Эллаппан Бабу, доктор философии, Сабариш Рамачандран, доктор философии, Вина Кутан-Кандасвами, доктор философии, Селвакумар Элангован, доктор философии, Путтур Д. Прасад, доктор философии, Вадивел Ганапати, доктор философии, и Мутхусами Тхангараджу, доктор философии.
Кафедра биохимии и молекулярной биологии, Медицинский колледж Джорджии, Университет наук о здоровье Джорджии, Огаста, Джорджия, США
Пользователи могут просматривать, распечатывать, копировать, загружать и обрабатывать содержание таких документов в целях академических исследований при условии соблюдения всех условий использования:
http://www.nature.com/authors/editorial_policies/license.html#terms
Корреспонденция: М. Тангараджу, доктор философии, кафедра биохимии и молекулярной биологии, Медицинский колледж Джорджии, Огаста, штат Джорджия, 30912, США. [email protected].
Конфликт интересов: Авторы заявляют об отсутствии конкурирующих финансовых интересов в связи с работой, описанной в данной рукописи.
Опубликовано в окончательной редакции как: Oncogene. 2011 September 22; 30(38): 4026-4037. doi:10.1038/onc.2011.113.
Ключевые слова: SLC5A8; дихлорацетат; противораковый препарат; эффект Варбурга; киназа пируватдегидрогеназы; митохондриальное окисление при раке
Аннотация
Среди общественности и ученых растет интерес к дихлорацетату как к потенциальному противораковому препарату. Существуют убедительные доказательства противоопухолевой активности этого соединения, но для значительного терапевтического эффекта необходимы высокие концентрации. К сожалению, такие высокие концентрации вызывают пагубные побочные эффекты, затрагивающие нервную систему, что исключает его использование для лечения рака. Механистической основой противоопухолевой активности соединения является его способность активировать пируватдегидрогеназный комплекс путем ингибирования киназы пируватдегидрогеназы. Поскольку соединение ингибирует киназу в микромолярных концентрациях, неизвестно, почему для подавления роста опухоли требуются терапевтически запретительные высокие дозы. Мы предположили, что в основе этого явления может лежать отсутствие эффективных механизмов проникновения дихлорацетата в опухолевые клетки. Здесь мы показали, что SLC5A8 переносит дихлорацетат очень эффективно и с высокой аффинностью. Этот транспортер экспрессируется в нормальных клетках, но его экспрессия заглушается в опухолевых клетках посредством эпигенетических механизмов. Отсутствие транспортера делает опухолевые клетки устойчивыми к противоопухолевой активности дихлорацетата. Однако если транспортер экспрессируется в опухолевых клетках эктопически, клетки становятся чувствительными к препарату в низких концентрациях. Это наблюдается в клетках рака молочной железы, рака толстой кишки и рака предстательной железы. Нормальные клетки, которые конститутивно экспрессируют транспортер, однако, не подвергаются воздействию соединения, что указывает на селективную терапевтическую активность в отношении опухолевых клеток. Механизм противоопухолевой активности соединения по-прежнему заключается в его способности ингибировать киназу пируватдегидрогеназы и заставлять митохондриальное окисление пирувата. Поскольку глушение SLC5A8 в опухолях связано с метилированием ДНК, а его экспрессия может быть индуцирована лечением ингибиторами метилирования ДНК, наши результаты позволяют предположить, что сочетание дихлорацетата с ингибитором метилирования ДНК позволит снизить дозы дихлорацетата, чтобы избежать вредных эффектов, связанных с высокими дозами, но без ущерба для противоопухолевой активности.
ВВЕДЕНИЕ
В настоящее время дихлорацетат используется для лечения врожденного молочнокислого ацидоза (Stacpoole et al., 2003, 2008). Терапевтическая эффективность этого препарата обусловлена его способностью активировать пируватдегидрогеназный комплекс (ПДК) в митохондриальном матриксе. Однако ферментный комплекс не является непосредственной мишенью для препарата. Дихлорацетат является ингибитором киназы пируватдегидрогеназы (PDK), которая фосфорилирует субъединицу E1α PDC и инактивирует комплекс (Stacpoole et al. 2003, 2008). Ингибируя PDK, дихлорацетат предотвращает фосфорилирование E1α и таким образом поддерживает PDC в активной форме. Вызванная препаратом активация PDC облегчает митохондриальный метаболизм пирувата. Поскольку цитозольный пируват может либо транспортироваться в митохондрии и метаболизироваться, либо превращаться в лактат в цитоплазме под действием лактатдегидрогеназы, активация PDC дихлорацетатом и обусловленный этим метаболизм пирувата в митохондриях смещает равновесие между пируватом и лактатом в сторону пирувата. Это способствует преобразованию лактата в пируват, тем самым снижая уровень лактата.
В последние годы наблюдается значительный интерес к дихлорацетату как к потенциальному противораковому препарату (Michelakis et al., 2008). Представление о том, что этот препарат может обладать способностью убивать опухолевые клетки, имеет рациональное обоснование. Опухолевые клетки получают большую часть энергии из аэробного гликолиза, а не из митохондриального окисления. Этот метаболический сдвиг в опухолевых клетках был впервые отмечен Варбургом (Warburg, 1956) и стал известен как эффект Варбурга (Gatenby and Gillies, 2004; Kim and Dang, 2006; Chen et al., 2007; Brahimi-Horn et al., 2007; Vander Heiden et al., 2009; Ganapathy et al., 2009; Mathupala et al., 2010). Учитывая, что аэробный гликолиз, превращающий глюкозу в лактат, генерирует только 2 АТФ, тогда как митохондриальное окисление пирувата, образующегося в результате гликолиза, генерирует 30 АТФ, кажется загадкой, что опухолевые клетки предпочитают менее эффективный путь метаболизма для получения энергии. Однако опухолевые клетки не страдают от дефицита АТФ; на самом деле они вырабатывают больше энергии, чем нормальные клетки, для поддержания своего усиленного роста и пролиферации. Это достигается за счет активации гликолиза в несколько раз. Действительно, митохондриальное окисление генерирует больше АТФ, чем цитоплазматический гликолиз, но в то же время митохондриальное окисление производит реактивные виды кислорода, которые могут оказаться губительными для клеток. Очевидно, опухолевые клетки осознают этот негативный аспект митохондриального окисления и поэтому выбирают гликолиз в качестве основного источника АТФ. Гликолиз не требует кислорода и не генерирует реактивные виды кислорода. Кроме того, благодаря получению энергии преимущественно за счет гликолиза, опухолевые клетки могут размножаться в анеробных условиях, которые часто существуют в солидных опухолях. Однако подавление функции митохондрий в опухолях является обратимым. Интересно, что одним из механизмов, с помощью которого опухолевые клетки подавляют митохондриальное окисление, является индуцирование PDK, тем самым инактивируя PDC (Semenza, 2010). Следовательно, окисление пирувата в митихондриях может быть индуцировано в опухолевых клетках путем обращения вспять связанного с раком подавления активности PDC. Дихлорацетат делает именно это, ингибируя PDK. Несмотря на рациональное обоснование дихлорацетата как потенциального противоракового препарата, в литературе существуют значительные разногласия относительно клинической пользы этого соединения для лечения рака у людей. Хотя исследование Bonnet et al (2007) продемонстрировало противоопухолевую эффективность дихлорацетата in vitro и in vivo на животных, недавнее исследование Stockwin et al (2010) показало, что очень высокие концентрации дихлорацетата необходимы для того, чтобы вызвать гибель клеток опухоли, и что при таких концентрациях соединение не обладает избирательностью в отношении опухолевых клеток.
