Serena Vella1*, Matteo Conti2*, Roberta Tasso1, Ranieri Cancedda1,3 y Aldo Pagano1,3
1 Departamento de Oncología, Biología y Genética (DOBiG), Universidad de Génova, Génova-Italia
2 Laboratorio de Farmacología Clínica y Toxicología, Ospedale S. Maria delle Croci, 48100 Ravenna-Italia
3 Instituto Nacional para la Investigación del Cáncer (IST) Génova, Largo R. Benzi, 10, 16132 Génova-Italia
La pequeña molécula soluble en agua Dicloroacetato (DCA) está despertando recientemente un vivo interés en el campo de la terapia del cáncer, ya que ha demostrado ser capaz de inhibir el crecimiento de tumores humanos actuando específicamente sobre las mitocondrias de las células cancerosas sin perturbar la fisiología de las células no malignas. El neuroblastoma era uno de los tipos tumorales en los que el DCA se consideraba ineficaz, ya que está compuesto por células con pocas anomalías mitocondriales reconocidas. Sin embargo, el neuroblastoma está compuesto por diferentes tipos celulares en términos de metabolismo, fenotipo y potencial maligno. A pesar de la predicción anterior, en este trabajo demostramos que (i) el DCA exhibe un efecto anticancerígeno inesperado en células tumorales de NB y (ii) este efecto se dirige selectivamente a células de NB muy malignas, mientras que las células de NB más diferenciadas/menos malignas son refractarias al tratamiento con DCA. Este resultado apoya la necesidad de una investigación detallada de las propiedades anticancerígenas del DCA contra este tipo de tumor con el objetivo final de su posible uso como agente terapéutico.
La pequeña molécula/medicamento huérfano DCA ha saltado recientemente a la palestra por su capacidad para restringir el crecimiento tumoral del glioblastoma multiforme (GBM) a dosis compatibles sin efectos secundarios.1-4 Así, considerando su toxicidad bien tolerada junto con su bajo coste, el DCA está despertando un vivo interés por su potencial uso en la terapia del cáncer y en la curación de ciertos tipos tumorales.5 De hecho, aunque el DCA ha demostrado su eficacia en el carcinoma pulmonarmicrocítico6, el cáncer demama7, el cáncer de próstata8 y el cáncer de endometrio9, así como en líneas celulares deglioblastoma2, la eficacia de esta pequeña molécula como tratamiento anticanceroso sólo se ha demostrado clínicamente hasta ahora en el GBM humano, por lo que la eficacia demostrada del DCA en otros tumores malignos aún está porevaluar10 En detalle, debido a su mecanismo de acción, se espera que el DCA sea ineficaz en aquellos tumores caracterizados por una baja polarización mitocondrial como el cáncer de pulmón de células de avena, linfomas, neuroblastoma (NB) y sarcomas.5 El DCA, como inhibidor de la enzima mitocondrial piruvato deshidrogenasa cinasa (PDK), activa la piruvato deshidrogenasa (PDH), una enzima portera que regula el flujo de piruvato hacia la mitocondria, aumentando la proporción de oxidación de glucosa respecto a la glucólisis.4-6 Bonnet et al. demostraron que este aumento de la fosforilación oxidativa es selectivamente proapoptótico en las células cancerosas, lo que conduce a una disminución de su hiperpolarización mitocondrial típica asociada a la resistencia a la apoptosis.6 A pesar de que el NB se consideró inicialmente un tipo de tumor en el que el DCA es probablemente ineficaz, debido a su característica específica de célula pequeña y a la presunta ausencia de hiperpolarización de la membrana mitocondrial,5 las células proliferantes del NB se sustentan en un fenotipo glucolítico.11 Estudiamos la posible eficacia del tratamiento con DCA en la inhibición del crecimiento de nódulos de NB humano generados en ratones NOD-SCID. Sorprendentemente, observamos que el DCA restringe significativamente el crecimiento tumoral in vivo. En los tumores NB humanos, existen tres tipos celulares distintos: Las células madre de tipo I, los precursores neuroblásticos/neuroendocrinos de tipo N y los precursores Schwannianos/melanoblásticos de tipo S. Estas células tienen una morfología y una estructura celular distintas. Estas células tienen propiedades morfológicas, bioquímicas y tumorigénicas distintas y estadios de diferenciación variables.12 Curiosamente, aprovechando las células NB SKNBE2 modificadas genéticamente y caracterizadas por un marcado compromiso neuronal y un potencial maligno muy bajo, descubrimos que el efecto del DCA se restringe a las células indiferenciadas, muy malignas y completamente cicladoras, mientras que no afecta a la tasa de proliferación de las células más diferenciadas y poco malignas. Los experimentos aquí descritos sugieren ampliar la posible eficacia del DCA como fármaco anticancerígeno al NB actuando diferencialmente sobre células fuertemente y/o pobremente malignas.
Palabras clave: dicloroacetato, neuroblastoma, mitocondrias *S.V. y M.C. contribuyeron a partes iguales en este trabajo.
Patrocinador de la subvención: Ministerio italiano de Universidad e Investigación-MIUR (Programa PRIN prot. 2007); Número de subvención: 2007945BZN; Patrocinador de la subvención: Associazione Italiana Ricerca sul Cancro (Programa AIRC 2009); Número de subvención: IG9378; Patrocinador de la subvención: Ministerio italiano de Universidad e Investigación-MIUR (2007 International FIRB Program), Associazione Italiana per la Lotta al Neuroblastoma (Génova, Italia)
DOI: 10.1002/ijc.26173
Historial: Recibido el 25 de febrero de 2011; Aceptado el 20 de abril de 2011; En línea el 9 de
mayo de 2011
Correspondencia a: Aldo Pagano, Departamento de Oncología, Biología y Genética (DOBiG), Universidad de Génova, Génova, Italia, Tel.: þ/39/ 010-5737241, Fax: þ/39/010-5737257, E-mail: [email protected]
Int. J. Cancer: 130, 1484-1493 (2012) VC 2011 UICC
Material y Métodos
Ratones
Los ratones homocigotos NOD-SCID (NOD.CB17-Prkdcscid) se adquirieron en el Jackson Laboratory (Bar Harbor, MA). Se utilizaron ratones de entre 5 y 8 semanas de edad. Todos los animales fueron criados y mantenidos en el animalario de la institución del Instituto Nacional para la Investigación del Cáncer, Génova, Italia. El cuidado y la utilización de los animales se ajustaron a las leyes del Ministerio de Sanidad italiano y a las directrices de la Comunidad Europea.
