Serena Vella1*, Matteo Conti2*, Roberta Tasso1, Ranieri Cancedda1,3 e Aldo Pagano1,3
1 Dipartimento di Oncologia, Biologia e Genetica (DOBiG), Università di Genova, Genova
2 Laboratorio di Farmacologia Clinica e Tossicologia, Ospedale S. Maria delle Croci, 48100 Ravenna
3 Istituto Nazionale per la Ricerca sul Cancro (IST) Genova, Largo R. Benzi, 10, 16132 Genova
La piccola molecola idrosolubile dicloroacetato (DCA) sta recentemente suscitando un vivo interesse nel campo della terapia del cancro, in quanto ha dimostrato di essere in grado di inibire la crescita dei tumori umani agendo specificamente sui mitocondri delle cellule tumorali senza perturbare la fisiologia delle cellule non maligne. Il neuroblastoma era uno dei tipi di tumore su cui il DCA era considerato inefficace, in quanto composto da cellule con poche anomalie mitocondriali riconosciute. Il neuroblastoma, tuttavia, è composto da tipi di cellule diverse in termini di metabolismo, fenotipo e potenziale maligno. Nonostante la previsione di cui sopra, in questo lavoro dimostriamo che (i) il DCA mostra un effetto antitumorale inaspettato sulle cellule tumorali NB e (ii) questo effetto è selettivamente diretto alle cellule NB molto maligne, mentre le cellule NB più differenziate/meno maligne sono refrattarie al trattamento con DCA. Questo risultato supporta la necessità di un’indagine dettagliata delle proprietà antitumorali del DCA contro questo tipo di tumore, con l’obiettivo finale di un suo possibile utilizzo come agente terapeutico.
Il DCA, piccolo farmaco orfano, è recentemente salito alla ribalta per la sua capacità di limitare la crescita tumorale del glioblastoma multiforme (GBM) a dosaggi compatibili con l’assenza di effetti collaterali.1-4 Quindi, considerando la sua tossicità ben tollerata e il suo basso costo, il DCA sta suscitando un vivo interesse per il suo potenziale utilizzo nella terapia del cancro e nella cura di alcuni tipi di tumori.5 Infatti, sebbene il DCA si sia dimostrato efficace nel carcinoma polmonare a piccole cellule,6 nel tumore della mammella,7 della prostata8 e dell’endometrio9 e nelle linee cellulari di glioblastoma,2 l’efficacia di questa piccola molecola come trattamento antitumorale è stata finora dimostrata clinicamente solo nel GBM umano, per cui l’efficacia dimostrata del DCA su altri tumori maligni rimane ancora da valutare.10 In particolare, a causa del suo meccanismo d’azione, ci si aspetta che il DCA sia inefficace su quei tumori caratterizzati da una bassa polarizzazione mitocondriale, come il carcinoma polmonare a cellule di avena, i linfomi, il neuroblastoma (NB) e i sarcomi.5 Il DCA, come inibitore dell’enzima mitocondriale piruvato deidrogenasi chinasi (PDK), attiva la piruvato deidrogenasi (PDH), un enzima guardiano che regola il flusso di piruvato nei mitocondri, aumentando il rapporto tra ossidazione del glucosio eglicolisi4 -Bonnet et al. hanno dimostrato che questo aumento della fosforilazione ossidativa è selettivamente pro-apoptotico nelle cellule tumorali, portando a una diminuzione della loro tipica iperpolarizzazione mitocondriale associata alla resistenza all’apoptosi.6 Nonostante il NB sia stato inizialmente considerato un tipo di tumore su cui il DCA è probabilmente inefficace, a causa della sua specifica caratteristica di piccola cellula e della presunta assenza di iperpolarizzazione mitocondriale di membrana,5 le cellule NB proliferanti sono sostenute da un fenotipo glicolitico.11 Abbiamo studiato la possibile efficacia del trattamento con DCA nell’inibire la crescita di noduli NB umani generati in topi NOD-SCID. Sorprendentemente, abbiamo osservato che il DCA limita significativamente la crescita tumorale in vivo. Nei tumori NB umani sono presenti tre tipi di cellule distinte: Le cellule staminali di tipo I, i precursori neuroblastici/neuroendocrini di tipo N e i precursori schwanniani/melanoblastici di tipo S, simili a staminali. Queste cellule hanno proprietà morfologiche, biochimiche e tumorigeniche distinte e stadi di differenziazione variabili.12 È interessante notare che, sfruttando cellule NB SKNBE2 geneticamente ingegnerizzate, caratterizzate da un marcato impegno neuronale e da un potenziale maligno molto basso, abbiamo scoperto che l’effetto del DCA è limitato alle cellule indifferenziate, molto maligne e completamente cicliche, mentre non influisce sul tasso di proliferazione delle cellule più differenziate e scarsamente maligne. Gli esperimenti qui riportati suggeriscono di estendere la possibile efficacia del DCA come farmaco antitumorale alle NB che agiscono in modo differenziato su cellule fortemente e/o scarsamente maligne.
Parole chiave: dicloroacetato, neuroblastoma, mitocondri *S.V. e M.C. hanno contribuito in egual misura a questo lavoro.
Sponsor della sovvenzione: Ministero dell’Università e della Ricerca-MIUR (Programma PRIN 2007 prot.); Numero di sovvenzione: 2007945BZN; Sponsor della sovvenzione: Associazione Italiana Ricerca sul Cancro (Programma AIRC 2009); Grant number: IG9378; Grant sponsor: Ministero dell’Università e della Ricerca-MIUR (Programma Internazionale FIRB 2007), Associazione Italiana per la Lotta al Neuroblastoma (Genova, Italia)
DOI: 10.1002/ijc.26173
Storia: Ricevuto il 25 febbraio 2011; Accettato il 20 aprile 2011; Online il 9
maggio 2011
Corrispondenza a: Aldo Pagano, Dipartimento di Oncologia, Biologia e Genetica (DOBiG), Università di Genova, Genova, Italia, Tel.: þ/39/ 010-5737241, Fax: þ/39/010-5737257, E-mail: [email protected]
Int. J. Cancer: 130, 1484-1493 (2012) VC 2011 UICC
Materiale e metodi
Topi
I topi omozigoti NOD-SCID (NOD.CB17-Prkdcscid) sono stati acquistati dal Jackson Laboratory (Bar Harbor, MA). I topi sono stati utilizzati tra le 5 e le 8 settimane di età. Tutti gli animali sono stati allevati e mantenuti presso la struttura dell’Istituto Nazionale per la Ricerca sul Cancro, Genova, Italia. La cura e l’uso degli animali sono stati conformi alle leggi del Ministero della Salute italiano e alle linee guida della Comunità Europea.
