Balkrishna Chaube1, Parmanand Malvi1, Shivendra Vikram Singh1, Naoshad Mohammad1, Avtar Singh Meena1,2 y Manoj Kumar Bhat1
1 Centro Nacional de Ciencia Celular, Campus de la Universidad Savitribai Phule Pune, Ganeshkhind, Pune, India
2 Dirección actual: Departamento de Fisiología, Centro de Ciencias de la Salud de la Universidad de Tennessee, Memphis, EE.UU
Correspondencia: Manoj Kumar Bhat, correo electrónico: [email protected]
Recibido: 16 de junio de 2015
Aceptado: 23 de septiembre de 2015
Publicado: 15 de octubre de 2015
Resumen
El melanoma es una neoplasia cutánea en gran medida incurable debido a la heterogeneidad molecular y metabólica subyacente confundida por el desarrollo de resistencia. Las células cancerosas tienen flexibilidad metabólica para elegir entre la fosforilación oxidativa (OXPHOS) o la glucólisis para la generación de ATP en función de la disponibilidad de nutrientes en el microambiente tumoral. En este estudio, investigamos la implicación del complejo respiratorio I y la lactato deshidrogenasa (LDH) en la progresión del melanoma. Demostramos que la inhibición del complejo I mediante metformina promueve el crecimiento del melanoma en ratones a través de la elevación de los niveles de lactato y VEGF. Por el contrario, conduce a la detención del crecimiento in vitro debido a una mayor acidificación extracelular como resultado del aumento de la glucólisis. La inhibición de la LDH o de la generación de lactato provoca una disminución de la glucólisis con la consiguiente detención del crecimiento tanto in vitro como in vivo. El bloqueo de la generación de lactato en células de melanoma tratadas con metformina provoca una disminución de la proliferación celular y de la progresión tumoral en ratones. Curiosamente, la inhibición de la LDH o del complejo I por separado no induce la apoptosis, mientras que la inhibición conjunta de ambos provoca el agotamiento de la reserva celular de ATP, lo que da lugar a una apoptosis inducida por catástrofe metabólica. En general, nuestro estudio sugiere que la LDH y el complejo I desempeñan funciones distintas en la regulación de la glucólisis y la proliferación celular. La inhibición de ambos aumenta la letalidad sintética en el melanoma.
Palabras clave: melanoma; complejo I; LDH; catástrofe metabólica; letalidad sintética
INTRODUCCIÓN
El melanoma maligno es una de las formas más agresivas de cáncer de piel, con un elevado potencial metastásico y resistencia a muchos agentes citotóxicos [1, 2]. A pesar de las numerosas investigaciones realizadas y de los éxitos parciales obtenidos con el uso de los fármacos disponibles en la actualidad, no existe ningún tratamiento eficaz contra el melanoma maligno [1-3]. Los casos de melanoma aumentan cada año y representan alrededor del 75% de las muertes relacionadas con el cáncer de piel en todo el mundo [2,
]. La escasa respuesta a las opciones terapéuticas actualmente disponibles y el desarrollo de resistencia a la terapia justifican la exploración de nuevas estrategias para tratar el melanoma.
El aumento de la glucólisis aeróbica es un rasgo característico de muchos tipos de cáncer [3-5]. Se ha descrito que las células de melanoma, debido a la mutación BRAF, dependen principalmente de la glucólisis para la generación de ATP y presentan una fosforilación oxidativa disfuncional [6, 7]. Las células cancerosas obtienen ATP, intermediarios biosintéticos y equivalentes reductores mediante la participación inusual en vías bioquímicas como la glucólisis, la glutaminólisis y la vía de las pentosas fosfato [5]. Las células normales (no cancerosas) obtienen ATP principalmente a través de la OXPHOS mitocondrial, mientras que las células cancerosas dependen principalmente de la glucólisis aeróbica para generar ATP e intermediarios glucolíticos que facilitan el crecimiento rápido [4,
5]. El aumento de la generación de lactato se ha correlacionado con la agresividad del cáncer. Numerosos estudios han identificado la lactato deshidrogenasa (LDH), que cataliza la conversión de piruvato en lactato, como el marcador más consistente de los cánceres agresivos y de crecimiento rápido [8-11]. La LDH desempeña un papel importante en la regulación de la glucólisis, el mantenimiento del estado redox celular, la fisiología mitocondrial y el mantenimiento de los tumores [12]. La alteración del metabolismo de las células cancerosas puede estar asociada a la disfunción mitocondrial, que implica la inhibición de la OXPHOS, el aumento de las especies reactivas del oxígeno (ROS) y la promoción de un crecimiento incontrolado, que a su vez favorece el desarrollo de un fenotipo metastásico [13,14]. El complejo respiratorio I es la enzima más grande y compleja que cataliza la oxidación del NADH en la cadena de transporte de electrones [15]. El complejo I desempeña un papel importante en la generación de ATP en las células normales y es uno de los principales lugares de generación de ROS. La mayoría de los informes sugieren que la actividad del complejo I está suprimida en las células cancerosas y que su inhibición favorece la proliferación y la metástasis [16-18]. A la inversa, también se ha informado de que las células de melanoma presentan un aumento de la función OXPHOS que provoca resistencia a los fármacos [19-21]. Debido a la flexibilidad metabólica en la elección de varias vías metabólicas, en particular el metabolismo de la glutamina [22], las células de melanoma pueden escapar de la perturbación de una molécula en una vía metabólica [23]. Por lo tanto, es necesario atacar más de una molécula de diferentes vías metabólicas para obtener un resultado eficaz y curativo.