Ensayo de tumorigenicidad in vivo
Se inyectó por vía subcutánea una suspensión celular de células SKNBE2 en PBS (107 células) en 37 ratones NOD/SCID. Los ratones se dividieron en cuatro grupos: -grupo de control (n ¼ 13 ratones): agua esterilizada; -grupo tratado con DCA (25 mg/kg/dosis) (n ¼ 14 ratones); -grupo tratado con DCA (2,5 mg/kg/dosis) (n ¼ 5 ratones); -grupo no tratado (n ¼ 5 ratones): ratones inyectados subcutáneamente con la suspensión celular, pero sacrificados antes de cualquier tipo de tratamiento. Se administró DCA, así como agua esterilizada, por vía intragástrica, una vez al día/5 días a la semana/durante 4 semanas. Los tratamientos comenzaron cuando la neoplasia alcanzó un diámetro umbral de 5 mm. Se observó semanalmente a los ratones para detectar la aparición de tumores en los puntos de inyección; el tamaño de los tumores se midió cada semana con calibradores en todos los grupos. Se recogieron imágenes de cada ratón en cada punto temporal considerado.
Cultivos celulares
Las células SKNBE2 wt, Mock y S1 se mantuvieron en medio RPMI 1640 (Sigma-Aldrich, Milán, Italia), 10% FBS (GIBCO, S.Giuliano Milanese, Milán, Italia), L-glutamina (2 mM; EuroClone, Devon, Reino Unido), penicilina-estreptomicina (100 U/ml/ 100 lg/ml; EuroClone) (medio estándar). Las células U2-OS se mantuvieron en medio Eagles modificado de Dulbecco (DMEM) (Sigma-Aldrich), 10% FBS (GIBCO), L-glutamina (2 mM; EuroClone) y penicilina-estreptomicina (100 U/ml/ 100 lg/ml; EuroClone). El dicloroacetato se preparó como solución acuosa y se añadió al medio. Se lavaron porciones de cada tumor considerado en PBS y se digirieron con 12,5 U/ml de colagenasa tipo I (Biochrom AG, Berlín, Alemania) y 12 U/ml de dispasa (Roche, Alemania) en PBS durante 20 min a 37oC. Se utilizaron células recién aisladas tanto para el análisis flujocitométrico como para la expansión in vitro en medio estándar. Todos los cultivos celulares se mantuvieron a 37oCen una atmósfera del 95% de aire/5% de CO2 con una humedad del 100%.
Análisis histológico e inmunohistoquímica
Para el examen histológico, los tumores derivados de cada grupo experimental se extirparon quirúrgicamente y se fijaron en formalina tamponada neutra al 10%, se deshidrataron y se incrustaron en parafina utilizando técnicas histológicas estándar. Se cortaron secciones seriadas de 4 lm y se tiñeron con hematoxilina y eosina (H/E) para examinar las características morfológicas o se procesaron para inmunohistoquímica. Tras la hidratación, la recuperación de antígenos se indujo con calor en tampón citrato con pH 6,0 y las peroxidasas endógenas se bloquearon con H2O2 al 3% en agua. La unión inespecífica se inhibió incubando los portaobjetos en suero de cabra normal al 10% (Sigma-Aldrich, Milán, Italia). Se utilizó el siguiente anticuerpo primario: monoclonal anti Ki-67 humano (clon K-3; Oncogene, San Diego, CA). Se realizaron en paralelo controles negativos con suero preinmune. Tras un lavado exhaustivo en solución salina tamponada con Tris, se incubaron los portaobjetos con un anticuerpo secundario antirratón (BioSpa, Milán, Italia). Tras el lavado, se añadió estreptavidina conjugada con peroxidasa de rábano picante (BioSpa) y se incubó durante 30 minutos. A continuación, los portaobjetos se tiñeron con cromógeno de diaminobencidina (Lab Vision, Fremont, California). La contratinción se realizó con hematoxilina. Las imágenes se capturaron mediante microscopía de luz transmitida utilizando un microscopio Zeiss Axiovert 200M equipado con una cámara 3CCD Zeiss Axio-Cam MRc de color refrigerado (Zeiss, Wetzlar, Alemania). Los portaobjetos teñidos con H/E se observaron con el mismo microscopio de luz transmitida. El tamaño de las células se analizó utilizando el software de dominio público ImageJ (Wayne Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ ij/). Se fijó el umbral para diferenciar el contorno de cada célula del fondo, y cada imagen se convirtió a binario (blanco y negro). Se utilizó la separación »Watershed» para separar las células individuales de los grupos. Las áreas se calcularon utilizando »analizar partículas» con un umbral de corte de tamaño de partícula de 100 píxeles. Los cambios en el volumen celular se determinaron dividiendo el recuento de píxeles con tratamiento farmacológico por el recuento de píxeles sin tratamiento farmacológico.
Análisis de apoptosis
La apoptosis se analizó por flujocitometría, utilizando Annexin V según las instrucciones del fabricante (Annexin VFITC Apoptosis Kit, Immunological Sciences, Roma, Italia; Annexin V-APC Apoptosis Detection Kit I; BD Biosciences, Oxford, Reino Unido; DAPI, Sigma-Aldrich, Milán, Italia). Brevemente, las células aisladas de las masas tumorales se lavaron en PBS y se resuspendieron en medio libre de suero. Se añadieron Annexin V-FITC y yoduro de propidio (PI) a las preparaciones celulares (105 células) y se incubaron durante 15 minutos en la oscuridad a temperatura ambiente. Las células Mock y S1 se tripsinizaron, se lavaron con PBS y se resuspendieron en medio libre de suero. Se añadieron Annexin V-APC y DAPI a las preparaciones celulares (105 células) y se incubaron durante 15 minutos en la oscuridad a temperatura ambiente. Las muestras se analizaron utilizando un citofluorímetro Cyan ADP (Beckman-Coulter, Brea CA). Para cada muestra se adquirieron 20.000 eventos. Los datos se analizaron utilizando el software Summit 4.3.1 (DakoCytomation, U.K.).
Ensayos de proliferación celular
(i) Para los estudios de recuento celular, se sembraron células Mock y S1 a 5 ×105 células en placas de cultivo tisular de 10 cm, se incubaron en medio estándar, con o sin adición de DCA (5 y 50 mM) y se contaron aprovechando un hemocitómetro tras 48 h de tratamiento. (ii) La proliferación celular también se evaluó mediante el sistema xCELLigence RTCA MP (Roche, Alemania) que Carcinogénesis Vella et al. 1485 monitoriza los eventos celulares en tiempo real midiendo la impedancia eléctrica a través de microelectrodos de oro interdigitados integrados en el fondo de las placas de cultivo tisular. La medición de la impedancia proporciona información cuantitativa sobre el estado biológico de las células, incluido el número de células, la viabilidad y la morfología.13 La impedancia del sensor celular se expresa como una unidad arbitraria denominada Índice Celular (IC). Para determinar el índice celular, se sembraron células aisladas de cada masa tumoral en 100 ll de medio estándar en placas de microtitulación de 96 (EPlate-Roche, Alemania). Se determinó la impedancia de fondo utilizando 100 ll de medio estándar. La adhesión, la extensión y la proliferación de las células se controlaron cada 30 minutos con el sistema xCELLigence. La proliferación celular se controló durante 72 horas. Los resultados experimentales se realizaron utilizando el software RTCA 1.2, que calculó la duplicación de la población ajustando la curva a una ecuación exponencial.