Saggio di tumorigenicità in vivo
Una sospensione cellulare di cellule SKNBE2 in PBS (107 cellule) è stata iniettata per via sottocutanea in 37 topi NOD/SCID. I topi sono stati divisi in quattro gruppi: -gruppo di controllo (n ¼ 13 topi): acqua sterilizzata; -gruppo trattato con DCA (25 mg/kg/dose) (n ¼ 14 topi); -gruppo trattato con DCA (2,5 mg/kg/dose) (n ¼ 5 topi); -gruppo pretrattato (n ¼ 5 topi): topi iniettati sottocute con la sospensione cellulare, ma sacrificati prima di qualsiasi tipo di trattamento. Il DCA, così come l’acqua sterilizzata, è stato somministrato per via intragastrica, una volta al giorno per 5 giorni alla settimana per 4 settimane. I trattamenti sono iniziati quando la neoplasia ha raggiunto un diametro soglia di 5 mm. I topi sono stati osservati settimanalmente per la comparsa di tumori nei siti di iniezione; le dimensioni del tumore sono state misurate ogni settimana con un calibro in tutti i gruppi. Le immagini di ciascun topo sono state raccolte a ogni punto temporale considerato.
Colture cellulari
Le cellule SKNBE2 wt, Mock e S1 sono state mantenute su terreno RPMI 1640 (Sigma-Aldrich, Milano, Italia), 10% FBS (GIBCO, S.Giuliano Milanese, Milano, Italia), L-glutammina (2 mM; EuroClone, Devon, UK), penicillina-streptomicina (100 U/ml/ 100 lg/ml; EuroClone) (terreno standard). Le cellule U2-OS sono state mantenute in terreno Dulbecco’s modified Eagles medium (DMEM) (Sigma-Aldrich), 10% FBS (GIBCO), L-glutamina (2 mM; EuroClone) e penicillina-streptomicina (100 U/ml/ 100 lg/ml; EuroClone). Il dicloroacetato è stato preparato come soluzione acquosa e aggiunto al terreno di coltura. Porzioni di ciascun tumore considerato sono state lavate in PBS e digerite con 12,5 U/ml di collagenasi di tipo I (Biochrom AG, Berlino, Germania) e 12 U/ml di dispasi (Roche, Germania) in PBS per 20 minuti a 37oC. Le cellule appena isolate sono state utilizzate sia per l’analisi flussimetrica che per l’espansione in vitro in terreno standard. Tutte le colture cellulari sono state mantenute a 37oCin un’atmosfera al 95% di aria/5% di CO2 con un’umidità del 100%.
Analisi istologica e immunoistochimica
Per l’esame istologico, i tumori derivati da ciascun gruppo sperimentale sono stati rimossi chirurgicamente e fissati in formalina tamponata neutra al 10%, disidratati e incorporati in paraffina utilizzando tecniche istologiche standard. Sezioni seriali di 4 lm sono state tagliate e colorate con ematossilina ed eosina (H/E) per esaminare le caratteristiche morfologiche o processate per l’immunoistochimica. Dopo l’idratazione, il recupero dell’antigene è stato indotto a caldo in tampone citrato a pH 6.0 e le perossidasi endogene sono state bloccate con H2O2 al 3% in acqua. Il legame aspecifico è stato inibito incubando i vetrini con il 10% di siero normale di capra (Sigma-Aldrich, Milano, Italia). È stato utilizzato il seguente anticorpo primario: anticorpo monoclonale Ki-67 umano (clone K-3; Oncogene, San Diego, CA). I controlli negativi con siero preimmune sono stati eseguiti in parallelo. Dopo un ampio lavaggio in soluzione salina tamponata Tris, i vetrini sono stati incubati con un anticorpo secondario antimouse (BioSpa, Milano, Italia). Dopo il lavaggio, è stata aggiunta streptavidina coniugata con perossidasi di rafano (BioSpa) e incubata per 30 minuti. I vetrini sono stati quindi colorati con il cromogeno diaminobenzidina (Lab Vision, Fremont, CA). La controcolorazione è stata eseguita con ematossilina. Le immagini sono state acquisite in microscopia a luce trasmessa utilizzando un microscopio Zeiss Axiovert 200M dotato di una fotocamera 3CCD raffreddata a colori Zeiss Axio-Cam MRc (Zeiss, Wetzlar, Germania). I vetrini colorati con H/E sono stati osservati con lo stesso microscopio a luce trasmessa. Le dimensioni delle cellule sono state analizzate utilizzando il software di dominio pubblico ImageJ (Wayne Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ ij/). La soglia è stata impostata in modo da differenziare il contorno di ogni cellula dallo sfondo e ogni immagine è stata convertita in binario (bianco e nero). La separazione ”Watershed” è stata utilizzata per separare le singole cellule dagli ammassi. Le aree sono state calcolate utilizzando ”analizza particelle” con una soglia di taglio delle dimensioni delle particelle di 100 pixel. Le variazioni del volume cellulare sono state determinate dividendo il numero di pixel con il trattamento farmacologico per il numero di pixel senza trattamento farmacologico.
Analisi dell’apoptosi
L’apoptosi è stata analizzata mediante citometria a flusso, utilizzando l’Annexina V secondo le istruzioni del produttore (Annexin VFITC Apoptosis Kit, Immunological Sciences, Roma, Italia; Annexin V-APC Apoptosis Detection Kit I; BD Biosciences, Oxford, UK; DAPI, Sigma-Aldrich, Milano, Italia). In breve, le cellule isolate dalle masse tumorali sono state lavate in PBS e risospese in terreno privo di siero. L’annessina V-FITC e lo ioduro di propidio (PI) sono stati aggiunti ai preparati cellulari (105 cellule) e incubati per 15 minuti al buio a temperatura ambiente. Le cellule Mock e S1 sono state tripsinizzate, lavate in PBS e risospese in terreno privo di siero. L’annessina V-APC e la DAPI sono state aggiunte alle preparazioni cellulari (105 cellule) e incubate per 15 minuti al buio a temperatura ambiente. I campioni sono stati analizzati con un citofluorimetro Cyan ADP (Beckman-Coulter, Brea CA). Per ogni campione sono stati acquisiti 20.000 eventi. I dati sono stati analizzati con il software Summit 4.3.1 (DakoCytomation, U.K.).
Saggi di proliferazione cellulare
(i) Per gli studi di conteggio cellulare, le cellule Mock e S1 sono state seminate a 5 ×105 cellule in piastre di coltura tissutale da 10 cm, incubate in terreno standard, con o senza aggiunta di DCA (5 e 50 mM) e contate con un emocitometro dopo 48 ore di trattamento. (ii) La proliferazione cellulare è stata valutata anche con il sistema xCELLigence RTCA MP (Roche, Germania), che monitora gli eventi cellulari in tempo reale misurando l’impedenza elettrica attraverso microelettrodi d’oro interdigitati integrati sul fondo delle piastre di coltura tissutale. La misurazione dell’impedenza fornisce informazioni quantitative sullo stato biologico delle cellule, tra cui il numero di cellule, la vitalità e la morfologia.13 L’impedenza del sensore cellulare è espressa come unità arbitraria chiamata Cell Index (CI). Per determinare l’indice cellulare, le cellule isolate da ciascuna massa tumorale sono state seminate in 100 ll di terreno standard in 96 piastre per microtitolazione (EPlate-Roche, Germania). L’impedenza di fondo è stata determinata utilizzando 100 ll di terreno standard. L’attaccamento, la diffusione e la proliferazione delle cellule sono stati monitorati ogni 30 minuti con il sistema xCELLigence. La proliferazione cellulare è stata monitorata per 72 ore. I risultati sperimentali sono stati eseguiti utilizzando il software RTCA 1.2 che ha calcolato il raddoppio della popolazione adattando la curva a un’equazione esponenziale.
