Serena Vella1*, Matteo Conti2*, Roberta Tasso1, Ranieri Cancedda1,3 und Aldo Pagano1,3
1 Abteilung Onkologie, Biologie und Genetik (DOBiG), Universität Genua, Genua-Italien2
Labor für klinische Pharmakologie und Toxikologie, Ospedale S. Maria delle Croci, 48100 Ravenna-Italien3
Nationales Institut für Krebsforschung (IST) Genua, Largo R. Benzi, 10, 16132 Genua-Italien
Das kleine, wasserlösliche Molekül Dichloracetat (DCA) weckt in letzter Zeit reges Interesse im Bereich der Krebstherapie, da es nachweislich in der Lage ist, das Wachstum menschlicher Tumore zu hemmen, indem es spezifisch auf die Mitochondrien von Krebszellen einwirkt, ohne die Physiologie nicht bösartiger Zellen zu stören. Das Neuroblastom war eine der Tumorarten, bei denen DCA als unwirksam angesehen wurde, da es aus Zellen mit wenigen anerkannten mitochondrialen Anomalien besteht. Das Neuroblastom setzt sich jedoch aus verschiedenen Zelltypen zusammen, was den Stoffwechsel, den Phänotyp und das bösartige Potenzial betrifft. Trotz der obigen Vorhersage zeigen wir in dieser Arbeit, dass (i) DCA eine unerwartete krebshemmende Wirkung auf NB-Tumorzellen zeigt und (ii) diese Wirkung selektiv auf sehr bösartige NB-Zellen gerichtet ist, während die differenzierteren/weniger bösartigen NB-Zellen gegenüber der DCA-Behandlung refraktär sind. Dieses Ergebnis unterstreicht die Notwendigkeit einer detaillierten Untersuchung der krebshemmenden Eigenschaften von DCA bei diesem Tumortyp mit dem Ziel, es als therapeutisches Mittel einzusetzen.
Das niedermolekulare/orphan drug DCA ist vor kurzem ins Rampenlicht getreten, weil es in der Lage ist, das Wachstum von Tumoren des Glioblastoma multiforme (GBM) in Dosierungen zu hemmen, die mit keinerlei Nebenwirkungen verbunden sind.1-4 In Anbetracht seiner gut verträglichen Toxizität und seiner geringen Kosten weckt DCA daher reges Interesse an seinem potenziellen Einsatz in der Krebstherapie und bei der Heilung bestimmter Tumorarten.5 Zwar hat sich DCA bei kleinzelligem Lungenkarzinom als wirksam erwiesen,6 brust,7 prostata8 und Endometrium9 endometriumkarzinom und Glioblastom-Zelllinien gezeigt,2 wurde die Wirksamkeit dieses kleinen Moleküls als Krebstherapie bisher nur bei menschlichem GBM klinisch nachgewiesen, so dass die nachgewiesene Wirksamkeit von DCA bei anderen bösartigen Erkrankungen noch zu bewerten ist.10 Aufgrund seines Wirkmechanismus ist davon auszugehen, dass DCA bei Tumoren, die durch eine geringe mitochondriale Polarisierung gekennzeichnet sind, wie z. B. Haferzell-Lungenkrebs, Lymphome, Neuroblastome (NB) und Sarkome, unwirksam ist.5 DCA als Inhibitor des mitochondrialen Enzyms Pyruvat-Dehydrogenase-Kinase (PDK) aktiviert die Pyruvat-Dehydrogenase (PDH), ein Gatekeeper-Enzym, das den Fluss von Pyruvat in die Mitochondrien reguliert und das Verhältnis von Glukoseoxidation zu Glykolyse erhöht.4-6 Bonnet et al. zeigten, dass diese Steigerung der oxidativen Phosphorylierung in Krebszellen selektiv pro-apoptotisch ist und zu einer Verringerung der typischen mitochondrialen Hyperpolarisation führt, die mit einer Apoptoseresistenz einhergeht.6 Obwohl NB zunächst als ein Tumortyp angesehen wurde, bei dem DCA aufgrund seiner spezifischen kleinzelligen Beschaffenheit und der vermutlich fehlenden mitochondrialen Membranhyperpolarisation höchstwahrscheinlich unwirksam ist,5 proliferierende NB-Zellen werden durch einen glykolytischen Phänotyp aufrechterhalten.11 Wir untersuchten die mögliche Wirksamkeit einer DCA-Behandlung bei der Hemmung des Wachstums menschlicher NB-Knoten, die in NOD-SCID-Mäusen erzeugt wurden. Überraschenderweise stellten wir fest, dass DCA das Tumorwachstum in vivo deutlich einschränkt. In menschlichen NB-Tumoren gibt es drei verschiedene Zelltypen: Stammzellen vom Typ I, neuroblastische/neuroendokrine Vorläuferzellen vom Typ N und stammähnliche schwannianische/melanoblastische Vorläuferzellen vom Typ S. Diese Zellen weisen unterschiedliche morphologische, biochemische und tumorerzeugende Eigenschaften und variable Differenzierungsstadien auf.12 Interessanterweise haben wir mit Hilfe gentechnisch hergestellter SKNBE2-NB-Zellen, die sich durch eine ausgeprägte neuronenähnliche Entwicklung und ein sehr geringes malignes Potenzial auszeichnen, herausgefunden, dass die DCA-Wirkung auf undifferenzierte, sehr maligne, vollständig zyklische Zellen beschränkt ist, während sie die Proliferationsrate von stärker differenzierten, wenig malignen Zellen nicht beeinflusst. Die hier berichteten Experimente legen nahe, die mögliche Wirksamkeit von DCA als Krebsmedikament auf BS auszudehnen, die in unterschiedlicher Weise auf stark und/oder schwach maligne Zellen wirken.
Schlüsselwörter: Dichloracetat, Neuroblastom, Mitochondrien *S.V. und M.C. haben gleichermaßen zu dieser Arbeit beigetragen.
Zuschussgeber: Italienisches Ministerium für Universität und Forschung-MIUR (2007 PRIN Program prot.); Förderungsnummer: 2007945BZN; Förderungsgeber: Associazione Italiana Ricerca sul Cancro (2009 AIRC Program); Förderungsnummer: IG9378; Förderungsgeber: Italienisches Ministerium für Universität und Forschung-MIUR (2007 International FIRB Program), Associazione Italiana per la Lotta al Neuroblastoma (Genua, Italien)
DOI: 10.1002/ijc.26173
Geschichte: Erhalten 25 Feb 2011; Angenommen 20 Apr 2011; Online 9May
2011
Korrespondenz mit: Aldo Pagano, Abteilung Onkologie, Biologie und Genetik (DOBiG), Universität Genua, Genua, Italien, Tel.: þ/39/ 010-5737241, Fax: þ/39/010-5737257, E-Mail: [email protected]
Int. J. Cancer: 130, 1484-1493 (2012) VC 2011 UICC
Material und Methoden
Mäuse
Homozygote NOD-SCID-Mäuse (NOD.CB17-Prkdcscid) wurden vom Jackson Laboratory (Bar Harbor, MA) erworben. Die Mäuse wurden im Alter zwischen 5 und 8 Wochen verwendet. Alle Tiere wurden in der Tieranlage des Nationalen Instituts für Krebsforschung in Genua, Italien, gezüchtet und gehalten. Die Pflege und der Einsatz der Tiere entsprachen den Gesetzen des italienischen Gesundheitsministeriums und den Leitlinien der Europäischen Gemeinschaft.