Se sabe que las biguanidas como la metformina y la fenformina inhiben el complejo respiratorio I. La metformina, una terapia de primera línea para pacientes con diabetes mellitus tipo 2 (DMT2), es un miembro de las biguanidas y recientemente se ha descubierto que tiene actividad antitumorigénica [24, 25]. A nivel fisiológico, la metformina ejerce su actividad biológica disminuyendo la glucogénesis hepática, aumentando la sensibilidad a la insulina, elevando la captación periférica de glucosa y reduciendo la absorción de glucosa del tracto gastrointestinal [26]. A nivel celular, la metformina actúa principalmente inhibiendo el complejo mitocondrial I, lo que impide la fosforilación oxidativa y provoca una disminución del nivel de ATP que conduce a la activación de la AMPK [27-30]. Además, se ha demostrado que la metformina disminuye la respiración mitocondrial acoplada a la generación de ATP, provocando así un aumento de la glucólisis [31, 32]. La acción anticancerígena de la metformina se ha demostrado en varios tipos de cáncer con mecanismos distintos [33, 34]. Sin embargo, el papel de la metformina en el melanoma no está muy claro. A diferencia de otros tipos de cáncer, se ha demostrado que la metformina tiene actividad tanto promotora como antitumoral en el melanoma [35-37]. Además, se ha demostrado que la metformina, en combinación con inhibidores de BRAF, suprime el crecimiento del melanoma [38].
En el presente estudio, con la especulación de que la interrupción de las rutas de generación de ATP mediante la inhibición conjunta del complejo I y la LDH podría ser sintéticamente letal para las células de melanoma, utilizamos metformina o fenformina y oxamato/dicloro acetato (DCA) para inhibir estas dos enzimas utilizando modelos celulares y animales. Demostramos que la inhibición de la actividad del complejo I y de la LDH tienen efectos distintos sobre el crecimiento y la proliferación celular. Curiosamente, la inhibición del complejo I por la metformina promueve aún más la glucólisis aeróbica, lo que resulta en un mayor crecimiento tumoral en ratones. La supresión de la generación de lactato inducida por la metformina mediante oxamato o DCA provoca citostasis y/o apoptosis en células de melanoma.
RESULTADOS
Lametformina exhibe distintas acciones in vitro e invivosobre el crecimiento del melanoma
La metformina suprime el crecimiento tumoral mediante la inhibición del complejo I, que está influenciado por la glucosa [30]. Además, se sabe que la glucosa altera la actividad de las enzimas respiratorias [39]. Por lo tanto, para explorar las consecuencias de la inhibición del complejo I y la influencia de la glucosa en la acción de la metformina sobre la progresión del melanoma, monitorizamos la progresión del isoinjerto/xenoinjerto en ratones hiperglucémicos inducidos por estreptozotocina (STZ). Observamos que la metformina promovía la progresión del isoinjerto derivado de B16F10 en ratones hiperglucémicos en comparación con el control no tratado (Figura 1A, 1B y 1C). Asimismo, la metformina influyó positivamente en la progresión del tumor en ratones C57BL/6J normoglucémicos (Figura 1D, 1E y 1F). Del mismo modo, la administración oral de metformina favoreció el crecimiento del xenoinjerto A375 en ratones NOD/SCID hiperglucémicos y normoglucémicos en comparación con el control no tratado (figuras 1G, 1H y 1I).