Análisis cuantitativo de RT-PCR en tiempo real
Los ARN totales de las muestras se extrajeron utilizando el reactivo TRIzol (Invitrogen, Carlsbad, CA) según el protocolo del fabricante y se sometieron a transcripción inversa mediante el Transcriptor First Strand cDNA Synthesis Kit (Roche, Alemania) siguiendo las instrucciones del fabricante. El ARN total de las muestras se midió mediante RTPCR cuantitativa en tiempo real utilizando PE ABI PRISM@ 7700 Sequence Detection System (Perkin Elmer Corp./Applied Biosystems, Foster City, CA) y el método Sybr Green siguiendo las instrucciones del fabricante. Las secuencias de los cebadores directo e inverso fueron las siguientes:
NF-68: para 5′ -CAAGGACGAGGTGTCCGAG-3′ , rev 5′ – CCCGGCATGCTTCGA-3′ ;
NDM29: para 5′ -GGCAGGCGGGTTCGTT-3′ , rev 5′ – CCACGCCTGGCTAAGTTTTG-3′ ;
c-Kit: para 5′ -GCAAGTCAGTGCTGTCGGAA-3′ , rev 5′ – AAGATAGCTTGTTGACACAGA-3′ . Para el control endógeno, se examinó la expresión de la Gliceraldehído 3 fosfato deshidrogenasa (GAPDH), ya que se demostró que el DCA no afecta a la actividad celular de la GAPDH.14 Las secuencias para los cebadores de la GAPDH humana fueron 5′ – GAAGGTGAAGGTCGGAGTC-3′ y 5′ – GAAGATGGTGATGGGATTTC-3′ . Los niveles relativos de transcripción se determinaron a partir de la curva estándar relativa construida a partir de diluciones stock de ADNc y se dividieron por la cantidad objetivo del calibrador siguiendo las instrucciones del fabricante.
Ensayo de potencial de membrana mitocondrial (∆Ψm)
El potencial de membrana mitocondrial (∆Ψm) se estudió en células vivas utilizando JC-1 (Cayman Chemical Company, Ann Arbor, MI). Para el análisis del potencial, SKNBE2 wt, Mock, S1 y U2-OS wt se sembraron a una densidadde 106 células/pocillo con o sin DCA (50 mM). Tras 72 h de tratamiento, las células se trataron con solución JC-1 y se incubaron a 37oCdurante 15-30 min. Las células se observaron con el microscopio Axiophot Zeiss (Zeiss, Jena, Alemania) [(Texas Red: excitación/emisión 590/610nm) (FITC: excitación/emisión 485/ 535nm)]. La cuantificación de la fluorescencia se determinó con el software ImageJ (Wayne Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/).
Análisis estadístico
La significación estadística de las diferencias observadas entre los distintos grupos experimentales se calculó mediante una prueba t de dos colas. Los valores p < 0,05 se consideraron estadísticamente significativos.
Resultados
ElDCA es eficaz en células de neuroblastoma humano
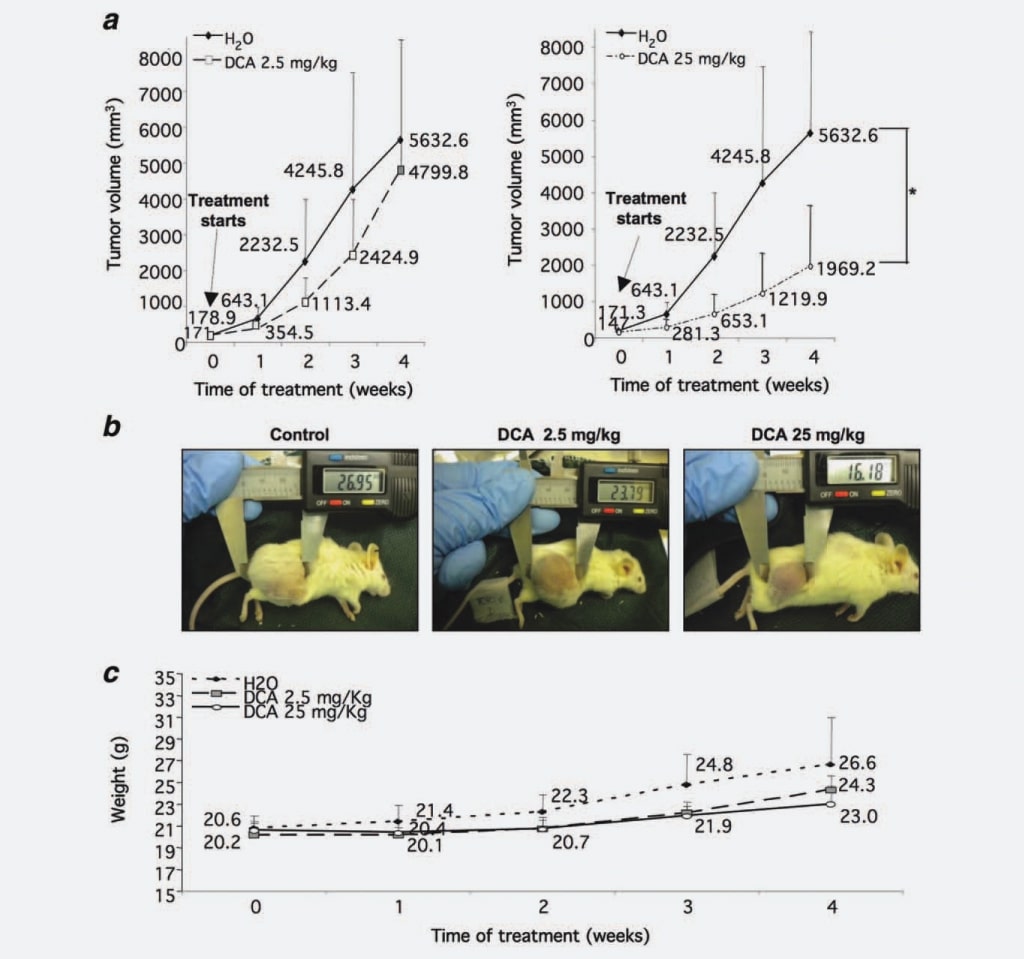
En primer lugar, para verificar si el DCA afecta al crecimiento de los nódulos de NB como se observa en el GBM, comprobamos su capacidad para limitar el crecimiento de la masa tumoral de NB in vivo. Inyectamos 37 ratones NOD-SCID con células SKNBE2, una línea celular humana de NB amplificada con N-myc caracterizada por un alto potencial maligno,17 con el fin de generar nódulos tumorales para ser tratados con DCA. Cuando los nódulos tumorales alcanzaron un diámetro umbral de 5 mm, cinco ratones fueron sacrificados antes del tratamiento, cinco ratones fueron tratados con 2,5 mg/kg/dosis de DCA, 14 ratones fueron tratados con 25 mg/kg/dosis de DCA, mientras que los 13 animales restantes fueron tratados con agua como control. Elegimos la administración intragástrica de DCA, ya que algunos datos de la literatura informaban de que la administración parenteral no era eficaz para disminuir el crecimiento tumoral.16 Cada semana se medían las masas tumorales, mostrando que los volúmenes tumorales derivados de los ratones tratados con 2,5 mg/kg/dosis de DCA se reducían en un 30% si se comparaban con el grupo control. Esta inhibición del crecimiento llegó a ser estadísticamente significativa (p ¼ 0,0008) cuando los ratones fueron tratados con 25 mg/kg/dosis de DCA (55% de reducción en comparación con el grupo control) (Figs. 1a y 1b). Para evaluar la tolerabilidad del tratamiento con DCA, evaluamos semanalmente el peso de los ratones. Los animales tratados con DCA (2,5 y 25 mg/kg/dosis) no presentaron diferencias estadísticamente significativas respecto al grupo control, aunque el peso del grupo control no tratado aumentó ligeramente, muy probablemente debido a las grandes masas tumorales (Fig. 1c). Asimismo, la ingesta de agua no afectó a los animales tratados (datos no mostrados).
