Ellappan Babu, Ph. D., Sabarish Ramachandran, Ph. D., Veena Coothan-Kandaswamy, Ph. D., Selvakumar Elangovan, Ph. D., Puttur D. Prasad, Ph. D, Vadivel Ganapathy, Ph. D, und Muthusamy Thangaraju, Ph. D.
Abteilung für Biochemie und Molekularbiologie, Medical College of Georgia, Georgia Health Sciences University, Augusta, Georgia, USA
Die Nutzer können den Inhalt dieser Dokumente für akademische Forschungszwecke ansehen, ausdrucken, kopieren, herunterladen sowie Texte und Daten auswerten, wobei stets die vollständigen Nutzungsbedingungen zu beachten sind:
http://www.nature.com/authors/editorial_policies/license.html#terms
Korrespondenz: M. Thangaraju, Ph. D., Abteilung für Biochemie und Molekularbiologie, Medical College of Georgia, Augusta, GA 30912, USA., [email protected].
Interessenkonflikt: Die Autoren erklären, dass es keine konkurrierenden finanziellen Interessen im Zusammenhang mit der in diesem Manuskript beschriebenen Arbeit gibt.
Veröffentlicht in der endgültigen, redigierten Form als: Oncogene. 2011 September 22; 30(38): 4026–4037. doi:10.1038/onc.2011.113.
Schlüsselwörter: SLC5A8; Dichloracetat; Krebsmedikament; Warburg-Effekt; Pyruvat-Dehydrogenase-Kinase; mitochondriale Oxidation bei Krebs
Zusammenfassung
In der Öffentlichkeit und in der Wissenschaft ist ein wachsendes Interesse an Dichloracetat als potenziellem Krebsmedikament zu verzeichnen. Es gibt glaubwürdige Beweise für die Antitumoraktivität dieser Verbindung, aber für eine signifikante therapeutische Wirkung sind hohe Konzentrationen erforderlich. Leider führen diese hohen Konzentrationen zu schädlichen Nebenwirkungen, die das Nervensystem betreffen, so dass die Verwendung zur Krebsbehandlung nicht möglich ist. Die mechanistische Grundlage der Antitumoraktivität der Verbindung ist ihre Fähigkeit, den Pyruvat-Dehydrogenase-Komplex durch Hemmung der Pyruvat-Dehydrogenase-Kinase zu aktivieren. Da die Verbindung die Kinase in mikromolaren Konzentrationen hemmt, ist nicht bekannt, warum therapeutisch untragbar hohe Dosen für die Unterdrückung des Tumorwachstums erforderlich sind. Wir stellten die Hypothese auf, dass ein Mangel an wirksamen Mechanismen für den Eintritt von Dichloracetat in Tumorzellen diesem Phänomen zugrunde liegen könnte. Hier zeigen wir, dass SLC5A8 Dichloracetat sehr effektiv und mit hoher Affinität transportiert. Dieser Transporter wird in normalen Zellen exprimiert, aber die Expression wird in Tumorzellen durch epigenetische Mechanismen unterdrückt. Das Fehlen des Transporters macht Tumorzellen resistent gegen die antitumorale Wirkung von Dichloracetat. Wird der Transporter jedoch ektopisch in Tumorzellen exprimiert, werden die Zellen schon bei niedrigen Konzentrationen empfindlich für das Medikament. Dies ist bei Brustkrebszellen, Dickdarmkrebszellen und Prostatakrebszellen zu beobachten. Normale Zellen, die den Transporter konstitutiv exprimieren, werden von der Substanz jedoch nicht beeinflusst, was auf eine selektive therapeutische Wirkung auf Tumorzellen hinweist. Der Mechanismus der Antitumoraktivität der Verbindung ist nach wie vor ihre Fähigkeit, die Pyruvat-Dehydrogenase-Kinase zu hemmen und die mitochondriale Oxidation von Pyruvat zu erzwingen. Da das Silencing von SLC5A8 in Tumoren mit DNA-Methylierung einhergeht und seine Expression durch die Behandlung mit DNA-Methylierungsinhibitoren induziert werden kann, deuten unsere Ergebnisse darauf hin, dass die Kombination von Dichloracetat mit einem DNA-Methylierungsinhibitor ein Mittel zur Verringerung der Dichloracetat-Dosen darstellen würde, um die mit hohen Dosen verbundenen schädlichen Auswirkungen zu vermeiden, ohne jedoch die Antitumoraktivität zu beeinträchtigen.
EINLEITUNG
Dichloracetat wird derzeit zur Behandlung der kongenitalen Laktatazidose eingesetzt (Stacpoole et al., 2003, 2008). Die therapeutische Wirksamkeit dieses Arzneimittels ist auf seine Fähigkeit zurückzuführen, den Pyruvatdehydrogenase-Komplex (PDC) in der mitochondrialen Matrix zu aktivieren. Der Enzymkomplex ist jedoch nicht das direkte Ziel des Medikaments. Dichloracetat ist ein Inhibitor der Pyruvatdehydrogenase-Kinase (PDK), die die E1α-Untereinheit von PDC phosphoryliert und den Komplex inaktiviert (Stacpoole et al. 2003, 2008). Durch die Hemmung der PDK verhindert Dichloracetat die Phosphorylierung von E1α und hält so PDC in aktiver Form aufrecht. Die medikamenteninduzierte Aktivierung von PDC erleichtert den mitochondrialen Pyruvat-Stoffwechsel. Da zytosolisches Pyruvat entweder in die Mitochondrien transportiert und dort verstoffwechselt oder im Zytoplasma durch Laktatdehydrogenase in Laktat umgewandelt werden kann, verschiebt sich durch die Aktivierung von PDC durch Dichloracetat und den daraus resultierenden Metabolismus von Pyruvat in den Mitochondrien das Gleichgewicht zwischen Pyruvat und Laktat in Richtung Pyruvat. Dadurch wird die Umwandlung von Laktat in Pyruvat gefördert, wodurch der Laktatspiegel sinkt.
In den letzten Jahren wurde erhebliches Interesse an Dichloracetat als potenziellem Krebsmedikament geäußert (Michelakis et al., 2008). Die Vorstellung, dass dieses Medikament in der Lage sein könnte, Tumorzellen abzutöten, hat eine rationale Grundlage. Tumorzellen beziehen den Großteil ihrer Energie aus der aeroben Glykolyse und nicht aus der mitochondrialen Oxidation. Diese Stoffwechselverschiebung in Tumorzellen wurde erstmals von Warburg (Warburg, 1956) erkannt und ist daher als Warburg-Effekt bekannt geworden (Gatenby und Gillies, 2004; Kim und Dang, 2006; Chen et al., 2007; Brahimi-Horn et al., 2007; Vander Heiden et al., 2009; Ganapathy et al., 2009; Mathupala et al., 2010). In Anbetracht der Tatsache, dass die aerobe Glykolyse, bei der Glukose in Laktat umgewandelt wird, nur 2 ATP erzeugt, während die mitochondriale Oxidation des aus der Glykolyse stammenden Pyruvats 30 ATP erzeugt, erscheint es rätselhaft, dass Tumorzellen den weniger effizienten Stoffwechselweg zur Energiegewinnung vorziehen. Tumorzellen leiden jedoch nicht unter ATP-Mangel, sondern erzeugen sogar mehr Energie als normale Zellen, um ihr verstärktes Wachstum und ihre Vermehrung zu unterstützen. Dies wird durch die Aktivierung der Glykolyse um ein Vielfaches erreicht. Zwar wird bei der mitochondrialen Oxidation mehr ATP erzeugt als bei der zytoplasmatischen Glykolyse, doch gleichzeitig entstehen bei der mitochondrialen Oxidation reaktive Sauerstoffspezies, die sich für die Zellen als schädlich erweisen könnten. Offenbar erkennen Tumorzellen diesen negativen Aspekt der mitochondrialen Oxidation und entscheiden sich daher für die Glykolyse als primäre ATP-Quelle. Bei der Glykolyse wird kein Sauerstoff benötigt und es entstehen keine reaktiven Sauerstoffspezies. Da die Energieerzeugung überwiegend durch Glykolyse erfolgt, können sich Tumorzellen auch unter anaeroben Bedingungen vermehren, die bei soliden Tumoren häufig gegeben sind. Die Unterdrückung der Mitochondrienfunktion in Tumoren ist jedoch reversibel. Interessanterweise besteht einer der Mechanismen, mit denen Tumorzellen die mitochondriale Oxidation unterdrücken, in der Induktion von PDK, wodurch PDC inaktiviert wird (Semenza, 2010). Daher kann die Oxidation von Pyruvat in Mitichondrien in Tumorzellen durch Umkehrung der krebsbedingten Unterdrückung der PDC-Aktivität induziert werden. Dichloracetat tut genau das, indem es die PDK hemmt. Trotz dieser rationalen Grundlage für Dichloracetat als potenzielles Krebsmedikament gibt es in der Literatur erhebliche Kontroversen über den klinischen Nutzen dieser Verbindung für die Krebsbehandlung beim Menschen. Obwohl die Studie von Bonnet et al. (2007) die Antitumor-Wirksamkeit von Dichloracetat in vitro und in vivo bei Tieren nachwies, hat eine neuere Studie von Stockwin et al. (2010) gezeigt, dass sehr hohe Konzentrationen von Dichloracetat erforderlich sind, um den Zelltod in Tumorzellen auszulösen, und dass die Verbindung bei diesen Konzentrationen keine Selektivität für Tumorzellen besitzt.