In-vivo-Tumorigenitätstest
Eine Zellsuspension von SKNBE2-Zellen in PBS (107 Zellen) wurde subkutan in 37 NOD/SCID-Mäuse injiziert. Die Mäuse wurden in vier Gruppen eingeteilt: -Kontrollgruppe (n ¼ 13 Mäuse): sterilisiertes Wasser; -DCA (25 mg/kg/Dosis) behandelte Gruppe (n ¼ 14 Mäuse); -DCA (2,5 mg/kg/Dosis) behandelte Gruppe (n ¼ 5 Mäuse); -vorbehandelte Gruppe (n ¼ 5 Mäuse): Mäuse, denen die Zellsuspension subkutan injiziert wurde, die aber vor jeder Art von Behandlung getötet wurden. DCA sowie sterilisiertes Wasser wurden 4 Wochen lang einmal täglich/5 Tage pro Woche intragastrisch verabreicht. Die Behandlungen begannen, wenn die Neoplasie einen Schwellendurchmesser von 5 mm erreichte. Die Mäuse wurden wöchentlich auf das Auftreten von Tumoren an den Injektionsstellen beobachtet; die Tumorgröße wurde in allen Gruppen jede Woche mit einem Messschieber gemessen. Die Bilder jeder Maus wurden zu jedem betrachteten Zeitpunkt gesammelt.
Zellkulturen
SKNBE2 wt-, Mock- und S1-Zellen wurden in RPMI 1640-Medium (Sigma-Aldrich, Mailand, Italien), 10 % FBS (GIBCO, S.Giuliano Milanese, Mailand, Italien), L-Glutamin (2 mM; EuroClone, Devon, UK), Penicillin-Streptomycin (100 U/ml/ 100 lg/ml; EuroClone) (Standardmedium) gehalten. U2-OS-Zellen wurden in Dulbecco’s modified Eagles medium (DMEM) (Sigma-Aldrich), 10% FBS (GIBCO), L-Glutamin (2 mM; EuroClone) und Penicillin-Streptomycin (100 U/ml/ 100 lg/ml; EuroClone) gehalten. Dichloracetat wurde als wässrige Lösung hergestellt und dem Medium zugesetzt. Teile jedes betrachteten Tumors wurden in PBS gewaschen und mit 12,5 U/ml Typ-I-Kollagenase (Biochrom AG, Berlin, Deutschland) und 12 U/ml Dispase (Roche, Deutschland) in PBS für 20 Minuten bei 37 °C verdaut. Frisch isolierte Zellen wurden sowohl für die durchflusszytometrische Analyse als auch für die In-vitro-Expansion in Standardmedium verwendet. Alle Zellkulturen wurden bei 37 °C in einer Atmosphäre von 95 % Luft/5 % CO2 und 100 % Luftfeuchtigkeit gehalten.
Histologische Analyse und Immunhistochemie
Für die histologische Untersuchung wurden die Tumoren jeder Versuchsgruppe chirurgisch entfernt und in 10 % neutral gepuffertem Formalin fixiert, dehydriert und unter Verwendung histologischer Standardverfahren in Paraffin eingebettet. Zur Untersuchung der morphologischen Merkmale wurden serielle 4-lm-Schnitte geschnitten und mit Hämatoxylin und Eosin (H/E) gefärbt oder für die Immunhistochemie aufbereitet. Nach der Hydratisierung wurde das Antigen-Retrieval in Citratpuffer pH 6,0 hitzeinduziert und endogene Peroxidasen wurden mit 3%H2O2in Wasser blockiert. Die unspezifische Bindung wurde durch Inkubation der Objektträger mit 10 % normalem Ziegenserum (Sigma-Aldrich, Mailand, Italien) gehemmt. Es wurde der folgende primäre Antikörper verwendet: monoklonaler anti-humaner Ki-67 (Klon K-3; Oncogene, San Diego, CA). Parallel dazu wurden Negativkontrollen mit Präimmunserum durchgeführt. Nach ausgiebigem Waschen in Tris-gepufferter Kochsalzlösung wurden die Objektträger mit einem Antimaus-Sekundärantikörper (BioSpa, Mailand, Italien) inkubiert. Nach dem Waschen wurde Meerrettichperoxidase-konjugiertes Streptavidin (BioSpa) hinzugefügt und 30 Minuten lang inkubiert. Die Objektträger wurden dann mit Diaminobenzidin-Chromogen (Lab Vision, Fremont, CA) gefärbt. Die Gegenfärbung wurde mit Hämatoxylin durchgeführt. Die Bilder wurden mittels Durchlichtmikroskopie mit einem Zeiss Axiovert 200M Mikroskop aufgenommen, das mit einer gekühlten 3CCD-Farbkamera Zeiss Axio-Cam MRc (Zeiss, Wetzlar, Deutschland) ausgestattet war. Die H/E-gefärbten Objektträger wurden unter demselben Durchlichtmikroskop betrachtet. Die Größe der Zellen wurde mit der öffentlich zugänglichen Software ImageJ (Wayne Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ ij/) analysiert. Der Schwellenwert wurde so festgelegt, dass die Umrisse jeder Zelle vom Hintergrund unterschieden werden konnten, und jedes Bild wurde in ein Binärbild (schwarz/weiß) umgewandelt. Die “Watershed“-Trennung wurde verwendet, um die einzelnen Zellen von den Klumpen zu trennen. Die Flächen wurden mit der Methode „Partikel analysieren“ berechnet, wobei eine Partikelgröße von 100 Pixeln als Grenzwert festgelegt wurde. Änderungen des Zellvolumens wurden bestimmt, indem die Pixelzahl mit Arzneimittelbehandlung durch die Pixelzahl ohne Arzneimittelbehandlung dividiert wurde.
Apoptose-Analyse
Die Apoptose wurde mittels Durchflusszytometrie unter Verwendung von Annexin V gemäß den Anweisungen des Herstellers analysiert (Annexin VFITC Apoptosis Kit, Immunological Sciences, Rom, Italien; Annexin V-APC Apoptosis Detection Kit I; BD Biosciences, Oxford, UK; DAPI, Sigma-Aldrich, Mailand, Italien). Die aus den Tumormassen isolierten Zellen wurden in PBS gewaschen und in serumfreiem Medium resuspendiert. Annexin V-FITC und Propidiumiodid (PI) wurden zu den Zellpräparaten (105 Zellen) gegeben und 15 Minuten lang im Dunkeln bei Raumtemperatur inkubiert. Mock- und S1-Zellen wurden trypsinisiert, in PBS gewaschen und in serumfreiem Medium resuspendiert. Annexin V-APC und DAPI wurden den Zellpräparationen (105 Zellen) zugesetzt und 15 Minuten lang im Dunkeln bei Raumtemperatur inkubiert. Die Proben wurden mit einem Cyan-ADP-Zytofluorimeter (Beckman-Coulter, Brea CA) analysiert. Für jede Probe wurden 20.000 Ereignisse erfasst. Die Daten wurden mit der Summit 4.3.1 Software (DakoCytomation, U.K.) ausgewertet.
Assays zur Zellproliferation
(i) Für Studien zur Zellzählung wurden Mock- und S1-Zellen mit 5 ×105 Zellen in 10-cm-Gewebekulturschalen ausgesät, in Standardmedium mit oder ohne Zugabe von DCA (5 und 50 mM) inkubiert und nach 48 Stunden Behandlung mit einem Hämozytometer gezählt. (ii) Die Zellproliferation wurde auch mit dem xCELLigence RTCA MP System (Roche, Deutschland) bewertet, das die zellulären Vorgänge in Echtzeit durch Messung der elektrischen Impedanz über ineinandergreifende Goldmikroelektroden am Boden der Gewebekulturplatten überwacht. Die Impedanzmessung liefert quantitative Informationen über den biologischen Status der Zellen, einschließlich Zellzahl, Lebensfähigkeit und Morphologie.13 Die Zellsensor-Impedanz wird als willkürliche Einheit ausgedrückt, die als Zellindex (CI) bezeichnet wird. Zur Bestimmung des Zellindexes wurden aus jeder Tumormasse isolierte Zellen in 96 Mikrotiterplatten (EPlate-Roche, Deutschland) in 100 ml Standardmedium ausgesät. Die Hintergrundimpedanz wurde mit 100 Litern Standardmedium bestimmt. Zellanhaftung, Ausbreitung und Proliferation wurden alle 30 Minuten mit dem xCELLigence-System überwacht. Die Zellproliferation wurde 72 Stunden lang überwacht. Die experimentellen Ergebnisse wurden mit der RTCA-Software 1.2 durchgeführt, die die Populationsverdopplung durch Anpassung der Kurve an eine Exponentialgleichung berechnete.