En conjunto, los datos anteriores evidencian un efecto potente y dosis-dependiente del DCA sobre el crecimiento tumoral del NB.
El DCAreduce la proliferación de células cancerosas
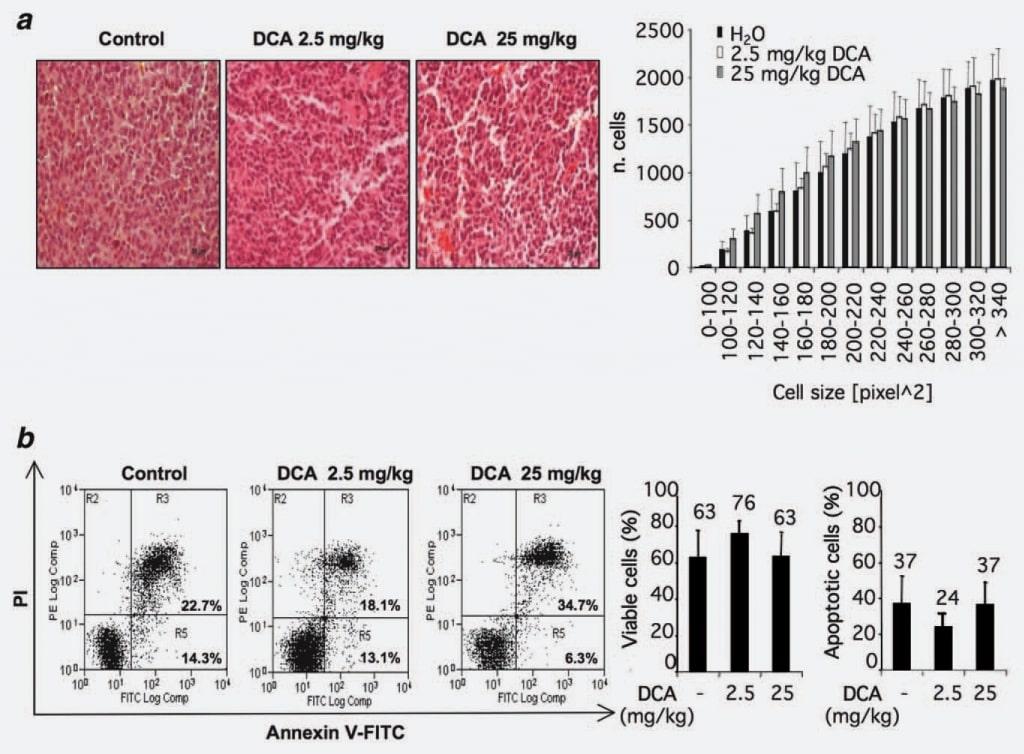
Para identificar posibles mecanismos de acción del DCA, evaluamos si la reducción observada del crecimiento tumoral podía atribuirse a una pérdida de volumen celular. Para ello, analizamos la morfología y/o el tamaño de las células dentro de los nódulos tumorales tratados o no tratados con DCA. Se consideraron 12 secciones del grupo de control y 12 de los grupos tratados con DCA (2,5 y 25 mg/kg). No se observaron diferencias estadísticamente significativas en los grupos tratados con DCA en comparación con el grupo control (Fig. 2a).
Dado que los informes publicados que estudian los efectos proapoptóticos del DCA son contradictorios y considerando las recientes observaciones de una posible especificidad del tipo celular del efecto proapoptótico del DCA,15 investigamos este fenómeno en nuestro entorno experimental mediante análisis flujocitométrico de células procedentes de tumores tratados con DCA y/o no tratados. Los resultados mostraron que el porcentaje de células anexina V-positivas es similar en los grupos considerados, lo que indica que el tratamiento con DCA no provocó un aumento de la tasa de apoptosis en la mayor parte de las células de los nódulos tumorales (Fig. 2b).