Dichloracetat ist ionisiert und kann die Plasmamembran nicht durch Diffusion passieren. Dies wirft die Frage auf, wie diese Verbindung in die Zellen gelangt und Zugang zu PDK in der mitochondrialen Matrix erhält. Soweit uns bekannt ist, gibt es nur einen einzigen Bericht über den Transport von Dichloracetat in Säugetierzellen, der zeigt, dass Monocarboxylat-Transporter in Hepatozyten und Ehrlich-Letter-Tumorzellen den zellulären Eintritt dieser Verbindung vermitteln (Jackson und Halestrap, 1996). Da die Monocarboxylat-Transporter elektroneutral sind, können die meisten Zellen, einschließlich Tumorzellen, die diese Transporter exprimieren, diesen Wirkstoff nicht konzentrieren. Vor kurzem haben wir und andere einen neuen Transporter für Monocarboxylate identifiziert, der eine ähnliche Substratselektivität wie die Monocarboxylat-Transporter aufweist, aber Na+-gekoppelt und elektrogen ist (Coady et al., 2004; Miyauchi et al., 2004). Dieser Transporter, der nach der Nomenklatur der Human Genome Organization als Natrium-gekoppelter Monocarboxylat-Transporter (SMCT1) oder SLC5A8 bezeichnet wird, ist in der Lage, seine Substrate gegen einen Konzentrationsgradienten zu konzentrieren, da der transmembrane Na+-Gradient und das Membranpotential als treibende Kräfte beteiligt sind. SLC5A8 transportiert Acetat, Propionat, Butyrat, Laktat, Pyruvat, 3-Bromopyruvat, Nicotinat, β-Hydroxybutyrat und Pyroglutamat (Miyauchi et al., 2004, 2010; Gopal et al., 2004, 2005; Martin et al., 2006; Thangaraju et al., 2006, 2008, 2009a). Wir fragten uns, ob dieser stark energiegekoppelte Transporter Dichloracetat als Substrat akzeptieren würde. Diese Frage ist für die Antitumoraktivität dieses Medikaments direkt relevant, da Tumorzellen diesen Transporter durch epigenetische Mechanismen zum Schweigen bringen (Ganapathy et al., 2005, 2008, 2009; Gupta et al., 2006). Daher haben wir die vorliegende Studie durchgeführt, um zwei Fragen zu beantworten: (a) Transportiert SLC5A8 Dichloracetat? (b) Hängt die Antitumoraktivität des Medikaments von der Expression des Transporters in Tumorzellen ab? Die Ergebnisse der Studie zeigen, dass SLC5A8 für die antitumorale Aktivität von Dichloracetat obligatorisch ist.
Ergebnisse
SLC5A8 transportiert Dichloracetat in einer Na+-gekoppelten Weise
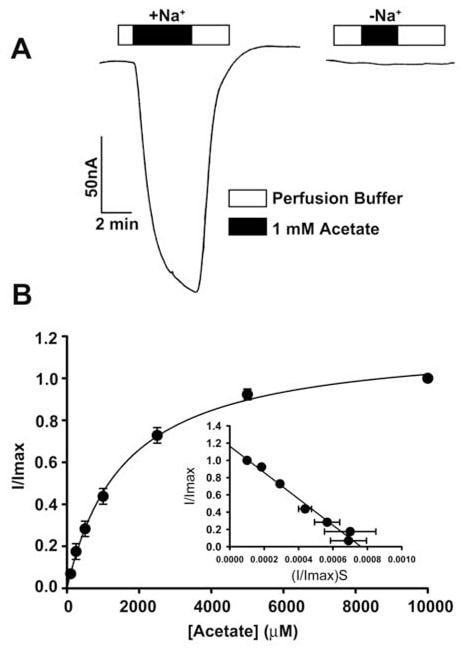
Der Transport von Acetat und seinen Chlorderivaten durch den menschlichen SLC5A8 wurde mit Hilfe des Oozyten-Expressionssystems von X. laevis untersucht. Der menschliche Transporter wurde in Oozyten durch Injektion von SLC5A8 cRNA heterolog exprimiert. Die Transportfunktion wurde elektrophysiologisch mit der Zwei-Mikroelektroden-Voltage-Clamp-Technik überwacht. SLC5A8 funktioniert als Na+-gekoppelter Transporter für Monocarboxylate mit einer Na+: Monocarboxylat-Stöchiometrie von 2:1. Der Transportprozess ist daher elektrogen, verbunden mit der Übertragung einer positiven Nettoladung in die Zellen pro Transportzyklus. Die sich daraus ergebende Depolarisierung der Membran kann als Einwärtsstrom unter Spannungsklemmenbedingungen überwacht werden. Wie in Abb. 1A zu sehen ist, induzierte die Exposition von SLC5A8-exprimierenden Oocyten gegenüber Acetat (1 mM) Einwärtsströme, wenn sie in Gegenwart von Na+ im Perfusionsmedium beobachtet wurden (129 ± 9 nA; n = 3 Oocyten). Diese Ströme wurden jedoch in Abwesenheit von Na+ nicht beobachtet. Diese Daten zeigen, dass SLC5A8 den Acetattransport in einer Na+-gekoppelten Weise vermittelt. Die Sättigungskinetik ergab, dass die Michaelis-Konstante (Kt) für den Transportprozess 1,6 ± 0,1 mM beträgt. Nach dem Nachweis der funktionellen Aktivität des klonierten menschlichen SLC5A8 unter Verwendung von Acetat als Positivkontrolle untersuchten wir den Transport von Dichloracetat. Die Exposition von SLC5A8-exprimierenden Eizellen gegenüber Dichloracetat (0,25 mM) induzierte deutliche Einwärtsströme in Gegenwart von Na+ (153 ± 28 nA; n = 5 Eizellen) (Abb. 2A). Solche Ströme wurden in Abwesenheit von Na+ nicht beobachtet. Der Transportprozess war sättigbar mit einem Kt von 36 ± 7 μM (Abb. 2B). Somit ist die Affinität von Dichloracetat für den Transporter ~45-mal höher als die von Acetat. Die durch Dichloracetat (0,25 mM) induzierten Ströme stiegen mit zunehmender Konzentration von Na+ im Perfusionsmedium an (Abb. 2C). Die Beziehung war sigmoidal, was auf die Beteiligung von mehr als einem Na+ am Aktivierungsprozess hinweist. Die Analyse der Daten nach der Hill-Gleichung ergab einen Wert von 2,1 ± 0,2 für den Hill-Koeffizienten. Diese Daten zeigen, dass die Na+: Dichloroaceat-Stöchiometrie für den Transportprozess 2:1 beträgt.
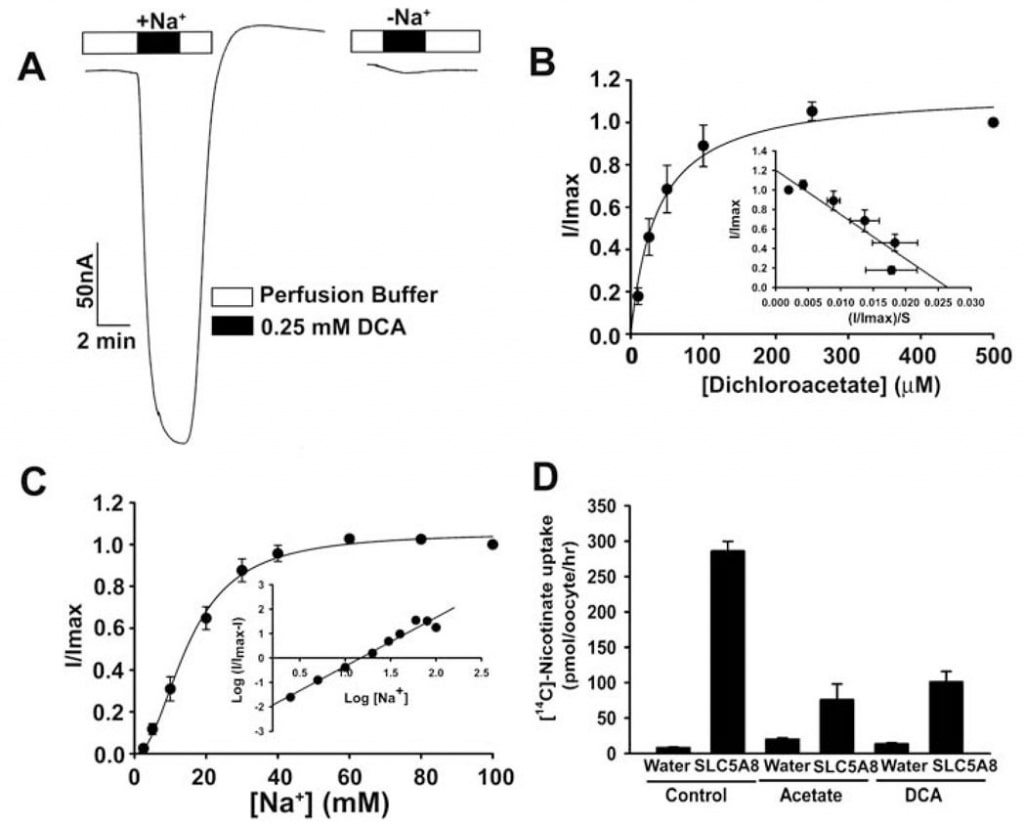
Wir verwendeten auch eine alternative Methode zur Bewertung des Transports von Dichloracetat über SLC5A8. Bei dieser Methode wurde[14C]-Nicotinat als Substrat für SLC5A8 verwendet. Mit Wasser injizierte Eizellen exprimierten den Transporter nicht und wurden daher als Kontrollen verwendet. Die Aufnahme von[14C]-Nikotinat (50 μM) war in SLC5A8-exprimierenden Oozyten 60-mal höher als in wasserinjizierten Oozyten (Abb. 2D). Die SLC5A8-spezifische Aufnahme von Nicotinat wurde in Gegenwart von Acetat (0,25 mM) oder Dichloracetat (0,25 mM) zu >80 % gehemmt, was darauf hindeutet, dass Acetat und Dichloracetat mit Nicotinat um die Aufnahme über SLC5A8 konkurrieren.