Дихлорацетат ионизирован и не может пройти через плазматическую мембрану путем диффузии. В связи с этим возникает вопрос, как это соединение попадает в клетки и получает доступ к PDK в митохондриальном матриксе. Насколько нам известно, о транспорте дихлорацетата в клетки млекопитающих имеется только одно сообщение, в котором показано, что монокарбоксилатные транспортеры в гепатоцитах и опухолевых клетках Эрлиха-Леттре опосредуют проникновение этого соединения в клетки (Jackson and Halestrap, 1996). Поскольку монокарбоксилатные транспортеры электронейтральны, большинство клеток, включая опухолевые, которые экспрессируют эти транспортеры, не способны концентрировать этот препарат. Недавно мы и другие исследователи выявили новый транспортер монокарбоксилатов, который обладает субстратной селективностью, сходной с таковой у транспортеров монокарбоксилатов, но является Na+-связанным и электрогенным (Coady et al., 2004; Miyauchi et al., 2004). Этот транспортер, известный как натрий-связанный монокарбоксилатный транспортер (SMCT1) или SLC5A8 по номенклатуре Организации генома человека, обладает способностью концентрировать свои субстраты против градиента концентрации благодаря участию трансмембранного градиента Na+ и мембранного потенциала в качестве движущих сил. SLC5A8 переносит ацетат, пропионат, бутират, лактат, пируват, 3-бромпируват, никотинат, β-гидроксибутират и пироглутамат (Miyauchi et al., 2004, 2010; Gopal et al., 2004, 2005; Martin et al., 2006; Thangaraju et al., 2006, 2008, 2009a). Мы задались вопросом, будет ли этот высокоэнергетический транспортер принимать дихлорацетат в качестве субстрата. Этот вопрос имеет непосредственное отношение к противоопухолевой активности данного препарата, поскольку опухолевые клетки заглушают этот транспортер с помощью эпигенетических механизмов (Ganapathy et al., 2005, 2008, 2009; Gupta et al., 2006). Поэтому мы предприняли настоящее исследование, чтобы ответить на два вопроса: (а) Транспортирует ли SLC5A8 дихлорацетат? (b) Зависит ли противоопухолевая активность препарата от экспрессии транспортера в опухолевых клетках? Результаты исследования показывают, что SLC5A8 является обязательным для противоопухолевой активности дихлорацетата.
Результаты
SLC5A8 переносит дихлорацетат Na+-связанным образом
Транспорт ацетата и его хлорпроизводных человеческим SLC5A8 был изучен с помощью системы экспрессии в ооцитах X. laevis. Человеческий транспортер был гетерологично экспрессирован в ооцитах путем введения кРНК SLC5A8. Транспортная функция контролировалась электрофизиологически с помощью двухмикроэлектродного метода зажима напряжения. SLC5A8 функционирует как Na+-связанный транспортер для монокарбоксилатов со стехиометрией Na+: монокарбоксилат 2:1. Таким образом, процесс транспорта является электрогенным, связанным с переносом одного чистого положительного заряда в клетки за цикл транспорта. Результирующую деполяризацию мембраны можно наблюдать в виде входящего тока в условиях вольтамперометрии. Как видно на рис. 1А, воздействие ацетата (1 мМ) на SLC5A8-экспрессирующие ооциты вызывало входящие токи при мониторинге в присутствии Na+ в перфузионной среде (129 ± 9 нА; n = 3 ооцита). Однако эти токи не наблюдались в отсутствие Na+. Эти данные показывают, что SLC5A8 опосредует транспорт ацетата Na+-связанным образом. Кинетика насыщения показала, что константа Михаэлиса (Kt) для транспортного процесса составляет 1,6 ± 0,1 мМ. После установления функциональной активности клонированной SLC5A8 человека с использованием ацетата в качестве положительного контроля, мы исследовали транспорт дихлорацетата. Воздействие дихлорацетата (0,25 мМ) на SLC5A8-экспрессирующие ооциты вызвало выраженные входящие токи в присутствии Na+ (153 ± 28 нА; n = 5 ооцитов) (рис. 2А). Такие токи не наблюдались в отсутствие Na+. Процесс транспорта был насыщаемым с Кт 36 ± 7 мкМ (рис. 2В). Таким образом, сродство дихлорацетата к транспортеру в ~45 раз выше, чем у ацетата. Индуцированные дихлорацетатом (0,25 мМ) токи возрастали по мере увеличения концентрации Na+ в перфузионной среде (рис. 2C). Зависимость была сигмоидальной, что указывает на участие более чем одного Na+ в процессе активации. Анализ данных в соответствии с уравнением Хилла дал значение 2,1 ± 0,2 для коэффициента Хилла. Эти данные показывают, что стехиометрия Na+: дихлорацеат для процесса переноса составляет 2:1.
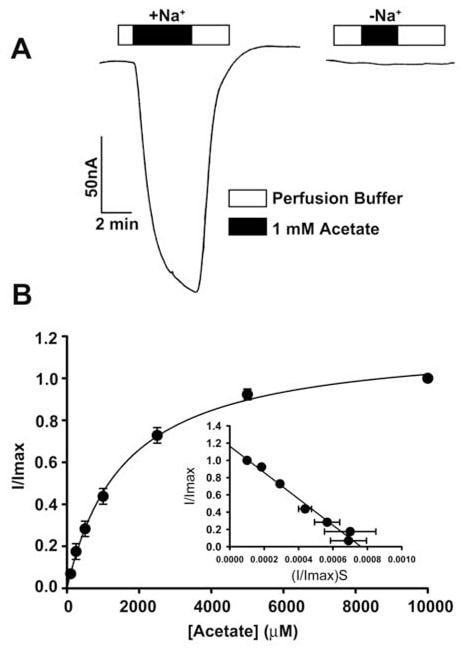
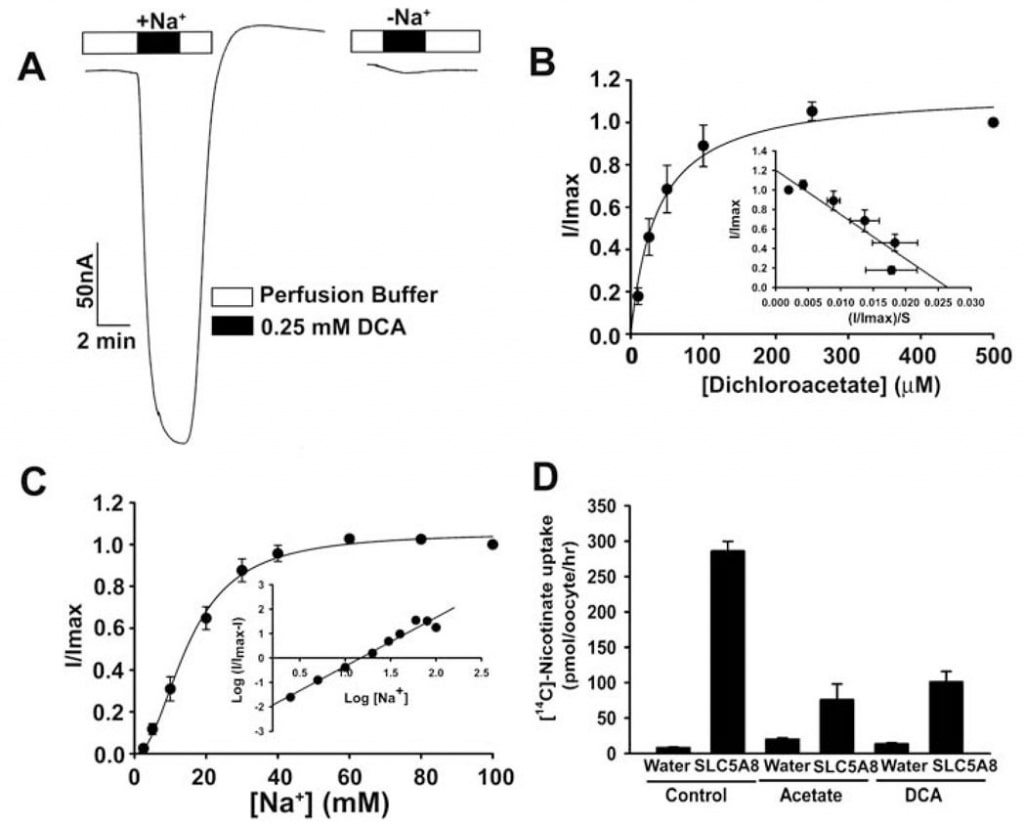
Мы также использовали альтернативный метод для оценки транспорта дихлорацетата через SLC5A8. Этот метод использовал [14C]-никотинат в качестве субстрата для SLC5A8. Инъецированные водой ооциты не экспрессировали транспортер и поэтому использовались в качестве контроля. Поглощение [14C]-никотината (50 мкМ) было в 60 раз выше в ооцитах, экспрессирующих SLC5A8, чем в ооцитах, инъецированных водой (рис. 2D). SLC5A8-специфическое поглощение никотината ингибировалось на >80% в присутствии ацетата (0,25 мМ) или дихлорацетата (0,25 мМ), что указывает на то, что ацетат и дихлорацетат конкурируют с никотинатом за поглощение через SLC5A8.