Quantitative RT-PCR-Analyse in Echtzeit
Die Gesamt-RNA aus den Proben wurde mit TRIzol-Reagenz (Invitrogen, Carlsbad, CA) gemäß dem Herstellerprotokoll extrahiert und einer reversen Transkription mit dem Transcriptor First Strand cDNA Synthesis Kit (Roche, Deutschland) gemäß den Anweisungen des Herstellers unterzogen. Die Gesamt-RNA aus den Proben wurde mittels quantitativer Echtzeit-RTPCR unter Verwendung des PE ABI PRISM@ 7700 Sequence Detection System (Perkin Elmer Corp./Applied Biosystems, Foster City, CA) und der Sybr Green-Methode nach den Anweisungen des Herstellers gemessen. Die Sequenzen der Vorwärts- und Rückwärtsprimer waren wie folgt:
NF-68: für 5′ -CAAGGACGAGGTGTCCGAG-3′ , rev 5′ – CCCGGCATGCTTCGA-3′ ;
NDM29: für 5′ -GGCAGGCGGTTCGTT-3′ , rev 5′ – CCACGCCTGGCTAAGTTTTG-3′ ;
c-Kit: für 5′ -GCAAGTCAGTGCTGTCGGAA-3′ , rev 5′ – AAGATAGCTTGCTTTGGACACAGA-3′ . Zur endogenen Kontrolle wurde die Expression von Glyceraldehyd-3-Phosphat-Dehydrogenase (GAPDH) untersucht, da nachgewiesen wurde, dass DCA die zelluläre GAPDH-Aktivität nicht beeinträchtigt.14 Die Sequenzen für humane GAPDH-Primer waren 5′ – GAAGGTGAAGGTCGGAGTC-3′ und 5′ – GAAGATGGTGATGGGATTTC-3′ . Die relativen Transkriptmengen wurden anhand der relativen Standardkurve bestimmt, die aus cDNA-Stammverdünnungen erstellt und durch die Zielmenge des Kalibrators gemäß den Anweisungen des Herstellers dividiert wurde.
Test des mitochondrialen Membranpotenzials (∆Ψm)
Das mitochondriale Membranpotenzial (∆Ψm) wurde in lebenden Zellen mit JC-1 (Cayman Chemical Company, Ann Arbor, MI) untersucht. Für die Analyse des Potenzials wurden SKNBE2 wt, Mock, S1 und U2-OS wt in einer Dichte von106 Zellen/Vertiefung mit oder ohne DCA (50 mM) plattiert. Nach 72 Stunden Behandlung wurden die Zellen mit JC-1-Lösung behandelt und 15-30 Minuten bei 37 °C inkubiert. Die Zellen wurden mit dem Axiophot Zeiss Mikroskop (Zeiss, Jena, Deutschland) beobachtet [(Texas Red: Anregung/Emission 590/610nm) (FITC: Anregung/Emission 485/ 535nm)]. Die Quantifizierung der Fluoreszenz wurde mit der Software ImageJ (Wayne Rasband, NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/) bestimmt.
Statistische Auswertung
Die statistische Signifikanz der beobachteten Unterschiede zwischen den verschiedenen Versuchsgruppen wurde anhand eines zweiseitigen t-Tests berechnet. p-Werte < 0,05 wurden als statistisch signifikant angesehen.
Ergebnisse
DCA ist wirksam auf menschliche Neuroblastomzellen
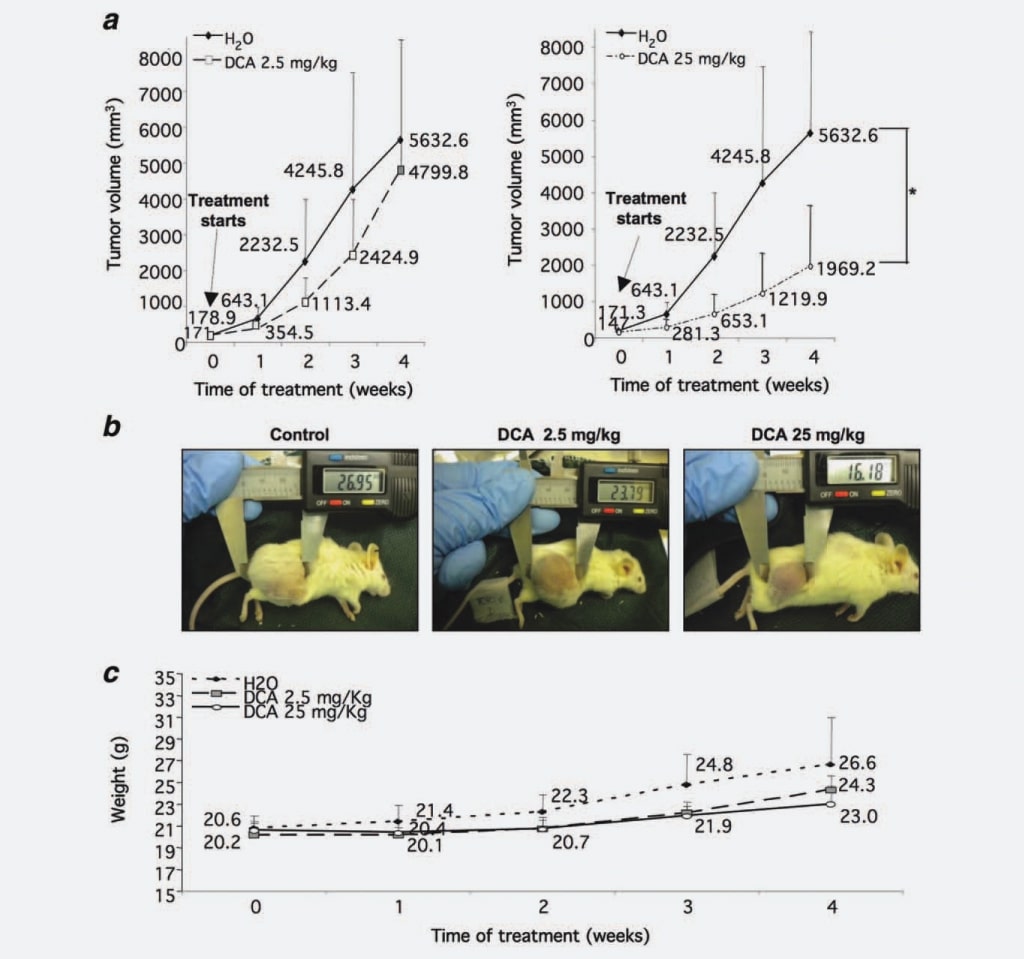
Um zu überprüfen, ob DCA das Wachstum von NB-Knoten beeinflusst, wie es bei GBM beobachtet wurde, haben wir zunächst seine Fähigkeit getestet, das Wachstum der NB-Tumormasse in vivo zu begrenzen. Wir injizierten 37 NOD-SCID-Mäuse mit SKNBE2-Zellen, einer menschlichen N-myc-verstärkten NB-Zelllinie, die sich durch ein hohes malignes Potenzial auszeichnet,17 um Tumorknötchen zu erzeugen, die mit DCA behandelt werden sollten. Wenn die Tumorknötchen einen Schwellendurchmesser von 5 mm erreicht hatten, wurden fünf Mäuse vor der Behandlung getötet, fünf Mäuse wurden mit 2,5 mg/kg/Dosis DCA behandelt, 14 Mäuse wurden mit 25 mg/kg/Dosis DCA behandelt, während die restlichen 13 Tiere mit Wasser als Kontrolle behandelt wurden. Wir entschieden uns für die intragastrische Verabreichung von DCA, da einige Literaturdaten darauf hinwiesen, dass die parenterale Verabreichung das Tumorwachstum nicht wirksam verringerte.16 Wöchentlich wurden die Tumormassen gemessen, und es zeigte sich, dass die Tumorvolumina von Mäusen, die mit 2,5 mg/kg/Dosis DCA behandelt wurden, im Vergleich zur Kontrollgruppe um 30 % reduziert waren. Diese Wachstumshemmung wurde statistisch signifikant (p ¼ 0,0008), wenn die Mäuse mit 25 mg/kg/Dosis DCA behandelt wurden (55% Reduktion im Vergleich zur Kontrollgruppe) (Abb. 1a und 1b). Um die Verträglichkeit der DCA-Behandlung zu beurteilen, wurde wöchentlich das Gewicht der Mäuse ermittelt. Die mit DCA (2,5 und 25 mg/kg/Dosis) behandelten Tiere wiesen keine statistisch signifikanten Unterschiede zur Kontrollgruppe auf, obwohl das Gewicht der unbehandelten Kontrollgruppe leicht erhöht war, was höchstwahrscheinlich auf die großen Tumormassen zurückzuführen ist (Abb. 1c). Auch die Wasseraufnahme hatte keinen Einfluss auf die behandelten Tiere (Daten nicht gezeigt).