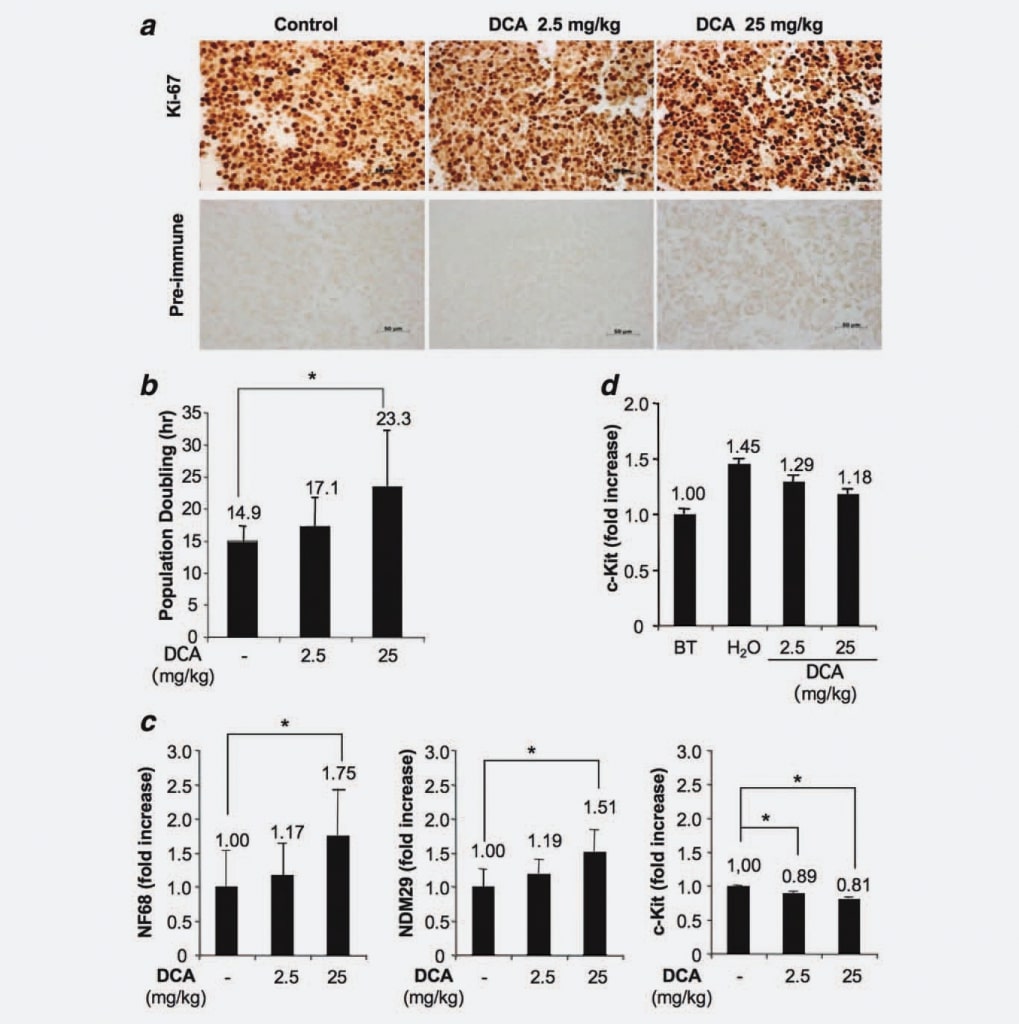
Dado que los tumores generados en nuestro modelo experimental derivaban de la inyección de una línea celular homogénea y considerando que demostramos que el tratamiento con DCA no repercutía ni en una reducción del volumen de células tumorales ni en un aumento de la apoptosis, planteamos la hipótesis de un posible efecto del DCA sobre la capacidad proliferativa de las células. Para probar esta hipótesis, analizamos células de nódulos tratados y no tratados mediante inmunohistoquímica aprovechando un anticuerpo monoclonal específico anti-Ki67. Los resultados no mostraron diferencias significativas en cuanto a la fracción de células proliferantes/cicladoras entre los grupos. Por lo tanto, este resultado indica que el tratamiento con DCA no reduce el número de células cicladoras, en consonancia con la falta de erradicación completa del tumor tras los tratamientos con DCA (Fig. 3a). A continuación, utilizamos el sistema xCELLigence (Roche, Alemania) para detectar un posible aumento dependiente de la dosis de la duplicación de la población inducida por el tratamiento con DCA. Encontramos que las células tumorales derivadas de ratones tratados (25 mg/kg) se duplican en 23,3 h, las células de animales tratados con 2,5 mg/kg se duplican en 17,1 h, mientras que las células derivadas de ratones control muestran un tiempo de duplicación poblacional de 14,9 h (Fig. 3b). Dado que las diferencias fueron estadísticamente significativas (DCA 25 mg/kg vs. grupo control; p ¼ 0,0476), estos resultados demuestran que el tratamiento con DCA induce un retraso del ciclo celular dependiente de la dosis. A continuación, investigamos si el retraso del ciclo celular de las células de los tumores tratados estaba asociado a un aumento de la diferenciación/compromiso. Para ello, medimos mediante RT-PCR en tiempo real los marcadores de diferenciación de las células NB [Neuroblastoma Differentiation Marker 29 (NDM29), Neurofilamento 68 (NF68)] y c-Kit, una proteína expresada específicamente por las células stem-like/Tumor Initiating, en tejidos tumorales tratados y no tratados con DCA. Como se muestra en la Figura 3c, se observó un aumento general de la síntesis de marcadores de diferenciación de células NB en células procedentes de tumores tratados con DCA. De hecho, el DCA utilizado a la concentración más alta indujo un aumento de la expresión de NF68 (75%, p < 0,001), mientras que la dosis más baja indujo un aumento modesto (17%, no estadísticamente significativo). Del mismo modo, el aumento de la expresión de NDM29 fue estadísticamente significativo en los tumores tratados con DCA 25 mg/kg (aumento del 51%, en comparación con el grupo de control; p < 0,001), mientras que sólo se observó una ligera diferencia entre las células de tumores tratados con 2,5 mg/kg y las células de tumores no tratados. Curiosamente, las muestras tratadas con DCA también se caracterizaron por una disminución de la expresión de c-Kit (11% de reducción comparando las muestras de DCA 2,5 mg/kg frente a las de control y 19% de reducción comparando las muestras de DCA 25 mg/kg frente a las de control, p < 0,001), lo que indica que el DCA induce una disminución del potencial stem-like/iniciador de tumores en las células tumorales proliferantes. De nuevo, estos resultados apoyan una disminución de la velocidad de proliferación y, posiblemente, una disminución del potencial maligno de las células tratadas con DCA.
A continuación, para evaluar si el DCA puede afectar a la fracción de células proliferantes indiferenciadas, medimos la expresión de c-Kit en tejidos tumorales de ratones sacrificados antes del tratamiento con DCA y de nódulos de tumores tratados. Encontramos que, aunque la expresión de c-Kit (y, presumiblemente, el potencial maligno) aumenta progresivamente durante el crecimiento de los nódulos (ver H2Ovs. Antes del tratamiento), la administración del fármaco DCA tiende a limitar este fenómeno confirmando en los tejidos de las masas tumorales lo ya observado en las células y proporcionando una razón para la falta de erradicación tumoral en los ratones tratados con DCA (Fig. 3d).