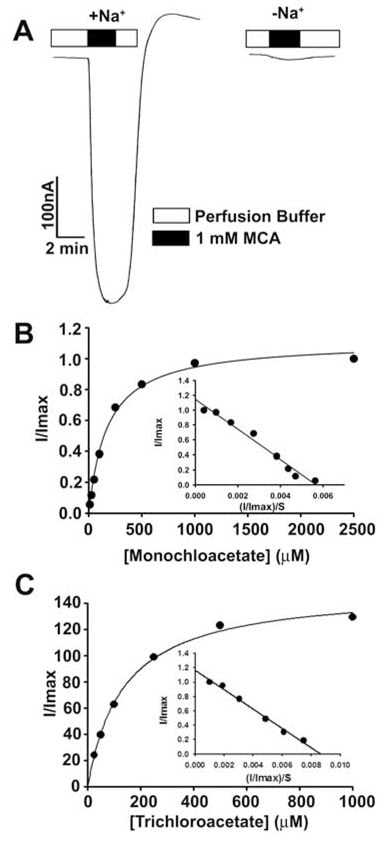
Diese Ergebnisse zeigten, dass Dichloracetat ein Substrat mit hoher Affinität für menschliches SLC5A8 ist. Anschließend untersuchten wir zum Vergleich den Transport von Monochloracetat und Trichloracetat (Abb. 3). Beide Verbindungen wurden über SLC5A8 in einer Na+-gekoppelten und sättigbaren Weise transportiert. Der Kt-Wert betrug 177 ± 16μM für Monochloracetat und 134 ± 11 μM für Trichloracetat.
Dichloracetat-induzierte Apoptose in Krebszellen erfordert SLC5A8
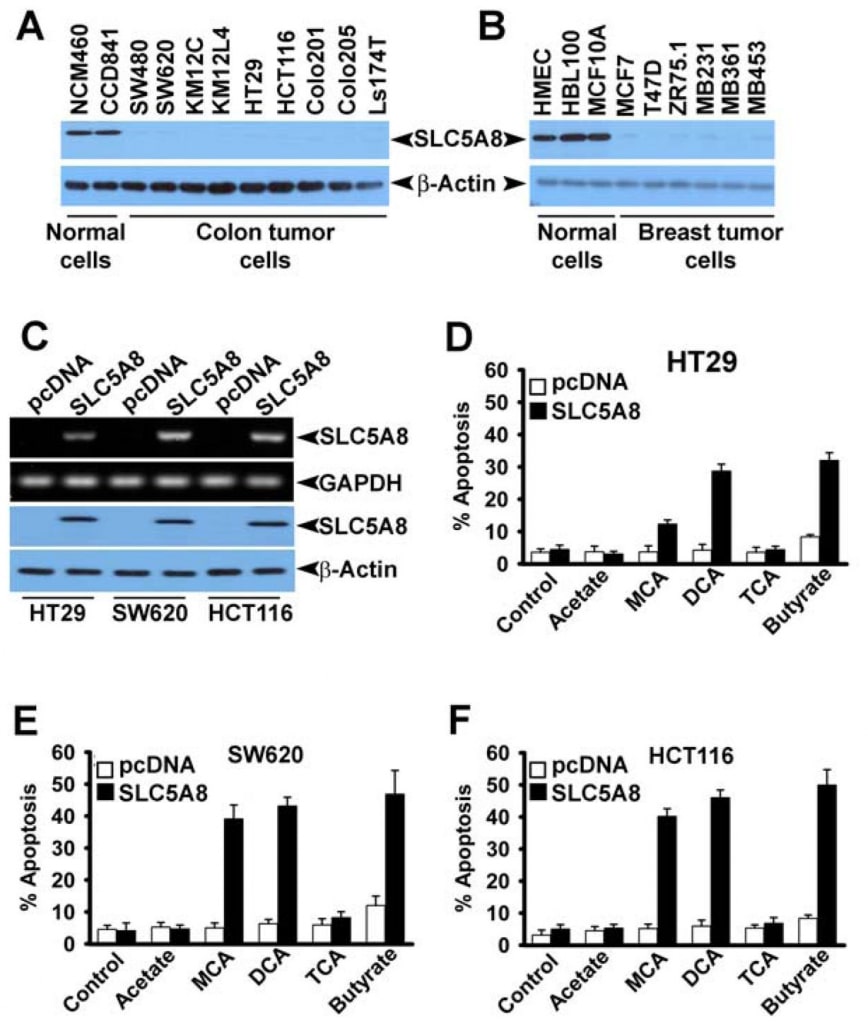
Obwohl mehrere Studien gezeigt haben, dass Dichloracetat in einer Vielzahl von Krebszelllinien Apoptose auslöst (Bonnet et al., 2007; Wong et al., 2008; Cao et al., 2008), konnte eine aktuelle Untersuchung diese Ergebnisse nicht bestätigen (Stockwin et al., 2010). Die Studien von Bonnet et al. (2007) zeigten, dass Dichloracetat in einer Konzentration von 0,5 mM in der Lage war, metabolische Veränderungen speziell in Krebszellen und nicht in normalen Zellen zu induzieren. Zu diesen Veränderungen gehörten die Depolarisierung der Mitochondrienmembran, die Unterdrückung der Glykolyse, die Verstärkung der mitochondrialen Oxidation, die Produktion reaktiver Sauerstoffspezies, die Induktion des Plasmamembran-Kaliumkanals Kv1.5 und die Freisetzung von pro-apoptotischen Faktoren aus den Mitochondrien. Wong et al. (2008) zeigten anschließend, dass Dichloracetat bei Endometriumkrebszellen Apoptose verursacht, und Cao et al. (2008) wiesen nach, dass die Verbindung Prostatakrebszellen für Strahlung sensibilisiert. Im Gegensatz dazu zeigten die Studien von Stockwin et al. (2010), dass Dichloracetat zwar in der Lage war, eine mitochondriale Depolarisierung und die Bildung reaktiver Sauerstoffspezies zu induzieren, diese Veränderungen jedoch sowohl in Krebszellen als auch in normalen Zellen auftraten. Außerdem war eine sehr hohe Konzentration der Verbindung (≥25 mM) erforderlich, um die Apoptose auszulösen. Ausgehend von den Erkenntnissen unserer Studie, dass SLC5A8 den energiegekoppelten aktiven Eintritt von Dichloracetat in die Zellen vermittelt, und der Tatsache, dass Krebszellen den Transporter zum Schweigen bringen, fragten wir uns, ob das Fehlen des Transporters in Krebszellen der Grund für das Fehlen einer nachweisbaren Apoptose bei niedrigen Konzentrationen der Verbindung ist, die von Stockwin et al (2010) beobachtet wurde. Wir untersuchten diese Frage anhand von drei verschiedenen menschlichen Dickdarmkrebs-Zelllinien (HCT116, SW620 und HT29). Diese drei Zelllinien exprimieren SLC5A8 nicht (Thangaraju et al., 2008).
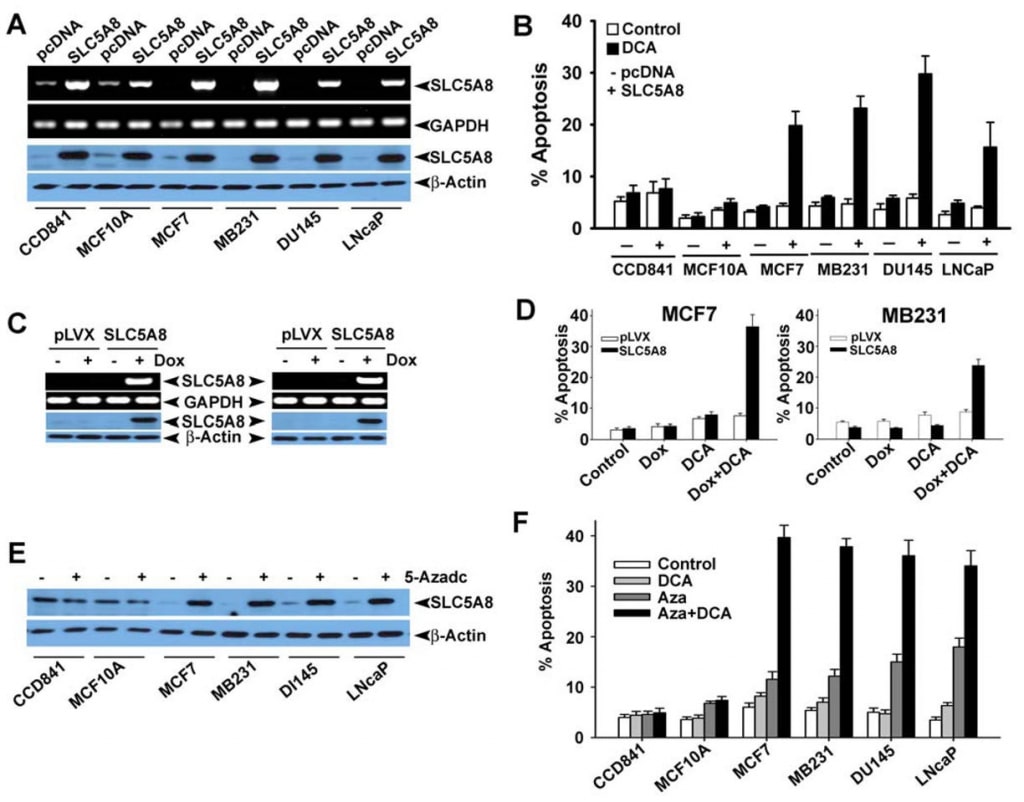
Wir bestätigten auch das Fehlen der SLC5A8-Expression in menschlichen Darm- und Brustkrebszelllinien auf Proteinebene (Abb. 4A und B). Wir exprimierten SLC5A8 in HCT116-, SW620- und HT29-Zelllinien durch transiente Transfektion eines Säugetier-Expressionskonstrukts und bestätigten die Expression durch RT-PCR und Western-Blot-Analyse (Abb. 4C). Zellen, die mit leerem Vektor transfiziert wurden, dienten als Kontrolle. Anschließend setzten wir Kontrollzellen und SLC5A8-exprimierende Zellen 48 Stunden lang Dichloracetat (1 mM) aus und überwachten die Apoptose (Abb. 4D-F). Die Ergebnisse waren interessant. In Kontrollzellen, die den Transporter nicht exprimierten, hatte Dichloracetat keine signifikante Wirkung. Bei SLC5A8-exprimierenden Zellen kam es jedoch unter den gleichen Bedingungen zu einer deutlichen Apoptose. Dieses Phänomen wurde bei allen drei Dickdarmkrebs-Zelllinien beobachtet. Monochloracetat verhielt sich auch bei HCT116- und SW620-Zellen ähnlich wie Dichloracetat, nicht jedoch bei HT29-Zellen, wo die Fähigkeit von Monochloracetat, Apoptose auszulösen, deutlich geringer war als die von Dichloracetat. Acetat und Trichloracetat hatten keine nennenswerte Wirkung. Wir verwendeten Butyrat als Positivkontrolle in diesen Experimenten, da wir bereits berichtet hatten, dass die Induktion der Apoptose durch Butyrat in Dickdarmkrebszelllinien zwingend von der Expression von SLC5A8 abhängt (Thangaraju et al., 2008).