Insgesamt belegen die oben genannten Daten eine starke, dosisabhängige Wirkung von DCA auf das Tumorwachstum von NB.
DCA reduziert die Vermehrung von Krebszellen
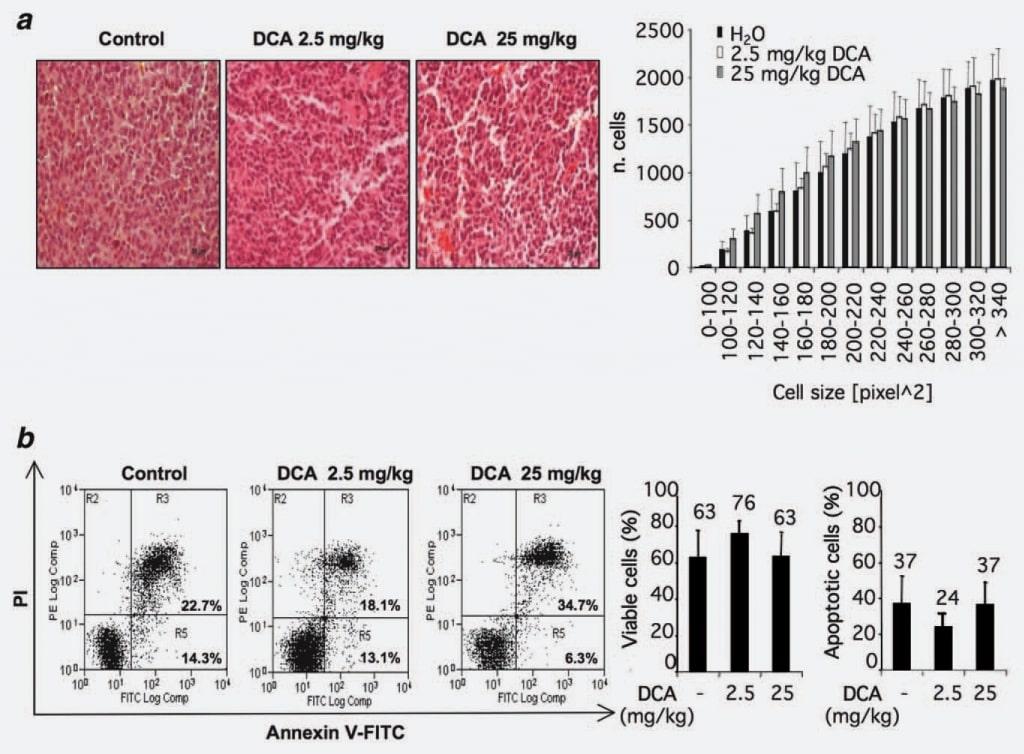
Um mögliche Wirkmechanismen von DCA zu identifizieren, untersuchten wir, ob die beobachtete Verringerung des Tumorwachstums auf einen Verlust des Zellvolumens zurückgeführt werden kann. Zu diesem Zweck analysierten wir die Morphologie und/oder die Größe der Zellen in den mit DCA behandelten oder unbehandelten Tumorknoten. Zwölf Schnitte aus der Kontrollgruppe und 12 aus den mit DCA behandelten Gruppen (2,5 und 25 mg/kg) wurden untersucht. In den DCA-behandelten Gruppen wurden keine statistisch signifikanten Unterschiede im Vergleich zur Kontrollgruppe festgestellt (Abb. 2a).
Da die veröffentlichten Berichte über die pro-apoptotische Wirkung von DCA widersprüchlich sind und in Anbetracht der jüngsten Beobachtungen über eine mögliche Zelltypspezifität der pro-apoptotischen Wirkung vonDCA15, untersuchten wir dieses Phänomen in unserer Versuchsanordnung durch eine durchflusszytometrische Analyse von Zellen aus DCA-behandelten und/oder unbehandelten Tumoren. Die Ergebnisse zeigten, dass der Prozentsatz der Annexin V-positiven Zellen in den betrachteten Gruppen ähnlich ist, was darauf hindeutet, dass die DCA-Behandlung keinen Anstieg der Apoptoserate in der Masse der Zellen der Tumorknötchen hervorruft (Abb. 2b).
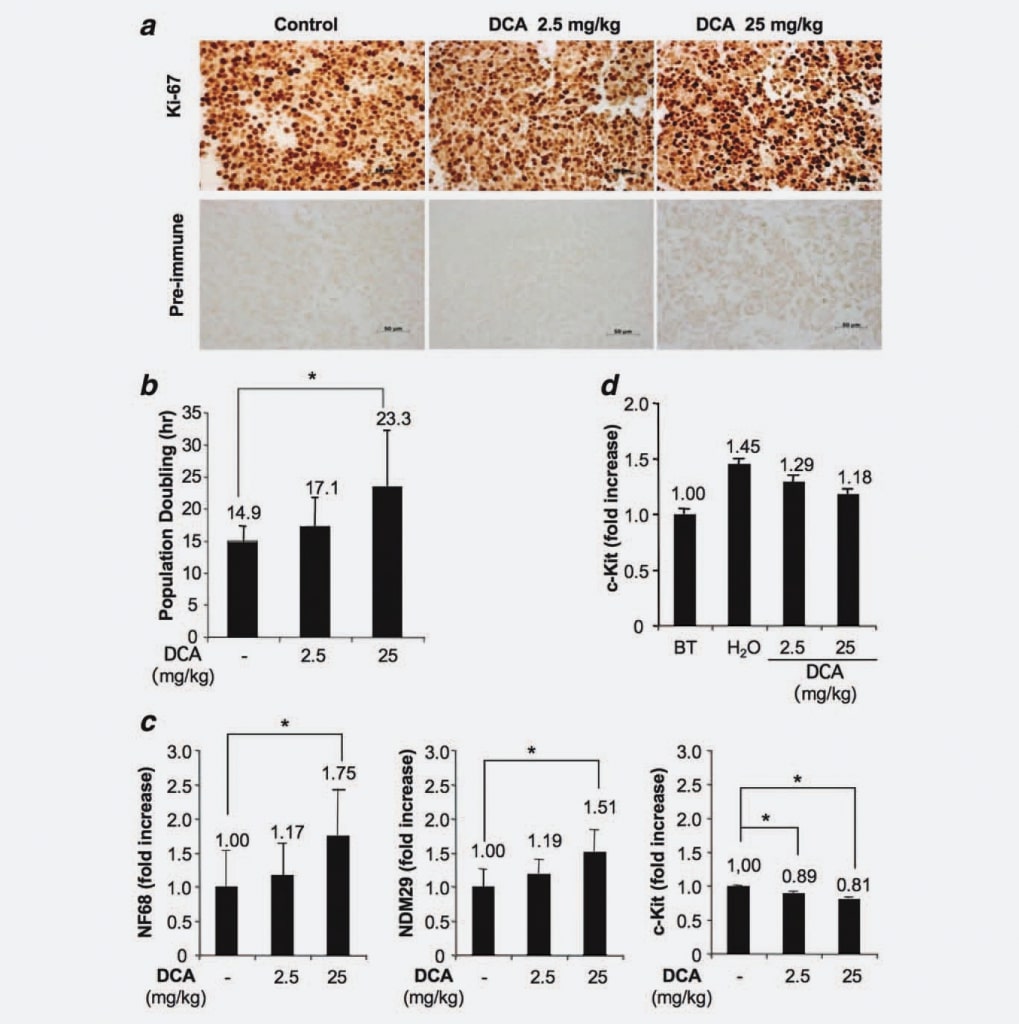
Da die in unserem Versuchsmodell erzeugten Tumore aus der Injektion einer homogenen Zelllinie stammen und wir gezeigt haben, dass die DCA-Behandlung weder eine Verringerung des Tumorzellvolumens noch eine Erhöhung der Apoptose zur Folge hatte, stellten wir die Hypothese auf, dass DCA möglicherweise eine Auswirkung auf die Proliferationsfähigkeit der Zellen hat. Um diese Hypothese zu testen, untersuchten wir Zellen aus behandelten und unbehandelten Knoten mittels Immunhistochemie unter Verwendung eines spezifischen monoklonalen Anti-Ki67-Antikörpers. Die Ergebnisse zeigten keine signifikanten Unterschiede in Bezug auf den Anteil der proliferierenden/zyklierenden Zellen zwischen den Gruppen. Dieses Ergebnis deutet also darauf hin, dass die DCA-Behandlung die Anzahl der zyklischen Zellen nicht verringert, was mit der fehlenden vollständigen Ausrottung des Tumors nach DCA-Behandlungen übereinstimmt (Abb. 3a). Als Nächstes verwendeten wir das xCELLigence-System (Roche, Deutschland), um einen möglichen dosisabhängigen Anstieg der durch die DCA-Behandlung induzierten Populationsverdopplung nachzuweisen. Wir fanden heraus, dass sich Tumorzellen von behandelten Mäusen (25 mg/kg) in 23,3 Stunden verdoppeln, Zellen von Tieren, die mit 2,5 mg/kg behandelt wurden, verdoppeln sich in 17,1 