Analisi RT-PCR quantitativa in tempo reale
Gli RNA totali dei campioni sono stati estratti con il reagente TRIzol (Invitrogen, Carlsbad, CA) secondo il protocollo del produttore e sottoposti a trascrizione inversa con Transcriptor First Strand cDNA Synthesis Kit (Roche, Germania) seguendo le istruzioni del produttore. L’RNA totale dei campioni è stato misurato mediante RTPCR quantitativa in tempo reale utilizzando il sistema di rivelazione di sequenze PE ABI PRISM@ 7700 (Perkin Elmer Corp./Applied Biosystems, Foster City, CA) e il metodo Sybr Green secondo le istruzioni del produttore. Le sequenze dei primer forward e reverse erano le seguenti:
NF-68: per 5′ -CAAGGACGAGGTGTCCGAG-3′ , rev 5′ – CCCGGCATGCTTCGA-3′ ;
NDM29: per 5′ -GGCAGGCGGTTCGTT-3′ , rev 5′ – CCACGCCTGGCTAAGTTG-3′ ;
c-Kit: per 5′ -GCAAGTCAGTGCTGTCGGAA-3′ , rev 5′ – AAGATAGCTTGTGGACAGA-3′ . Per il controllo endogeno, è stata esaminata l’espressione della gliceraldeide 3 fosfato deidrogenasi (GAPDH), poiché è stato dimostrato che il DCA non influisce sull’attività della GAPDH cellulare.14 Le sequenze dei primer per la GAPDH umana erano 5′ – GAAGGTGAAGGTCGGAGTC-3′ e 5′ – GAAGATGGTGATGATTTC-3′ . I livelli relativi di trascrizione sono stati determinati dalla curva standard relativa costruita da diluizioni stock di cDNA e divisa per la quantità target del calibratore secondo le istruzioni del produttore.
Analisi del potenziale di membrana mitocondriale (∆Ψm)
Il potenziale di membrana mitocondriale (∆Ψm) è stato studiato in cellule vive utilizzando JC-1 (Cayman Chemical Company, Ann Arbor, MI). Per l’analisi del potenziale, SKNBE2 wt, Mock, S1 e U2-OS wt sono state piastrate a una densità di106 cellule/pozzetto con o senza DCA (50 mM). Dopo 72 ore di trattamento, le cellule sono state trattate con la soluzione JC-1 e incubate a 37oCper 15-30 minuti. Le cellule sono state osservate al microscopio Axiophot Zeiss (Zeiss, Jena, Germania) [(Texas Red: eccitazione/emissione 590/610nm) (FITC: eccitazione/emissione 485/535nm)]. La quantificazione della fluorescenza è stata determinata con il software ImageJ (Wayne Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/).
Analisi statistica
La significatività statistica delle differenze osservate tra i diversi gruppi sperimentali è stata calcolata utilizzando un test t a due code. I valori p < 0,05 sono stati considerati statisticamente significativi.
Risultati
IlDCA è efficace sulle cellule di neuroblastoma umano
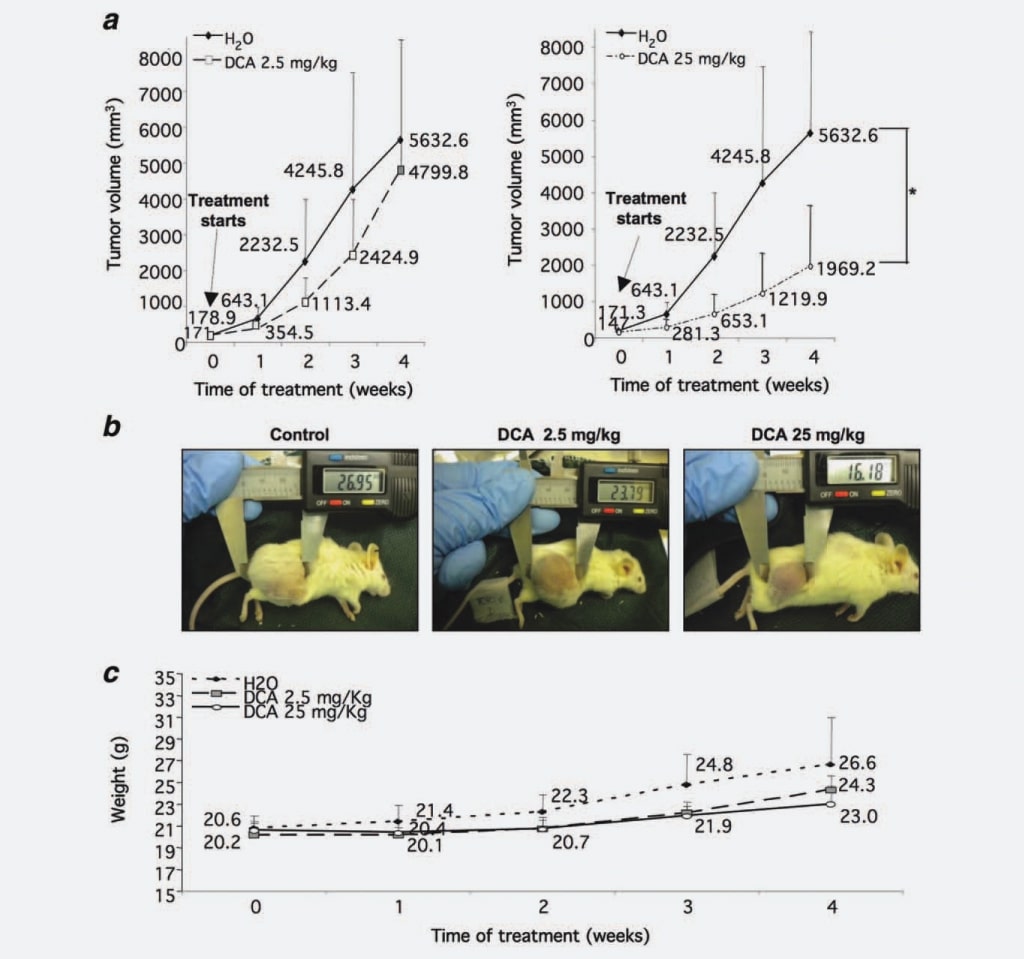
In primo luogo, per verificare se il DCA influisce sulla crescita dei noduli di NB come osservato nel GBM, abbiamo testato la sua capacità di limitare la crescita della massa tumorale di NB in vivo. Abbiamo iniettato 37 topi NOD-SCID con cellule SKNBE2, una linea cellulare umana di NB amplificata con N-myc e caratterizzata da un elevato potenziale maligno,17 al fine di generare noduli tumorali da trattare con DCA. Quando i noduli tumorali hanno raggiunto un diametro soglia di 5 mm, cinque topi sono stati sacrificati prima del trattamento, cinque topi sono stati trattati con 2,5 mg/kg/dose di DCA, 14 topi sono stati trattati con 25 mg/kg/dose di DCA, mentre i restanti 13 animali sono stati trattati con acqua come controllo. Abbiamo scelto la somministrazione intragastrica di DCA, poiché alcuni dati della letteratura riportano che la somministrazione parenterale non è efficace nel ridurre la crescita tumorale.16 Ogni settimana sono state misurate le masse tumorali, dimostrando che i volumi tumorali derivati dai topi trattati con 2,5 mg/ kg/dose di DCA erano ridotti del 30% rispetto al gruppo di controllo. Questa inibizione della crescita è diventata statisticamente significativa (p ¼ 0,0008) quando i topi sono stati trattati con 25 mg/kg/dose di DCA (55% di riduzione rispetto al gruppo di controllo) (Figg. 1a e 1b). Per valutare la tollerabilità del trattamento con DCA, abbiamo valutato settimanalmente il peso dei topi. Gli animali trattati con DCA (2,5 e 25 mg/kg/dose) non presentavano differenze statisticamente significative rispetto al gruppo di controllo, sebbene il peso del gruppo di controllo non trattato fosse leggermente aumentato, molto probabilmente a causa delle grandi masse tumorali (Fig. 1c). Anche l’assunzione di acqua non ha avuto alcun impatto sugli animali trattati (dati non mostrati).
