Serena Vella1*, Matteo Conti2*, Roberta Tasso1, Ranieri Cancedda1,3 et Aldo Pagano1,3
1 Département d’oncologie, de biologie et de génétique (DOBiG), Université de Gênes, Gênes-Italie
2 Laboratoire de pharmacologie et de toxicologie cliniques, Ospedale S. Maria delle Croci, 48100 Ravenne-Italie
3 Institut national de recherche sur le cancer (IST) Gênes, Largo R. Benzi, 10, 16132 Gênes-Italie
La petite molécule hydrosoluble Dichloroacetate (DCA) a récemment suscité un vif intérêt dans le domaine de la thérapie du cancer car il a été démontré qu’elle était capable d’inhiber la croissance des tumeurs humaines en agissant spécifiquement sur les mitochondries des cellules cancéreuses sans perturber la physiologie des cellules non malignes. Le neuroblastome était l’un des types de tumeurs sur lequel le DCA était considéré comme inefficace car il est composé de cellules présentant peu d’anomalies mitochondriales reconnues. Le neuroblastome, cependant, est composé de différents types de cellules en termes de métabolisme, de phénotype et de potentiel malin. Malgré les prédictions ci-dessus, nous montrons dans ce travail que (i) le DCA présente un effet anticancéreux inattendu sur les cellules tumorales du NB et (ii) que cet effet est sélectivement dirigé vers les cellules NB très malignes, alors que les cellules NB plus différenciées/moins malignes sont réfractaires au traitement par le DCA. Ce résultat confirme la nécessité d’une étude détaillée des propriétés anticancéreuses du DCA sur ce type de tumeur, dans le but de l’utiliser éventuellement comme agent thérapeutique.
Le DCA, une petite molécule/médicament orphelin, est récemment apparu sous les feux de la rampe pour sa capacité à limiter la croissance des tumeurs du glioblastome multiforme (GBM) à des doses compatibles avec l’absence d’effets secondaires.1-4 Ainsi, compte tenu de sa toxicité bien tolérée et de son faible coût, le DCA suscite un vif intérêt pour son utilisation potentielle dans la thérapie du cancer et dans la guérison de certains types de tumeur.5 En effet, bien que le DCA se soit révélé efficace dans le carcinome pulmonaire à petitescellules6, les cancers dusein7, de la prostate8 et de l’endomètre9 ainsi que dans les lignées cellulaires deglioblastome2, l’efficacité de cette petite molécule en tant que traitement anticancéreux n’a jusqu’à présent été démontrée cliniquement que dans le GBM humain, de sorte que l’efficacité prouvée du DCA sur d’autres tumeurs malignes reste encore àévaluer10 En détail, en raison de son mécanisme d’action, le DCA devrait être inefficace sur les tumeurs caractérisées par une faible polarisation mitochondriale, comme le cancer du poumon à cellules d’avoine, les lymphomes, les neuroblastomes (NB) et lessarcomes5 Le DCA, en tant qu’inhibiteur de l’enzyme mitochondriale pyruvate déshydrogénase kinase (PDK), active la pyruvate déshydrogénase (PDH), une enzyme gardienne qui régule le flux de pyruvate dans la mitochondrie, augmentant ainsi le rapport entre l’oxydation du glucose et la glycolyse.4 -Bonnet et al. ont montré que cette stimulation de la phosphorylation oxydative est sélectivement pro-apoptotique dans les cellules cancéreuses, entraînant une diminution de leur hyperpolarisation mitochondriale typique associée à la résistance à l’apoptose6 Bien que le NB soit initialement considéré comme un type de tumeur sur lequel le DCA est très probablement inefficace, en raison de sa caractéristique spécifique de petites cellules et de l’absence présumée d’hyperpolarisation de la membrane mitochondriale,5 les cellules NB en prolifération sont soutenues par un phénotype glycolytique.11 Nous avons étudié l’efficacité éventuelle du traitement par DCA pour inhiber la croissance de nodules de NB humains générés chez des souris NOD-SCID. De manière surprenante, nous avons observé que le DCA limite de manière significative la croissance tumorale in vivo. Dans les tumeurs NB humaines, il existe trois types de cellules distinctes : Les cellules souches de type I, les précurseurs neuroblastiques/neuroendocriniens de type N et les précurseurs schwanniens/mélanoblastiques de type S. Ces cellules présentent des caractéristiques morphologiques, des caractéristiques de fonctionnement et des propriétés de conservation distinctes. Ces cellules ont des propriétés morphologiques, biochimiques et tumorigènes distinctes et des stades de différenciation variables.12 Il est intéressant de noter qu’en tirant parti de cellules NB SKNBE2 génétiquement modifiées, caractérisées par un engagement neuronal marqué et un très faible potentiel de malignité, nous avons constaté que l’effet du DCA est limité aux cellules indifférenciées, très malignes et à cycle complet, alors qu’il n’affecte pas le taux de prolifération des cellules plus différenciées et peu malignes. Les expériences rapportées ici suggèrent d’étendre l’efficacité possible du DCA comme médicament anticancéreux au NB agissant de manière différentielle sur les cellules fortement et/ou faiblement malignes.
Mots clés : dichloroacétate, neuroblastome, mitochondries *S.V. et M.C. ont contribué à parts égales à ce travail.
Sponsor de la subvention : Ministère italien de l’Université et de la Recherche-MIUR (2007 PRIN Program prot.) ; Numéro de subvention : 2007945BZN ; Sponsor de subvention : Associazione Italiana Ricerca sul Cancro (2009 AIRC Program) ; Numéro de subvention : IG9378 ; Subvention sponsorisée : Ministère italien de l’Université et de la Recherche-MIUR (2007 International FIRB Program), Associazione Italiana per la Lotta al Neuroblastoma (Gênes, Italie)
DOI : 10.1002/ijc.26173
Historique : Reçu le 25 février 2011 ; Accepté le 20 avril 2011 ; En ligne le 9
mai 2011
Correspondance à : Aldo Pagano, Département d’Oncologie, de Biologie et de Génétique (DOBiG), Université de Gênes, Gênes, Italie, Tél. : þ/39/ 010-5737241, Fax : þ/39/010-5737257, E-mail : [email protected]
Int. J. Cancer : 130, 1484-1493 (2012) VC 2011 UICC
Matériel et méthodes
Souris
Des souris homozygotes NOD-SCID (NOD.CB17-Prkdcscid) ont été achetées au Jackson Laboratory (Bar Harbor, MA). Les souris ont été utilisées entre 5 et 8 semaines d’âge. Tous les animaux ont été élevés et maintenus dans l’animalerie de l’Institut national pour la recherche sur le cancer, à Gênes, en Italie. Les soins et l’utilisation des animaux étaient conformes aux lois du ministère italien de la santé et aux directives de la Communauté européenne.
