Biol Chem. 1996 ; 271:861-868. [PubMed : 8557697]
17KimJW, Dang CV. Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res. 2006 ; 66:8927-8930. [PubMed : 16982728]
19LiH, Myeroff L, Smiraglia D, Romero MF, Pretlow TP, Kasturi L, et al. SLC5A8, un transporteur de sodium, est un gène suppresseur de tumeur réduit au silence par méthylation dans les foyers de crypte aberrants et les cancers du côlon humain. Proc Natl Acad Sci USA. 2003 ; 100:8412-8417. [PubMed : 12829793]
20MadhokBM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Le dichloroacétate induit l’apoptose et l’arrêt du cycle cellulaire dans les cellules cancéreuses colorectales. Br J Cancer. 2010 ; 102:1746-1752. [PubMed : 20485289]
21MartinPM, Gopal E, Ananth S, Zhuang L, Itagaki S, Prasad BM, et al. Identity of SMCT1 (SLC5A8) as a neuron-specific Na+-coupled transporter for active uptake of L-lactate and ketone bodies in the brain. J Neurochem. 2006 ; 98:279-288. [PubMed : 16805814]
22MathupalaSP, KOYH, Pedersen PL. The pivotal roles of mitochondria in cancer : Warburg and beyond and encouraging prospects for effective therapies. Biochim Biophys Acta. 2010 ; 1797:1225-1230. [PubMed : 20381449]
23MichelakisED, Webster L, Mackey JR. Le dichloroacétate (DCA) comme thérapie potentielle de ciblage métabolique pour le cancer. Br J Cancer. 2008 ; 99:989-994. [PubMed : 18766181]
24MichelakisED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010 ; 2:31-34.
25MiyauchiS, Gopal E, Fei YJ, Ganapathy V. Functional identification of SLC5A8, a tumor suppressor downregulated in colon cancer, as a Na+-coupled transporter for short-chain fatty acids. J Biol Chem. 2004 ; 279:13293-13296. [PubMed : 14966140]
26MiyauchiS, Gopal E, Babu E, Sonne SR, Kubo Y, Umapathy NS, et al. Sodium-coupled electrogenic transport of pyroglutamate (5-oxoproline) via SLC5A8, a monocarboxylate transporter. Biochim Biophys Acta. 2010 ; 1798:1164-1171. [PubMed : 20211600]
27ParkJY, Zheng W, Kim D, Cheng JQ, Kumar N, Ahmad N, et al. Le gène suppresseur de tumeur candidat SLC5A8 est fréquemment régulé à la baisse par l’hyperméthylation du promoteur dans le cancer de la prostate. Cancer Detect Prev. 2007 ; 31:359-365. [PubMed : 18037591]
28ParkJY, Helm JF, Zheng W, Ly QP, Hodul PJ, Centeno BA, et al. Silencing of the candidate tumor suppressor gene solute carrier family 5 member 8 (SLC5A8) in human pancreatic cancer. Pancréas. 2008 ; 36:e32-e39. [PubMed : 18437076]
29PearsonH. Cancer patients opt for un médicament non approuvé. Nature. 2007 ; 446:474-475. [PubMed : 17392750]
30PorraV, Ferraro-Peyret C, Durand C, Selmi-Ruby S, Giroud H, Berger-Dutrieux N, et al. Silencing of the tumor suppressor gene SLC5A8 is associated with BRAF mutations is classical papillary thyroid carcinomas. J Clin Endocrinol Metab. 2005 ; 90:3028-3035. [PubMed : 15687339]
31SemenzaGL. HIF-1 : en amont et en aval du métabolisme du cancer. Curr Opin Genet Dev. 2010 ; 20:51-56. [PubMed : 19942427]
32StacpoolePW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacocinétique, métabolisme et toxicologie du dichloroacétate. Drug Metab Rev. 1998 ; 30:499-539. [PubMed : 9710704]
33StacpoolePW, Nagaraja NV, Hutson AD. Efficacité du dichloroacétate comme médicament pour abaisser le taux de lactate. J Clin Pharmacol. 2003 ; 43:683-691. [PubMed : 12856382]
34StacpoolePW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008 ; 60:1478-1487. [PubMed : 18647626]
35StockwinLH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Le dichloroacétate de sodium (DCA) cible sélectivement les cellules présentant des défauts dans l’ETC mitochondrial. Int J Cancer. 2010 sous presse.
36SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. L’inversion du phénotype glycolytique par le dichloroacétate inhibe la croissance des cellules métastatiques du cancer du sein in vitro et in vivo. Breast Cancer Res Treat. 2010 ; 120:253-260. [PubMed : 19543830]
37ThangarajuM, Gopal E, Martin PM, Ananth S, Smith SB, Prasad PD, et al. SLC5A8 déclenche l’apoptose des cellules tumorales par l’inhibition des histones désacétylases en fonction du pyruvate. Cancer Res. 2006 ; 66:11560-11564. [PubMed : 17178845]
38ThangarajuM, Cresci G, Itagaki S, Mellinger J, Browning DD, Berger FG, et al. Sodium-coupled transport of the short-chain fatty acid butyrate by SLC5A8 and its relevance to colon cancer. J Gastrointest Surg. 2008 ; 12:1773-1782. [PubMed : 18661192]
39ThangarajuM, Karunakaran S, Itagaki S, Gopal E, Elangovan S, Prasad PD, et al. Le transport via SLC5A8 avec l’inhibition subséquente des histone désacétylases HDAC1 et HDAC3 sous-tend l’activité antitumorale du 3-bromopyruvate. Cancer. 2009a ; 115:4655-4666. [PubMed : 19637353]
40ThangarajuM, Carswell KN, Prasad PD, Ganapathy V. Les cellules du cancer du côlon maintiennent de faibles niveaux de pyruvate pour éviter la mort cellulaire causée par l’inhibition de HDAC1/HDAC3. Biochem J. 2009b ; 417:379-389. [PubMed : 18789002]
41UenoM, Toyota M, Akino K, Suzuki H, Kusano M, Satoh A, et al. Aberrant methylation and histone deacetylation associated with silencing of SLC5A8 in gastric cancer. Tumour Biol. 2004 ; 25:134- 140. [PubMed : 15361710]
42VanderHeiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect : the metabolic requirements of cell proliferation. Science. 2009 ; 324:1029-1033. [PubMed : 19460998]
43WarburgO. On the origin of cancer cells. Science. 1956 ; 123:309-314. [PubMed : 13298683]
44WeimerLH, Sachdev N. Update on medication-induced peripheral neuropathy. Curr Neurol Neurosci Rep. 2009 ; 9:69-75. [PubMed : 19080756]
45WhitehouseS, Cooper RH, Randle PJ. Mécanisme d’activation de la pyruvate déshydrogénase par le dichloroacétate et d’autres acides carboxyliques halogénés. Biochem J. 1974 ; 141:761-774. [PubMed : 4478069]
46WongJY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induit l’apoptose dans les cellules cancéreuses de l’endomètre. Gynecol Oncol. 2008 ; 109:394-402. [PubMed : 18423823]
47XiaoL, Li X, Niu N, Qian J, Xie G, Wang Y. Le dichloroacétate (DCA) améliore la mort des cellules tumorales en combinaison avec un adénovirus oncolytique armé de MDA-7/IL-24. Mol Cell Biochem. 2010 ; 340:31-40. [PubMed : 20165905]