Anschließend wollten wir feststellen, ob die durch Dichloracetat induzierte Apoptose eine Selektivität für Tumorzellen aufweist und ob der Effekt auch bei Krebszelllinien anderer Gewebe als dem Dickdarm zu beobachten ist. Zu diesem Zweck wählten wir CCD841 und MCF10A als Vertreter normaler Zelllinien (CCD841, Dickdarm; MCF10A, Brustepithel) und vier menschliche Krebszelllinien: MCF7 (eine Östrogenrezeptor-positive Brustkrebszelllinie), MB231 (eine Östrogenrezeptor-negative Brustkrebszelllinie), DU145 (eine Androgen-unempfindliche Prostatakrebszelllinie) und LNCaP (eine Androgen-empfindliche Prostatakrebszelllinie). Wie bereits berichtet (Thangaraju et al., 2006, 2008), exprimierten die normalen Zelllinien CCD841 und MCF10A nachweisbare Mengen von SLC5A8 sowohl auf mRNA- als auch auf Proteinebene (Abb. 5A). Im Gegensatz dazu exprimierte keine der vier hier untersuchten Krebszelllinien den Transporter. Die Expression wurde in den Krebszelllinien nach transienter Transfektion eines Säugetier-Expressionskonstrukts deutlich. In normalen Zellen stieg die Expression nach der Transfektion an. Anhand dieser Zelllinien verglichen wir die Fähigkeit von Dichloracetat, Apoptose zwischen normalen und Krebszelllinien zu induzieren (Abb. 5B). Wir fanden keinen signifikanten Unterschied in der Apoptose bei normalen Zelllinien mit und ohne Behandlung mit Dichloracetat (1 mM). Auch die erhöhte Expression von SLC5A8 hatte keinen Einfluss auf das Ausmaß der Apoptose. Im Gegensatz dazu konnte Dichloracetat bei den vier Krebszelllinien eine deutliche Apoptose auslösen, allerdings nur, wenn die Zellen den Transporter exprimierten. Ohne die Expression des Transporters durchliefen die Krebszelllinien bei der Behandlung mit Dichloracetat keine Apoptose. Diese Ergebnisse zeigen, dass Dichloracetat selbst bei Konzentrationen von nur 1 mM selektiv die Apoptose von Krebszellen auslösen kann, allerdings nur, wenn SLC5A8 exprimiert wird. Diese Beobachtungen wurden auch mit der durch Lenti-Viren vermittelten stabilen Expression von SLC5A8 (Doxycylin-induzierbar) in zwei Brustkrebszelllinien, MCF7 und MB231, bestätigt (Abb. 5C, D). Es ist bekannt, dass das Silencing von SLC5A8 mit DNA-Methylierung zusammenhängt und dass die Behandlung von Krebszellen mit DNA-Demethylierungsmitteln die SLC5A8-Expression wieder aktiviert. Daher behandelten wir normale und Krebszelllinien mit 5′-aza-2-Desoxycytidin (5-Azadc), einem DNA-Demethylierungsmittel, und die Reaktivierung von SLC5A8 wurde durch Immunoblotting-Analyse bestätigt (Abb. 5E). die Behandlung mit 5-Azadc veränderte die SLC5A8-Proteinexpression in normalen Dickdarm- und Brustepithelzellen nicht, während sie die SLC5A8-Expression in allen Krebszelllinien reaktivierte. Darüber hinaus löste die Behandlung dieser Zellen mit 5-Azadc selbst eine signifikante Apoptose aus; die Behandlung dieser Zellen mit Dichloracetat (1 mM) verstärkte jedoch die 5-Azadc-induzierte Apoptose dramatisch (Abb. 5F).
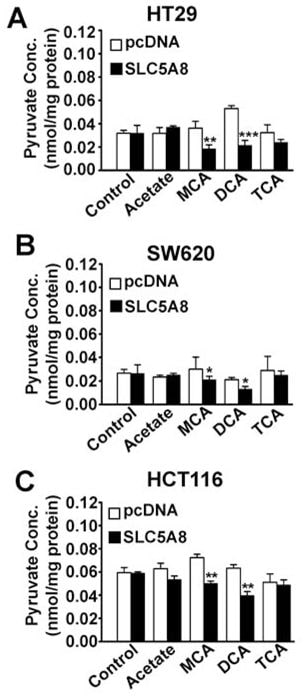
Wirkung von Dichloracetat auf den intrazellulären Pyruvatspiegel
Welcher Mechanismus ist für die Induktion der Apoptose durch SLC5A8/Dichloracetat in Krebszellen verantwortlich? SLC5A8 ist ein aktiver Transporter für Dichloracetat mit einem Kt von 36 ± 7μM. Daher wird die Verbindung in Krebszellen konzentriert, wenn der Transporter exprimiert wird. Dies würde erklären, warum SLC5A8 bei niedrigen Konzentrationen von Dichloracetat zwingend erforderlich ist, um in diesen Zellen Apoptose auszulösen. Wenn der Transporter nicht exprimiert wird, kann es sein, dass die Zellen die Verbindung nicht in ausreichender Menge anreichern, um Apoptose auszulösen. Wie wirkt Dichloracetat, sobald es sich in den Krebszellen angesammelt hat, um die Apoptose auszulösen? Der einzige bekannte Wirkmechanismus von Dichloracetat ist seine Fähigkeit, PDC durch Hemmung von PDK zu aktivieren (Stacpoole et al., 1998, 2003, 2008). Dies würde zu einer Stimulierung der mitochondrialen Oxidation, zur Bildung reaktiver Sauerstoffspezies, zur Depolarisierung des mitochondrialen Membranpotenzials und zur Induktion der Apoptose führen. Die Aktivierung von PDC durch Dichloracetat in intakten Zellen würde den intrazellulären Pyruvatspiegel senken. Auf dieser Grundlage stellten wir die Hypothese auf, dass Dichloracetat den Pyruvatspiegel in Krebszellen senken würde und dass dieser Effekt zwingend von der SLC5A8-Expression abhängig wäre. Wir testeten diese Hypothese in drei verschiedenen Krebszelllinien (HT29, SW620 und HCT116) mit und ohne exogene Expression von SLC5A8 (Abb. 6). Die Expression des Transporters an sich hatte keinen Einfluss auf die intrazellulären Pyruvatspiegel. Dichloracetat war in der Lage, die Pyruvatkonzentration in allen drei Krebszelllinien in erheblichem Maße zu senken, allerdings nur, wenn der Transporter exprimiert wurde. Interessanterweise hatte auch Monochloracetat eine ähnliche Wirkung, wiederum nur in Anwesenheit von SLC5A8; im Gegensatz dazu waren Trichloracetat und Acetat nicht in der Lage, den Pyruvatspiegel zu beeinflussen.