El DCA actúa específicamente sobre células de NB malignas y es ineficaz en células de NB diferenciadas y poco malignas
Cada vez hay más pruebas experimentales que sugieren que el pronóstico del NB y su respuesta a los tratamientos anticancerosos dependen del estadio de diferenciación de los nódulos de NB. De hecho, una característica peculiar del NB es la notable heterogeneidad de las células que pueden derivar de diferentes linajes de la cresta neural en estadios variables.12,17 En este contexto, es razonable plantear la hipótesis de una posible susceptibilidad variable a la DCA de los diferentes tipos celulares. Para verificar esta hipótesis, utilizamos un modelo in vitro de diferenciación de células NB basado en la expresión de un ARNnc transcrito por pol III. De hecho, recientemente aislamos un nuevo ARN no codificante (nc) transcrito por la ARN polimerasa (pol) III (concretamente NDM29) cuya expresión desencadena la diferenciación neuronal de las células NB (NB) reduciendo fuertemente su potencial maligno y la expresión de marcadores de iniciación tumoral/células madre. Integrando copias adicionales de la unidad transcripcional NDM29 en células tumorales SKNBE2, generamos una línea celular SKNBE2 sobreexpresante de NDM29 (en adelante S1) y la correspondiente Mock negativa que expresan NDM29 a su nivel endógeno. Las células S1 exhiben un fenotipo parcialmente diferenciado/neuronal, son poco malignas y se caracterizan por un marcado retraso del ciclo celular.18,19
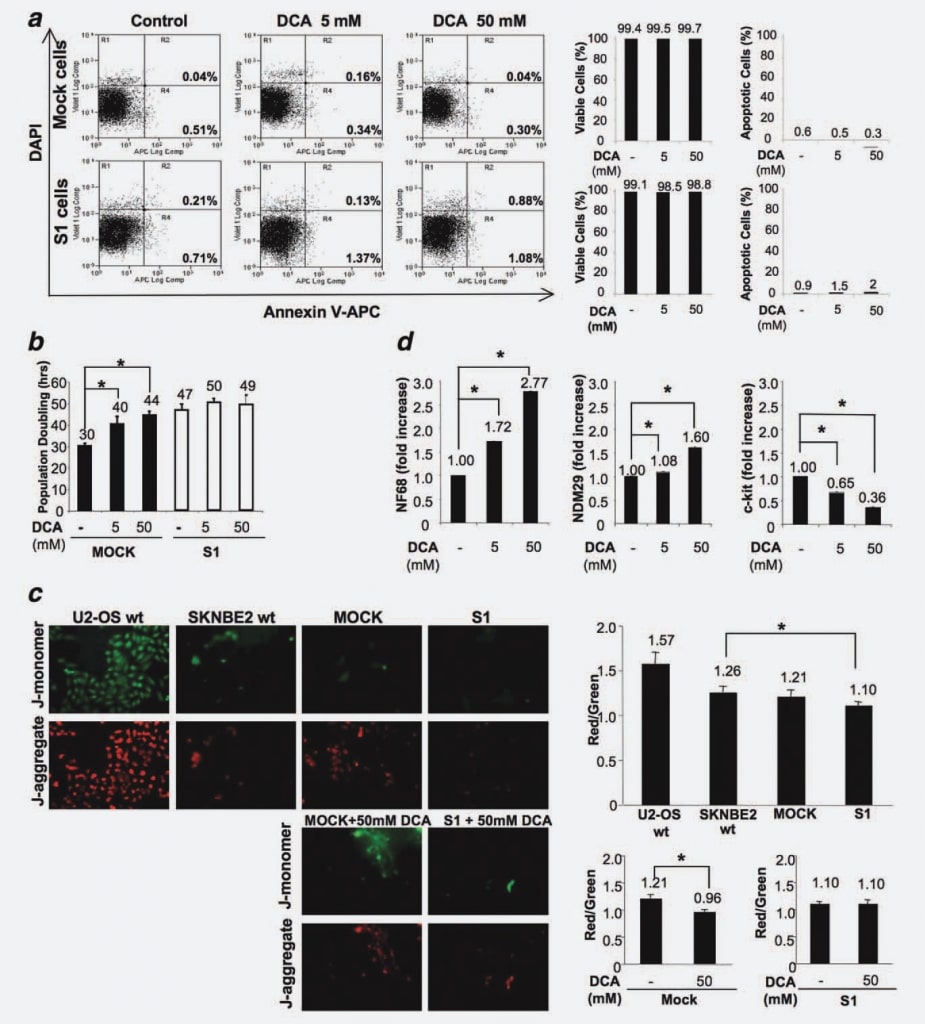
Para excluir que el tratamiento con DCA pudiera inducir apoptosis in vitro, como se demostró en los experimentos in vivo, medimos el porcentaje de células apoptóticas en las líneas celulares Mock y S1, tratadas y/o no tratadas con el fármaco durante 48 hr. No se observaron diferencias entre las líneas celulares (no tratadas o tratadas con diferentes dosis de DCA), lo que confirma que el DCA no provoca apoptosis en las células NB (Fig. 4a).
Para identificar los efectos sobre la proliferación de células Mock frente a S1, tratamos las células con 5 mM y 50 mM de DCA y medimos el tiempo de duplicación de la población celular (PD) en cuatro experimentos independientes. La concentración de 50 mM se seleccionó como la dosis máxima, ya que diferentes estudios previos informaron de que el DCA parecía ser relativamente inactivo en diferentes líneas celulares cuando se utilizaba a dosis más bajas.16,20 Como se muestra en la Figura 4b, el tiempo de PD aumenta significativamente con el tratamiento con DCA de forma dependiente de la dosis en las células Mock. De hecho, en ausencia de tratamiento, Mock se duplicó en 30 h, mientras que la administración de 5 mM de DCA indujo un retraso del ciclo celular de 10 h y la dosis más alta de 50 mM de DCA condujo a un aumento de 14 h (p < 0,001). Estos resultados demuestran que el DCA induce un retraso del ciclo celular en células NB indiferenciadas/de proliferación completa. Por el contrario, la PD de las células S1 permaneció aproximadamente inalterada, pasando de las 47 h de las células no tratadas a las 50 h de las células tratadas con 5 mM de DCA y a las 49 h de las células tratadas con 50 mM de DCA (Fig. 4b). Este resultado demuestra que la respuesta al tratamiento con DCA está directamente correlacionada con el estadio de diferenciación/proliferación de las células NB, ya que las células diferenciadas con muy bajo potencial maligno responden poco al tratamiento, mientras que sus homólogas en plena proliferación/muy malignas se ven fuertemente afectadas por el tratamiento con DCA.
Para comprender mejor este fenómeno y teniendo en cuenta que los efectos del DCA están relacionados con la inhibición de las PDK, la activación de la fosforilación oxidativa mitocondrial y la reducción específica de la hiperpolarización mitocondrial,6 analizamos las células Mock y S1 en busca de posibles cambios en la hiperpolarización mitocondrial como base de la susceptibilidad diferencial de estas células al DCA. Para ello, tratamos las células con JC-1 (5,5′ ,6,6′ -tetracloro-1,1′ ,3,3′ -tetraetilbenzimidazolocarbocianina yoduro), un colorante fluorescente cuya captación es proporcional al potencial mitocondrial. El JC-1 puede penetrar selectivamente en las mitocondrias y cambia reversiblemente de color verde a rojo a medida que aumenta el potencial mitocondrial, de modo que una disminución de la relación de fluorescencia rojo/verde pone de manifiesto una reactivación mitocondrial. Comparamos la polarización de la membrana mitocondrial de las células de osteosarcoma U2-OS (células de control hiperpolarizadas21), SKNBE2 wt, Mock y S1. Encontramos que las células Mock presentan un grado de polarización similar si se comparan con las células SKNBE2 wt, mientras que las células S1 se caracterizan por un grado de polarización mitocondrial disminuido (p ¼ 0,0184) (Fig. 4c). Los resultados, referidos a tres réplicas, mostraron que, en las células Mock, se produce una reactivación mitocondrial impulsada por el DCA (25% de reducción de la relación fluorescencia roja/verde; p ¼ 0,0077), mientras que en las células S1 la actividad mitocondrial no se modifica por la presencia de DCA (Fig. 4c). Por lo tanto, estos resultados demuestran que la susceptibilidad de las células malignas/cicladoras completas está asociada a un estado mitocondrial sensible a la acción del DCA, mientras que las células comprometidas/diferenciadas, poco cicladoras, son refractarias a la acción de este fármaco.