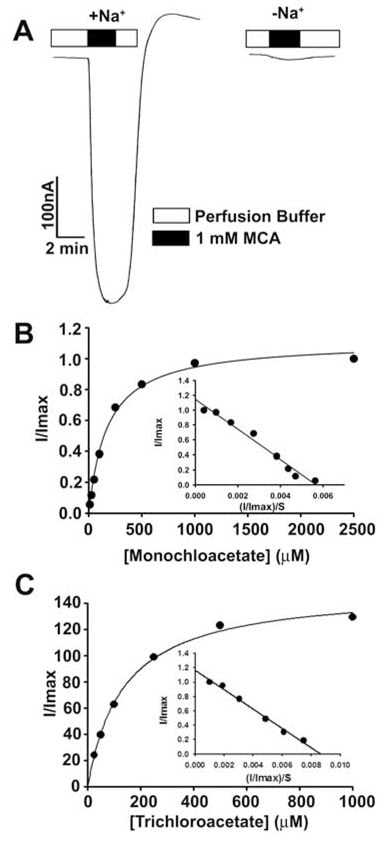
Эти результаты показали, что дихлорацетат является высокоаффинным субстратом для SLC5A8 человека. Затем мы изучили транспорт монохлорацетата и трихлорацетата для сравнения (рис. 3). Оба соединения переносились через SLC5A8 в Na+-связанном и насыщаемом виде. Кт составил 177 ± 16 мкМ для монохлорацетата и 134 ± 11 мкМ для трихлорацетата.
Индуцированный дихлорацетатом апоптоз в раковых клетках требует участия SLC5A8
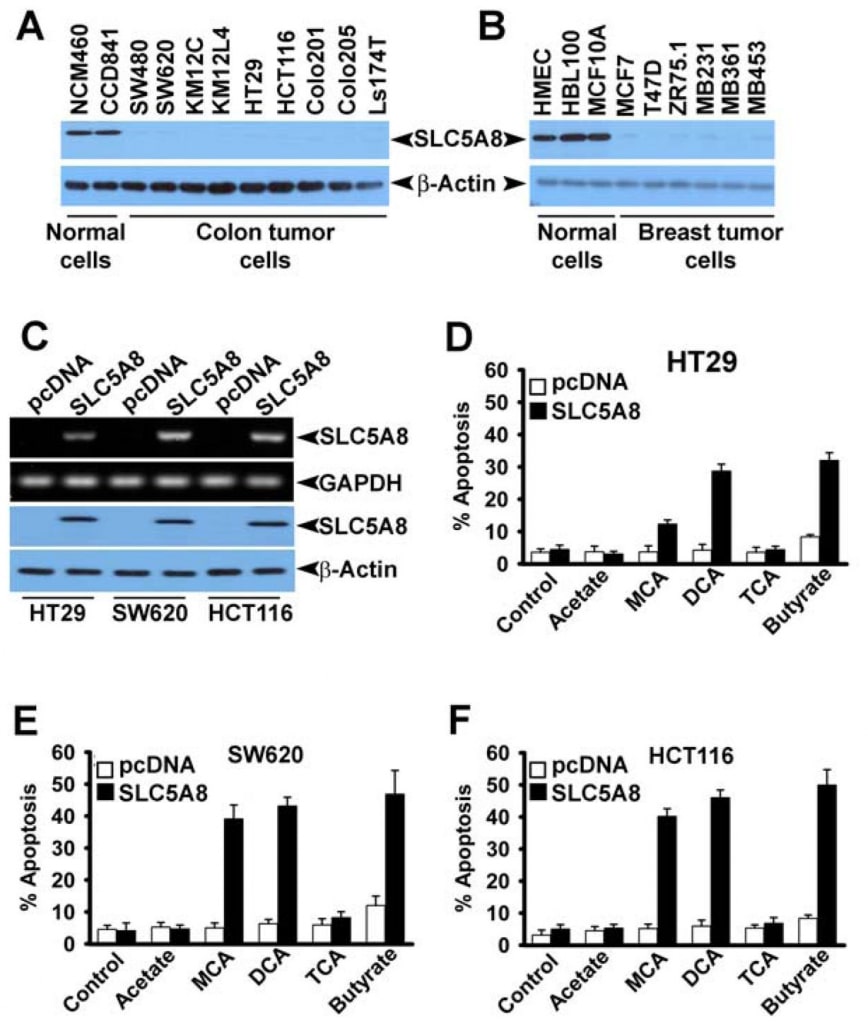
Несмотря на то, что несколько исследований показали, что дихлорацетат индуцирует апоптоз в различных линиях раковых клеток (Bonnet et al., 2007; Wong et al., 2008; Cao et al., 2008), недавнее исследование не смогло подтвердить эти результаты (Stockwin et al., 2010). Исследования Bonnet et al. (2007) показали, что дихлорацетат в концентрации 0,5 мМ способен вызывать метаболические изменения именно в раковых клетках, а не в нормальных. Эти изменения включали деполяризацию митохондриальной мембраны, подавление гликолиза, усиление митохондриального окисления, производство реактивных видов кислорода, индукцию калиевого канала Kv1.5 плазматической мембраны и высвобождение проапоптотических факторов из митохондрий. Впоследствии Вонг и др. (2008) показали, что дихлорацетат вызывает апоптоз в клетках рака эндометрия, а Цао и др. (2008) показали, что это соединение сенсибилизирует клетки рака простаты к радиации. В отличие от этого, исследования Stockwin et al (2010) показали, что хотя дихлорацетат способен вызывать деполяризацию митохондрий и генерацию реактивных видов кислорода, эти изменения происходят как в раковых, так и в нормальных клетках. Кроме того, для индукции апоптоза требовалась очень высокая концентрация соединения (≥25 мМ). Исходя из результатов нашего исследования, согласно которым SLC5A8 опосредует энергетически связанный активный вход дихлорацетата в клетки, а также того факта, что раковые клетки выключают этот транспортер, мы задались вопросом, является ли отсутствие транспортера в раковых клетках причиной отсутствия обнаруживаемого апоптоза при низких концентрациях соединения, наблюдаемого Stockwin et al. (2010). Мы рассмотрели этот вопрос, используя три различные клеточные линии рака толстой кишки человека (HCT116, SW620 и HT29). Эти три клеточные линии не экспрессируют SLC5A8 (Thangaraju et al., 2008).