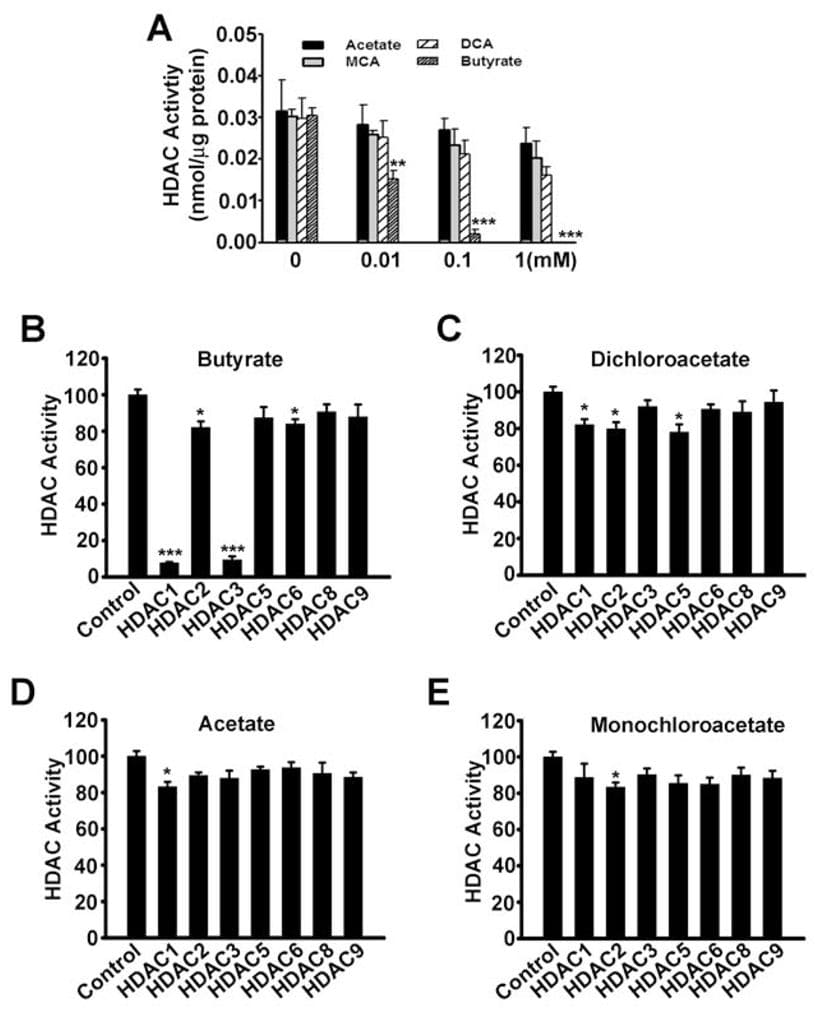
Bisher war der einzige Mechanismus, durch den SLC5A8 als Tumorsuppressor fungiert, seine Fähigkeit, Butyrat und Pyruvat, die Inhibitoren von Histon-Deacetylasen sind, in den Zellen zu konzentrieren. In der vorliegenden Studie haben wir gezeigt, dass Dichloracetat auch die Apoptose in Krebszellen auslöst und dass dieser Effekt von der Expression von SLC5A8 abhängt. Diese Ergebnisse ähneln denen mit Butyrat und Pyruvat. Daher fragten wir uns, ob die Hemmung von Histon-Deacetylasen an der durch Dichloracetat ausgelösten Apoptose in Krebszellen beteiligt ist. Dies schien jedoch unwahrscheinlich, da Acetat kein Inhibitor von Histondeacetylasen ist (Thangaraju et al., 2006). Wir konnten jedoch nicht ausschließen, dass Dichloracetat als Inhibitor von Histon-Deacetylasen fungieren könnte. Daher untersuchten wir den Einfluss von Acetat, Monochloracetat, Dichloracetat und Butyrat auf die Aktivität von Histondeacetylasen. Zunächst verwendeten wir Lysate von SW620-Zellen als Quelle für Histon-Deacetylasen (Abb. 7A). Butyrat wurde bei diesen Experimenten als Positivkontrolle verwendet. Wie erwartet, hemmte Butyrat die Aktivität der Histondeacetylasen. Unter identischen Bedingungen war Dichloracetat bei der Hemmung von Histondeacetylasen mindestens 100-mal weniger wirksam als Butyrat. Anschließend haben wir rekombinante menschliche Histondeacetylase-Isoformen verwendet, um festzustellen, ob bestimmte Isoformen des Enzyms durch Dichloracetat gehemmt werden können. Unsere früheren Studien haben gezeigt, dass Butyrat ein spezifischer Inhibitor der Histon-Deacetylase-Isoformen HDAC1 und HDAC3 ist (Thangaraju et al., 2009b). In der vorliegenden Studie fanden wir dasselbe mit Butyrat (Abb. 7B). Dichloracetat hemmte ebenfalls die Isoformen HDAC1, HDAC2 und HDAC5 in signifikantem Ausmaß, aber die Wirksamkeit war sehr gering (~20 % Hemmung bei einer Konzentration von 1 mM) (Abb. 7C). Acetat (Abb. 7D) und Monochloracetat (Abb. 7E) hatten auf keine der in der Studie untersuchten Isoformen eine Wirkung.
Diskussion
Seit der Veröffentlichung des Artikels in Cancer Cell von Bonnet et al. (2007), in dem gezeigt wurde, dass Dichloracetat die Apoptose in Krebszelllinien in vitro fördert und das Krebswachstum in Mäuse-Xenografts in vivo hemmt, hat das öffentliche Interesse an der potenziellen Nützlichkeit dieser Verbindung als Krebsmedikament stetig zugenommen. Dichloroetat wurde als Medikament zur Behandlung von kongenitaler Laktatazidose eingesetzt, und daher wurden Einzelheiten der Pharmakokinetik und des Toxizitätsprofils des Medikaments ausgearbeitet (Stacpoole at al., 1998). Die jüngsten Erkenntnisse, dass das Medikament auch bei der Krebsbehandlung nützlich sein könnte, haben viele Krebspatienten dazu veranlasst, dieses nicht zugelassene Medikament ohne die Empfehlung ihres Arztes zu verwenden (Pearson, 2007). Interessanterweise gibt es keine kontrollierten klinischen Studien, die die therapeutische Wirksamkeit von Dichloracetat als Krebsmedikament belegen. Da die Struktur von Dichloracetat nicht patentiert werden kann, waren die Pharmaunternehmen offenbar nicht daran interessiert, das Medikament für die Krebstherapie durch klinische Studien zu entwickeln, was den Hauptautor der ursprünglichen Veröffentlichung in Cancer Cell dazu veranlasste, Mittel für seine eigenen kleinen klinischen Studien aufzubringen (Pearson, 2007). Die Ergebnisse der klinischen Studie wurden vor kurzem veröffentlicht (Machelakis et al., 2010), aus denen hervorging, dass das Medikament bei Patienten mit Gliobastom wirksam ist. Obwohl das Medikament in vivo bei der Behandlung von Glioblastomen wirksam war, mussten leider hohe Konzentrationen des Medikaments verwendet werden, um therapeutisch wirksam zu sein, was zu peripherer Neuropathie führte. Dies war die einzige unerwünschte Nebenwirkung, die die Verwendung höherer Dosierungen des Medikaments für eine größere therapeutische Wirksamkeit einschränkte. Es gab keine Hinweise auf eine hämatologische, hepatische, renale oder kardiale Toxizität. Mehrere Studien haben neurologische Komplikationen bei hohen Dosen von Dichloracetat bei Tieren und Menschen dokumentiert (Calcutt et al., 2009; Wiemer und Sachdev, 2009; Brandsma et al., 2010). Die neurologischen Komplikationen treten nur bei sehr hohen Dosen von Dichloracetat auf: 12.5 mg/kg/Tag für 3 Monate (Bonnet et al., 2007) oder 15 mg/kg/Tag für 4 Wochen (Brandsma et al., 2010). Die Notwendigkeit hoher Dichloracetat-Dosen, um in vivo therapeutisch wirksam zu sein, stimmt mit den meisten der kürzlich veröffentlichten In-vitro-Daten überein, die zeigen, dass Konzentrationen von 10 mM oder höher erforderlich sind, um bei Krebszelllinien Apoptose auszulösen (Stockwin et al., 2010; Madhok et al., 2010; Xiao et al., 2010; Heshe et al., 2010; Sun et al., 2010).
Es gibt überzeugende Beweise dafür, dass Dichloracetat in der Lage ist, den metabolischen Phänotyp von Krebszellen umzukehren und ihren Tod zu fördern, aber die Antitumorwirkung tritt nur bei sehr hohen Konzentrationen sowohl in vitro als auch in vivo auf. Da bei diesen therapeutisch wirksamen Konzentrationen neurologische Toxizität auftritt, ist der Nutzen der Verbindung als Krebsmedikament beim Menschen begrenzt. Interessanterweise ist das einzige molekulare Ziel für die Antitumoraktivität von Dichloracetat die PDK, die von der Verbindung bei mikromolaren Konzentrationen gehemmt wird (Hemmkonstante 50-100 μM) (Whitehouse et al., 1974; Cooper et al., 1974). Warum sind dann in vitro hohe millimolare Konzentrationen von Dichloracetat erforderlich, um Krebszellen abzutöten? Diese Frage muss beantwortet werden, um das Potenzial dieser Verbindung als Krebsmedikament nutzen zu können. Die vorliegenden Studien liefern eine zufriedenstellende Antwort auf diese wichtige Frage. Krebszellen exprimieren kein Transportsystem, das die Verbindung wirksam aus dem extrazellulären Medium an ihr intrazelluläres Ziel bringen kann. Monocarboxylat-Transporter, die in Krebszellen exprimiert werden, transportieren Dichloracetat mit einem Kt von 0,6 mM (Jackson und Halestrap, 1996). Dies ist ein Transportprozess mit geringer Affinität. Außerdem sind Monocarboxylat-Transporter nicht hochkonzentriert. Darüber hinaus erzeugen Tumorzellen große Mengen an Laktat, das ebenfalls ein Substrat für Monocarboxylat-Transporter mit einer vergleichbaren Affinität wie Dichloracetat ist und somit eine Konkurrenz für den Transportprozess darstellt. Die vorliegenden Studien zeigen, dass SLC5A8 Dichloracetat mit einer deutlich höheren Affinität transportiert als Monocarboxylat-Transporter (Kt : 36 μM versus 600 μM). Noch wichtiger ist die Tatsache, dass SLC5A8 beim Transport von Dichloracetat viel effektiver ist als die Monocarboxylat-Transporter, weil der erstere durch einen transmembranen elektrochemischen Na+-Gradienten angeregt wird. Interessant ist, dass sich die Affinität für den Transporter durch die Chlorierung des zweiten Kohlenstoffs in Acetat drastisch ändert (Kt : 1572 ± 101 μM für Acetat; 177 ± 16 μM für Monochloracetat; 36 ± 7 μM für Dichloracetat; 134 ± 11 μM für Trichloracetat). Unsere Ergebnisse lassen den Schluss zu, dass Tumorzellen gegen die durch Dichloracetat ausgelöste Apoptose resistent sind, weil diese Zellen keine wirksamen Transportsysteme für das Medikament besitzen. Dies ist der wahrscheinlichste Grund dafür, dass sehr hohe Konzentrationen der Verbindung erforderlich sind, um den Zelltod in Tumorzellen auszulösen. Es ist auch möglich, dass der in Tumorzellen bei diesen hohen Konzentrationen beobachtete Zelltod nicht auf die Aktivierung von PDC zurückzuführen ist, da ähnliche Effekte auch in normalen Zellen beobachtet werden (Stockwin et al., 2010).