Test de tumorigénicité in vivo
Une suspension cellulaire de cellules SKNBE2 dans du PBS (107 cellules) a été injectée par voie sous-cutanée à 37 souris NOD/SCID. Les souris ont été divisées en quatre groupes : -groupe témoin (n ¼ 13 souris) : eau stérilisée ; -groupe traité au DCA (25 mg/kg/dose) (n ¼ 14 souris) ; -groupe traité au DCA (2,5 mg/kg/dose) (n ¼ 5 souris) ; -groupe prétraité (n ¼ 5 souris) : souris injectées par voie sous-cutanée avec la suspension cellulaire, mais sacrifiées avant tout type de traitement. Le DCA, ainsi que de l’eau stérilisée, ont été administrés par voie intragastrique, une fois par jour/5 jours par semaine/pendant 4 semaines. Les traitements ont commencé lorsque la néoplasie a atteint un diamètre seuil de 5 mm. Les souris ont été observées chaque semaine pour l’apparition de tumeurs aux sites d’injection ; la taille des tumeurs a été mesurée chaque semaine avec des compas dans tous les groupes. L’imagerie de chaque souris a été recueillie à chaque point temporel considéré.
Cultures cellulaires
Les cellules SKNBE2 wt, Mock et S1 ont été maintenues dans un milieu RPMI 1640 (Sigma-Aldrich, Milan, Italie), 10% de FBS (GIBCO, S.Giuliano Milanese, Milan, Italie), L-glutamine (2 mM ; EuroClone, Devon, UK), pénicilline-streptomycine (100 U/ml/ 100 lg/ml ; EuroClone) (milieu standard). Les cellules U2-OS ont été maintenues sur du milieu Dulbecco’s modified Eagles (DMEM) (Sigma-Aldrich), 10% de FBS (GIBCO), L-glutamine (2 mM ; EuroClone) et pénicilline-streptomycine (100 U/ml/ 100 lg/ml ; EuroClone). Le dichloroacétate a été préparé sous forme de solution aqueuse et ajouté au milieu. Des portions de chaque tumeur considérée ont été lavées dans du PBS et digérées avec 12,5 U/ml de Collagénase de type I (Biochrom AG, Berlin, Allemagne) et 12 U/ml de Dispase (Roche, Allemagne) dans du PBS pendant 20 min à 37oC. Des cellules fraîchement isolées ont été utilisées pour l’analyse cytométrique en flux et l’expansion in vitro dans un milieu standard. Toutes les cultures cellulaires ont été maintenues à 37oCdans une atmosphère à 95% d’air/5% de CO2 et à 100% d’humidité.
Analyse histologique et immunohistochimie
Pour l’examen histologique, les tumeurs dérivées de chaque groupe expérimental ont été enlevées chirurgicalement et fixées dans du formol tamponné neutre à 10%, déshydratées et incorporées dans de la paraffine en utilisant des techniques histologiques standard. Des sections sérielles de 4 mm ont été coupées et colorées à l’hématoxyline et à l’éosine (H/E) pour examiner les caractéristiques morphologiques ou traitées pour l’immunohistochimie. Après hydratation, la récupération de l’antigène a été induite par la chaleur dans un tampon au citrate à pH 6,0 et les peroxydases endogènes ont été bloquées avec 3 % de H2O2 dans l’eau. La liaison non spécifique a été inhibée en incubant les lames dans du sérum de chèvre normal à 10 % (Sigma-Aldrich, Milan, Italie). L’anticorps primaire suivant a été utilisé : anticorps monoclonal anti-Ki-67 humain (clone K-3 ; Oncogene, San Diego, CA). Des contrôles négatifs avec du sérum pré-immun ont été effectués en parallèle. Après un lavage approfondi dans une solution saline tamponnée au Tris, les lames ont été incubées avec un anticorps secondaire anti-souris (BioSpa, Milan, Italie). Après lavage, de la streptavidine conjuguée à de la peroxydase de raifort (BioSpa) a été ajoutée et incubée pendant 30 minutes. Les lames ont ensuite été colorées avec du chromogène diaminobenzidine (Lab Vision, Fremont, CA). Une contre-coloration a été effectuée avec de l’hématoxyline. Les images ont été capturées par microscopie à lumière transmise à l’aide d’un microscope Zeiss Axiovert 200M équipé d’une caméra 3CCD couleur refroidie Zeiss Axio-Cam MRc (Zeiss, Wetzlar, Allemagne). Les lames colorées au H/E ont été observées sous le même microscope à lumière transmise. La taille des cellules a été analysée à l’aide du logiciel ImageJ du domaine public (Wayne Rasband, NIH, Bethesda, MD ; http://rsb.info.nih.gov/ ij/). Le seuil a été fixé de manière à différencier le contour de chaque cellule du fond, et chaque image a été convertie en binaire (noir et blanc). La séparation »Watershed » a été utilisée pour séparer les cellules individuelles des amas. Les surfaces ont été calculées en utilisant l’option « analyser les particules » avec un seuil de coupure de la taille des particules de 100 pixels. Les changements de volume cellulaire ont été déterminés en divisant le nombre de pixels avec traitement médicamenteux par le nombre de pixels sans traitement médicamenteux.
Analyse de l’apoptose
L’apoptose a été analysée par cytométrie en flux, en utilisant l’Annexin V conformément aux instructions du fabricant (kit d’apoptose Annexin VFITC, Immunological Sciences, Rome, Italie ; kit de détection de l’apoptose Annexin V-APC I ; BD Biosciences, Oxford, Royaume-Uni ; DAPI, Sigma-Aldrich, Milan, Italie). En bref, les cellules isolées des masses tumorales ont été lavées dans du PBS et remises en suspension dans un milieu sans sérum. L’Annexin V-FITC et l’iodure de propidium (PI) ont été ajoutés aux préparations cellulaires (105 cellules) et incubés pendant 15 minutes dans l’obscurité à température ambiante. Les cellules Mock et S1 ont été trypsinées, lavées dans du PBS et remises en suspension dans un milieu sans sérum. L’Annexin V-APC et le DAPI ont été ajoutés aux préparations cellulaires (105 cellules) et incubés pendant 15 minutes dans l’obscurité à température ambiante. Les échantillons ont été analysés à l’aide d’un cytofluorimètre Cyan ADP (Beckman-Coulter, Brea CA). Pour chaque échantillon, 20 000 événements ont été acquis. Les données ont été analysées à l’aide du logiciel Summit 4.3.1 (DakoCytomation, Royaume-Uni).
Essais de prolifération cellulaire
(i) Pour les études de comptage cellulaire, les cellules Mock et S1 ont été ensemencées à 5 ×105 cellules dans des boîtes de culture tissulaire de 10 cm, incubées dans un milieu standard, avec ou sans ajout de DCA (5 et 50 mM) et comptées à l’aide d’un hémocytomètre après 48 heures de traitement. (ii) La prolifération cellulaire a également été évaluée par le système xCELLigence RTCA MP (Roche, Allemagne) qui Carcinogenèse Vella et al. 1485 suit les événements cellulaires en temps réel en mesurant l’impédance électrique à travers des microélectrodes en or interdigitées intégrées au fond des plaques de culture de tissus. La mesure de l’impédance fournit des informations quantitatives sur l’état biologique des cellules, notamment leur nombre, leur viabilité et leur morphologie.13 L’impédance du capteur cellulaire est exprimée sous la forme d’une unité arbitraire appelée indice cellulaire (IC). Pour déterminer l’indice cellulaire, les cellules isolées de chaque masse tumorale ont été ensemencées dans 100 ll de milieu standard dans 96 plaques de microtitration (EPlate-Roche, Allemagne). L’impédance de fond a été déterminée en utilisant 100 ll de milieu standard. La fixation, l’étalement et la prolifération des cellules ont été suivis toutes les 30 minutes en utilisant le système xCELLigence. La prolifération cellulaire a été surveillée pendant 72 heures. Les résultats expérimentaux ont été réalisés à l’aide du logiciel RTCA 1.2 qui a calculé le doublement de la population en ajustant la courbe à une équation exponentielle.