Stunden, während Zellen von Kontrollmäusen eine Populationsverdopplungszeit von 14,9 Stunden aufweisen (Abb. 3b). Da die Unterschiede statistisch signifikant waren (DCA 25 mg/kg vs. Kontrollgruppe; p ¼ 0,0476), zeigen diese Ergebnisse, dass die DCA-Behandlung eine dosisabhängige Verzögerung des Zellzyklus bewirkt. Als nächstes untersuchten wir, ob die Zellzyklusverzögerung von Zellen aus behandelten Tumoren mit einer verstärkten Differenzierung/Bindung verbunden war. Zu diesem Zweck haben wir mittels Real Time RT-PCR NB-Zelldifferenzierungsmarker [Neuroblastoma Differentiation Marker 29 (NDM29), Neurofilament 68 (NF68)] und c-Kit, ein Protein, das spezifisch von stammähnlichen/Tumor initiierenden Zellen exprimiert wird, in DCA-behandeltem und unbehandeltem Tumorgewebe gemessen. Wie in Abbildung 3c dargestellt, wurde in Zellen aus DCA-behandelten Tumoren ein allgemeiner Anstieg der Synthese von NB-Zelldifferenzierungsmarkern beobachtet. Die höhere DCA-Konzentration führte zu einer verstärkten Expression von NF68 (75 %, p < 0,001), während die niedrigere Dosis einen bescheidenen Anstieg (17 %, statistisch nicht signifikant) bewirkte. Ebenso war der Anstieg der Expression von NDM29 in Tumoren, die mit DCA 25 mg/kg behandelt wurden, statistisch signifikant (51 % Anstieg im Vergleich zur Kontrollgruppe; p < 0,001), während nur ein geringer Unterschied zwischen Zellen aus Tumoren, die mit 2,5 mg/kg behandelt wurden, und Zellen aus unbehandelten Tumoren beobachtet wurde. Interessanterweise zeichneten sich DCA-behandelte Proben auch durch eine verringerte Expression von c-Kit aus (11% Reduktion im Vergleich von DCA 2,5 mg/kg zu Kontrollproben und 19% Reduktion im Vergleich von DCA 25 mg/kg zu Kontrollproben, p < 0,001), was darauf hindeutet, dass DCA eine Verringerung des stammartigen/tumorinitiierenden Potenzials in proliferierenden Tumorzellen bewirkt. Auch diese Ergebnisse sprechen für eine Verlangsamung der Proliferationsgeschwindigkeit und möglicherweise für eine Verringerung des malignen Potenzials der mit DCA behandelten Zellen.
Um festzustellen, ob DCA den Anteil undifferenzierter proliferierender Zellen beeinflussen kann, haben wir als Nächstes die c-Kit-Expression in Tumorgeweben von Mäusen, die vor der DCA-Behandlung geopfert wurden, und in Knoten behandelter Tumoren gemessen. Wir fanden heraus, dass, obwohl die c-Kit-Expression (und vermutlich das bösartige Potenzial) während des Wachstums der Knoten progressiv ansteigt (sieheH2Ovs. vor der Behandlung), die Verabreichung des DCA-Medikaments dieses Phänomen tendenziell einschränkt, was in den Geweben der Tumormassen bestätigt, was bereits in den Zellen beobachtet wurde und eine Erklärung für die fehlende Tumorausrottung in DCA-behandelten Mäusen liefert (Abb. 3d).