Complessivamente, i dati sopra riportati dimostrano un potente effetto dose-dipendente del DCA sulla crescita del tumore NB.
IlDCA riduce la proliferazione delle cellule tumorali
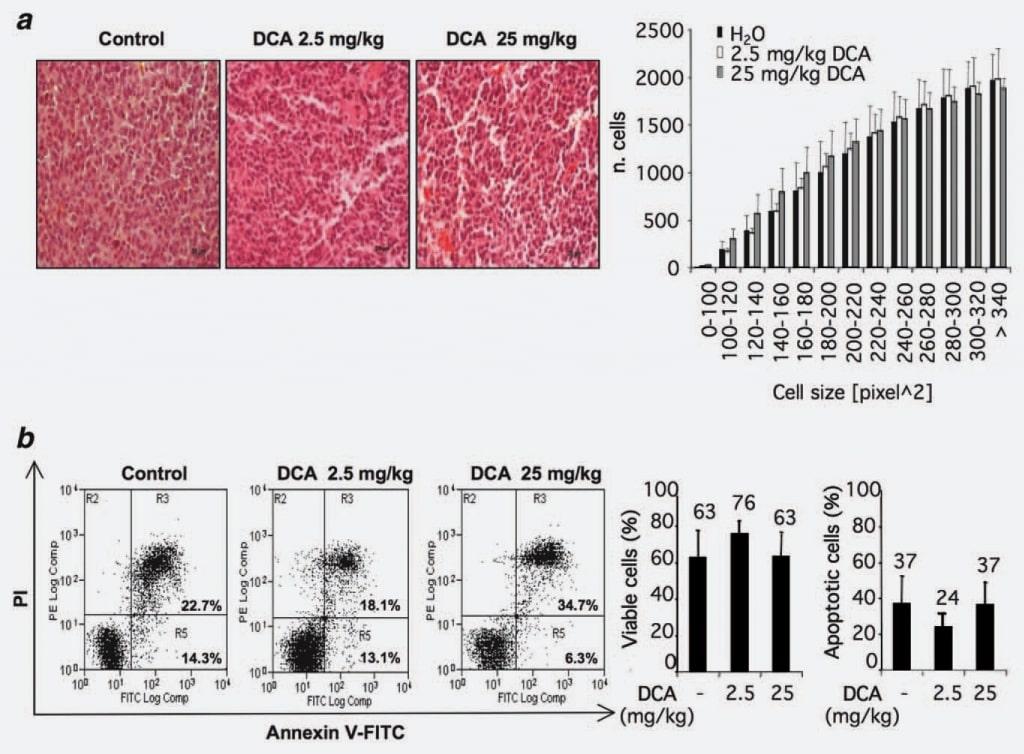
Per identificare i possibili meccanismi d’azione del DCA, abbiamo valutato se la riduzione osservata della crescita tumorale potesse essere attribuita a una perdita di volume delle cellule. A questo scopo, abbiamo analizzato la morfologia e/o le dimensioni delle cellule all’interno dei noduli tumorali trattati o non trattati con DCA. Sono state considerate 12 sezioni del gruppo di controllo e 12 dei gruppi trattati con DCA (2,5 e 25 mg/kg). Non sono state osservate differenze statisticamente significative nei gruppi trattati con DCA rispetto al gruppo di controllo (Fig. 2a).
Poiché i rapporti pubblicati che studiano gli effetti pro-apoptotici del DCA sono contraddittori e considerando le recenti osservazioni di una possibile specificità del tipo di cellula dell’effetto pro-apoptotico del DCA,15 abbiamo indagato questo fenomeno nel nostro setting sperimentale mediante analisi flussimetrica delle cellule provenienti da tumori trattati con DCA e/o non trattati. I risultati hanno mostrato che la percentuale di cellule annexin V-positive è simile nei gruppi considerati, indicando che il trattamento con DCA non ha provocato un aumento del tasso di apoptosi nella massa di cellule dei noduli tumorali (Fig. 2b).
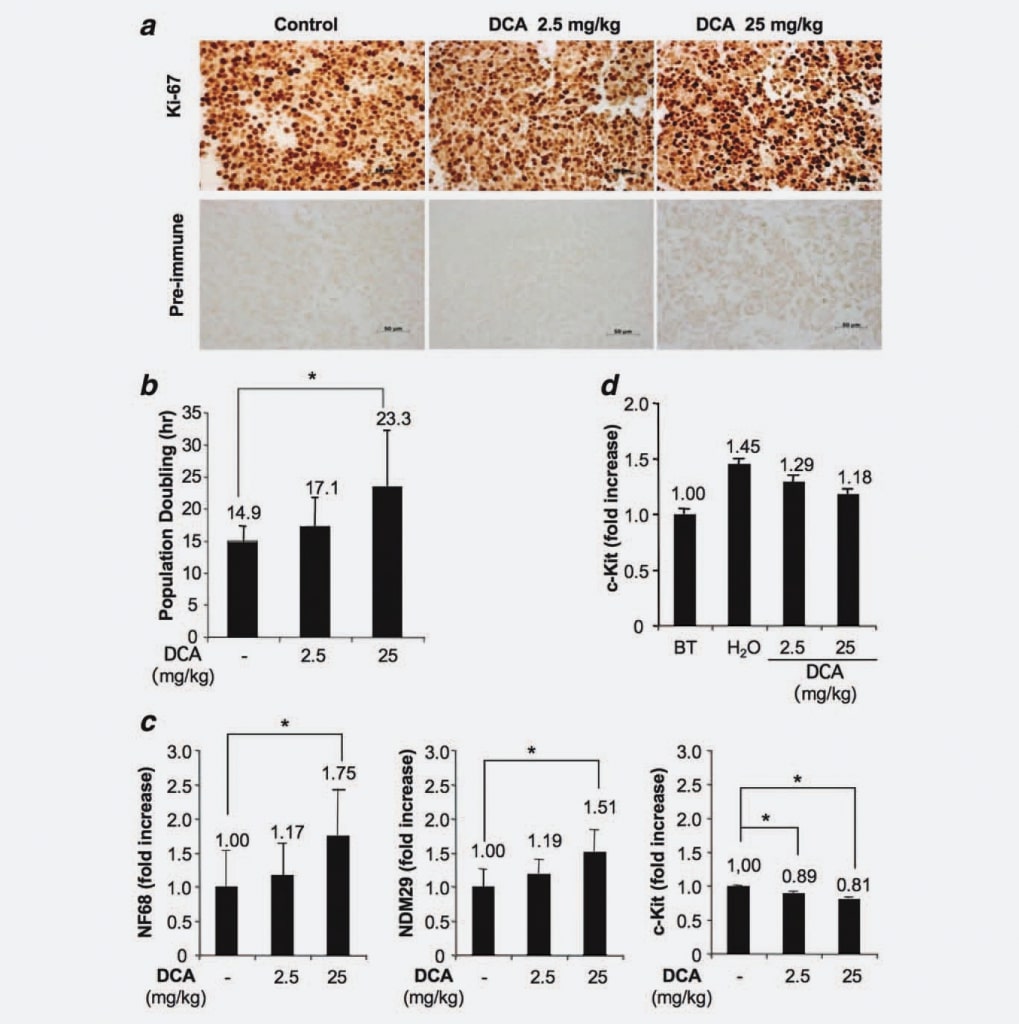
Poiché i tumori generati nel nostro modello sperimentale derivavano dall’iniezione di una linea cellulare omogenea e considerando che abbiamo dimostrato che il trattamento con DCA non ha avuto alcun impatto né sulla riduzione del volume delle cellule tumorali né sull’aumento dell’apoptosi, abbiamo ipotizzato un possibile effetto del DCA sulla capacità proliferativa delle cellule. Per verificare questa ipotesi, abbiamo analizzato le cellule dei noduli trattati e non trattati mediante immunoistochimica, utilizzando un anticorpo monoclonale specifico anti-Ki67. I risultati non hanno mostrato differenze significative in termini di frazione di cellule proliferanti/cicatrizzanti tra i gruppi. Pertanto, questo risultato indica che il trattamento con DCA non riduce il numero di cellule cicliche, coerentemente con la mancanza di eradicazione completa del tumore dopo il trattamento con DCA (Fig. 3a). Successivamente, abbiamo utilizzato il sistema xCELLigence (Roche, Germania) per rilevare un possibile aumento dose-dipendente del raddoppio della popolazione indotto dal trattamento con DCA. Abbiamo riscontrato che le cellule tumorali derivate da topi trattati (25 mg/kg) si duplicano in 23,3 ore, le cellule di animali trattati con 2,5 mg/kg si duplicano in 17,1 ore, mentre le cellule derivate da