Analyse RT-PCR quantitative en temps réel
Les ARN totaux des échantillons ont été extraits à l’aide du réactif TRIzol (Invitrogen, Carlsbad, CA) selon le protocole du fabricant et soumis à une transcription inverse par le kit de synthèse d’ADNc de premier brin Transcriptor (Roche, Allemagne) selon les instructions du fabricant. L’ARN total des échantillons a été mesuré par RTPCR quantitative en temps réel à l’aide du système de détection de séquences PE ABI PRISM@ 7700 (Perkin Elmer Corp./Applied Biosystems, Foster City, CA) et de la méthode Sybr Green selon les instructions du fabricant. Les séquences des amorces avant et arrière étaient les suivantes :
NF-68 : pour 5′ -CAAGGACGAGGTGTCCGAG-3′ , rev 5′ – CCCGGCATGCTTCGA-3′ ;
NDM29 : pour 5′ -GGCAGGCGGGTTCGTT-3′ , rev 5′ – CCACGCCTGGCTAAGTTTTG-3′ ;
c-Kit : pour 5′ -GCAAGTCAGTGCTGTCGGAA-3′ , rev 5′ – AAGATAGCTTGCTTTGGACACAGA-3′ . Pour le contrôle endogène, l’expression de la Glyceraldehyde 3 phosphate déshydrogénase (GAPDH) a été examinée, car il a été démontré que le DCA n’affecte pas l’activité cellulaire de la GAPDH.14 Les séquences des amorces de la GAPDH humaine étaient 5′ – GAAGGTGAAGGTCGGAGTC-3′ et 5′ – GAAGATGGTGATGGGATTTC-3′ . Les niveaux de transcription relatifs ont été déterminés à partir de la courbe standard relative construite à partir de dilutions d’ADNc stock et divisée par la quantité cible du calibrateur selon les instructions du fabricant.
Test du potentiel de la membrane mitochondriale (∆Ψm)
Le potentiel de la membrane mitochondriale (∆Ψm) a été étudié dans des cellules vivantes à l’aide de JC-1 (Cayman Chemical Company, Ann Arbor, MI). Pour l’analyse du potentiel, SKNBE2 wt, Mock, S1 et U2-OS wt ont été placées à une densité de106 cellules/puits avec ou sans DCA (50 mM). Après 72 heures de traitement, les cellules ont été traitées avec la solution JC-1 et incubées à 37oCpendant 15-30 minutes. Les cellules ont été observées avec le microscope Axiophot Zeiss (Zeiss, Jena, Allemagne) [(Texas Red : excitation/émission 590/610nm) (FITC : excitation/émission 485/ 535nm)]. La quantification de la fluorescence a été déterminée avec le logiciel ImageJ (Wayne Rasband, NIH, Bethesda, MD ; http://rsb.info.nih.gov/ij/).
Analyse statistique
La signification statistique des différences observées entre les différents groupes expérimentaux a été calculée à l’aide d’un test t bilatéral. Les valeurs p < 0,05 ont été considérées comme statistiquement significatives.
Résultats
Le DCA est efficace sur les cellules de neuroblastome humain
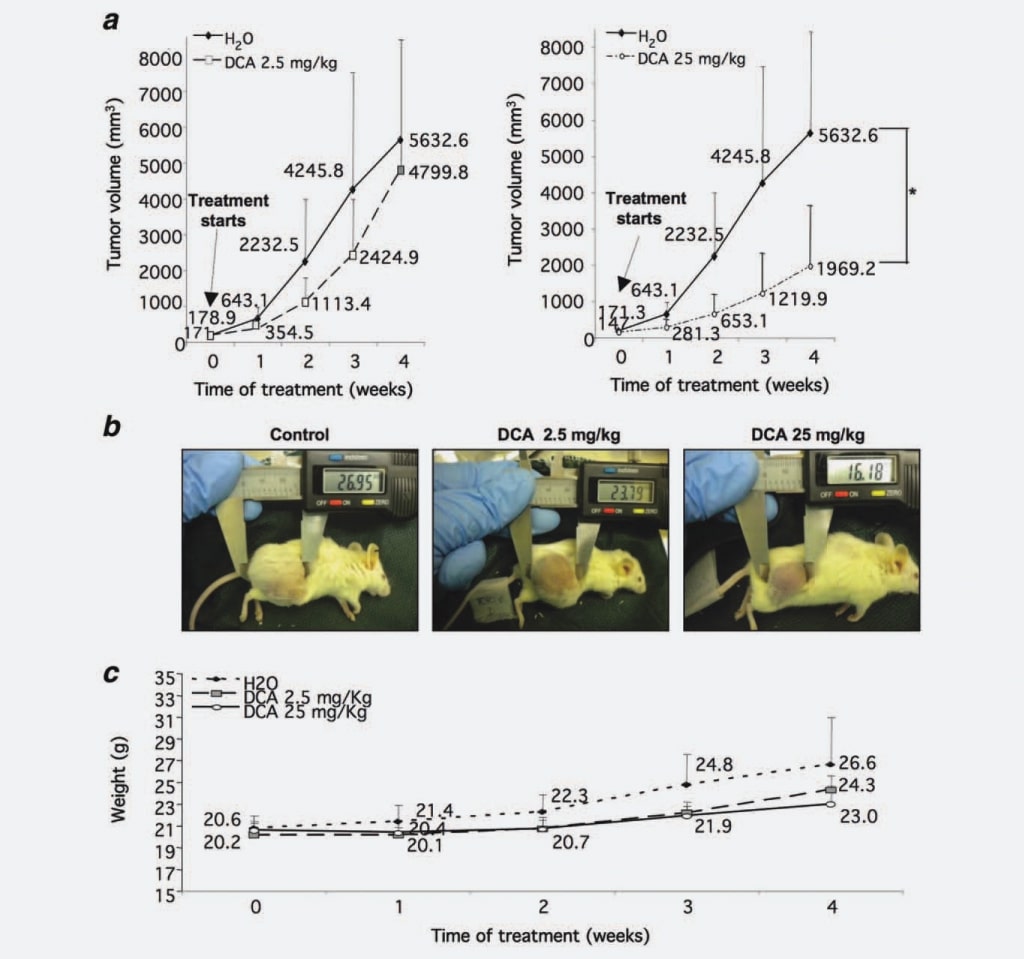
Tout d’abord, pour vérifier si le DCA affecte la croissance des nodules du NB comme cela est observé dans les GBM, nous avons testé sa capacité à limiter la croissance de la masse tumorale du NB in vivo. Nous avons injecté à 37 souris NOD-SCID des cellules SKNBE2, une lignée cellulaire NB humaine amplifiée par N-myc et caractérisée par un fort potentielmalin17, afin de générer des nodules tumoraux à traiter avec le DCA. Lorsque les nodules tumoraux ont atteint un diamètre seuil de 5 mm, cinq souris ont été sacrifiées avant le traitement, cinq souris ont été traitées avec 2,5 mg/kg/dose de DCA, 14 souris ont été traitées avec 25 mg/kg/dose de DCA, tandis que les 13 animaux restants ont été traités avec de l’eau comme contrôle. Nous avons choisi l’administration intragastrique de DCA, car certaines données de la littérature ont rapporté que l’administration parentérale n’était pas efficace pour diminuer la croissance tumorale.16 Chaque semaine, les masses tumorales ont été mesurées, ce qui a montré que les volumes tumoraux des souris traitées avec 2,5 mg/ kg/dose de DCA étaient réduits de 30 % par rapport au groupe témoin. Cette inhibition de la croissance est devenue statistiquement significative (p ¼ 0,0008) lorsque les souris ont été traitées avec 25 mg/kg/dose de DCA (55 % de réduction par rapport au groupe témoin) (figures 1a et 1b). Pour évaluer la tolérance du traitement au DCA, nous avons évalué chaque semaine le poids des souris. Les animaux traités au DCA (2,5 et 25 mg/kg/dose) n’ont pas présenté de différences statistiquement significatives par rapport au groupe témoin, bien que le poids du groupe témoin non traité ait légèrement augmenté, très probablement en raison des grandes masses tumorales (Fig. 1c). De même, la consommation d’eau n’a pas eu d’impact sur les animaux traités (données non présentées).