DCA wirkt spezifisch auf bösartige NB-Zellen und ist bei differenzierten, wenig bösartigen NB-Zellen unwirksam
Immer mehr experimentelle Belege deuten darauf hin, dass die Prognose von BS und das Ansprechen auf Krebsbehandlungen vom Differenzierungsstadium der BS-Knötchen abhängen. Ein besonderes Merkmal der NB ist die bemerkenswerte Heterogenität der Zellen, die sich in unterschiedlichen Stadien aus verschiedenen Neuralleisten ableiten können.12,17 In diesem Zusammenhang liegt die Hypothese nahe, dass die verschiedenen Zelltypen möglicherweise unterschiedlich empfindlich auf DCA reagieren. Um diese Hypothese zu überprüfen, haben wir ein In-vitro-Modell der NB-Zelldifferenzierung verwendet, das auf der Expression einer Pol-III-transkribierten ncRNA basiert. In der Tat haben wir vor kurzem eine neuartige, von der RNA-Polymerase (pol) III umgeschriebene nichtcodierende (nc) RNA (nämlich NDM29) isoliert, deren Expression die neuronale Differenzierung von NB-Zellen auslöst und ihr malignes Potenzial sowie die Expression von tumorinitiierenden/stammähnlichen Zellmarkern stark reduziert. Durch die Integration zusätzlicher Kopien der NDM29-Transkriptionseinheit in SKNBE2-Tumorzellen haben wir eine NDM29-überexprimierende SKNBE2-Zelllinie (im Folgenden als S1 bezeichnet) und die entsprechende Mock-Negative erzeugt, die NDM29 auf endogenem Niveau exprimieren. S1-Zellen weisen einen teilweise differenzierten/neuronenähnlichen Phänotyp auf, sind wenig bösartig und zeichnen sich durch eine deutliche Verzögerung des Zellzyklus aus.18,19
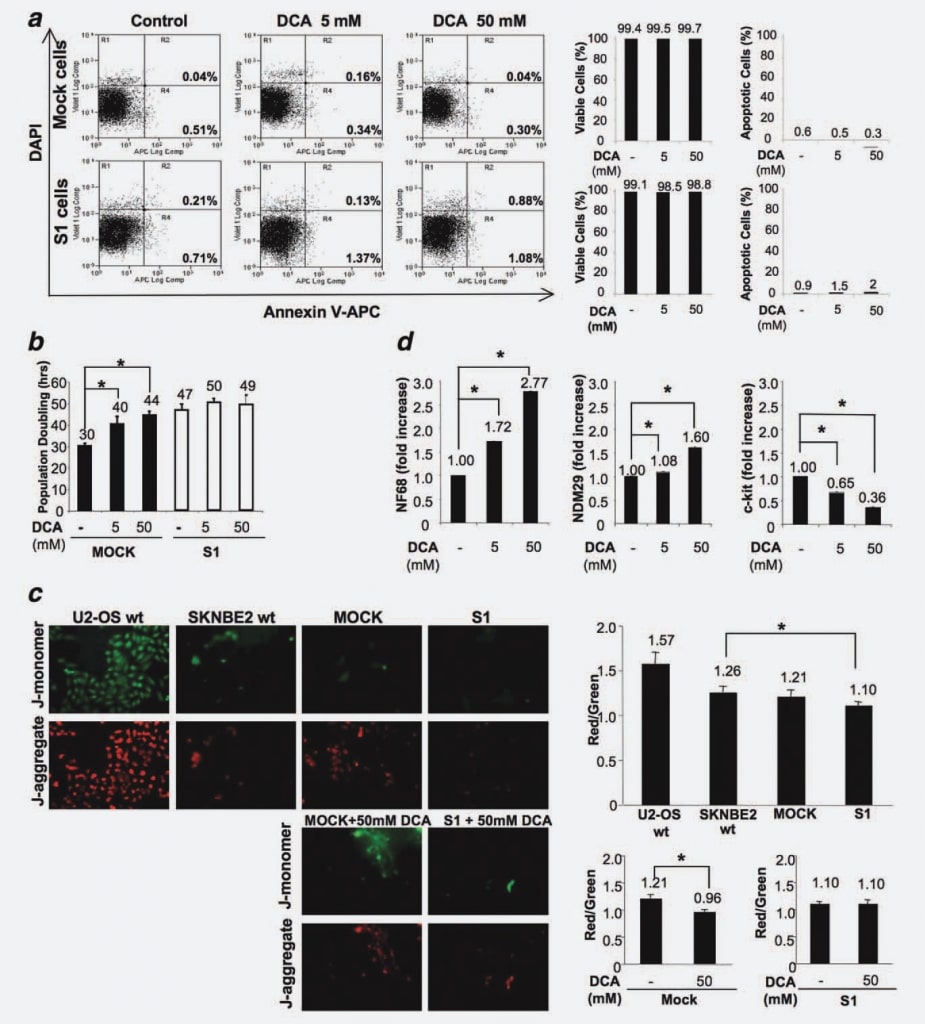
Um auszuschließen, dass eine DCA-Behandlung in vitro Apoptose auslösen könnte, wie in den In-vivo-Experimenten gezeigt wurde, haben wir den Prozentsatz apoptotischer Zellen in Mock- und S1-Zelllinien gemessen, die 48 Stunden lang mit dem Medikament behandelt bzw. unbehandelt waren. Es wurden keine Unterschiede zwischen den Zelllinien (unbehandelt oder mit unterschiedlichen DCA-Dosen behandelt) festgestellt, was bestätigt, dass DCA keine Apoptose in NB-Zellen auslöst (Abb. 4a).
Um die Auswirkungen auf die Proliferation von Mock- und S1-Zellen zu ermitteln, behandelten wir die Zellen mit 5 mM und 50 mM DCA und maßen die Verdopplungszeit der Zellpopulation (PD) in vier unabhängigen Experimenten. Die Konzentration von 50 mM wurde als Höchstdosis gewählt, da verschiedene frühere Studien berichteten, dass DCA bei verschiedenen Zelllinien relativ inaktiv zu sein scheint, wenn es in niedrigeren Dosen verwendet wird.16,20 Wie aus Abbildung 4b hervorgeht, wird die PD-Zeit durch eine DCA-Behandlung in Mock-Zellen dosisabhängig deutlich verlängert. Ohne Behandlung verdoppelte sich die Mock-Zelle innerhalb von 30 Stunden, während die Verabreichung von 5 mM DCA eine Verzögerung des Zellzyklus um 10 Stunden bewirkte und die höhere Dosis von 50 mM DCA zu einem Anstieg um 14 Stunden führte (p < 0,001). Diese Ergebnisse zeigen, dass DCA eine Verzögerung des Zellzyklus in undifferenzierten/stark proliferierenden NB-Zellen hervorruft. Im Gegensatz dazu blieb die PD für S1-Zellen annähernd unverändert und verschob sich von 47 Stunden bei unbehandelten Zellen auf 50 Stunden bei mit 5 mM DCA behandelten Zellen und auf 49 Stunden bei mit 50 mM DCA behandelten Zellen (Abb. 4b). Dieses Ergebnis zeigt, dass die Reaktion auf die DCA-Behandlung direkt mit dem Differenzierungs-/Proliferationsstadium der BS-Zellen korreliert, da differenzierte Zellen mit sehr geringem malignen Potenzial kaum auf die Behandlung ansprechen, während ihre vollständig proliferierenden/sehr malignen Gegenstücke stark von der DCA-Behandlung betroffen sind.
Um dieses Phänomen besser zu verstehen und in Anbetracht der Tatsache, dass die Wirkungen von DCA mit der PDK-Hemmung, der Aktivierung der oxidativen Phosphorylierung in den Mitochondrien und der spezifischen Verringerung der mitochondrialen Hyperpolarisation zusammenhängen,6 haben wir Mock- und S1-Zellen auf mögliche Veränderungen der mitochondrialen Hyperpolarisation als Grundlage für die unterschiedliche Anfälligkeit dieser Zellen für DCA untersucht. Zu diesem Zweck behandelten wir die Zellen mit JC-1 (5,5′ ,6,6′ -Tetrachlor-1,1′ ,3,3′ -Tetraethylbenzimidazolocarbocyaninjodid), einem fluoreszierenden Farbstoff, dessen Aufnahme proportional zum mitochondrialen Potenzial ist. JC-1 kann selektiv in die Mitochondrien eindringen und ändert seine Farbe reversibel von grün zu rot, wenn das mitochondriale Potenzial ansteigt, so dass ein verringertes Verhältnis von roter zu grüner Fluoreszenz eine mitochondriale Reaktivierung anzeigt. Wir verglichen die mitochondriale Membranpolarisation von U2-OS-Osteosarkomzellen (hyperpolarisierte Kontrollzellen21), SKNBE2 wt, Mock und S1 Zellen. Wir fanden heraus, dass Mock-Zellen im Vergleich zu SKNBE2 wt-Zellen einen ähnlichen Polarisationsgrad aufweisen, während S1-Zellen durch einen geringeren Grad an mitochondrialer Polarisation gekennzeichnet sind (p ¼ 0,0184) (Abb. 4c). Die Ergebnisse, die sich auf drei Wiederholungen beziehen, zeigen, dass in Mock-Zellen eine DCA-bedingte mitochondriale Reaktivierung stattfindet (25%ige Verringerung des Verhältnisses von roter zu grüner Fluoreszenz; p ¼ 0,0077), während in S1-Zellen die mitochondriale Aktivität durch die Anwesenheit von DCA nicht verändert wird (Abb. 4c). Diese Ergebnisse zeigen also, dass die Anfälligkeit von malignen/voll zyklischen Zellen mit einem mitochondrialen Status verbunden ist, der empfindlich auf die Wirkung von DCA reagiert, während engagierte/differenzierte, schlecht zyklische Zellen refraktär gegenüber der Wirkung dieses Medikaments sind.