topi di controllo mostrano un tempo di raddoppiamento della popolazione di 14,9 ore (Fig. 3b). Poiché le differenze erano statisticamente significative (DCA 25 mg/kg vs. gruppo di controllo; p ¼ 0,0476), questi risultati dimostrano che il trattamento con DCA induce un ritardo del ciclo cellulare dose-dipendente. Successivamente, abbiamo indagato se il ritardo del ciclo cellulare delle cellule dei tumori trattati fosse associato a un aumento della differenziazione/impegno. A tal fine, abbiamo misurato mediante Real Time RT-PCR i marcatori di differenziazione delle cellule NB [Neuroblastoma Differentiation Marker 29 (NDM29), Neurofilament 68 (NF68)] e c-Kit, una proteina espressa specificamente dalle cellule simil-staminali/iniziative tumorali, nei tessuti tumorali trattati con DCA e non trattati. Come mostrato nella Figura 3c, è stato osservato un aumento generale della sintesi dei marcatori di differenziazione delle cellule NB nelle cellule dei tumori trattati con DCA. Infatti, il DCA utilizzato alla concentrazione più alta ha indotto un aumento dell’espressione di NF68 (75%, p < 0,001), mentre la dose più bassa ha indotto un aumento modesto (17%, non statisticamente significativo). Analogamente, l’aumento dell’espressione di NDM29 è stato statisticamente significativo nei tumori trattati con DCA 25 mg/kg (aumento del 51%, rispetto al gruppo di controllo; p < 0,001), mentre è stata osservata solo una lieve differenza tra le cellule di tumori trattati con 2,5 mg/kg e le cellule di tumori non trattati. È interessante notare che i campioni trattati con DCA erano caratterizzati anche da una minore espressione di c-Kit (riduzione dell’11% rispetto ai campioni di controllo con DCA 2,5 mg/kg e del 19% rispetto ai campioni di controllo con DCA 25 mg/kg, p < 0,001), a indicare che il DCA induce una riduzione del potenziale staminale/iniziatore tumorale nelle cellule tumorali proliferanti. Ancora una volta, questi risultati supportano un rallentamento della velocità di proliferazione e, possibilmente, una diminuzione del potenziale maligno delle cellule trattate con DCA.
Successivamente, per valutare se il DCA può influenzare la frazione di cellule proliferanti indifferenziate, abbiamo misurato l’espressione di c-Kit nei tessuti tumorali di topi sacrificati prima del trattamento con DCA e nei noduli dei tumori trattati. Abbiamo riscontrato che, sebbene l’espressione di c-Kit (e, presumibilmente, il potenziale maligno) aumenti progressivamente durante la crescita dei noduli (vediH2Ovs. Prima del trattamento), la somministrazione del farmaco DCA tende a limitare questo fenomeno confermando nei tessuti delle masse tumorali quanto già osservato nelle cellule e fornendo un razionale per la mancata eradicazione del tumore nei topi trattati con DCA (Fig. 3d).