A continuación, para detectar posibles efectos del DCA sobre el potencial de diferenciación, medimos mediante RT-PCR en tiempo real el nivel de expresión de NDM29, NF68 y c-Kit en células Mock tratadas y no tratadas con DCA. Realizamos este análisis específicamente en las células Mock, ya que las células S1 eran refractarias a la disminución de la proliferación provocada por el DCA. Como se muestra en la Figura 4d, el tratamiento con DCA produjo un aumento de la expresión del marcador de diferenciación NF68 (células Mock tratadas con 5 mM o 50 mM de DCA frente a células no tratadas, p < 0,0001). Del mismo modo, la expresión de NDM29 aumentó significativamente en las células Mock tratadas con respecto al control (p < 0,001). Por el contrario, la expresión de c-Kit disminuyó de forma dependiente de la dosis en las células tratadas (p < 0,0001). En conjunto, estos resultados sugieren que en las células NB el efecto antitumoral del DCA se debe principalmente a una acción antiproliferativa/prodiferenciativa y no se traduce en un aumento de la tasa apoptótica.
Discusión
El DCA es una pequeña molécula que se ha utilizado durante más de 30 años en humanos para tratar la acidosis láctica congénita y adquirida.22 De hecho, está bien descrito que el tratamiento con DCA reduce los niveles de lactato en la circulación al cambiar el metabolismo celular de la glucólisis a la oxidación de la glucosa, sin ningún efecto deletéreo en las células normales.6 Por su capacidad de favorecer el metabolismo de la glucosa mediante la glucólisis aeróbica, el DCA se utiliza con éxito en ensayos clínicos para enfermedades cardíacas, incluida la insuficiencia cardíaca congestiva y la cardiopatía isquémica, ya que la disfunción postisquémica de los corazones hipertrofiados se asocia con tasas bajas de oxidación de la glucosa y tasas glucolíticas altas.23
Una propiedad muy atractiva del DCA reside en su tolerabilidad y seguridad. De hecho, más de 40 ensayos clínicos con DCA informan de que el efecto adverso más significativo de la administración de DCA a largo plazo es una neuropatía periférica reversible.24,25
En los últimos años, existen pruebas sustanciales de modelos in vitro e in vivo de que el DCA también podría ser útil para tratar algunos tipos de cáncer en humanos.6 De hecho, el DCA es capaz de revertir el efecto Warburg inhibiendo la PDK, restaurando el potencial de membrana mitocondrial y aumentando la producción de ROS. Por esta razón, el DCA fue celebrado como la bala mágica contra el cáncer,1 aunque en la actualidad todavía no esté aprobado para el tratamiento del cáncer.26 A pesar de que el neuroblastoma se ha considerado hasta ahora un tipo de cáncer para el que el tratamiento con DCA debería ser probablemente ineficaz, debido a la presunta ausencia de hiperpolarización de la membrana mitocondrial en las células que componen los nódulos NB,27 se ha descrito que las células NB proliferantes se mantienen con un fenotipo glucolítico,11 y que incluso en condiciones aeróbicas, las células NB convierten cantidades notables de glucosa en lactato en lugar de utilizar la oxidación de la glucosa.28 Sobre la base de este hallazgo, el objetivo de este estudio era determinar si el DCA podría tener un efecto beneficioso en este tipo de tumor. Nuestros resultados sacan a la luz un efecto positivo imprevisto del DCA sobre el crecimiento tumoral del NB tanto en modelos in vivo como in vitro. En efecto, el DCA fue eficaz en tumores NB establecidos, sin provocar ningún efecto secundario evidente en los animales tratados.
En detalle, el análisis realizado en células derivadas de los tumores tratados demostró que el DCA provoca un retraso proliferativo sin signos de aumento de la apoptosis o muerte celular. Esto es coherente con la disminución observada de los volúmenes de las masas tumorales, no acompañada de una erradicación completa del cáncer. Los mecanismos de acción subyacentes del DCA siguen siendo controvertidos. En este contexto, nuestros resultados contrastan con estudios previos en los que se trataron células de cáncer de endometrio, próstata, colorrectal y pulmón con DCA.6,9,20 En esos casos, se informó de un aumento de la apoptosis sin efecto sobre la
del ciclo celular9,21 o de un aumento de la apoptosis acompañado de una disminución de la proliferación8. Por otro lado, otros informes indican que el DCA inhibe la proliferación de las células cancerosas, pero no aumenta la apoptosis ni la muerte celular.7 Este último mecanismo estaría respaldado por un análisis proteómico en el que el tratamiento con DCA induce cambios en los marcadores de proliferación celular y no en la cantidad de proteínas relacionadas con la apoptosis.16 Este comportamiento dual del DCA podría depender del tipo celular, probablemente debido a diferencias en la expresión de las isoenzimas PDK en las células cancerosas examinadas. Se están realizando estudios para correlacionar la expresión de PDK con la sensibilidad al DCA.
Curiosamente, aquí mostramos que un aumento de la expresión de algunos marcadores de diferenciación, como NF68 y NDM29, acompañado de una reducción de la expresión de c-kit, un marcador de células madre/células iniciadoras de tumores, se correlacionó con la disminución observada de la proliferación de células cancerosas. Así pues, la inducción dependiente del DCA de un retraso del ciclo celular concomitante a un aumento de la diferenciación celular representa un mecanismo de acción novedoso, aún no descrito, de este fármaco que vincula los efectos del DCA al estadio de diferenciación de las células cancerosas.