In der vorliegenden Studie haben wir gezeigt, dass Dichloracetat, wenn SLC5A8 in Tumorzellen exprimiert wird, den Zelltod bei einer relativ niedrigen Konzentration (1 mM) in einer tumorzellselektiven Weise auslöst. Normale Zellen (CCD841 und MCF10A) sind unter gleichen Bedingungen nicht betroffen. Die Resistenz normaler Zellen gegenüber Dichloracetat ist nicht auf das Fehlen eines Transportsystems für den Wirkstoff zurückzuführen. Normale Zellen exprimieren SLC5A8 konstitutiv, und wir haben gezeigt, dass die Zellen weder mit noch ohne zusätzliche Expression des Transporters apoptotisch werden. Dies bestätigt die tumorzellspezifische Wirkung von Dichloracetat. Unsere Studien haben auch gezeigt, dass die Wirksamkeit von Dichloracetat als Auslöser der Apoptose nicht nur bei Dickdarmkrebs-Zelllinien, sondern auch bei Brustkrebs-Zelllinien und Prostatakrebs-Zelllinien zu beobachten ist. Der Mechanismus des Dichloracetat-induzierten Zelltods in Krebszellen ist hauptsächlich auf die Aktivierung der mitochondrialen Oxidation von Pyruvat zurückzuführen. Dies wird durch die Feststellung gestützt, dass der intrazelluläre Pyruvatspiegel in Krebszellen als Reaktion auf die Behandlung mit Dichloracetat in einer SLC5A8-abhängigen Weise abnimmt, was darauf hindeutet, dass der konzentrative Eintritt des Medikaments in die Krebszellen, vermittelt durch das exogen exprimierte SLC5A8, für diesen Effekt verantwortlich ist. Dichloracetat hemmt keine Histon-Deacetylasen, was eine Hemmung von Histon-Deacetylasen als möglichen Mechanismus der durch die Verbindung induzierten Apoptose in Tumorzellen ausschließt.
Die Feststellung, dass Dichloracetat bei niedrigen Konzentrationen selektiv den Zelltod in Tumorzellen auslöst, aber nur, wenn SLC5A8 exprimiert wird, hat klinische und therapeutische Bedeutung. Die Fähigkeit von Dichloracetat, PDC über die Hemmung von PDK in Krebszellen zu aktivieren, liefert eine mechanistisch rationale Grundlage für die Antitumoraktivität der Verbindung. Krebszellen sind jedoch aufgrund des Fehlens eines wirksamen Transporters für das Medikament resistent gegen das Mittel, so dass hohe Konzentrationen des Mittels erforderlich sind, um den Zelltod herbeizuführen, was leider schädliche Nebenwirkungen wie Neuropathie verursacht. In der vorliegenden Studie haben wir gezeigt, dass SLC5A8 als aktiver Transporter für Dichloracetat dient. Da jedoch die Expression des Transporters in Tumorzellen unterdrückt wird, stellt sich die Frage, inwiefern die vorliegenden Ergebnisse für den potenziellen therapeutischen Einsatz des Medikaments relevant sind Die Stilllegung von SLC5A8 in Krebszellen erfolgt über epigenetische Mechanismen, an denen die DNA-Methylierung beteiligt ist; die Behandlung von Krebszellen mit 5′-Azacytidin, einem Inhibitor der DNA-Methylierung, reaktiviert die Expression des Gens (Li et al., 2003; Ueno et al., 2004; Hong et al., 2005; Porra et al., 2005; Thangaraju et al., 2006; Park et al., 2007, 2008). Die DNA-Methylierung spielt eine entscheidende Rolle bei der Stilllegung von Tumorsuppressorgenen in einer Reihe von Krebsarten, und DNA-Methylierungsinhibitoren sind vielversprechende Krebsmedikamente (Baylin, 2005). Zwei Wirkstoffe, die die DNA-Methylierung hemmen, werden bereits klinisch zur Behandlung von hämatologischen Malignomen eingesetzt. Dabei handelt es sich um 5′-aza-2′-Desoxycytidin, auch bekannt als Decitabin (Handelsname Dacogen) und 5′-Azacytidin (Handelsname Vidaza). In-vitro-Studien haben gezeigt, dass die Behandlung einer Reihe von Krebszelllinien mit diesen Verbindungen die Expression von SLC5A8 reaktiviert. Wir vermuten, dass das gleiche Phänomen auch in vivo auftreten würde. Daher ist eine Kombination aus einem DNA-Methylierungsinhibitor und Dichloracetat wahrscheinlich für die Behandlung von Krebs wirksam, da der DNA-Methylierungsinhibitor die Expression von SLC5A8 in Tumoren induzieren würde, die dann Dichloracetat effektiv in die Tumorzellen transportieren würde, um seine Antitumoraktivität auszulösen. Diese Art der Behandlung würde die Konzentration von Dichloracetat, die für eine In-vivo-Wirksamkeit als Krebsmittel erforderlich ist, erheblich verringern und somit möglicherweise eine Tumorselektivität bewirken und auch die schädlichen Nebenwirkungen wie Neuropathie vermeiden. Die Ergebnisse der vorliegenden Studie liefern eine rationale Grundlage für eine solche Kombinationstherapie.
Materialien und Methoden
Materialien
[ 14C]-Nikotinat wurde von American Radiolabeled Chemicals (St. Louis, MO) erworben. Acetat und seine Chlorderivate wurden von Sigma (St. Louis, MO) bezogen. SLC5A8 wurde ursprünglich aus dem menschlichen Darm kloniert (Miyauchi et al., 2004).
X. laevis Eizellen-Expressionssystem
Capped cRNA aus humaner SLC5A8 cDNA (kloniert in pGH19, einem X. laevis Eizellen-Expressionsvektor) wurde mit dem mMESSAGE-mMACHINE-Kit (Ambion, Austin, TX) synthetisiert. Reife Oozyten von X. laevis wurden durch Behandlung mit Kollagenase A (1,6 mg/ml) isoliert, manuell defollikuliert und bei 18 °C in modifiziertem Barth’schen Medium, ergänzt mit 25 μg/ml Gentamicin, gehalten. Die Oozyten wurden mit 50 ng cRNA injiziert. Wasserinjizierte Oozyten dienten als Kontrollen. Elektrophysiologische Untersuchungen wurden mit der Zwei-Mikroelektroden-Voltage-Clamp-Methode durchgeführt. Die Oozyten wurden mit einem NaCl-haltigen Puffer (100 mM NaCl, 2 mM KCl, 1 mM MgCl2, 1 mM CaCl2, 10 mM Hepes, pH 7,5) perifundiert, gefolgt von demselben Puffer, der Acetat oder seine Chlorderivate enthält. Das Membranpotenzial wurde bei -50 mV abgeklemmt. Die Unterschiede zwischen den in Gegenwart und Abwesenheit von Substraten gemessenen stationären Strömen wurden als substratinduzierte Ströme betrachtet. Bei der Analyse der Sättigungskinetik der substratinduzierten Ströme wurde der kinetische Parameter Kt (d. h. die für die Induktion des halbmaximalen Stroms erforderliche Substratkonzentration) durch Anpassung der Michaelis-Menten-Gleichung an die Werte der substratinduzierten Ströme berechnet. Die Na+-Aktivierungskinetik der substratinduzierten Ströme wurde durch Messung der substratspezifischen Ströme in Gegenwart steigender Na+-Konzentrationen analysiert. Die Daten für die Na+-abhängigen Ströme wurden nach der Hill-Gleichung analysiert, um den Hill-Koeffizienten (die Anzahl der am Aktivierungsprozess beteiligten Na+-Ionen) zu bestimmen. Da die Expressionsniveaus von Eizelle zu Eizelle erheblich variierten, wurden die kinetischen Analysen durch Normalisierung der Expressionsniveaus durchgeführt. Dazu wurde der maximal induzierte SLC5A8-spezifische Strom in jedem kinetischen Experiment in einzelnen Eizellen als 1 genommen. Jedes Experiment wurde mit 3 oder 4 verschiedenen Eizellen wiederholt. Die Aktivität des heterolog exprimierten SLC5A8 in den Eizellen wurde auch durch die Aufnahme von[14C]-Nicotinat überwacht. Die Konzentration von Nikotinat in diesen Experimenten betrug 50 μM und die Inkubationszeit 1 h.