IlDCA agisce specificamente sulle cellule NB maligne ed è inefficace sulle cellule NB differenziate e scarsamente maligne
Un numero crescente di evidenze sperimentali suggerisce che la prognosi del NB e la sua risposta ai trattamenti antitumorali dipendono dallo stadio di differenziazione dei noduli NB. Infatti, una caratteristica peculiare del NB è la notevole eterogeneità delle cellule che possono derivare da diversi lignaggi della cresta neurale in stadi variabili.12,17 In questo contesto, è ragionevole ipotizzare una possibile suscettibilità variabile ai DCA di diversi tipi di cellule. Per verificare questa ipotesi, abbiamo utilizzato un modello in vitro di differenziazione delle cellule NB basato sull’espressione di un ncRNA trascritto da pol III. Abbiamo recentemente isolato un nuovo RNA polimerasi (pol) III trascritto come RNA non codificante (NDM29), la cui espressione innesca il differenziamento neuronale delle cellule NB, riducendone fortemente il potenziale maligno e l’espressione di marcatori di cellule tumorali iniziali/staminali. Integrando copie aggiuntive dell’unità trascrizionale NDM29 nelle cellule tumorali SKNBE2, abbiamo generato una linea cellulare SKNBE2 sovraesprimente NDM29 (di seguito denominata S1) e la corrispondente Mock negativa che esprime NDM29 al suo livello endogeno. Le cellule S1 presentano un fenotipo parzialmente differenziato/neuronale, sono scarsamente maligne e sono caratterizzate da un marcato ritardo del ciclo cellulare.18,19
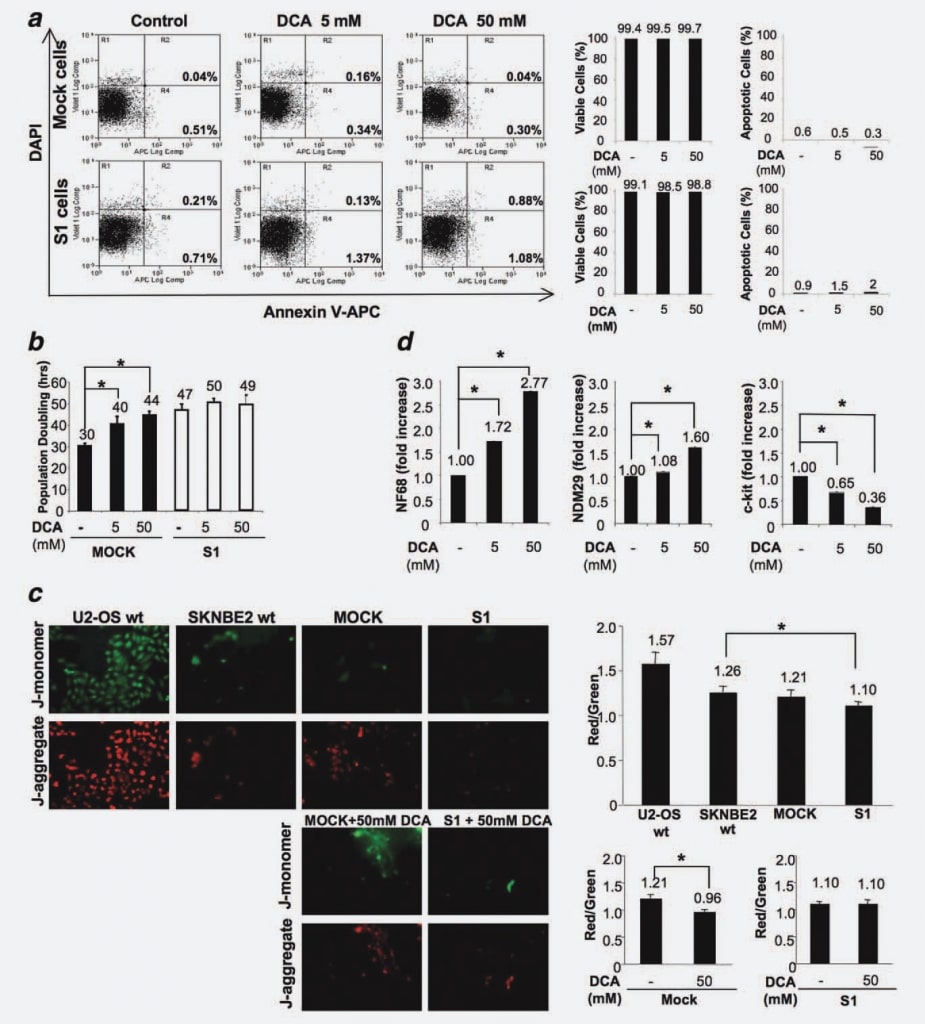
Per escludere che il trattamento con DCA possa indurre apoptosi in vitro, come dimostrato negli esperimenti in vivo, abbiamo misurato la percentuale di cellule apoptotiche nelle linee cellulari Mock e S1, trattate e/o non trattate con il farmaco per 48 ore. Non sono state osservate differenze tra le linee cellulari (non trattate o trattate con dosi diverse di DCA), confermando che il DCA non provoca apoptosi nelle cellule NB (Fig. 4a).
Per identificare gli effetti sulla proliferazione delle cellule Mock rispetto a quelle S1, abbiamo trattato le cellule con 5 mM e 50 mM di DCA e abbiamo misurato il tempo di raddoppio della popolazione cellulare (PD) in quattro esperimenti indipendenti. La concentrazione di 50 mM è stata scelta come dose massima poiché diversi studi precedenti hanno riportato che il DCA sembrava essere relativamente inattivo in diverse linee cellulari quando veniva utilizzato a dosi inferiori.16,20 Come mostrato nella Figura 4b, il tempo di raddoppiamento della popolazione cellulare è significativamente aumentato dal trattamento con DCA in modo dose-dipendente nelle cellule Mock. Infatti, in assenza di trattamento, le Mock si sono duplicate in 30 ore, mentre la somministrazione di 5 mM DCA ha indotto un ritardo del ciclo cellulare di 10 ore e la dose più elevata di 50 mM DCA ha portato a un aumento di 14 ore (p < 0,001). Questi risultati dimostrano che il DCA induce un ritardo del ciclo cellulare nelle cellule NB indifferenziate/pienamente proliferanti. Al contrario, la PD per le cellule S1 è rimasta approssimativamente inalterata, passando da 47 ore delle cellule non trattate a 50 ore per le cellule trattate con 5 mM DCA a 49 ore per le cellule trattate con 50 mM DCA (Fig. 4b). Questo risultato dimostra che la risposta al trattamento con DCA è direttamente correlata allo stadio di differenziazione/proliferazione delle cellule NB, poiché le cellule differenziate con un potenziale maligno molto basso sono poco reattive al trattamento, mentre le loro controparti completamente proliferanti/molto maligne sono fortemente influenzate dal trattamento con DCA.
Per comprendere meglio questo fenomeno e considerando che gli effetti del DCA sono legati all’inibizione della PDK, all’attivazione della fosforilazione ossidativa mitocondriale e alla riduzione specifica dell’iperpolarizzazione mitocondriale,6 abbiamo analizzato le cellule Mock e S1 per verificare le possibili alterazioni dell’iperpolarizzazione mitocondriale alla base della diversa suscettibilità di queste cellule al DCA. A questo scopo, abbiamo trattato le cellule con JC-1 (5,5′ ,6,6′ -tetracloro-1,1′ ,3,3′ -tetraetilbenzimidazolocarbocianina ioduro), un colorante fluorescente la cui captazione è proporzionale al potenziale mitocondriale. JC-1 può entrare selettivamente nei mitocondri e cambia reversibilmente colore da verde a rosso quando il potenziale mitocondriale aumenta, in modo che una diminuzione del rapporto di fluorescenza rosso/verde evidenzi una riattivazione mitocondriale. Abbiamo confrontato la polarizzazione della membrana mitocondriale di cellule osteosarcoma U2-OS (cellule di controllo iperpolarizzate21), SKNBE2 wt, Mock e S1. Abbiamo riscontrato che le cellule Mock presentano un grado di polarizzazione simile rispetto alle cellule SKNBE2 wt, mentre le cellule S1 sono caratterizzate da un grado di polarizzazione mitocondriale ridotto (p ¼ 0,0184) (Fig. 4c). I risultati, riferiti a tre repliche, hanno mostrato che, nelle cellule Mock, si verifica una riattivazione mitocondriale guidata dal DCA (riduzione del 25% del rapporto di fluorescenza rosso/verde; p ¼ 0,0077), mentre nelle cellule S1 l’attività mitocondriale non viene modificata dalla presenza del DCA (Fig. 4c). Pertanto, questi risultati dimostrano che la suscettibilità delle cellule maligne/pienamente cicliche è associata a uno stato mitocondriale sensibile all’azione del DCA, mentre le cellule impegnate/differenziate, scarsamente cicliche, sono refrattarie all’azione di questo farmaco.