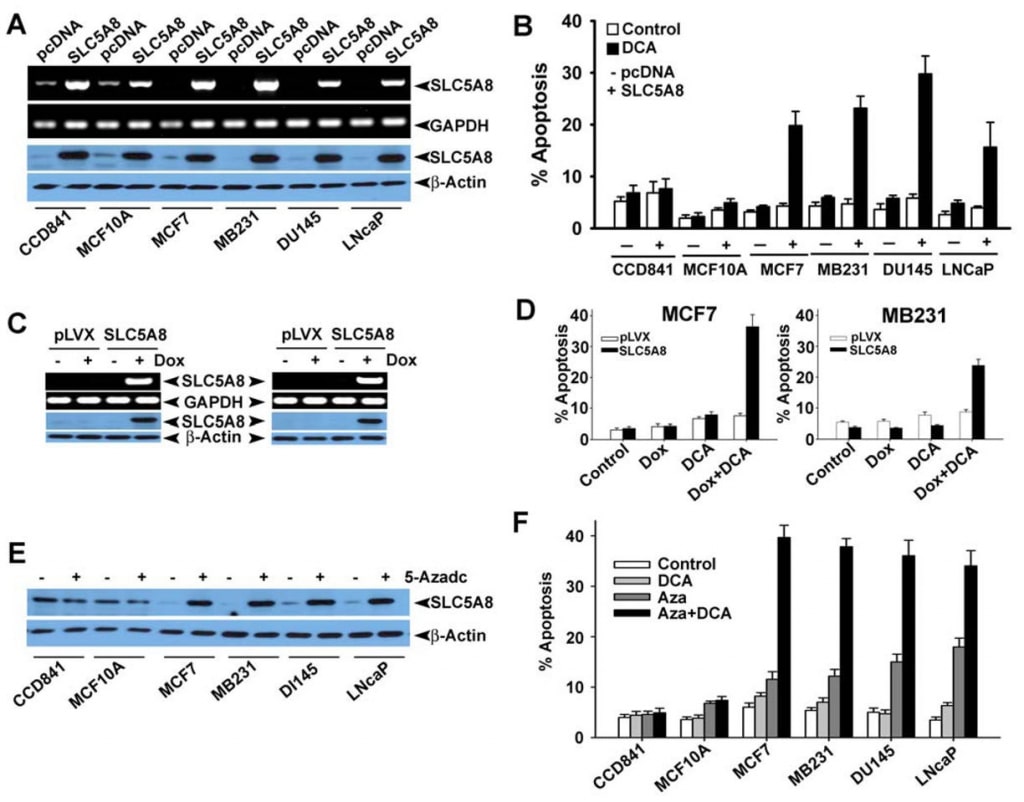
Мы также подтвердили отсутствие экспрессии SLC5A8 в клеточных линиях рака толстой кишки и молочной железы человека на уровне белка (рис. 4A и B). Мы экспрессировали SLC5A8 в клеточных линиях HCT116, SW620 и HT29 путем транзиторной трансфекции экспрессирующей конструкции млекопитающих и подтвердили экспрессию с помощью RT-PCR и Вестерн-блот анализа (рис. 4C). Клетки, трансфецированные пустым вектором, служили контролем. Затем мы подвергли контрольные и экспрессирующие SLC5A8 клетки воздействию дихлорацетата (1 мМ) в течение 48 ч и проследили за апоптозом (рис. 4D-F). Результаты оказались интересными. В контрольных клетках, не экспрессирующих транспортер, дихлорацетат не оказывал существенного влияния. Однако в идентичных условиях клетки, экспрессирующие SLC5A8, подвергались апоптозу в значительной степени. Это явление наблюдалось во всех трех клеточных линиях рака толстой кишки. Монохлорацетат также вел себя аналогично дихлорацетату в клетках HCT116 и SW620, но не в клетках HT29, где способность монохлорацетата вызывать апоптоз была значительно ниже, чем у дихлорацетата. Ацетат и трихлорацетат не оказывали заметного эффекта. Мы использовали бутират в качестве положительного контроля в этих экспериментах на основании нашего предыдущего сообщения о том, что индукция апоптоза бутиратом в клеточных линиях рака толстой кишки обязательным образом зависит от экспрессии SLC5A8 (Thangaraju et al., 2008).
Затем мы хотели определить, обладает ли апоптоз, вызванный дихлорацетатом, избирательностью в отношении опухолевых клеток, а также наблюдается ли этот эффект в клеточных линиях раковых клеток другого тканевого происхождения, чем толстая кишка. Для этого мы выбрали CCD841 и MCF10A в качестве представителей нормальных клеточных линий (CCD841 — толстая кишка; MCF10A — эпителий молочной железы) и четыре линии раковых клеток человека: MCF7 (клеточная линия рака молочной железы с рецептором эстрогена), MB231 (клеточная линия рака молочной железы с отрицательным рецептором эстрогена), DU145 (клеточная линия рака простаты с нечувствительностью к андрогенам) и LNCaP (клеточная линия рака простаты с чувствительностью к андрогенам). Как сообщалось ранее (Thangaraju et al., 2006, 2008), нормальные клеточные линии CCD841 и MCF10A экспрессировали обнаруживаемые уровни SLC5A8, как на уровне мРНК, так и на уровне белка (рис. 5A). В отличие от них, ни одна из четырех исследованных здесь линий раковых клеток не экспрессировала этот транспортер. Экспрессия стала очевидной в линиях раковых клеток после транзиторной трансфекции экспрессирующей конструкции млекопитающих. В нормальных клетках уровень экспрессии увеличивался после трансфекции. Используя эти клеточные линии, мы сравнили способность дихлорацетата вызывать апоптоз между нормальными и раковыми клеточными линиями (рис. 5B). Мы не обнаружили существенной разницы в апоптозе в нормальных клеточных линиях при обработке и без обработки дихлорацетатом (1 мМ). Повышение уровня экспрессии SLC5A8 также не оказало никакого влияния на степень апоптоза. Напротив, дихлорацетат был способен вызывать выраженный апоптоз в четырех линиях раковых клеток, но только если клетки экспрессировали транспортер. Без экспрессии транспортера линии раковых клеток не подвергались апоптозу при обработке дихлорацетатом. Эти результаты показывают, что дихлорацетат способен вызывать апоптоз даже при концентрации 1 мМ в селективной для раковых клеток концентрации, но только при условии экспрессии SLC5A8. Эти наблюдения были также подтверждены с помощью опосредованной вирусом lenti стабильной экспрессии SLC5A8 (индуцируемой доксицилином) в двух клеточных линиях рака молочной железы, MCF7 и MB231 (рис. 5C, D). Хорошо известно, что сайленсинг SLC5A8 связан с метилированием ДНК и что лечение раковых клеток агентами, метилирующими ДНК, восстанавливает экспрессию SLC5A8. Поэтому мы обработали нормальные и раковые клеточные линии 5′-аза-2-дезоксицитидином (5-Azadc), ДНК-деметилирующим агентом, и повторная активация SLC5A8 была подтверждена иммуноблотинговым анализом (рис. 5E). обработка 5-Azadc не изменила экспрессию белка SLC5A8 в нормальных эпителиальных клетках толстой кишки и молочной железы, в то время как она вновь активировала экспрессию SLC5A8 во всех линиях раковых клеток. Более того, обработка этих клеток 5-азадком сама по себе вызывала значительный апоптоз; однако обработка этих клеток дихлорацетатом (1 мМ) резко усиливала апоптоз, вызванный 5-азадком (рис. 5F).
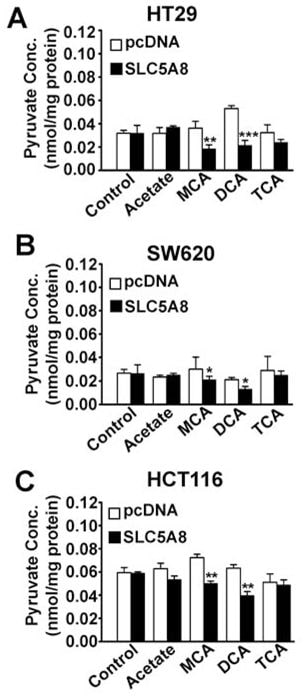
Влияние дихлорацетата на внутриклеточный уровень пирувата
Каков механизм, ответственный за индукцию апоптоза SLC5A8/дихлорацетатом в раковых клетках? SLC5A8 является активным транспортером дихлорацетата с Кт 36 ± 7 мкМ. Следовательно, соединение будет концентрироваться в раковых клетках, если транспортер экспрессирован. Это объясняет обязательное требование SLC5A8 к низким концентрациям дихлорацетата, чтобы вызвать апоптоз в этих клетках. Если транспортер не экспрессирован, клетки могут не накапливать соединение до уровня, достаточного для того, чтобы вызвать апоптоз. Как дихлорацетат, накопившись в раковых клетках, вызывает апоптоз? Единственным известным механизмом действия дихлорацетата является его способность активировать PDC через ингибирование PDK (Stacpoole et al., 1998, 2003, 2008). Это приводит к стимуляции митохондриального окисления, генерации реактивных форм кислорода, деполяризации мембранного потенциала митохондрий и индукции апоптоза. Активация PDC дихлорацетатом в интактных клетках снижает внутриклеточный уровень пирувата. Исходя из этого, мы предположили, что дихлорацетат будет снижать уровень пирувата в раковых клетках и что этот эффект будет зависеть в обязательном порядке от экспрессии SLC5A8. Мы проверили эту гипотезу на трех различных линиях раковых клеток (HT29, SW620 и HCT116) с экзогенной экспрессией SLC5A8 и без нее (рис. 6). Экспрессия транспортера сама по себе не оказывала никакого влияния на внутриклеточные уровни пирувата. Дихлорацетат был способен снижать уровень пирувата в значительной степени во всех трех линиях раковых клеток, но только при экспрессии транспортера. Интересно, что монохлорацетат также оказывал аналогичный эффект, но только в присутствии SLC5A8; в отличие от него, трихлорацетат и ацетат не влияли на уровень пирувата.