Dans l’ensemble, les données ci-dessus mettent en évidence un effet puissant et dose-dépendant du DCA sur la croissance des tumeurs du NB.
LeDCA réduit la prolifération des cellules cancéreuses
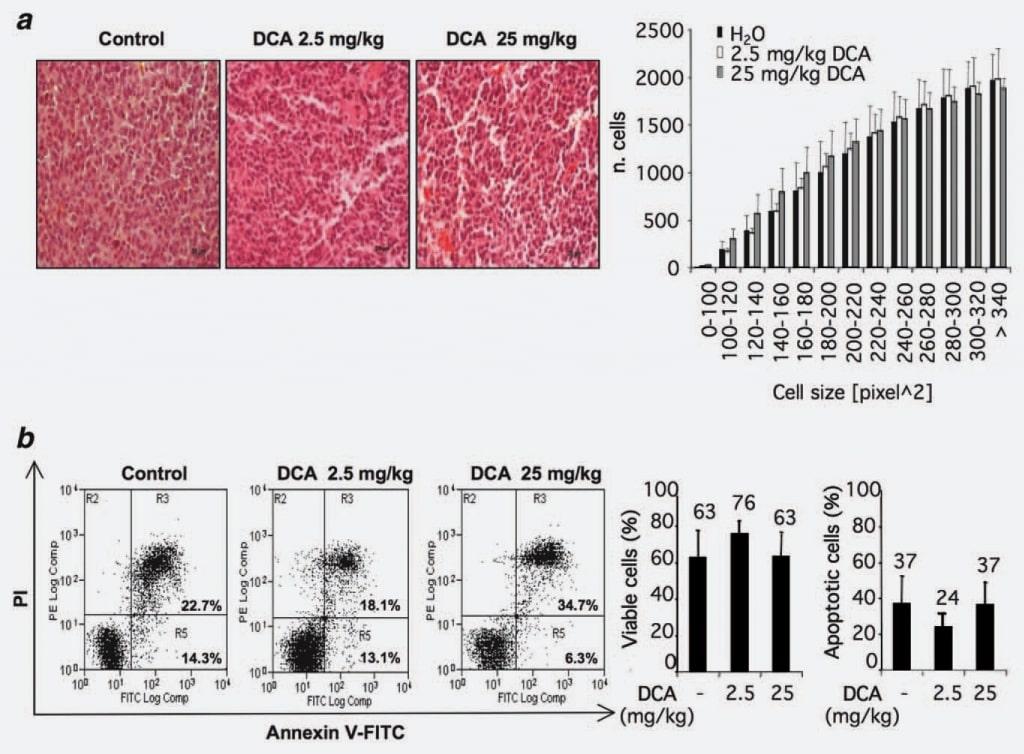
Pour identifier les mécanismes d’action possibles du DCA, nous avons évalué si la réduction observée de la croissance tumorale pouvait être attribuée à une perte de volume cellulaire. Dans ce but, nous avons analysé la morphologie et/ou la taille des cellules dans les nodules tumoraux traités ou non par le DCA. Douze sections du groupe témoin et 12 sections des groupes traités au DCA (2,5 et 25 mg/kg) ont été considérées. Aucune différence statistiquement significative n’a été observée dans les groupes traités au DCA par rapport au groupe témoin (figure 2a).
Étant donné que les rapports publiés étudiant les effets pro-apoptotiques du DCA sont contradictoires et compte tenu des observations récentes d’une possible spécificité de type cellulaire de l’effet pro-apoptotique duDCA15, nous avons étudié ce phénomène dans notre cadre expérimental par une analyse cytométrique en flux des cellules provenant de tumeurs traitées et/ou non traitées au DCA. Les résultats montrent que le pourcentage de cellules positives à l’annexine V est similaire dans les groupes considérés, ce qui indique que le traitement au DCA n’a pas provoqué d’augmentation du taux d’apoptose dans la masse des cellules des nodules tumoraux (Fig. 2b).
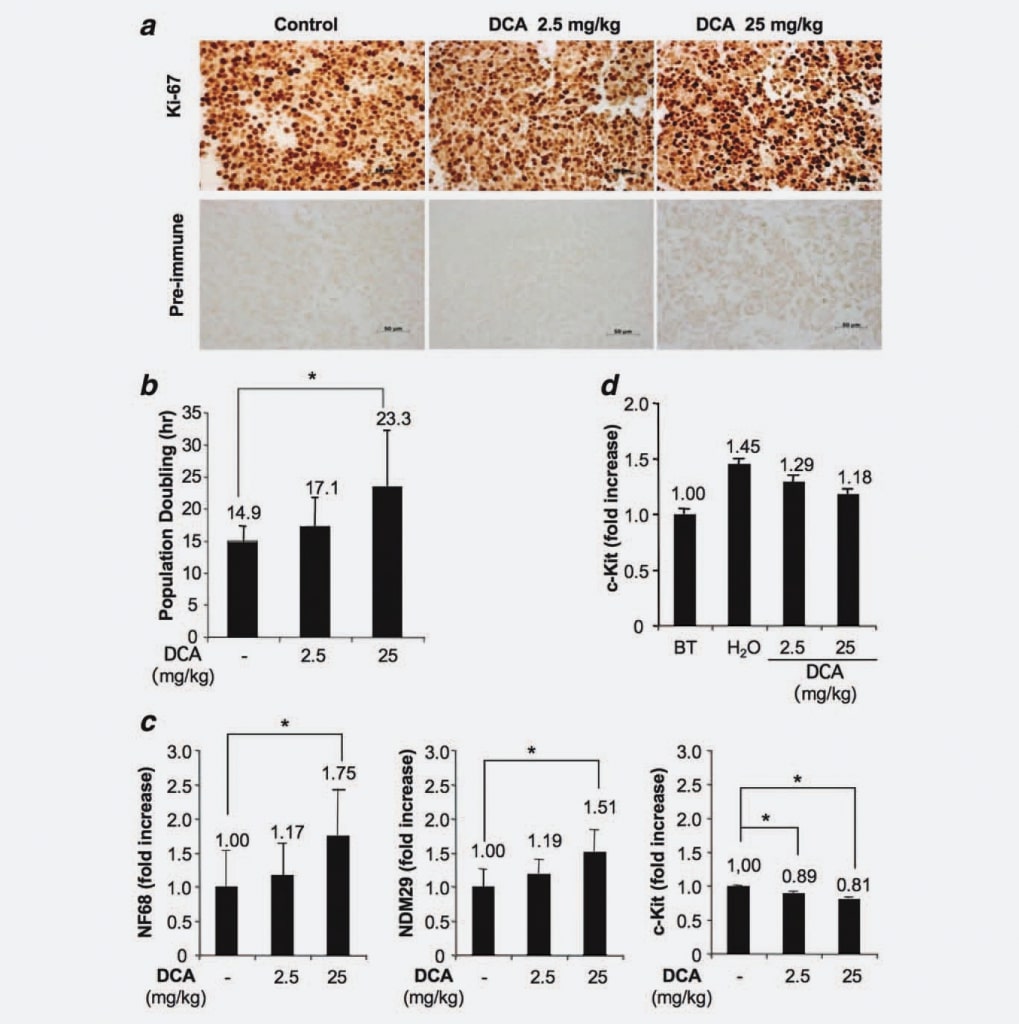
Puisque les tumeurs générées dans notre modèle expérimental proviennent de l’injection d’une lignée cellulaire homogène et considérant que nous avons démontré que le traitement au DCA n’avait pas d’impact ni sur une réduction du volume des cellules tumorales ni sur une augmentation de l’apoptose, nous avons émis l’hypothèse d’un possible effet du DCA sur la capacité proliférative des cellules. Pour tester cette hypothèse, nous avons analysé des cellules provenant de nodules traités et non traités par immunohistochimie à l’aide d’un anticorps monoclonal spécifique anti-Ki67. Les résultats n’ont montré aucune différence significative en termes de fraction de cellules en prolifération/cyclage entre les groupes. Ce résultat indique donc que le traitement au DCA ne réduit pas le nombre de cellules cyclées, ce qui est cohérent avec l’absence d’éradication complète de la tumeur après les traitements au DCA (Fig. 3a). Ensuite, nous avons utilisé le système xCELLigence (Roche, Allemagne) pour détecter une éventuelle augmentation dose-dépendante du doublement de la population induite par le traitement au DCA. Nous avons constaté que les cellules tumorales dérivées des souris traitées (25 mg/kg) doublent en 23,3 heures, les cellules des animaux traités avec 2,5 mg/kg doublent en 17,1 heures, tandis que les cellules dérivées des souris témoins présentent un temps de doublement de la population de 14,9 heures (Fig. 