Successivamente, per individuare i possibili effetti del DCA sul potenziale differenziativo, abbiamo misurato mediante Real Time RT-PCR il livello di espressione di NDM29, NF68 e c-Kit in cellule Mock trattate e non trattate con DCA. Abbiamo eseguito questa analisi specificamente sulle cellule Mock, poiché le cellule S1 erano refrattarie alla diminuzione della proliferazione indotta dal DCA. Come mostrato nella Figura 4d, il trattamento con DCA ha determinato un aumento dell’espressione del marcatore di differenziazione NF68 (cellule Mock trattate con 5 mM o 50 mM di DCA rispetto alle cellule non trattate, p < 0,0001). Analogamente, l’espressione di NDM29 è aumentata significativamente nelle cellule Mock trattate rispetto al controllo (p < 0,001). Al contrario, l’espressione di c-Kit è diminuita in modo dosedipendente nelle cellule trattate (p < 0,0001). Complessivamente, questi risultati suggeriscono che nelle cellule NB l’effetto antitumorale del DCA è dovuto principalmente a un’azione antiproliferativa/prodifferenziale e non risulta in un aumento del tasso apoptotico.
Discussione
Il DCA è una piccola molecola utilizzata da oltre 30 anni nell’uomo per il trattamento dell’acidosi lattica congenita e acquisita.22 Infatti, è ben descritto che il trattamento con DCA riduce i livelli di lattato in circolo spostando il metabolismo cellulare dalla glicolisi all’ossidazione del glucosio, senza alcun effetto deleterio sulle cellule normali.6 Per la sua capacità di favorire il metabolismo del glucosio attraverso la glicolisi aerobica, il DCA viene utilizzato con successo negli studi clinici per le malattie cardiache, tra cui l’insufficienza cardiaca congestizia e la cardiopatia ischemica, poiché la disfunzione postischemica dei cuori ipertrofizzati è associata a bassi tassi di ossidazione del glucosio e ad alti tassi glicolitici.23
Una proprietà molto interessante del DCA risiede nella sua tollerabilità e sicurezza. Infatti, più di 40 studi clinici sul DCA riportano che l’effetto avverso più significativo della somministrazione di DCA a lungo termine è una neuropatia periferica reversibile.24,25
Negli ultimi anni, ci sono prove sostanziali, sia in vitro che in modelli in vivo, che il DCA potrebbe essere utile anche per trattare alcuni tipi di cancro nell’uomo.6 Infatti, il DCA è in grado di invertire l’effetto Warburg inibendo la PDK, ripristinando il potenziale di membrana mitocondriale e aumentando la produzione di ROS. Per questo motivo, il DCA è stato celebrato come il proiettile magico contro il cancro,1 anche se attualmente non è ancora approvato per il trattamento del cancro.26 Nonostante il neuroblastoma sia stato finora considerato un tipo di cancro per il quale il trattamento con DCA dovrebbe essere molto probabilmente inefficace, a causa della presunta assenza di iperpolarizzazione della membrana mitocondriale nelle cellule che compongono i noduli di NB,27 è stato descritto che le cellule di NB proliferanti sono sostenute da un fenotipo glicolitico,11 e che anche in condizioni aerobiche, le cellule di NB convertono notevoli quantità di glucosio in lattato invece di utilizzare l’ossidazione del glucosio.28 Sulla base di questi dati, lo scopo di questo studio è stato quello di determinare se il DCA potesse avere un effetto benefico su questo tipo di tumore. I nostri risultati portano alla luce un effetto positivo non previsto del DCA sulla crescita del tumore NB sia in modelli in vivo che in vitro. Infatti, il DCA è risultato efficace nei tumori NB stabiliti, senza provocare alcun effetto collaterale evidente negli animali trattati.
In particolare, l’analisi eseguita sulle cellule derivate dai tumori trattati ha dimostrato che il DCA determina un ritardo proliferativo senza segni di aumento dell’apoptosi o della morte cellulare. Ciò è coerente con la diminuzione osservata del volume delle masse tumorali, non accompagnata da una completa eradicazione del cancro. I meccanismi d’azione del DCA sono ancora controversi. In questo contesto, i nostri risultati contrastano con quelli di studi precedenti in cui sono state trattate con DCA cellule di cancro dell’endometrio, della prostata, del colon-retto e del polmone.6,9,20 In questi casi, è stato riportato un aumento dell’apoptosi senza effetti sulla
del ciclo cellulare9,21 o un aumento dell’apoptosi accompagnato da una diminuzione della proliferazione8. D’altra parte, altri rapporti indicano che il DCA inibisce la proliferazione delle cellule tumorali, ma non l’aumento dell’apoptosi o della morte cellulare.7 Quest’ultimo meccanismo sarebbe supportato da un’analisi proteomica in cui il trattamento con DCA induce cambiamenti nei marcatori di proliferazione cellulare piuttosto che nella quantità di proteine correlate all’apoptosi.16 Questo duplice comportamento del DCA potrebbe dipendere dal tipo di cellula, probabilmente a causa delle differenze nell’espressione degli isoenzimi PDK nelle cellule tumorali esaminate. Sono in corso studi per correlare l’espressione di PDK con la sensibilità al DCA.
È interessante notare che l’aumento dell’espressione di alcuni marcatori di differenziazione, come NF68 e NDM29, accompagnato da una riduzione dell’espressione di c-kit, un marcatore di cellule staminali/iniziative tumorali, è correlato alla diminuzione osservata della proliferazione delle cellule tumorali. Pertanto, l’induzione dipendente dal DCA di un ritardo del ciclo cellulare concomitante a un aumento della differenziazione cellulare rappresenta un meccanismo d’azione nuovo, non ancora descritto, di questo farmaco che collega gli effetti del DCA allo stadio di differenziazione delle cellule tumorali.
È stato documentato che i tumori NB umani sono caratterizzati da tipi di cellule distinte che hanno proprietà morfologiche, biochimiche e tumorigeniche diverse e stadi di differenziazione variabili.12 Per approfondire la possibile correlazione della suscettibilità ai DCA con il livello di differenziazione cellulare, abbiamo utilizzato un modello in vitro di differenziazione delle cellule NB basato sulla sovraespressione di un ncRNA trascritto da pol III (NDM29). Abbiamo generato una linea cellulare SKNBE2 sovraesprimente NDM29 (qui indicata come S1) e il corrispondente controllo negativo Mock.19 Queste linee cellulari sono state trattate con DCA 5 mM e 50 mM. La concentrazione di 50 mM è stata scelta come dose massima poiché diversi studi precedenti hanno riportato che il DCA è relativamente inattivo in diverse linee cellulari quando viene utilizzato a dosi inferiori,16,20 sebbene questa concentrazione non sia raggiungibile in vivo. Tuttavia, nel nostro contesto sperimentale, gli effetti del DCA sono evidenti alla dose più bassa (5 mM), che rappresenta quindi la concentrazione appropriata da utilizzare in vivo.
Come previsto, abbiamo riscontrato che anche nelle cellule Mock e S1 il trattamento con DCA non ha prodotto segni di apoptosi, confermando i nostri risultati in vivo. È interessante notare che l’effetto del DCA è limitato alle cellule indifferenziate, molto maligne e completamente cicliche (cellule Mock), mentre non influisce sul tasso di proliferazione delle cellule più differenziate e scarsamente maligne (cellule S1). Questo effetto potrebbe essere attribuito ai diversi gradi di polarizzazione mitocondriale di questi tipi di cellule. Infatti, abbiamo dimostrato che le cellule Mock presentano una maggiore polarizzazione mitocondriale rispetto alla loro controparte più differenziata S1.