Um mögliche Auswirkungen von DCA auf das Differenzierungspotenzial festzustellen, haben wir als Nächstes mittels Real Time RT-PCR die Expressionswerte von NDM29, NF68 und c-Kit in DCA-behandelten und unbehandelten Mock-Zellen gemessen. Wir führten diese Analyse speziell an Mock-Zellen durch, da S1-Zellen gegenüber dem DCA-bedingten Rückgang der Proliferation refraktär waren. Wie in Abbildung 4d gezeigt, führte die DCA-Behandlung zu einer erhöhten Expression des Differenzierungsmarkers NF68 (Mock-Zellen, die mit 5 mM oder 50 mM DCA behandelt wurden, im Vergleich zu unbehandelten Zellen, p < 0,0001). Auch die Expression von NDM29 war in den behandelten Mock-Zellen im Vergleich zu den Kontrollzellen signifikant erhöht (p < 0,001). Im Gegensatz dazu war die c-Kit-Expression in den behandelten Zellen dosisabhängig verringert (p < 0,0001). Insgesamt deuten diese Ergebnisse darauf hin, dass die antitumorale Wirkung von DCA in NB-Zellen hauptsächlich auf eine antiproliferative/prodifferenzierende Wirkung zurückzuführen ist und nicht zu einer erhöhten Apoptoserate führt.
Diskussion
DCA ist ein kleines Molekül, das seit mehr als 30 Jahren beim Menschen zur Behandlung von angeborener und erworbener Laktatazidose eingesetzt wird.22 Es ist bekannt, dass die Behandlung mit DCA den Laktatspiegel im Blutkreislauf senkt, indem der Zellstoffwechsel von der Glykolyse auf die Glukoseoxidation umgestellt wird, ohne dass sich dies nachteilig auf normale Zellen auswirkt.6 Aufgrund seiner Fähigkeit, den Glukosestoffwechsel durch aerobe Glykolyse zu begünstigen, wird DCA erfolgreich in klinischen Studien bei Herzerkrankungen, einschließlich kongestiver Herzinsuffizienz und ischämischer Herzerkrankungen, eingesetzt, da postischämische Funktionsstörungen hypertrophierter Herzen mit niedrigen Glukoseoxidations- und hohen Glykolyse-Raten verbunden sind.23
Eine sehr attraktive Eigenschaft von DCA liegt in seiner Verträglichkeit und Sicherheit. In mehr als 40 klinischen Studien mit DCA wurde berichtet, dass die wichtigste unerwünschte Wirkung einer langfristigen DCA-Gabe eine reversible periphere Neuropathie ist.24,25
In den letzten Jahren haben sowohl In-vitro- als auch In-vivo-Modelle deutliche Hinweise darauf geliefert, dass DCA auch für die Behandlung einiger Krebsarten beim Menschen nützlich sein könnte.6 Tatsächlich ist DCA in der Lage, den Warburg-Effekt umzukehren, indem es die PDK hemmt, das mitochondriale Membranpotenzial wiederherstellt und die ROS-Produktion erhöht. Aus diesem Grund wurde DCA als Wunderwaffe gegen Krebs gefeiert,1 auch wenn es derzeit noch nicht für die Krebsbehandlung zugelassen ist.26 Obwohl das Neuroblastom bisher als eine Krebsart galt, bei der eine DCA-Behandlung höchstwahrscheinlich unwirksam sein dürfte, da in den Zellen, aus denen sich die NB-Knoten zusammensetzen, vermutlich keine Hyperpolarisation der Mitochondrienmembran vorliegt,27 wurde beschrieben, dass proliferierende NB-Zellen durch einen glykolytischen Phänotyp aufrechterhalten werden,11 und dass NB-Zellen selbst unter aeroben Bedingungen beachtliche Mengen an Glukose in Laktat umwandeln, anstatt Glukose zu oxidieren.28 Auf der Grundlage dieser Erkenntnisse sollte in dieser Studie untersucht werden, ob DCA eine positive Wirkung auf diesen Tumortyp haben könnte. Unsere Ergebnisse zeigen eine unvorhergesehene positive Wirkung von DCA auf das Wachstum von BS-Tumoren sowohl in in vivo als auch in vitro Modellen. Tatsächlich war DCA bei etablierten NB-Tumoren wirksam, ohne bei den behandelten Tieren irgendwelche offensichtlichen Nebenwirkungen hervorzurufen.
Im Einzelnen zeigte die Analyse von Zellen aus behandelten Tumoren, dass DCA zu einer Verzögerung der Proliferation führt, ohne Anzeichen von erhöhter Apoptose oder Zelltod. Dies steht im Einklang mit der beobachteten Verringerung des Tumorvolumens, die nicht mit einer vollständigen Ausrottung des Krebses einhergeht. Die zugrundeliegenden Wirkmechanismen von DCA sind noch umstritten. In diesem Zusammenhang stehen unsere Ergebnisse im Gegensatz zu früheren Studien, in denen Endometrium-, Prostata-, Kolorektal- und Lungenkrebszellen mit DCA behandelt wurden.6,9,20 In diesen Fällen wurde über eine erhöhte Apoptose ohne Auswirkung auf die Zellzyklusverteilung9,21 oder eine erhöhte Apoptose bei verringerter Proliferation8 berichtet. Andererseits weisen andere Berichte darauf hin, dass DCA die Proliferation von Krebszellen hemmt, nicht aber die Apoptose oder den Zelltod erhöht.7 Dieser letzte Mechanismus wird durch eine proteomische Analyse gestützt, bei der eine DCA-Behandlung eher Veränderungen bei den Zellproliferationsmarkern als bei der Menge der Apoptose-bezogenen Proteine bewirkt.16 Dieses duale Verhalten von DCA könnte zelltypabhängig sein, wahrscheinlich aufgrund von Unterschieden in der Expression von PDK-Isoenzymen in den untersuchten Krebszellen. Studien zur Korrelation der PDK-Expression mit der DCA-Empfindlichkeit sind im Gange.
Interessanterweise zeigen wir hier, dass eine erhöhte Expression einiger Differenzierungsmarker wie NF68 und NDM29, begleitet von einer Verringerung der Expression von c-kit, einem Marker für stammesartige/tumorinitiierende Zellen, mit dem beobachteten Rückgang der Krebszellproliferation korreliert. Die DCA-abhängige Verzögerung des Zellzyklus, die mit einer verstärkten Zelldifferenzierung einhergeht, stellt somit einen neuen, noch nicht beschriebenen Wirkmechanismus dieses Medikaments dar, der die DCA-Effekte mit dem Differenzierungsstadium der Krebszellen verknüpft.
Es wurde dokumentiert, dass menschliche NB-Tumoren durch verschiedene Zelltypen mit unterschiedlichen morphologischen, biochemischen und tumorerzeugenden Eigenschaften und variablen Differenzierungsstadien gekennzeichnet sind.12 Um die mögliche Korrelation der DCA-Anfälligkeit mit dem Differenzierungsgrad der Zellen zu vertiefen, verwendeten wir ein In-vitro-Modell der NB-Zelldifferenzierung, das auf der Überexpression einer Pol-III-transkribierten ncRNA (NDM29) basiert. Wir erzeugten eine NDM29-überexprimierende SKNBE2-Zelllinie (hier als S1 bezeichnet) und die entsprechende Mock-Negativkontrolle.19 Diese Zelllinien wurden mit 5 mM und 50 mM DCA behandelt. Die Konzentration von 50 mM wurde als Höchstdosis gewählt, da verschiedene frühere Studien berichteten, dass DCA in verschiedenen Zelllinien relativ inaktiv ist, wenn es in niedrigeren Dosen verwendet wird,16,20 obwohl diese Konzentration in vivo nicht erreicht werden wird. In unserem Versuchsaufbau sind die Wirkungen von DCA jedoch bereits bei einer niedrigeren Dosis (5 mM) offensichtlich, so dass diese Konzentration für die Verwendung in vivo geeignet ist.