3b). Les différences étant statistiquement significatives (DCA 25 mg/kg par rapport au groupe témoin ; p ¼ 0,0476), ces résultats démontrent que le traitement au DCA induit un retard du cycle cellulaire proportionnel à la dose. Ensuite, nous avons cherché à savoir si le retard du cycle cellulaire des cellules issues des tumeurs traitées était associé à une augmentation de la différenciation/engagement. À cette fin, nous avons mesuré par RT-PCR en temps réel les marqueurs de différenciation des cellules du N.-B. [Neuroblastoma Differentiation Marker 29 (NDM29), Neurofilament 68 (NF68)] et c-Kit, une protéine spécifiquement exprimée par les cellules de type souche/de début de tumeur, dans les tissus tumoraux traités au DCA et non traités. Comme le montre la figure 3c, une augmentation générale de la synthèse des marqueurs de différenciation des cellules NB a été observée dans les cellules provenant de tumeurs traitées au DCA. En effet, le DCA utilisé à la plus forte concentration a induit une augmentation de l’expression de NF68 (75 %, p < 0,001), alors que la plus faible dose a induit une augmentation modeste (17 %, non statistiquement significative). De même, l’augmentation de l’expression de NDM29 était statistiquement significative dans les tumeurs traitées par 25 mg/kg de DCA (augmentation de 51 %, par rapport au groupe témoin ; p < 0,001), alors que seule une légère différence était observée entre les cellules provenant de tumeurs traitées par 2,5 mg/kg et les cellules provenant de tumeurs non traitées. Il est intéressant de noter que les échantillons traités au DCA étaient également caractérisés par une diminution de l’expression de c-Kit (réduction de 11 % en comparant les échantillons traités au DCA à 2,5 mg/kg par rapport aux échantillons témoins et de 19 % en comparant les échantillons traités au DCA à 25 mg/kg par rapport aux échantillons témoins, p < 0,001), ce qui indique que le DCA induit une diminution du potentiel d’initiation de tumeurs de type souche dans les cellules tumorales en prolifération. Une fois encore, ces résultats confirment un ralentissement de la vitesse de prolifération et, éventuellement, une diminution du potentiel malin des cellules traitées au DCA.
Ensuite, afin d’évaluer si le DCA peut affecter la fraction des cellules proliférantes indifférenciées, nous avons mesuré l’expression de c-Kit dans les tissus tumoraux de souris sacrifiées avant le traitement au DCA et dans les nodules des tumeurs traitées. Nous avons constaté que, bien que l’expression de c-Kit (et, vraisemblablement, le potentiel malin) augmente progressivement au cours de la croissance des nodules (voirH2Ovs. avant traitement), l’administration du DCA tend à limiter ce phénomène, confirmant dans les tissus des masses tumorales ce qui avait déjà été observé dans les cellules et fournissant une explication à l’absence d’éradication des tumeurs chez les souris traitées au DCA (Fig. 3d).