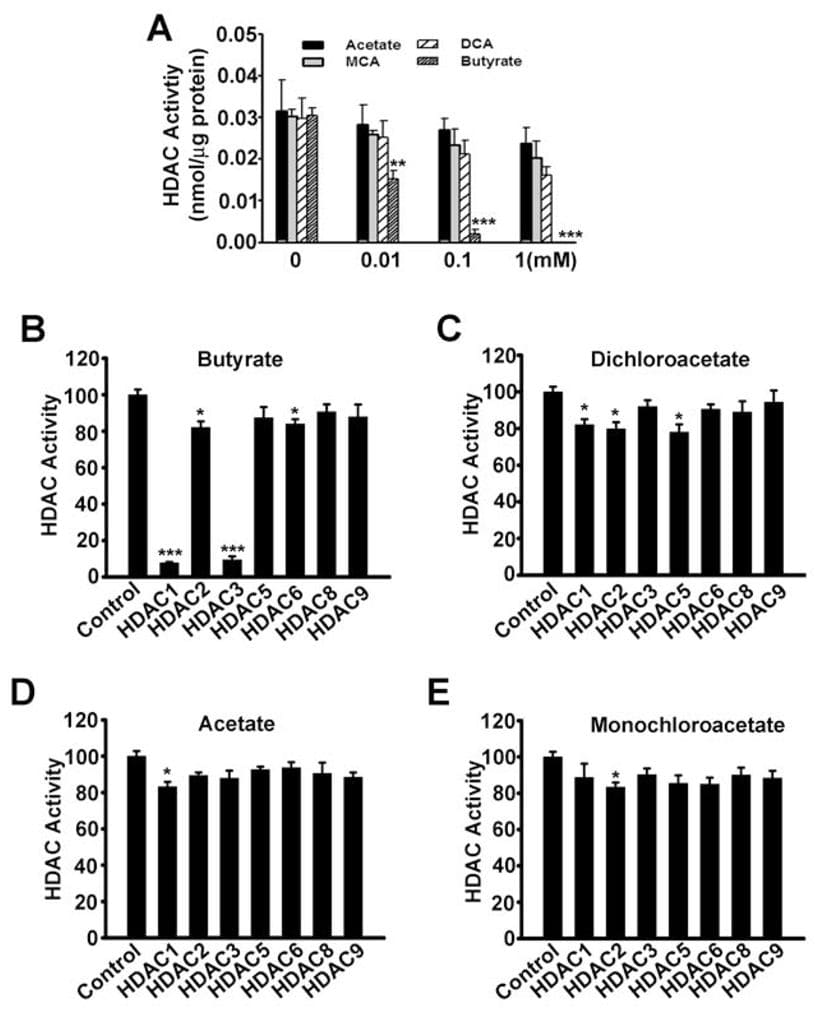
До сих пор единственным механизмом, с помощью которого SLC5A8 функционирует как опухолевый супрессор, была его способность концентрировать внутри клеток бутират и пируват, которые являются ингибиторами деацетилаз гистонов. В настоящем исследовании мы показали, что дихлорацетат также индуцирует апоптоз в раковых клетках и что этот эффект зависит от экспрессии SLC5A8. Эти результаты аналогичны результатам, полученным с бутиратом и пируватом. Поэтому мы задались вопросом, может ли ингибирование гистоновых деацетилаз участвовать в апоптозе раковых клеток, вызванном дихлорацетатом. Однако это казалось маловероятным, поскольку ацетат не является ингибитором деацетилаз гистонов (Thangaraju et al., 2006). Однако мы не могли исключить возможность того, что дихлорацетат может функционировать как ингибитор деацетилаз гистонов. Поэтому мы изучили влияние ацетата, монохлорацетата, дихлорацетата и бутирата на активность деацетилаз гистонов. Сначала мы использовали лизаты клеток SW620 в качестве источника деацетилаз гистонов (рис. 7А). В качестве положительного контроля в этих экспериментах использовался бутират. Как и ожидалось, бутират подавлял активность деацетилаз гистонов. В идентичных условиях дихлорацетат был, по крайней мере, в 100 раз менее эффективен по сравнению с бутиратом в ингибировании деацетилаз гистонов. Затем мы использовали рекомбинантные изоформы деацетилазы гистонов человека, чтобы определить, могут ли конкретные изоформы фермента ингибироваться дихлорацетатом. Наши предыдущие исследования показали, что бутират является специфическим ингибитором изоформ деацетилазы гистонов HDAC1 и HDAC3 (Thangaraju et al., 2009b). В настоящем исследовании мы обнаружили то же самое в отношении бутирата (рис. 7B). Дихлорацетат также в значительной степени ингибировал изоформы HDAC1, HDAC2 и HDAC5, но его эффективность была очень низкой (~20% ингибирования при концентрации 1 мМ) (рис. 7C). Ацетат (рис. 7D) и монохлорацетат (рис. 7E) не оказывали никакого влияния ни на одну из изоформ, изученных в исследовании.
Обсуждение
После публикации в журнале Cancer Cell статьи Bonnet et al. (2007), в которой было показано, что дихлорацетат способствует апоптозу в линиях раковых клеток in vitro и ингибирует рост рака в ксенотрансплантатах мыши in vivo, наблюдается постоянный рост общественного интереса к потенциальной полезности этого соединения в качестве противоракового препарата. Дихлорэтат использовался в качестве препарата для лечения врожденного молочнокислого ацидоза, поэтому фармакокинетика и профиль токсичности препарата были подробно изучены (Stacpoole at al., 1998). Недавние данные о том, что препарат может быть полезен и при лечении рака, побудили многих онкологических больных использовать этот неодобренный препарат без рекомендации врача (Pearson, 2007). Интересно, что контролируемых клинических исследований, подтверждающих терапевтическую эффективность дихлорацетата как противоракового препарата, не проводилось. Поскольку структура дихлорацетата не может быть запатентована, фармацевтические компании не были заинтересованы в разработке препарата для лечения рака путем проведения клинических испытаний, что побудило главного исследователя оригинальной публикации в журнале Cancer Cell собрать средства для проведения собственных небольших клинических испытаний (Pearson, 2007). Недавно были опубликованы результаты клинических испытаний (Machelakis et al., 2010), которые показали, что препарат эффективен у пациентов с глиобастомой. К сожалению, несмотря на то, что препарат был эффективен in vivo при лечении глиобластомы, для достижения терапевтической эффективности пришлось использовать высокие концентрации препарата, что привело к развитию периферической нейропатии. Это был единственный нежелательный побочный эффект, который ограничивал использование более высоких доз препарата для достижения большей терапевтической эффективности. Не было никаких доказательств гематологической, печеночной, почечной или сердечной токсичности. В нескольких исследованиях были зафиксированы неврологические осложнения при применении высоких доз дихлорацетата у животных и людей (Calcutt et al., 2009; Wiemer and Sachdev, 2009; Brandsma et al., 2010). Неврологические осложнения возникают только при очень высоких дозах дихлорацетата: 12.5 мг/кг/день в течение 3 месяцев (Bonnet et al., 2007) или 15 мг/кг/день в течение 4 недель (Brandsma et al., 2010). Необходимость высоких доз дихлорацетата для терапевтической эффективности in vivo согласуется с большинством недавно опубликованных данных in vitro, которые показывают, что концентрации 10 мМ и выше необходимы для того, чтобы вызвать апоптоз в линиях раковых клеток (Stockwin et al., 2010; Madhok et al., 2010; Xiao et al., 2010; Heshe et al., 2010; Sun et al., 2010).