Wie erwartet, haben wir festgestellt, dass die Behandlung mit DCA auch in Mock- und S1-Zellen keine Anzeichen von Apoptose hervorruft, was unsere In-vivo-Ergebnisse bestätigt. Interessanterweise stellten wir fest, dass die Wirkung von DCA auf undifferenzierte, sehr bösartige, vollständig zyklische Zellen (Mock-Zellen) beschränkt ist, während sie die Vermehrungsrate von stärker differenzierten, wenig bösartigen Zellen (S1-Zellen) nicht beeinträchtigt. Dieser Effekt könnte auf den unterschiedlichen Grad der mitochondrialen Polarisierung dieser Zelltypen zurückgeführt werden. In der Tat haben wir gezeigt, dass Mock-Zellen eine höhere mitochondriale Polarisierung aufweisen als ihr differenzierteres Gegenstück S1.
Daher haben wir hier gezeigt, dass die verschiedenen Zellen, aus denen sich die NB-Tumormassen zusammensetzen, unterschiedlich empfänglich für DCA sind, das selektiv auf die sehr bösartigen und vollständig proliferierenden Zellen wirkt, während es eine sehr geringe Wirkung auf schwach bösartige, stärker differenzierte Zellen ausübt. Eine ähnliche differentielle Wirkung von DCA wurde auch in einer Studie über Glioblastoma multiforme (GBM)2 berichtet, in der die Autoren zeigten, dass Krebsstammzellen im Vergleich zu den differenzierteren Gegenstücken, aus denen GBM besteht, denselben metabolischen und mitochondrialen Umbau aufweisen, jedoch in verstärktem Maße, da sie die meisten hyperpolarisierten Mitochondrien besitzen.2
Zusammenfassend lässt sich sagen, dass die hier beschriebenen Experimente die Ansicht stützen, dass DCA ein sehr selektiver Wirkstoff ist, der bei der Behandlung von NB-Krebs helfen kann, ohne dass es zu erheblichen Nebenwirkungen auf gesunde Zellen kommt. Daher erweitert diese vorläufige Studie den möglichen Einsatz von DCA in der Krebstherapie von BS und untermauert die Notwendigkeit einer detaillierten Untersuchung seiner krebshemmenden Eigenschaften bei diesem Tumortyp.
Danksagung
R.C. wurde vom italienischen Ministerium für Universität und ForschungMIUR (Internationales FIRB-Programm 2007) unterstützt. A.P. wurde vom italienischen Ministerium für Universität und Forschung MIUR (2007 PRIN Program prot. 2007945BZN), von der Associazione Italiana Ricerca sul Cancro (2009 AIRC Program no. IG9378) und von der Associazione Italiana per la Lotta al Neuroblastoma (Genua, Italien) unterstützt.
REFERENZEN
1 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. Dichloroacetate metabolically targeted therapy defeats cytotoxicity of standard anticancer drugs. Cancer Chemother Pharmacol 2011;67:647-55
2 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med; 2010;2:31ra4.
3 Stacpoole PW, Henderson GN, Yan Z, James MO. Klinische Pharmakologie und Toxikologie von Dichloracetat. Environ Health Perspect 1998;106 Suppl 4:989-94.
4 Stacpoole PW. Die Pharmakologie von Dichloracetat. Metabolism 1989;38: 1124-44.
5 Michelakis ED, Webster L, Mackey JR. Dichloracetat (DCA) als potenzielle metabolische Zieltherapie für Krebs. Br J Cancer 2008;99:989-94.
6 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle 2007;11:37-51.
7 Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. Die Umkehrung des glykolytischen Phänotyps durch Dichloracetat hemmt das Wachstum metastasierender Brustkrebszellen in vitro und in vivo. Breast Cancer Res Treat 2010;120:253-60.
8 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Dichloracetat (DCA) sensibilisiert sowohl Wildtyp- als auch überexprimierende Bcl-2-Prostatakrebszellen in vitro für Strahlung. Prostate 2008;68:1223-31.
9 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloracetat induziert Apoptose in Endometriumkrebszellen. Gynecol Oncol 2008;109:394-402.
10 Vander Heiden MG. Zielgerichteter Zellstoffwechsel bei Krebspatienten. Sci Transl Med 2009;2:31ed1.
11 Almeida A, Bolanos JP, Moncada S. Die E3-Ubiquitin-Ligase APC/C-Cdh1 ist für den Warburg-Effekt verantwortlich, indem sie die Glykolyse mit der Zellproliferation verknüpft. Proc Natl Acad Sci USA; 107:738-41.
12 Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, Biedler JL, Cheung NK, Ross RA. Merkmale von Stammzellen aus menschlichen Neuroblastom-Zelllinien und in Tumoren. Neoplasia 2004;6:838-45.
13 Vistejnova L, Dvorakova J, Hasova M, Muthny T, Velebny V, Soucek K, Kubala L. The comparison of impedance-based method of cell proliferation monitoring with commonly used metabolic-based techniques. Neuro Endocrinol Lett 2009;30 Suppl 1:121-7.
14 Schmidt MM, Rohwedder A, Dringen R. Effects of chlorinated acetates on the glutathione metabolism and on glycolysis of cultured astrocytes. Neurotox Res 2011; 19:628-37.
15 Papandreou I, Goliasova T, Denko NC. Krebsmedikamente, die auf den Stoffwechsel abzielen: Ist Dichloracetat das neue Paradigma? Int J Cancer 2011;128:1001-8.
16 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Natriumdichloracetat greift selektiv Zellen mit Defekten im mitochondrialen ETC an. Int J Cancer 2010;127: 2510-9.
17 George RE, Variend S, Cullinane C, Cotterill SJ, McGuckin AG, Ellershaw C, Lunec J, Pearson AD. Beziehung zwischen histopathologischen Merkmalen. MYCN-Amplifikation und Prognose: eine UKCCSG-Studie. United Kingdom Children Cancer Study Group. Med Pediatr Oncol 2001;36: 169-76.
18 Pagano A, Castelnuovo M, Tortelli F, Ferrari R, Dieci G, Cancedda R. New small nuclear RNA gene-like transcriptional units as sources of regulatory transcripts. PLoS Genet 2007;3:e1.
19 Castelnuovo M, Massone S, Tasso R, Fiorino G, Gatti M, Robello M, Gatta E, Berger A, Strub K, Florio T, Dieci G, Cancedda R, et al. An Alu-like RNA promotes cell differentiation and reduces malignancy of human neuroblastoma cells. FASEB J 2010;24:4033-46.
20 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Dichloracetat induziert Apoptose und Zellzyklus-Stillstand in kolorektalen Krebszellen. Br J Cancer 2010;102:1746-52.
21 Liu TJ, Lin SY, Chau YP. Inhibition of poly(ADP-ribose) polymerase activation attenuates beta-lapachone-induced necrotic cell death in human osteosarcoma cells. Toxicol Appl Pharmacol 2002;182:116-25.
22 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 2006;117: 1519-31.
23 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Myokardiale metabolische und hämodynamische Effekte von Dichloracetat bei koronarer Herzkrankheit. Am J Cardiol 1988;61: 65-70.
24 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Dichloroacetate causes toxic neuropathy in MELAS: a randomized, controlled clinical trial. Neurologie 2006;66: 324-30.
25 Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmakokinetik, Metabolismus und Toxikologie von Dichloroacetat. Drug Metab Rev 1998;30: 499-539.
26 Pearson H. Krebspatienten entscheiden sich für ein nicht zugelassenes Medikament. Nature 2007;446: 474-5.
27 Chen LB. Mitochondriales Membranpotential in lebenden Zellen. Annu Rev Cell Biol 1988;4:155-81.
28 Deubzer B, Mayer F, Kuci Z, Niewisch M, Merkel G, Handgretinger R, Bruchelt G. H(2)O(2)-mediated cytotoxicity of pharmacologic ascorbate concentrations to neuroblastoma cells: potential role of lactate and ferritin. Cell Physiol Biochem 2010;25:767-74.