LeDCA agit spécifiquement sur les cellules malignes du NB et est inefficace sur les cellules différenciées et peu malignes du NB
Un nombre croissant de preuves expérimentales suggère que le pronostic du NB et sa réponse aux traitements anticancéreux dépendent du stade de différenciation des nodules du NB. En effet, une caractéristique particulière du NB est la remarquable hétérogénéité des cellules qui peuvent dériver de différentes lignées de crête neurale à des stades variables.12,17 Dans ce contexte, il est raisonnable d’émettre l’hypothèse d’une possible sensibilité variable au DCA de différents types de cellules. Pour vérifier cette hypothèse, nous avons utilisé un modèle in vitro de différenciation des cellules NB basé sur l’expression d’un ARNnc transcrit par la pol III. En effet, nous avons récemment isolé un nouvel ARN non codant (nc) transcrit par l’ARN polymérase (pol) III (à savoir NDM29) dont l’expression déclenche la différenciation de type neuronal des cellules NB (NB), réduisant fortement leur potentiel malin et l’expression de marqueurs de cellules initiatrices de tumeurs/de type souche. En intégrant des copies supplémentaires de l’unité transcriptionnelle NDM29 dans les cellules tumorales SKNBE2, nous avons généré une lignée cellulaire SKNBE2 surexprimant NDM29 (ci-après dénommée S1) et le simili négatif correspondant qui exprime NDM29 à son niveau endogène. Les cellules S1 présentent un phénotype partiellement différencié/neuronique, sont peu malignes et se caractérisent par un retard marqué du cycle cellulaire.18,19
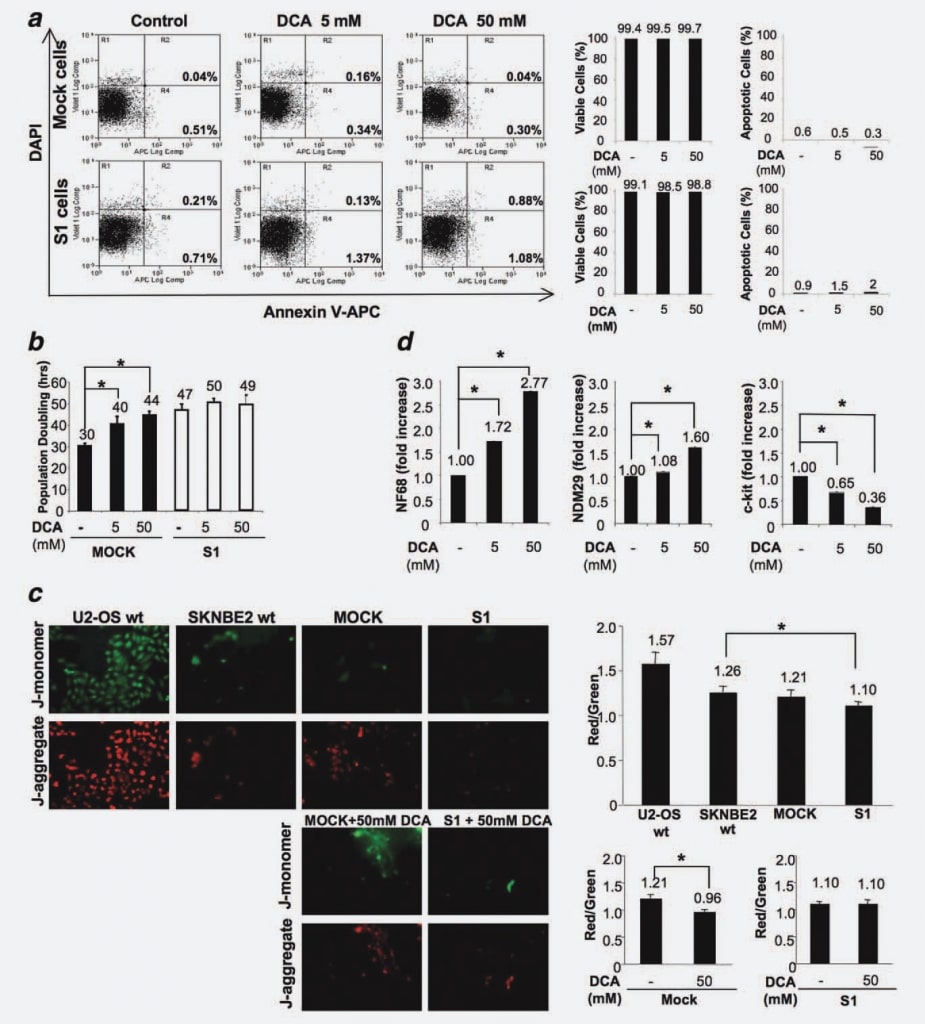
Pour exclure que le traitement au DCA puisse induire l’apoptose in vitro, comme cela a été démontré dans les expériences in vivo, nous avons mesuré le pourcentage de cellules apoptotiques dans les lignées cellulaires Mock et S1, traitées et/ou non traitées par le médicament pendant 48 heures. Aucune différence entre les lignées cellulaires (non traitées ou traitées avec différentes doses de DCA) n’a été observée, ce qui confirme que le DCA ne provoque pas d’apoptose dans les cellules NB (Fig. 4a).
Pour déterminer les effets sur la prolifération des cellules Mock et S1, nous avons traité les cellules avec 5 mM et 50 mM de DCA et mesuré le temps de doublement de la population cellulaire (PD) dans quatre expériences indépendantes. La concentration de 50 mM a été choisie comme dose maximale, car différentes études antérieures ont indiqué que le DCA semblait être relativement inactif dans différentes lignées cellulaires lorsqu’il était utilisé à des doses plus faibles.16,20 Comme le montre la figure 4b, le temps de doublement de la population est significativement augmenté par le traitement au DCA de manière dose-dépendante dans les cellules Mock. En effet, en l’absence de traitement, les cellules Mock se dupliquent en 30 h, alors que l’administration de 5 mM de DCA induit un retard du cycle cellulaire de 10 h et que la dose plus élevée de 50 mM de DCA entraîne une augmentation de 14 h (p < 0,001). Ces résultats démontrent que le DCA induit un retard du cycle cellulaire dans les cellules NB indifférenciées/en pleine prolifération. Au contraire, le DP des cellules S1 est resté à peu près inchangé, passant de 47 h pour les cellules non traitées à 50 h pour les cellules traitées par 5 mM de DCA et à 49 h pour les cellules traitées par 50 mM de DCA (Fig. 4b). Ce résultat démontre que la réponse au traitement par DCA est directement corrélée au stade de différenciation/prolifération des cellules NB, puisque les cellules différenciées à très faible potentiel malin répondent peu au traitement, alors que leurs homologues en pleine prolifération/très malignes sont fortement affectées par le traitement par DCA.
Pour mieux comprendre ce phénomène et compte tenu du fait que les effets du DCA sont liés à l’inhibition de la PDK, à l’activation de la phosphorylation oxydative mitochondriale et à la réduction spécifique de l’hyperpolarisationmitochondriale6, nous avons testé les cellules Mock et S1 afin de déterminer si l’hyperpolarisation mitochondriale pouvait être à l’origine de la sensibilité différentielle de ces cellules au DCA. À cette fin, nous avons traité les cellules avec du JC-1 (5,5′ ,6,6′ -tétrachloro-1,1′ ,3,3′ -tétraéthylbenzimidazolocarbocyanine iodure), un colorant fluorescent dont l’absorption est proportionnelle au potentiel mitochondrial. Le JC-1 peut entrer sélectivement dans les mitochondries et change réversiblement de couleur du vert au rouge lorsque le potentiel mitochondrial augmente, de sorte qu’une diminution du rapport de fluorescence rouge/vert met en évidence une réactivation mitochondriale. Nous avons comparé la polarisation de la membrane mitochondriale des cellules d’ostéosarcome U2-OS (cellules témoins hyperpolarisées21), des cellules SKNBE2 wt, Mock et S1. Nous avons constaté que les cellules Mock présentent un degré de polarisation similaire si on les compare aux cellules SKNBE2 wt, tandis que les cellules S1 sont caractérisées par une diminution du degré de polarisation mitochondriale (p ¼ 0,0184) (figure 4c). Les résultats, rapportés à trois répétitions, ont montré que, dans les cellules Mock, une réactivation mitochondriale induite par le DCA a lieu (25 % de réduction du rapport de fluorescence rouge/vert ; p ¼ 0,0077), alors que dans les cellules S1 l’activité mitochondriale n’est pas modifiée par la présence de DCA (Fig. 4c). Ces résultats démontrent donc que la sensibilité des cellules malignes/complètement cyclées est associée à un statut mitochondrial sensible à l’action du DCA alors que les cellules engagées/différenciées, peu cyclées sont réfractaires à l’action de ce médicament.