Существуют убедительные доказательства того, что дихлорацетат обладает способностью изменять метаболический фенотип раковых клеток и способствовать их гибели, однако противоопухолевый эффект проявляется только при очень высоких концентрациях как in vitro, так и in vivo. Поскольку при таких терапевтически эффективных концентрациях возникает неврологическая токсичность, применение этого соединения в качестве противоракового препарата у человека ограничено. Интересно, что единственной молекулярной мишенью противоопухолевой активности дихлорацетата является PDK, которая ингибируется соединением в микромолярных концентрациях (константа ингибирования 50-100 мкМ) (Whitehouse et al., 1974; Cooper et al., 1974). Почему же тогда для уничтожения раковых клеток in vitro требуются высокие миллимолярные концентрации дихлорацетата? На этот вопрос необходимо ответить, чтобы использовать потенциал этого соединения в качестве противоракового препарата. Наши исследования дают удовлетворительный ответ на этот важный вопрос. Раковые клетки не экспрессируют никакой транспортной системы, которая могла бы эффективно переносить соединение из внеклеточной среды к его внутриклеточной мишени. Монокарбоксилатные транспортеры, экспрессируемые в раковых клетках, переносят дихлорацетат с Кт 0,6 мМ (Jackson and Halestrap, 1996). Это низкоаффинный транспортный процесс. Кроме того, монокарбоксилатные транспортеры не являются высококонцентрированными. Кроме того, опухолевые клетки вырабатывают большое количество лактата, который также является субстратом для монокарбоксилатных транспортеров со сродством, сравнимым с дихлорацетатом, что вызывает конкуренцию в процессе транспорта. Настоящие исследования показывают, что SLC5A8 переносит дихлорацетат со сродством, значительно большим, чем монокарбоксилатные транспортеры (Kt: 36 мкМ против 600 мкМ). Еще более важным является тот факт, что SLC5A8 гораздо эффективнее монокарбоксилатных транспортеров переносит дихлорацетат благодаря тому, что первый заряжается энергией за счет трансмембранного электрохимического градиента Na+. Интересно отметить, что сродство к транспортерам резко меняется при хлорировании второго углерода в ацетате (Кт: 1572 ± 101 мкМ для ацетата; 177 ± 16 мкМ для монохлорацетата; 36 ± 7 мкМ для дихлорацетата; 134 ± 11 мкМ для трихлорацетата). Полученные нами данные позволяют сделать вывод, что опухолевые клетки устойчивы к апоптозу, вызванному дихлорацетатом, потому что эти клетки не обладают эффективными транспортными системами для этого препарата. Это наиболее вероятная причина, по которой для того, чтобы вызвать гибель клеток опухоли, необходимы очень высокие концентрации соединения. Возможно также, что гибель клеток, наблюдаемая в опухолевых клетках при таких высоких концентрациях, не связана с активацией PDC, поскольку подобные эффекты наблюдаются и в нормальных клетках (Stockwin et al., 2010).
В настоящем исследовании мы показали, что если SLC5A8 экспрессируется в опухолевых клетках, дихлорацетат вызывает гибель клеток при относительно низкой концентрации (1 мМ) селективным для опухолевых клеток образом. В идентичных условиях нормальные клетки (CCD841 и MCF10A) не поражаются. Устойчивость нормальных клеток к дихлорацетату не связана с отсутствием транспортной системы для препарата. Нормальные клетки конститутивно экспрессируют SLC5A8, и мы показали, что клетки не подвергаются апоптозу как при дополнительной экспрессии транспортера, так и без нее. Это подтверждает специфический для опухолевых клеток эффект дихлорацетата. Наши исследования также показали, что эффективность дихлорацетата как индуктора апоптоза проявляется не только на клеточных линиях рака толстой кишки, но и на клеточных линиях рака молочной железы и рака предстательной железы. Механизм индуцированной дихлорацетатом клеточной гибели раковых клеток в основном связан с активацией митохондриального окисления пирувата. Это подтверждается данными о том, что внутриклеточный уровень пирувата в раковых клетках снижается в ответ на лечение дихлорацетатом в SLC5A8-зависимой манере, что позволяет предположить, что за этот эффект отвечает концентрированное проникновение препарата в раковые клетки, опосредованное экзогенно экспрессированным SLC5A8. Дихлорацетат не ингибирует деацетилазы гистонов, что исключает ингибирование деацетилаз гистонов как потенциальный механизм апоптоза в опухолевых клетках, индуцированного этим соединением.
Выводы о том, что дихлорацетат вызывает гибель клеток избирательно в опухолевых клетках при низких концентрациях, но только при наличии экспрессии SLC5A8, имеют клиническое и терапевтическое значение. Способность дихлорацетата активировать PDC через ингибирование PDK в раковых клетках обеспечивает механистически рациональную основу для противоопухолевой активности этого соединения. Однако раковые клетки устойчивы к действию препарата из-за отсутствия эффективного транспортера для него, что приводит к необходимости применения высоких концентраций соединения, чтобы вызвать гибель клеток, что, к сожалению, вызывает такие пагубные побочные эффекты, как нейропатия. В настоящем исследовании мы показали, что SLC5A8 служит активным транспортером для дихлорацетата. Однако, поскольку экспрессия этого транспортера подавлена в опухолевых клетках, как полученные результаты могут быть связаны с потенциальным терапевтическим использованием препарата? Глушение SLC5A8 в раковых клетках происходит через эпигенетические механизмы, включающие метилирование ДНК; лечение раковых клеток 5′-азацитидином, ингибитором метилирования ДНК, вновь активирует экспрессию гена (Li et al., 2003; Ueno et al., 2004; Hong et al., 2005; Porra et al., 2005; Thangaraju et al., 2006; Park et al., 2007, 2008). Метилирование ДНК играет важную роль в глушении генов-супрессоров опухоли при различных видах рака, и ингибиторы метилирования ДНК перспективны в качестве противораковых препаратов (Baylin, 2005). Два соединения с активностью ингибирования метилирования ДНК используются в клинической практике для лечения гематологических злокачественных опухолей. Это 5′-аза-2′-дезоксицитидин, также известный как децитабин (торговое название Dacogen) и 5′-азацитидин (торговое название Vidaza). Исследования in vitro показали, что обработка различных линий раковых клеток этими соединениями вновь активирует экспрессию SLC5A8. Мы предполагаем, что такое же явление будет происходить и in vivo. Поэтому комбинация ингибитора метилирования ДНК и дихлорацетата, вероятно, будет эффективна для лечения рака, поскольку ингибитор метилирования ДНК вызывает экспрессию SLC5A8 в опухолях, который затем эффективно транспортирует дихлорацетат в опухолевые клетки для проявления его противоопухолевой активности. Такой способ лечения позволит значительно снизить концентрацию дихлорацетата, необходимую для эффективности противоракового препарата in vivo, что потенциально обеспечит селективность в отношении опухолей, а также позволит избежать вредных побочных эффектов, таких как нейропатия. Результаты настоящего исследования обеспечивают рациональную основу для такой комбинированной терапии.
Материалы и методы
Материалы
[ 14C]-никотинат был приобретен у компании American Radiolabeled Chemicals (Сент-Луис, МО). Ацетат и его хлорпроизводные были получены от Sigma (Сент-Луис, МО). SLC5A8 был первоначально клонирован из кишечника человека (Miyauchi et al., 2004).
Система экспрессии ооцитов X. laevis
Кэппированная кРНК из кДНК SLC5A8 человека (клонированная в pGH19, вектор экспрессии ооцитов X. laevis) была синтезирована с помощью набора mMESSAGE-mMACHINE (Ambion, Austin, TX). Зрелые ооциты X. laevis были выделены путем обработки коллагеназой А (1,6 мг/мл), дефолликулированы вручную и поддерживались при 18 °C в модифицированной среде Барта, дополненной 25 мкг/мл гентамицина. В ооциты вводили 50 нг кРНК. Контролем служили ооциты, инъецированные водой. Электрофизиологические исследования проводили методом двухмикроэлектродного зажима напряжения. Ооциты перифузировали буфером, содержащим NaCl (100 мМ NaCl, 2 мМ KCl, 1 мМ MgCl2, 1 мМ CaCl2, 10 мМ Hepes, pH 7,5), затем тем же буфером, содержащим ацетат или его хлорпроизводные. Мембранный потенциал зажимали при -50 мВ. Разница между стационарными токами, измеренными в присутствии и отсутствии субстратов, рассматривалась как субстрат-индуцированные токи. При анализе кинетики насыщения субстрат-индуцированных токов кинетический параметр Kt (т.е. концентрация субстрата, необходимая для индукции полумаксимального тока) рассчитывали путем подбора уравнения Михаэлиса-Ментен к значениям субстрат-индуцированных токов. Кинетика Na+-активации субстрат-индуцированных токов анализировалась путем измерения субстрат-специфических токов в присутствии возрастающих концентраций Na+. Данные для Na+-зависимых токов анализировали в соответствии с уравнением Хилла для определения коэффициента Хилла (количество ионов Na+, участвующих в процессе активации). Поскольку уровни экспрессии значительно различались в разных ооцитах, кинетический анализ проводили путем нормализации уровней экспрессии. Для этого максимальный индуцированный SLC5A8-специфический ток в каждом кинетическом эксперименте в отдельных ооцитах принимали за 1. Каждый эксперимент повторяли с 3-4 разными ооцитами. Активность гетерологично экспрессированного SLC5A8 в ооцитах также контролировалась по поглощению [14C]-никотината. Концентрация никотината в этих экспериментах составляла 50 мкМ, время инкубации — 1 ч.