Das Protokoll für die Verwendung von Fröschen in diesen Experimenten wurde vom Institutional Animal Care and Use Committee genehmigt.
Ektopische Expression von SLC5A8 in kultivierten Zelllinien Die
Zellen wurden in 35-mm-Kulturschalen ausgesät und in Abwesenheit von Pyruvat kultiviert. Die Zellen wurden mit pcDNA oder humaner SLC5A8 cDNA unter Verwendung von Fugene 6 und Opti-MEM transfiziert. pEGFP-N1 wurde zur Bestimmung der Transfektionseffizienz als Co-Transfektion verwendet. Nach 24 Stunden wurden die Zellen 24 Stunden lang mit oder ohne Acetat oder seinen Chlorderivaten (1 mM) behandelt. Für die FACS-Analyse wurden die Zellen in 50 %igem Ethanol fixiert, mit 0,1 % Natriumcitrat, 1 mg/ml RNase A und 50 μg/ml Propidiumiodid behandelt und anschließend einer fluoreszenzaktivierten Zellsortierung (FACS; FACS Caliber, Becton Dickinson) unterzogen.
Generierung von SLC5A8-exprimierenden stabilen Zelllinien
Wir generierten SLC5A8-exprimierende stabile Klone in zwei Zelllinien, MCF7 und MDA-MB231, um SLC5A8 bedingt zu exprimieren, indem wir SLC5A8 cDNA unter die Kontrolle des Tet-abhängigen Promotors (Tet-On Advanced, Clontech) stellten. Das System besteht aus zwei Elementen, dem Regulatorvektor (pLVX-Tet-On Advanced) und dem Reaktionsvektor (pLVX-Tight-Puro). Kurz gesagt, rekombinante Lenti-Viren wurden durch Co-Transfektion des Regulatorvektors (pLVX-Tet-On Advanced) und dreier anderer Hilfsvektoren, pLP-1, pLP-2 und pVSVG (Invitrogen), in 293FT-Zellen unter Verwendung des Transfektionsreagenzes Lipofectamine 2000 hergestellt. Der Lenti-Virus-Überstand wurde 72 Stunden nach der Transfektion geerntet und durch eine 0,45-μm-Membran filtriert. MCF7- und MDA-MB231-Zellen wurden 24 Stunden lang mit dem Lenti-Virus in einem Medium infiziert, das 8 μg/ml Polybren enthielt, und weitere 48 Stunden lang kultiviert. Die Zellen wurden mit G418 (2 mg/ml) selektiert, und die Expression des rTet R-Proteins wurde mittels Western Blot überprüft. SLC5A8 wurde in pLVX-Tight-Puro subkloniert, und SLC5A8 pLVX-Tight-Puro wurde in MCF7- und MDA-MB231-Zellen, die das rTetR-Protein exprimieren, mit demselben oben beschriebenen Verfahren transduziert. Die Zellen wurden mit 2 μg/ml Puromycin selektiert und in einem Medium mit Tetfree FBS, 0,25 μg/ml Puromycin und 250 μg/ml G418 gehalten. Die Induktion von SLC5A8 mRNA bei Zugabe von Doxycyclin wurde durch RT-PCR bestätigt.
Reaktivierung der SLC5A8-Expression durch DNA-Demethylierungsmittel
Die Zellen wurden in 10-cm-Schalen mit sehr geringer Dichte (0,5 ×106 Zellen/Schale) ausgesät und in dem entsprechenden Medium ohne Natriumpyruvat kultiviert. Nach 24 Stunden wurden die Zellen 72 Stunden lang mit 5′-Azadc (2 μg/ml) behandelt. Das Medium wurde alle 24 Stunden durch frisches Medium mit 5′-Azadc ersetzt. Nach der Behandlung mit 5′-Azadc wurden die Zellen 48 Stunden lang mit DCA (1 mM) behandelt. Nach der Behandlung wurden die Zellen geerntet und für die Proteinextraktion sowie für die FACS-Analyse verarbeitet.
Reverse Transkriptions-PCR Die
aus normalen und Krebszelllinien gewonnene RNA wurde für die semiquantitative RT-PCR verwendet. RNA (2 μg) wurde mit dem GeneAmp PCR-System (Roche) in cDNA umgeschrieben. Die PCR-Primer für spezifische Gene wurden auf der Grundlage der in GenBank verfügbaren Nukleotidsequenzen entworfen. Die Reverse Transkriptions-PCR (RT-PCR) wurde mit jeder RNA-Probe zweimal wiederholt. GAPDH (Glyceraldehyd-3-phosphat-Dehydrogenase) wurde als interne Kontrolle verwendet.
Western-Blot-Analyse
Fünfzig Mikrogramm Protein wurden durch SDS-PAGE fraktioniert, und die fraktionierten Proteine wurden auf eine Nitrocellulosemembran (Whatman GmbH) übertragen. Die Membranen wurden mit 5 % fettfreier Trockenmilch blockiert und dann bei 4 °C über Nacht mit dem primären Anti-SLC5A8-Antikörper (Kat.-Nr. ARP44110, Aviva System Biology) und anschließend mit den entsprechenden sekundären Antikörpern behandelt. Die Proteine wurden mit dem ECL SuperSignal Western System (GE Healthcare) sichtbar gemacht.
Analyse der Apoptose
Die Zellen wurden in 50 %igem Ethanol fixiert, mit 0,1 % Natriumcitrat, 1 mg/ml RNase A und 50 μg/ml Propidiumiodid behandelt und einer Fluoreszenz-aktivierten Zellsortierung (FACS, Becton Dickinson) unterzogen.
Messung der HDAC-Aktivität
Zur Bestimmung der HDAC-Aktivität wurde ein handelsübliches Kit (Biovision, Mountain View, CA) verwendet. Wenn das Zelllysat der Dickdarmkrebs-Zelllinie SW620 als Quelle für die HDAC-Aktivität verwendet wurde, wurden 100 μg des Proteins im Lysat in Gegenwart oder Abwesenheit von Acetat oder seinen Chlorderivaten (1 mM) zum Enzymtest hinzugefügt. Die Aktivität rekombinanter humaner HDAC-Isoformen wurde ebenfalls mit demselben Kit gemessen. Die rekombinanten HDAC-Isoformen wurden von der Cayman Chemical Company erworben.
Messung der intrazellulären Pyruvatkonzentration
Die Zellen wurden in 35-mm-Kulturschalen ausgesät und in Abwesenheit von Pyruvat kultiviert. Die Zellen wurden mit pcDNA oder humaner SLC5A8 cDNA unter Verwendung von Fugene 6 und Opti-MEM transfiziert. Nach 24 Stunden wurden die Zellen 24 Stunden lang mit oder ohne Acetat oder dessen Chlorderivaten (1 mM) behandelt. Die Zelllysate wurden dann zur Messung von Pyruvat mit einem kommerziell erhältlichen Kit (Biovision, Mountain View, CA) verwendet.
Danksagungen
Die hier beschriebene Arbeit wurde zum Teil durch den Zuschuss CA131402 der National Institutes of Health unterstützt.
REFERENZEN
1 Baylin SB. DNA-Methylierung und Gen-Silencing bei Krebs. Nat Clin Pract Oncol. 2005; 2:S4-S11. [PubMed: 16341240]
2 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, et al. A mitochondriaK + channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11:37-51. [PubMed: 17222789]
2Brahimi-HornMC, Chiche J, Pouyssegur J. Hypoxia signaling controls metabolic demand. Curr Opin Cell Biol. 2007; 19:223-229. [PubMed: 17303407]
2BrandsmaD, Dorlo TP, Haanen JH, Beijnen JH, Boogerd W. Severe encephalopathy and polyneuropathy induced by dichloroacetate. J Neurol. 2010 Im Druck.
2CalcuttNA, Lopez VL, Bautista AD, Mizisin LM, Torres BR, Shroads AL, et al. Periphere Neuropathie bei Ratten, die Dichloracetat ausgesetzt waren. J Neuropathol Exp Neurol. 2009; 68:985-993. [PubMed: 19680144]
3CaoW, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, et al. Dichloracetat (DCA) sensibilisiert sowohl Wildtyp- als auch überexprimierende Bcl-2-Prostatakrebszellen in vitro für Strahlung. Prostate. 2008; 68:1223-1231. [PubMed: 18465755]
4ChenZ, Lu W, Garcia-Prieto C, Huang P. The Warburg effect and its cancer therapeutic implications. J Bioenerg Biomembr. 2007; 39:267-274. [PubMed: 17551814]
5CoadyMJ, Chang MH, Charron FM, Plata C, Wallendorff B, Sah JF, et al. The tumour suppressor gene SLC5A8 expresses a Na+-monocarboxylate cotransporter. J Physiol. 2004; 557:719-731. [PubMed: 15090606]
6CooperRH, Randle PJ, Denton RM. Regulation der Herzmuskel-Pyruvatdehydrogenase-Kinase. Biochem J. 1974; 143:625-641. [PubMed: 4462746]
7GanapathyV, Gopal E, Miyauchi S, Prasad PD. Biologische Funktionen von SLC5A8, einem möglichen Tumorsuppressor. Biochem Soc Trans. 2005; 33:237-240. [PubMed: 15667316]
8GanapathyV, Thangaraju M, Gopal E, Itagaki S, Miyauchi S, Prasad PD. Natrium-gekoppelte Monocarboxylat-Transporter in normalem Gewebe und bei Krebs. AAPS J. 2008; 10:193-199. [PubMed: 18446519]
9GanapathyV, Thangaraju M, Prasad PD. Nährstofftransporter bei Krebs: Relevanz für die Warburg-Hypothese und darüber hinaus. Pharmacol Ther. 2009; 121:29-40. [PubMed: 18992769]
10GatenbyRA, Gillies RJ. Warum haben Krebserkrankungen eine hohe aerobe Glykolyse? Nat Rev Cancer. 2004; 4:891- 899. [PubMed: 15516961]
11GopalE, Fei YJ, Sugawara M, Miyauchi S, Zhuang L, Martin PM, et al. Expression von Slc5a8 in der Niere und seine Rolle beim Na+-gekoppelten Transport von Laktat. J Biol Chem. 2004; 279:44522-44532. [PubMed: 15322102]
12GopalE, Fei YJ, Miyauchi S, Zhuang L, Prasad PD, Ganapathy V. Sodium-coupled and electrogenic transport of B-complex vitamin nicotinic acid by Slc5a8, a member of the Na+/glucose cotransporter gene family. Biochem J. 2005; 388:309-316. [PubMed: 15651982]
13GuptaN, Martin PM, Prasad PD, Ganapathy V. SLC5A8 (SMCT1)-vermittelter Transport von Butyrat bildet die Grundlage für die tumorsuppressive Funktion des Transporters. Life Sci. 2006; 78:2419- 2425. [PubMed: 16375929]
14HesheD, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetat metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol. 2010 Im Druck.