Pertanto, abbiamo dimostrato che le diverse cellule che compongono le masse tumorali NB sono diversamente sensibili al DCA, che agisce selettivamente sulle cellule molto maligne e in piena proliferazione, mentre esercita un effetto molto ridotto sulle cellule poco maligne e più differenziate. Un simile effetto differenziale del DCA è stato riportato anche in uno studio sul glioblastoma multiforme (GBM)2 in cui gli autori hanno dimostrato che le cellule staminali tumorali, rispetto alla controparte più differenziata che compone il GBM, presentano lo stesso rimodellamento metabolico e mitocondriale, ma in misura maggiore, poiché hanno i mitocondri maggiormente iperpolarizzati.2
In conclusione, gli esperimenti qui descritti supportano la visione del DCA come un farmaco molto selettivo che può aiutare i trattamenti antitumorali NB senza fornire effetti collaterali considerevoli sulle cellule sane. Pertanto, questo studio preliminare amplia il possibile uso del DCA per la terapia del tumore del neurinoma, sostenendo la necessità di un’indagine dettagliata delle sue proprietà antitumorali contro questo tipo di tumore.
Ringraziamenti
R.C. è stato sostenuto dal Ministero dell’Università e della RicercaMIUR (Programma FIRB Internazionale 2007). A.P. è stato sostenuto dal Ministero dell’Università e della Ricerca-MIUR (Programma PRIN 2007 prot. 2007945BZN), dall’Associazione Italiana Ricerca sul Cancro (Programma AIRC 2009 n. IG9378) e dall’Associazione Italiana per la Lotta al Neuroblastoma (Genova, Italia).
RIFERIMENTI
1 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. La terapia a bersaglio metabolico con dicloroacetato sconfigge la citotossicità dei farmaci antitumorali standard. Cancer Chemother Pharmacol 2011;67:647-55
2 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Modulazione metabolica del glioblastoma con dicloroacetato. Sci Transl Med; 2010;2:31ra4.
3 Stacpoole PW, Henderson GN, Yan Z, James MO. Farmacologia clinica e tossicologia del dicloroacetato. Environ Health Perspect 1998;106 Suppl 4:989-94.
4 Stacpoole PW. La farmacologia del dicloroacetato. Metabolismo 1989;38: 1124-44.
5 Michelakis ED, Webster L, Mackey JR. Il dicloroacetato (DCA) come potenziale terapia a bersaglio metabolico per il cancro. Br J Cancer 2008;99:989-94.
6 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37-51.
7 Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. L’inversione del fenotipo glicolitico mediante dicloroacetato inibisce la crescita delle cellule metastatiche di cancro al seno in vitro e in vivo. Breast Cancer Res Treat 2010;120:253-60.
8 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Il dicloroacetato (DCA) sensibilizza alle radiazioni sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2 in vitro. Prostate 2008;68:1223-31.
9 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Il dicloroacetato induce l’apoptosi nelle cellule del cancro endometriale. Gynecol Oncol 2008;109:394-402.
10 Vander Heiden MG. Puntare sul metabolismo cellulare nei pazienti oncologici. Sci Transl Med 2009;2:31ed1.
11 Almeida A, Bolanos JP, Moncada S. La E3 ubiquitina ligasi APC/C-Cdh1 spiega l’effetto Warburg collegando la glicolisi alla proliferazione cellulare. Proc Natl Acad Sci USA; 107:738-41.
12 Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, Biedler JL, Cheung NK, Ross RA. Caratteristiche delle cellule staminali delle linee cellulari di neuroblastoma umano e dei tumori. Neoplasia 2004;6:838-45.
13 Vistejnova L, Dvorakova J, Hasova M, Muthny T, Velebny V, Soucek K, Kubala L. Il confronto tra il metodo di monitoraggio della proliferazione cellulare basato sull’impedenza e le tecniche metaboliche comunemente utilizzate. Neuro Endocrinol Lett 2009;30 Suppl 1:121-7.
14 Schmidt MM, Rohwedder A, Dringen R. Effetti degli acetati clorurati sul metabolismo del glutatione e sulla glicolisi degli astrociti in coltura. Neurotox Res 2011; 19:628-37.
15 Papandreou I, Goliasova T, Denko NC. Farmaci antitumorali che colpiscono il metabolismo: il dicloroacetato è il nuovo paradigma? Int J Cancer 2011;128:1001-8.
16 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Il dicloroacetato di sodio colpisce selettivamente le cellule con difetti nell’ETC mitocondriale. Int J Cancer 2010;127: 2510-9.
17 George RE, Variend S, Cullinane C, Cotterill SJ, McGuckin AG, Ellershaw C, Lunec J, Pearson AD. Relazione tra caratteristiche istopatologiche. Amplificazione di MYCN e prognosi: uno studio dell’UKCCSG. Gruppo di studio sul cancro infantile del Regno Unito. Med Pediatr Oncol 2001;36: 169-76.
18 Pagano A, Castelnuovo M, Tortelli F, Ferrari R, Dieci G, Cancedda R. New small nuclear RNA gene-like transcriptional units as sources of regulatory transcripts. PLoS Genet 2007;3:e1.
19 Castelnuovo M, Massone S, Tasso R, Fiorino G, Gatti M, Robello M, Gatta E, Berger A, Strub K, Florio T, Dieci G, Cancedda R, et al. An Alu-like RNA promotes cell differentiation and reduces malignancy of human neuroblastoma cells. FASEB J 2010;24:4033-46.
20 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Il dicloroacetato induce apoptosi e arresto del ciclo cellulare nelle cellule di cancro del colon-retto. Br J Cancer 2010;102:1746-52.
21 Liu TJ, Lin SY, Chau YP. L’inibizione dell’attivazione della poli(ADP-ribosio) polimerasi attenua la morte cellulare necrotica indotta dal beta-lapachone in cellule di osteosarcoma umano. Toxicol Appl Pharmacol 2002;182:116-25.
22 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, et al. Studio clinico controllato del dicloroacetato per il trattamento dell’acidosi lattica congenita nei bambini. Pediatria 2006;117: 1519-31.
23 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Effetti metabolici ed emodinamici del dicloroacetato nel miocardio nella malattia coronarica. Am J Cardiol 1988;61: 65-70.
24 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Il dicloroacetato causa neuropatia tossica nel MELAS: uno studio clinico randomizzato e controllato. Neurology 2006;66: 324-30.
25 Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Farmacocinetica, metabolismo e tossicologia del dicloroacetato. Drug Metab Rev 1998;30: 499-539.
26 Pearson H. I pazienti affetti da cancro scelgono un farmaco non approvato. Nature 2007;446: 474-5.
27 Chen LB. Potenziale di membrana mitocondriale nelle cellule viventi. Annu Rev Cell Biol 1988;4:155-81.
28 Deubzer B, Mayer F, Kuci Z, Niewisch M, Merkel G, Handgretinger R, Bruchelt G. H(2)O(2) – citotossicità mediata da concentrazioni farmacologiche di ascorbato nelle cellule di neuroblastoma: ruolo potenziale di lattato e ferritina. Cell Physiol Biochem 2010;25:767-74.