Протокол использования лягушек в этих экспериментах был одобрен Институциональным комитетом по уходу и использованию животных.
экспрессия SLC5A8 в культивируемых клеточных линиях
Клетки высевали в 35-миллиметровые культуральные чашки и культивировали в отсутствие пирувата. Клетки трансфецировали пкДНК или кДНК SLC5A8 человека с помощью Fugene 6 и Opti-MEM. pEGFP-N1 использовали для совместной трансфекции для определения эффективности трансфекции. Через 24 ч клетки обрабатывали ацетатом или его хлорпроизводными (1 мМ) или без них в течение 24 ч. Для анализа FACS клетки фиксировали в 50% этаноле, обрабатывали 0,1% цитратом натрия, 1 мг/мл RNase A и 50 мкг/мл пропидия йодида, а затем подвергали флуоресцентно-активированной сортировке клеток (FACS; FACS Caliber, Becton Dickinson).
Генерация стабильных линий клеток, экспрессирующих SLC5A8
Мы генерировали стабильные клоны, экспрессирующие SLC5A8, в двух линиях клеток, MCF7 и MDA-MB231, для условной экспрессии SLC5A8 путем помещения кДНК SLC5A8 под контроль Tet-зависимого промотора (Tet-On Advanced, Clontech). Система поставляется с двумя элементами, вектором регулятора (pLVX-Tet-On Advanced) и вектором ответа (pLVX-Tight-Puro). Вкратце, рекомбинантный ленти-вирус был получен путем совместной трансфекции в клетки 293FT вектора-регулятора (pLVX-Tet-On Advanced) и трех других векторов-помощников, pLP-1, pLP-2 и pVSVG (Invitrogen) с использованием реагента для трансфекции Lipofectamine 2000. Супернатант Lenti вирусов собирали через 72 ч после трансфекции и фильтровали через 0,45 мкм мембрану. Клетки MCF7 и MDA-MB231 инфицировали на 24 ч ленти-вирусом в среде, содержащей 8 мкг/мл полибрена, и культивировали еще 48 ч. Клетки отбирали с помощью G418 (2 мг/мл), а экспрессию белка rTet R проверяли с помощью Вестерн-блота. SLC5A8 был субклонирован в pLVX-Tight-Puro, и SLC5A8 pLVX-Tight-Puro был трансдуцирован в клетки MCF7 и MDA-MB231, экспрессирующие белок rTetR, с помощью той же процедуры, которая описана выше. Клетки отбирали с помощью 2 мкг/мл пуромицина и поддерживали в среде, содержащей Tetfree FBS, 0,25 мкг/мл пуромицина и 250 мкг/мл G418. Индукция мРНК SLC5A8 при добавлении доксициклина была подтверждена методом RT-PCR.
Повторная активация экспрессии SLC5A8 с помощью ДНК-деметилирующего агента
Клетки высевали в 10-сантиметровую посуду при очень низкой плотности (0,5 ×106 клеток/диск) и культивировали в соответствующей среде без пирувата натрия. Через 24 ч клетки обрабатывали 5′-азадком (2 мкг/мл) в течение 72 ч. Среду заменяли свежей средой, содержащей 5′-азадк, каждые 24 ч. После обработки 5′-азадком клетки обрабатывали DCA (1 мМ) в течение 48 ч. После обработки клетки собирали и обрабатывали для выделения белка, а также для анализа FACS.
Обратная транскрипция-ПЦР
РНК, полученная из нормальных и раковых клеток, использовалась для полуколичественной ПЦР. РНК (2 мкг) подвергалась обратной транскрипции в кДНК с помощью системы GeneAmp PCR (Roche). Праймеры для ПЦР для конкретных генов были разработаны на основе нуклеотидных последовательностей, доступных в GenBank. Обратную транскрипцию-ПЦР (RT-PCR) повторяли дважды с каждым образцом РНК. В качестве внутреннего контроля использовали GAPDH (глицеральдегид-3-фосфатдегидрогеназу).
Вестерн-блот анализ
Пятьдесят микрограммов белка фракционировали с помощью SDS-PAGE, и фракционированные белки переносили на нитроцеллюлозную мембрану (Whatman GmbH). Мембраны блокировали 5% обезжиренным сухим молоком и затем подвергали воздействию анти-SLC5A8 первичного антитела (кат. № ARP44110, Aviva System Biology) при 4 °C в течение ночи, после чего обрабатывали соответствующими вторичными антителами. Белки визуализировали с помощью ECL SuperSignal Western System (GE Healthcare).
Анализ апоптоза
Клетки фиксировали в 50% этаноле, обрабатывали 0,1% цитратом натрия, 1 мг/мл RNase A и 50 мкг/мл йодистого пропидия и подвергали анализу с помощью флуоресцентно-активированной сортировки клеток (FACS, Becton Dickinson).
Измерение активности HDAC
Для определения активности HDAC использовали коммерчески доступный набор (Biovision, Mountain View, CA). Если в качестве источника активности HDAC использовался лизат клеток линии клеток рака толстой кишки SW620, 100 мкг белка в лизате добавляли в ферментный анализ в присутствии или отсутствии ацетата или его хлорпроизводных (1 мМ). Активность рекомбинантных изоформ HDAC человека также измеряли с помощью того же набора. Рекомбинантные изоформы HDAC были приобретены у Cayman Chemical Company.
Измерение внутриклеточного уровня пирувата
Клетки высевали в 35-миллиметровые культуральные чашки и культивировали в отсутствие пирувата. Клетки трансфицировали пкДНК или кДНК SLC5A8 человека с помощью Fugene 6 и Opti-MEM. Через 24 ч клетки обрабатывали ацетатом или его хлорпроизводными (1 мМ) или без них в течение 24 ч. Затем лизаты клеток использовали для измерения пирувата с помощью коммерчески доступного набора (Biovision, Mountain View, CA).
Благодарности
Описанная здесь работа частично поддержана грантом Национальных институтов здравоохранения CA131402.
ССЫЛКИ
1 Baylin SB. Метилирование ДНК и сайленсинг генов при раке. Nat Clin Pract Oncol. 2005; 2:S4-S11. [PubMed: 16341240]
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondriaK + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11:37-51. [PubMed: 17222789]
2Brahimi-HornMC, Chiche J, Pouyssegur J. Hypoxia signaling controls metabolic demand. Curr Opin Cell Biol. 2007; 19:223-229. [PubMed: 17303407]
2BrandsmaD, Dorlo TP, Haanen JH, Beijnen JH, Boogerd W. Severe encephalopathy and polyneuropathy induced by dichloroacetate. J Neurol. 2010 В печати.
2CalcuttNA, Lopez VL, Bautista AD, Mizisin LM, Torres BR, Shroads AL, et al. Периферическая нейропатия у крыс, подвергшихся воздействию дихлорацетата. J Neuropathol Exp Neurol. 2009; 68:985-993. [PubMed: 19680144]
3CaoW, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Dichloroacetate (DCA) sensitizes both wild type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate. 2008; 68:1223-1231. [PubMed: 18465755]
4ChenZ, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007; 39:267-274. [PubMed: 17551814]
5CoadyMJ, Chang MH, Charron FM, Plata C, Wallendorff B, Sah JF, et al. Ген опухолевого супрессора SLC5A8 экспрессирует Na+-монокарбоксилатный котранспортер. J Physiol. 2004; 557:719-731. [PubMed: 15090606]
6CooperRH, Randle PJ, Denton RM. Регуляция киназы пируватдегидрогеназы сердечной мышцы. Biochem J. 1974; 143:625-641. [PubMed: 4462746]
7GanapathyV, Gopal E, Miyauchi S, Prasad PD. Биологические функции SLC5A8, кандидата в опухолевые супрессоры. Biochem Soc Trans. 2005; 33:237-240. [PubMed: 15667316]
8GanapathyV, Thangaraju M, Gopal E, Itagaki S, Miyauchi S, Prasad PD. Натрий-связанные монокарбоксилатные транспортеры в нормальных тканях и при раке. AAPS J. 2008; 10:193-199. [PubMed: 18446519]
9GanapathyV, Thangaraju M, Prasad PD. Транспортеры питательных веществ в раке: Релевантность гипотезы Варбурга и не только. Pharmacol Ther. 2009; 121:29-40. [PubMed: 18992769]
10GatenbyRA, Gillies RJ. Почему раковые опухоли имеют высокий аэробный гликолиз? Nat Rev Cancer. 2004; 4:891- 899. [PubMed: 15516961]
11GopalE, Fei YJ, Sugawara M, Miyauchi S, Zhuang L, Martin PM, et al. Expression of Slc5a8 in kidney and its role in Na+-coupled transport of lactate. J Biol Chem. 2004; 279:44522-44532. [PubMed: 15322102]
12GopalE, Fei YJ, Miyauchi S, Zhuang L, Prasad PD, Ganapathy V. Sodium-coupled and electrogenic transport of B-complex vitamin nicotinic acid by Slc5a8, a member of the Na+/glucose cotransporter gene family. Biochem J. 2005; 388:309-316. [PubMed: 15651982]
13GuptaN, Martin PM, Prasad PD, Ganapathy V. SLC5A8 (SMCT1)-опосредованный транспорт бутирата формирует основу для опухолеподавляющей функции транспортера. Life Sci. 2006; 78:2419- 2425. [PubMed: 16375929]
14HesheD, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol. 2010 В печати.