Ensuite, pour détecter les effets possibles du DCA sur le potentiel de différenciation, nous avons mesuré par RT-PCR en temps réel le niveau d’expression de NDM29, NF68 et c-Kit dans les cellules Mock traitées et non traitées au DCA. Nous avons effectué cette analyse spécifiquement sur les cellules Mock puisque les cellules S1 étaient réfractaires à la diminution de la prolifération induite par le DCA. Comme le montre la figure 4d, le traitement au DCA a entraîné une augmentation de l’expression du marqueur de différenciation NF68 (cellules Mock traitées avec 5 mM ou 50 mM de DCA par rapport aux cellules non traitées, p < 0,0001). De même, l’expression de NDM29 était significativement augmentée dans les cellules Mock traitées par rapport au contrôle (p < 0,001). Au contraire, l’expression de c-Kit a été diminuée de manière dosedépendante dans les cellules traitées (p < 0,0001). Dans l’ensemble, ces résultats suggèrent que, dans les cellules du N.-B., l’effet antitumoral du DCA est principalement dû à une action antiproliférative/prodifférentielle et ne se traduit pas par une augmentation du taux d’apoptose.
Discussion
Le DCA est une petite molécule qui est utilisée depuis plus de 30 ans chez l’homme pour traiter l’acidose lactique congénitale et acquise.22 En effet, il est bien décrit que le traitement au DCA réduit les niveaux de lactate dans la circulation en déplaçant le métabolisme cellulaire de la glycolyse vers l’oxydation du glucose, sans aucun effet délétère sur les cellules normales6 Pour sa capacité à favoriser le métabolisme du glucose par la glycolyse aérobie, le DCA est utilisé avec succès dans les essais cliniques pour les maladies cardiaques, y compris l’insuffisance cardiaque congestive et les cardiopathies ischémiques, puisque le dysfonctionnement postischémique des cœurs hypertrophiés est associé à de faibles taux d’oxydation du glucose et à des taux glycolytiques élevés.23
Une propriété très attrayante du DCA réside dans sa tolérance et son innocuité. En effet, plus de 40 essais cliniques du DCA indiquent que l’effet indésirable le plus important de l’administration à long terme de DCA est une neuropathie périphérique réversible.24,25
Ces dernières années, des preuves substantielles provenant de modèles in vitro et in vivo montrent que le DCA pourrait également être utile pour traiter certains types de cancer chez l’homme.6 En effet, le DCA est capable d’inverser l’effet Warburg en inhibant la PDK, en restaurant le potentiel de la membrane mitochondriale et en augmentant la production de ROS. Pour cette raison, le DCA a été célébré comme la balle magique contre lecancer1, même s’il n’est actuellement pas encore approuvé pour le traitement ducancer26 Bien que le neuroblastome ait été considéré jusqu’à présent comme un type de cancer pour lequel le traitement au DCA devrait être très probablement inefficace, en raison de l’absence présumée d’hyperpolarisation de la membrane mitochondriale dans les cellules composant les nodules de NB,27 il a été décrit que les cellules NB en prolifération sont soutenues par un phénotype glycolytique,11 et que même dans des conditions aérobies, les cellules NB convertissent des quantités remarquables de glucose en lactate au lieu d’utiliser l’oxydation du glucose.28 Sur la base de cette constatation, l’objectif de cette étude était de déterminer si le DCA pouvait avoir un effet bénéfique sur ce type de tumeur. Nos résultats mettent en évidence un effet positif imprévu du DCA sur la croissance des tumeurs du N.-B., tant dans les modèles in vivo qu’in vitro. En effet, le DCA a été efficace sur les tumeurs NB établies, sans provoquer d’effet secondaire évident chez les animaux traités.
En détail, l’analyse effectuée sur les cellules dérivées des tumeurs traitées a démontré que le DCA entraîne un retard de prolifération sans aucun signe d’augmentation de l’apoptose ou de la mort cellulaire. Ceci est cohérent avec la diminution observée des volumes des masses tumorales non accompagnée d’une éradication complète du cancer. Les mécanismes d’action sous-jacents du DCA sont encore controversés. Dans ce contexte, nos résultats contrastent avec ceux d’études antérieures dans lesquelles des cellules cancéreuses de l’endomètre, de la prostate, du colorectal et du poumon ont été traitées avec du DCA.6,9,20 Dans ces cas, on a signalé une augmentation de l’apoptose sans effet sur la
du cycle cellulaire9,21 ou une augmentation de l’apoptose accompagnée d’une diminution de la prolifération8. D’autre part, d’autres rapports indiquent que le DCA inhibe la prolifération des cellules cancéreuses, mais n’augmente pas l’apoptose ou la mort cellulaire.7 Ce dernier mécanisme serait soutenu par une analyse protéomique dans laquelle le traitement au DCA induit des changements dans les marqueurs de prolifération cellulaire plutôt que dans la quantité de protéines liées à l’apoptose.16 Ce double comportement du DCA pourrait dépendre du type de cellule, probablement en raison des différences d’expression des isoenzymes PDK dans les cellules cancéreuses examinées. Des études visant à corréler l’expression de la PDK avec la sensibilité au DCA sont en cours.
De manière intéressante, nous montrons ici qu’une augmentation de l’expression de certains marqueurs de différenciation, tels que NF68 et NDM29, accompagnée d’une réduction de l’expression de c-kit, un marqueur de cellules souches/de cellules initiatrices de tumeurs, est corrélée à la diminution observée de la prolifération des cellules cancéreuses. Ainsi, l’induction par le DCA d’un retard du cycle cellulaire concomitant à une augmentation de la différenciation cellulaire représente un nouveau mécanisme d’action, non encore décrit, de ce médicament qui lie les effets du DCA au stade de différenciation des cellules cancéreuses.
Il a été documenté que les tumeurs humaines du NB sont caractérisées par des types de cellules distincts qui ont des propriétés morphologiques, biochimiques et tumorigènes distinctes et des stades de différenciation variables.12 Pour approfondir la corrélation possible de la sensibilité au DCA avec le niveau de différenciation cellulaire, nous avons utilisé un modèle in vitro de différenciation des cellules du NB basé sur la surexpression d’un ARNnc transcrit par la pol III (NDM29). Nous avons généré une lignée cellulaire SKNBE2 surexprimant NDM29 (désignée ici par S1) et le contrôle négatif Mock correspondant.19 Ces lignées cellulaires ont été traitées avec 5 mM et 50 mM de DCA. Ces lignées cellulaires ont été traitées avec 5 mM et 50 mM de DCA. La concentration de 50 mM a été choisie comme dose maximale car différentes études antérieures ont signalé que le DCA est relativement inactif dans différentes lignées cellulaires lorsqu’il est utilisé à des doses plus faibles,16,20 bien que cette concentration ne soit pas atteinte in vivo. Néanmoins, dans notre contexte expérimental, les effets du DCA sont évidents à la dose la plus faible (5 mM), ce qui constitue la concentration appropriée à utiliser in vivo.
Comme prévu, nous avons constaté que dans les cellules Mock et S1, le traitement au DCA n’a pas produit de signes d’apoptose, ce qui confirme nos résultats in vivo. Il est intéressant de noter que l’effet du DCA est limité aux cellules indifférenciées, très malignes et à cycle complet (cellules Mock), alors qu’il n’affecte pas le taux de prolifération des cellules plus différenciées et peu malignes (cellules S1). Cet effet pourrait être attribué aux différents degrés de polarisation mitochondriale de ces types de cellules. En effet, nous avons démontré que les cellules Mock présentent une polarisation mitochondriale plus élevée que leur homologue S1 plus différenciée.