15HongC, Maunakea A, Jun P, Bollen AW, Hodgson JG, Goldenberg DD, et al. Gemeinsame epigenetische Mechanismen in menschlichen und Maus-Gliomen inaktivieren die Expression des Wachstumssuppressors SLC5A8. Cancer Res. 2005; 65:3617-3623. [PubMed: 15867356]
16JacksonVN, Halestrap AP. Kinetik, Substrat und Inhibitorspezifität des Monocarboxylat-(Laktat-)Transporters von Rattenleberzellen, bestimmt mit dem fluoreszierenden intrazellulären pH-Indikator 2′,7′-bis(Carboxyethyl)-5(6)-carboxyfluorescein. J Biol Chem. 1996; 271:861-868. [PubMed: 8557697]
17KimJW, Dang CV. Die molekulare Vorliebe von Krebs und der Warburg-Effekt. Cancer Res. 2006; 66:8927-8930. [PubMed: 16982728]
19LiH, Myeroff L, Smiraglia D, Romero MF, Pretlow TP, Kasturi L, et al. SLC5A8, ein Natriumtransporter, ist ein Tumorsuppressorgen, das durch Methylierung in aberranten Kryptenherden und Krebserkrankungen des menschlichen Dickdarms zum Schweigen gebracht wird. Proc Natl Acad Sci USA. 2003; 100:8412-8417. [PubMed: 12829793]
20MadhokBM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloracetat induziert Apoptose und Zellzyklus-Stillstand in kolorektalen Krebszellen. Br J Cancer. 2010; 102:1746-1752. [PubMed: 20485289]
21MartinPM, Gopal E, Ananth S, Zhuang L, Itagaki S, Prasad BM, et al. Identity of SMCT1 (SLC5A8) as a neuron-specific Na+-coupled transporter for active uptake of L-lactate and ketone bodies in the brain. J Neurochem. 2006; 98:279-288. [PubMed: 16805814]
22MathupalaSP, KOYH, Pedersen PL. Die zentrale Rolle der Mitochondrien bei Krebs: Warburg und darüber hinaus und ermutigende Aussichten für wirksame Therapien. Biochim Biophys Acta. 2010; 1797:1225-1230. [PubMed: 20381449]
23MichelakisED, Webster L, Mackey JR. Dichloracetat (DCA) als potenzielle metabolische Zieltherapie für Krebs. Br J Cancer. 2008; 99:989-994. [PubMed: 18766181]
24MichelakisED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010; 2:31-34.
25MiyauchiS, Gopal E, Fei YJ, Ganapathy V. Functional identification of SLC5A8, a tumor suppressor downregulated in colon cancer, as a Na+-coupled transporter for short-chain fatty acids. J Biol Chem. 2004; 279:13293-13296. [PubMed: 14966140]
26MiyauchiS, Gopal E, Babu E, Sonne SR, Kubo Y, Umapathy NS, et al. Natrium-gekoppelter elektrogener Transport von Pyroglutamat (5-Oxoprolin) über SLC5A8, einen Monocarboxylat-Transporter. Biochim Biophys Acta. 2010; 1798:1164-1171. [PubMed: 20211600]
27ParkJY, Zheng W, Kim D, Cheng JQ, Kumar N, Ahmad N, et al. Candidate tumor suppressor gene SLC5A8 is frequently down-regulated by promoter hypermethylation in prostate cancer. Cancer Detect Prev. 2007; 31:359-365. [PubMed: 18037591]
28ParkJY, Helm JF, Zheng W, Ly QP, Hodul PJ, Centeno BA, et al. Silencing of the candidate tumor suppressor gene solute carrier family 5 member 8 (SLC5A8) in human pancreatic cancer. Pancreas. 2008; 36:e32-e39. [PubMed: 18437076]
29PearsonH. Krebspatienten entscheiden sich für ein nicht zugelassenes Medikament. Nature. 2007; 446:474-475. [PubMed: 17392750]
30PorraV, Ferraro-Peyret C, Durand C, Selmi-Ruby S, Giroud H, Berger-Dutrieux N, et al. Silencing des Tumorsuppressorgens SLC5A8 steht in Verbindung mit BRAF-Mutationen in klassischen papillären Schilddrüsenkarzinomen. J Clin Endocrinol Metab. 2005; 90:3028-3035. [PubMed: 15687339]
31SemenzaGL. HIF-1: Upstream und Downstream des Krebsstoffwechsels. Curr Opin Genet Dev. 2010; 20:51-56. [PubMed: 19942427]
32StacpoolePW, Henderson GN, Yan Z, Cornett R, James MO. Pharmakokinetik, Metabolismus und Toxikologie von Dichloroacetat. Drug Metab Rev. 1998; 30:499-539. [PubMed: 9710704]
33StacpoolePW, Nagaraja NV, Hutson AD. Die Wirksamkeit von Dichloracetat als laktatsenkendes Medikament. J Clin Pharmacol. 2003; 43:683-691. [PubMed: 12856382]
34StacpoolePW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008; 60:1478-1487. [PubMed: 18647626]
35StockwinLH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Natriumdichloracetat (DCA) wirkt selektiv auf Zellen mit Defekten im mitochondrialen ETC. Int J Cancer. 2010 im Druck.
36SunRC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Die Umkehrung des glykolytischen Phänotyps durch Dichloracetat hemmt das Wachstum metastasierender Brustkrebszellen in vitro und in vivo. Breast Cancer Res Treat. 2010; 120:253-260. [PubMed: 19543830]
37ThangarajuM, Gopal E, Martin PM, Ananth S, Smith SB, Prasad PD, et al. SLC5A8 löst die Apoptose von Tumorzellen durch Pyruvat-abhängige Hemmung von Histon-Deacetylasen aus. Cancer Res. 2006; 66:11560-11564. [PubMed: 17178845]
38ThangarajuM, Cresci G, Itagaki S, Mellinger J, Browning DD, Berger FG, et al. Sodium-coupled transport of the short-chain fatty acid butyrate by SLC5A8 and its relevance to colon cancer. J Gastrointest Surg. 2008; 12:1773-1782. [PubMed: 18661192]
39ThangarajuM, Karunakaran S, Itagaki S, Gopal E, Elangovan S, Prasad PD, et al. Transport über SLC5A8 mit anschließender Hemmung der Histondeacetylasen HDAC1 und HDAC3 liegt der antitumoralen Aktivität von 3-Bromopyruvat zugrunde. Cancer. 2009a; 115:4655-4666. [PubMed: 19637353]
40ThangarajuM, Carswell KN, Prasad PD, Ganapathy V. Colon cancer cells maintain low levels of pyruvate to avoid cell death caused by inhibition of HDAC1/HDAC3. Biochem J. 2009b; 417:379-389. [PubMed: 18789002]
41UenoM, Toyota M, Akino K, Suzuki H, Kusano M, Satoh A, et al. Aberrant methylation and histone deacetylation associated with silencing of SLC5A8 in gastric cancer. Tumor Biol. 2004; 25:134- 140. [PubMed: 15361710]
42VanderHeiden MG, Cantley LC, Thompson CB. Den Warburg-Effekt verstehen: die metabolischen Anforderungen der Zellproliferation. Science. 2009; 324:1029-1033. [PubMed: 19460998]
43WarburgO. Über den Ursprung von Krebszellen. Science. 1956; 123:309-314. [PubMed: 13298683]
44WeimerLH, Sachdev N. Update on medication-induced peripheral neuropathy. Curr Neurol Neurosci Rep. 2009; 9:69-75. [PubMed: 19080756]
45WhitehouseS, Cooper RH, Randle PJ. Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J. 1974; 141:761-774. [PubMed: 4478069]
46WongJY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloracetat induziert Apoptose in Endometriumkrebszellen. Gynecol Oncol. 2008; 109:394-402. [PubMed: 18423823]
47XiaoL, Li X, Niu N, Qian J, Xie G, Wang Y. Dichloracetat (DCA) verstärkt den Tod von Tumorzellen in Kombination mit einem onkolytischen Adenovirus, das mit MDA-7/IL-24 ausgestattet ist. Mol Cell Biochem. 2010; 340:31-40. [PubMed: 20165905]