15HongC, Maunakea A, Jun P, Bollen AW, Hodgson JG, Goldenberg DD, et al. Общие эпигенетические механизмы в глиомах человека и мыши инактивируют экспрессию супрессора роста SLC5A8. Cancer Res. 2005; 65:3617-3623. [PubMed: 15867356]
16JacksonVN, Halestrap AP. Кинетика, субстрат и ингибиторная специфичность монокарбоксилатного (лактатного) транспортера клеток печени крысы, определенные с помощью флуоресцентного внутриклеточного индикатора pH, 2′, 7′-бис (карбоксиэтил)-5(6)-карбоксифлуоресцеина. J Biol Chem. 1996; 271:861-868. [PubMed: 8557697]
17KimJW, Dang CV. Молекулярная сладость рака и эффект Варбурга. Cancer Res. 2006; 66:8927-8930. [PubMed: 16982728]
19LiH, Myeroff L, Smiraglia D, Romero MF, Pretlow TP, Kasturi L, et al. SLC5A8, транспортер натрия, является геном-супрессором опухоли, заглушенным метилированием в аберрантных очагах крипт и раке толстой кишки человека. Proc Natl Acad Sci USA. 2003; 100:8412-8417. [PubMed: 12829793]
20MadhokBM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Дихлорацетат индуцирует апоптоз и остановку клеточного цикла в клетках колоректального рака. Br J Cancer. 2010; 102:1746-1752. [PubMed: 20485289]
21MartinPM, Gopal E, Ananth S, Zhuang L, Itagaki S, Prasad BM, et al. Identity of SMCT1 (SLC5A8) as a neuron-specific Na+-coupled transporter for active uptake of L-lactate and ketone bodies in the brain. J Neurochem. 2006; 98:279-288. [PubMed: 16805814]
22MathupalaSP, KOYH, Pedersen PL. Важнейшая роль митохондрий в раке: Варбург и далее и обнадеживающие перспективы для эффективной терапии. Biochim Biophys Acta. 2010; 1797:1225-1230. [PubMed: 20381449]
23MichelakisED, Webster L, Mackey JR. Дихлорацетат (DCA) как потенциальная метаболическая таргетная терапия рака. Br J Cancer. 2008; 99:989-994. [PubMed: 18766181]
24MichelakisED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010; 2:31-34.
25MiyauchiS, Gopal E, Fei YJ, Ganapathy V. Functional identification of SLC5A8, tumor suppressor downregulated in colon cancer, as a Na+-coupled transporter for short-chain fatty acids. J Biol Chem. 2004; 279:13293-13296. [PubMed: 14966140]
26MiyauchiS, Gopal E, Babu E, Sonne SR, Kubo Y, Umapathy NS, et al. Sodium-coupled electrogenic transport of pyroglutamate (5-oxoproline) via SLC5A8, a monocarboxylate transporter. Biochim Biophys Acta. 2010; 1798:1164-1171. [PubMed: 20211600]
27ParkJY, Zheng W, Kim D, Cheng JQ, Kumar N, Ahmad N, et al. Кандидат в опухолевые супрессоры ген SLC5A8 часто даун-регулируется гиперметилированием промотора при раке простаты. Cancer Detect Prev. 2007; 31:359-365. [PubMed: 18037591]
28ParkJY, Helm JF, Zheng W, Ly QP, Hodul PJ, Centeno BA, et al. Silencing of the candidate tumor suppressor gene solute carrier family 5 member 8 (SLC5A8) in human pancreatic cancer. Pancreas. 2008; 36:e32-e39. [PubMed: 18437076]
29PearsonH. Онкологические больные выбирают неодобренный препарат. Nature. 2007; 446:474-475. [PubMed: 17392750]
30PorraV, Ferraro-Peyret C, Durand C, Selmi-Ruby S, Giroud H, Berger-Dutrieux N, et al. Silencing of the tumor suppressor gene SLC5A8 is associated with BRAF mutations is classical papillary thyroid carcinomas. J Clin Endocrinol Metab. 2005; 90:3028-3035. [PubMed: 15687339]
31SemenzaGL. HIF-1: вверх и вниз по течению метаболизма рака. Curr Opin Genet Dev. 2010; 20:51-56. [PubMed: 19942427]
32StacpoolePW, Henderson GN, Yan Z, Cornett R, James MO. Фармакокинетика, метаболизм и токсикология дихлорацетата. Drug Metab Rev. 1998; 30:499-539. [PubMed: 9710704]
33StacpoolePW, Nagaraja NV, Hutson AD. Efficacy of dichloroacetate as a lactate-lowering drug. J Clin Pharmacol. 2003; 43:683-691. [PubMed: 12856382]
34StacpoolePW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008; 60:1478-1487. [PubMed: 18647626]
35StockwinLH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Дихлорацетат натрия (DCA) избирательно нацелен на клетки с дефектами в митохондриальном ЭТЦ. Int J Cancer. 2010 в печати.
36SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Обращение гликолитического фенотипа дихлорацетатом подавляет рост метастатических клеток рака молочной железы in vitro и in vivo. Breast Cancer Res Treat. 2010; 120:253-260. [PubMed: 19543830]
37ThangarajuM, Gopal E, Martin PM, Ananth S, Smith SB, Prasad PD, et al. SLC5A8 запускает апоптоз опухолевых клеток через пируват-зависимое ингибирование гистоновых деацетилаз. Cancer Res. 2006; 66:11560-11564. [PubMed: 17178845]
38ThangarajuM, Cresci G, Itagaki S, Mellinger J, Browning DD, Berger FG, et al. Sodium-coupled transport of the short-chain fatty acid butyrate by SLC5A8 and its relevance to colon cancer. J Gastrointest Surg. 2008; 12:1773-1782. [PubMed: 18661192]
39ThangarajuM, Karunakaran S, Itagaki S, Gopal E, Elangovan S, Prasad PD, et al. Транспорт через SLC5A8 с последующим ингибированием гистоновых деацетилаз HDAC1 и HDAC3 лежит в основе противоопухолевой активности 3-бромпирувата. Cancer. 2009a; 115:4655-4666. [PubMed: 19637353]
40ThangarajuM, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J. 2009b; 417:379-389. [PubMed: 18789002]
41UenoM, Toyota M, Akino K, Suzuki H, Kusano M, Satoh A, et al. Aberrant methylation and histone deacetylation associated with silencing of SLC5A8 in gastric cancer. Tumour Biol. 2004; 25:134- 140. [PubMed: 15361710]
42VanderHeiden MG, Cantley LC, Thompson CB. Понимание эффекта Варбурга: метаболические требования клеточной пролиферации. Science. 2009; 324:1029-1033. [PubMed: 19460998]
43ВарбургО. О происхождении раковых клеток. Science. 1956; 123:309-314. [PubMed: 13298683]
44WeimerLH, Sachdev N. Update on medication-induced peripheral neuropathy. Curr Neurol Neurosci Rep. 2009; 9:69-75. [PubMed: 19080756]
45WhitehouseS, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974; 141:761-774. [PubMed: 4478069]
46WongJY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008; 109:394-402. [PubMed: 18423823]
47XiaoL, Li X, Niu N, Qian J, Xie G, Wang Y. Дихлорацетат (DCA) усиливает гибель опухолевых клеток в сочетании с онколитическим аденовирусом, вооруженным MDA-7/IL-24. Mol Cell Biochem. 2010; 340:31-40. [PubMed: 20165905]