Se ha documentado que los tumores NB humanos se caracterizan por distintos tipos celulares que tienen propiedades morfológicas, bioquímicas y tumorigénicas distintas y estadios de diferenciación variables.12 Para profundizar en la posible correlación de la susceptibilidad al DCA con el nivel de diferenciación celular, utilizamos un modelo in vitro de diferenciación celular NB basado en la sobreexpresión de un ncRNA transcrito por pol III (NDM29). Generamos una línea celular SKNBE2 con sobreexpresión de NDM29 (denominada aquí S1) y el correspondiente control negativo Mock.19 Estas líneas celulares se trataron con 5 mM y 50 mM de DCA. Se seleccionó la concentración de 50 mM como dosis máxima ya que diferentes estudios previos informaron de que el DCA es relativamente inactivo en diferentes líneas celulares cuando se utiliza a dosis más bajas,16,20 aunque esta concentración no se va a alcanzar in vivo. No obstante, en nuestro entorno experimental, los efectos del DCA son evidentes a la dosis más baja (5 mM), por lo que es la concentración adecuada para ser utilizada in vivo.
Como era de esperar, también en las células Mock y S1, el tratamiento con DCA no produjo signos de apoptosis, confirmando nuestros resultados in vivo. Curiosamente, descubrimos que el efecto del DCA se limita a las células indiferenciadas, muy malignas y que ciclan completamente (células Mock), mientras que no afecta a la tasa de proliferación de las células más diferenciadas y poco malignas (células S1). Este efecto podría atribuirse a los diferentes grados de polarización mitocondrial de estos tipos celulares. De hecho, demostramos que las células Mock presentan una mayor polarización mitocondrial que su homóloga S1, más diferenciada.
Por lo tanto, aquí demostramos que las distintas células que componen las masas tumorales de NB son diferencialmente susceptibles al DCA, que actúa selectivamente sobre aquellas células muy malignas y en plena proliferación, mientras que ejerce un efecto muy bajo sobre las células poco malignas y más diferenciadas. Un efecto diferencial similar del DCA también se comunicó en un estudio sobre el glioblastoma multiforme (GBM)2 en el que los autores demostraron que las células madre cancerosas, en comparación con las células homólogas más diferenciadas que componen el GBM, presentan la misma remodelación metabólica y mitocondrial pero en mayor medida ya que tienen mitocondrias más hiperpolarizadas.2
En conclusión, los experimentos aquí descritos apoyan la visión del DCA como un fármaco muy selectivo que puede ayudar a los tratamientos anticancerígenos NB sin proporcionar efectos secundarios considerables en las células sanas. Por lo tanto, este estudio preliminar amplía el posible uso del DCA para la terapia del cáncer de NB apoyando la necesidad de una investigación detallada de sus propiedades anticancerígenas contra este tipo de tumor.
Agradecimientos
R.C. recibió el apoyo del Ministerio Italiano de Universidad e InvestigaciónMIUR (2007 International FIRB Program). A.P. recibió el apoyo del Ministerio Italiano de Universidad e Investigación-MIUR (2007 PRIN Program prot. 2007945BZN), de la Associazione Italiana Ricerca sul Cancro (2009 AIRC Program no. IG9378) y de la Associazione Italiana per la Lotta al Neuroblastoma (Génova, Italia).
REFERENCIAS
1 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 2011;67:647-55
2 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Modulación metabólica del glioblastoma con dicloroacetato. Sci Transl Med; 2010;2:31ra4.
3 Stacpoole PW, Henderson GN, Yan Z, James MO. Farmacología clínica y toxicología del dicloroacetato. Environ Health Perspect 1998;106 Suppl 4:989-94.
4 Stacpoole PW. The pharmacology of dichloroacetate. Metabolism 1989;38: 1124-44.
5 Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer 2008;99:989-94.
6 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37-51.
7 Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 2010;120:253-60.
8 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008;68:1223-31.
9 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol 2008;109:394-402.
10 Vander Heiden MG. Targeting cell metabolism in cancer patients. Sci Transl Med 2009;2:31ed1.
11 Almeida A, Bolanos JP, Moncada S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc Natl Acad Sci USA; 107:738-41.
12 Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, Biedler JL, Cheung NK, Ross RA. Características de las células madre de líneas celulares de neuroblastoma humano y en tumores. Neoplasia 2004;6:838-45.
13 Vistejnova L, Dvorakova J, Hasova M, Muthny T, Velebny V, Soucek K, Kubala L. The comparison of impedance-based method of cell proliferation monitoring with commonly used metabolic-based techniques. Neuro Endocrinol Lett 2009;30 Suppl 1:121-7.
14 Schmidt MM, Rohwedder A, Dringen R. Effects of chlorinated acetates on the glutathione metabolism and on glycolysis of cultured astrocytes. Neurotox Res 2011; 19:628-37.
15 Papandreou I, Goliasova T, Denko NC. Fármacos anticancerígenos dirigidos al metabolismo: ¿es el dicloroacetato el nuevo paradigma? Int J Cancer 2011;128:1001-8.
16 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer 2010;127: 2510-9.
17 George RE, Variend S, Cullinane C, Cotterill SJ, McGuckin AG, Ellershaw C, Lunec J, Pearson AD. Relationship between histopathological features. MYCN amplification, and prognosis: a UKCCSG study. Grupo de Estudio del Cáncer Infantil del Reino Unido. Med Pediatr Oncol 2001;36: 169-76.
18 Pagano A, Castelnuovo M, Tortelli F, Ferrari R, Dieci G, Cancedda R. New small nuclear RNA gene-like transcriptional units as sources of regulatory transcripts. PLoS Genet 2007;3:e1.
19 Castelnuovo M, Massone S, Tasso R, Fiorino G, Gatti M, Robello M, Gatta E, Berger A, Strub K, Florio T, Dieci G, Cancedda R, et al. An Alu-like RNA promotes cell differentiation and reduces malignancy of human neuroblastoma cells. FASEB J 2010;24:4033-46.
20 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloroacetate induces apoptosis and cell-cycle arrest in colorectal cancer cells. Br J Cancer 2010;102:1746-52.
21 Liu TJ, Lin SY, Chau YP. Inhibition of poly(ADP-ribose) polymerase activation attenuates beta-lapachone-induced necrotic cell death in human osteosarcoma cells. Toxicol Appl Pharmacol 2002;182:116-25.
22 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 2006;117: 1519-31.
23 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Myocardial metabolic and hemodynamic effects of dichloroacetate in coronary artery disease. Am J Cardiol 1988;61: 65-70.
24 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurology 2006;66: 324-30.
25 Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Farmacocinética, metabolismo y toxicología del dicloroacetato. Drug Metab Rev 1998;30: 499-539.
26 Pearson H. Cancer patients opt for unpproved drug. Nature 2007;446: 474-5.
27 Chen LB. Mitochondrial membrane potential in living cells. Annu Rev Cell Biol 1988;4:155-81.
28 Deubzer B, Mayer F, Kuci Z, Niewisch M, Merkel G, Handgretinger R, Bruchelt G. H(2)O(2)-mediated cytotoxicity of pharmacologic ascorbate concentrations to neuroblastoma cells: potential role of lactate and ferritin. Cell Physiol Biochem 2010;25:767-74.