Par conséquent, nous avons démontré ici que les différentes cellules qui composent les masses tumorales NB sont sensibles de manière différentielle au DCA qui agit sélectivement sur les cellules très malignes et en pleine prolifération, alors qu’il exerce un effet très faible sur les cellules peu malignes et plus différenciées. Un effet différentiel similaire du DCA a également été rapporté dans une étude sur le glioblastome multiforme (GBM)2 où les auteurs ont montré que les cellules souches cancéreuses, comparées à leurs homologues plus différenciées qui composent le GBM, présentent le même remodelage métabolique et mitochondrial mais dans une plus grande mesure puisqu’elles ont les mitochondries les plus hyperpolarisées.2
En conclusion, les expériences décrites ici soutiennent l’idée que le DCA est un médicament très sélectif qui peut aider les traitements anticancéreux du Nouveau-Brunswick sans avoir d’effets secondaires considérables sur les cellules saines. Par conséquent, cette étude préliminaire élargit l’utilisation possible du DCA pour le traitement du cancer du N.-B., soutenant la nécessité d’une étude détaillée de ses propriétés anticancéreuses contre ce type de tumeur.
Remerciements
R.C. a été soutenu par le ministère italien de l’Université et de la RechercheMIUR (programme international FIRB 2007). A.P. a été soutenu par le ministère italien de l’Université et de la Recherche-MIUR (2007 PRIN Program prot. 2007945BZN), par l’Associazione Italiana Ricerca sul Cancro (2009 AIRC Program no. IG9378) et par l’Associazione Italiana per la Lotta al Neuroblastoma (Gênes, Italie).
RÉFÉRENCES
1 Heshe D, Hoogestraat S, Brauckmann C, Karst U, Boos J, Lanvers-Kaminsky C. La thérapie métabolique ciblée par le dichloroacétate déjoue la cytotoxicité des médicaments anticancéreux standard. Cancer Chemother Pharmacol 2011;67:647-55
2 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med ; 2010;2:31ra4.
3 Stacpoole PW, Henderson GN, Yan Z, James MO. Pharmacologie clinique et toxicologie du dichloroacétate. Environ Health Perspect 1998;106 Suppl 4:989-94.
4 Stacpoole PW. La pharmacologie du dichloroacétate. Metabolism 1989;38 : 1124-44.
5 Michelakis ED, Webster L, Mackey JR. Le dichloroacétate (DCA) comme thérapie potentielle de ciblage métabolique pour le cancer. Br J Cancer 2008;99:989-94.
6 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-Kþ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11:37-51.
7 Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC. L’inversion du phénotype glycolytique par le dichloroacétate inhibe la croissance des cellules métastatiques du cancer du sein in vitro et in vivo. Breast Cancer Res Treat 2010;120:253-60.
8 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ. Le dichloroacétate (DCA) sensibilise in vitro les cellules cancéreuses de la prostate de type sauvage et sur-exprimant Bcl-2 aux radiations. Prostate 2008;68:1223-31.
9 Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Le dichloroacétate induit l’apoptose dans les cellules cancéreuses de l’endomètre. Gynecol Oncol 2008;109:394-402.
10 Vander Heiden MG. Cibler le métabolisme cellulaire chez les patients atteints de cancer. Sci Transl Med 2009;2:31ed1.
11 Almeida A, Bolanos JP, Moncada S. E3 ubiquitin ligase APC/C-Cdh1 accounts for the Warburg effect by linking glycolysis to cell proliferation. Proc Natl Acad Sci USA ; 107:738-41.
12 Walton JD, Kattan DR, Thomas SK, Spengler BA, Guo HF, Biedler JL, Cheung NK, Ross RA. Caractéristiques des cellules souches des lignées cellulaires de neuroblastomes humains et dans les tumeurs. Neoplasia 2004;6:838-45.
13 Vistejnova L, Dvorakova J, Hasova M, Muthny T, Velebny V, Soucek K, Kubala L. The comparison of impedance-based method of cell proliferation monitoring with commonly used metabolic-based techniques. Neuro Endocrinol Lett 2009;30 Suppl 1:121-7.
14 Schmidt MM, Rohwedder A, Dringen R. Effets des acétates chlorés sur le métabolisme du glutathion et sur la glycolyse des astrocytes en culture. Neurotox Res 2011 ; 19:628-37.
15 Papandreou I, Goliasova T, Denko NC. Les médicaments anticancéreux qui ciblent le métabolisme : le dichloroacétate est-il le nouveau paradigme ? Int J Cancer 2011;128:1001-8.
16 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL. Le dichloroacétate de sodium cible sélectivement les cellules présentant des défauts dans l’ETC mitochondrial. Int J Cancer 2010;127 : 2510-9.
17 George RE, Variend S, Cullinane C, Cotterill SJ, McGuckin AG, Ellershaw C, Lunec J, Pearson AD. Relation entre les caractéristiques histopathologiques. MYCN amplification, and prognosis : a UKCCSG study. Groupe d’étude britannique sur le cancer chez l’enfant. Med Pediatr Oncol 2001;36 : 169-76.
18 Pagano A, Castelnuovo M, Tortelli F, Ferrari R, Dieci G, Cancedda R. New small nuclear RNA gene-like transcriptional units as sources of regulatory transcripts. PLoS Genet 2007;3:e1.
19 Castelnuovo M, Massone S, Tasso R, Fiorino G, Gatti M, Robello M, Gatta E, Berger A, Strub K, Florio T, Dieci G, Cancedda R, et al. An Alu-like RNA promotes cell differentiation and reduces malignancy of human neuroblastoma cells. FASEB J 2010;24:4033-46.
20 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG. Le dichloroacétate induit l’apoptose et l’arrêt du cycle cellulaire dans les cellules cancéreuses colorectales. Br J Cancer 2010;102:1746-52.
21 Liu TJ, Lin SY, Chau YP. L’inhibition de l’activation de la poly(ADP-ribose) polymérase atténue la mort cellulaire nécrotique induite par la bêta-lapachone dans les cellules d’ostéosarcome humain. Toxicol Appl Pharmacol 2002;182:116-25.
22 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, et al. Controlled clinical trial of dichloroacetate for treatment of congenital lactic acidosis in children. Pediatrics 2006;117 : 1519-31.
23 Wargovich TJ, MacDonald RG, Hill JA, Feldman RL, Stacpoole PW, Pepine CJ. Effets métaboliques et hémodynamiques myocardiques du dichloroacétate dans la maladie coronarienne. Am J Cardiol 1988;61 : 65-70.
24 Kaufmann P, Engelstad K, Wei Y, Jhung S, Sano MC, Shungu DC, Millar WS, Hong X, Gooch CL, Mao X, Pascual JM, Hirano M, et al. Dichloroacetate causes toxic neuropathy in MELAS : a randomized, controlled clinical trial. Neurology 2006;66 : 324-30.
25 Stacpoole PW, Henderson GN, Yan Z, Cornett R, James MO. Pharmacocinétique, métabolisme et toxicologie du dichloroacétate. Drug Metab Rev 1998;30 : 499-539.
26 Pearson H. Cancer patients opt for un médicament non approuvé. Nature 2007;446 : 474-5.
27 Chen LB. Potentiel de la membrane mitochondriale dans les cellules vivantes. Annu Rev Cell Biol 1988;4:155-81.
28 Deubzer B, Mayer F, Kuci Z, Niewisch M, Merkel G, Handgretinger R, Bruchelt G. H(2)O(2)-mediated cytotoxicity of pharmacologic ascorbate concentrations to neuroblastoma cells : potential role of lactate and ferritin. Cell Physiol Biochem 2010;25:767-74.