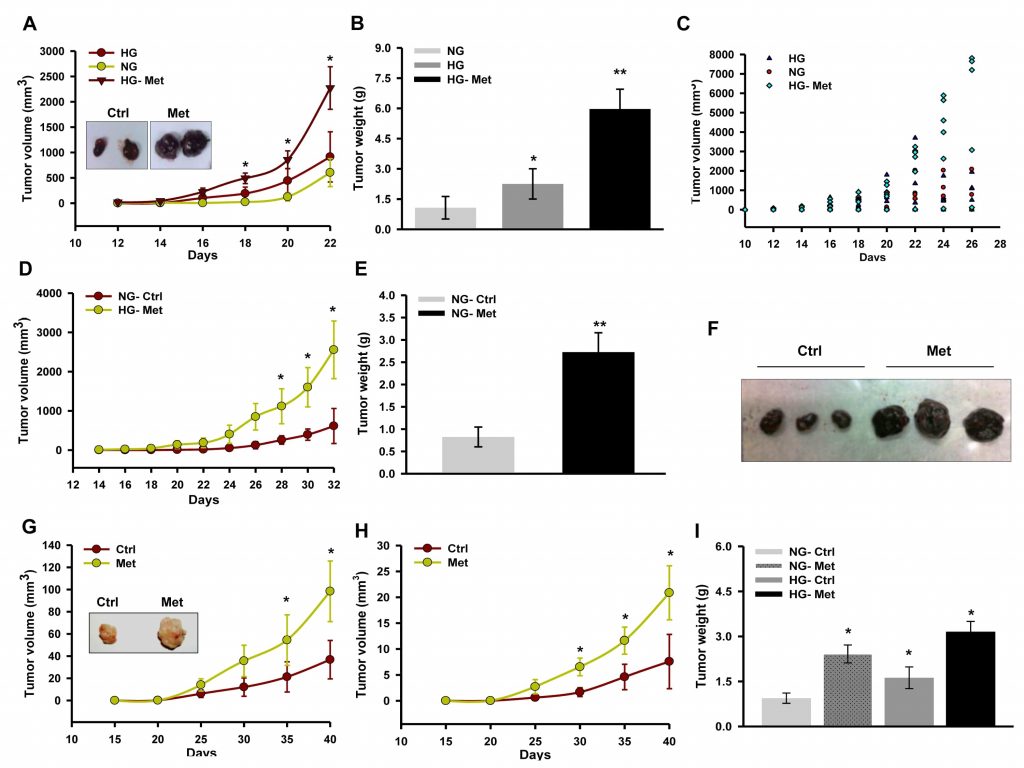
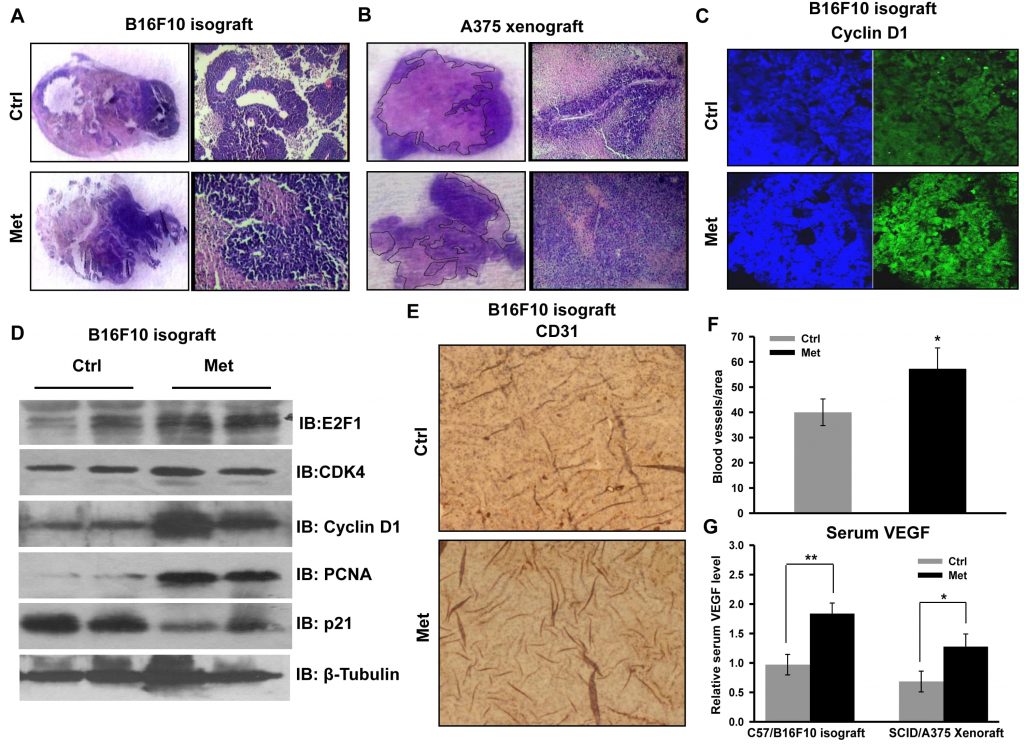
Para comprobar los eventos celulares y moleculares asociados a una mayor progresión tumoral, se examinaron secciones tumorales para su análisis histopatológico. En las secciones de ambos tipos de tumor (isoinjerto derivado de B16F10 y xenoinjerto derivado de A375) procedentes de ratones administrados con metformina se observó una elevada densidad celular y una reducción de la necrosis (Figura 2A y 2B). Observamos que el aumento de la proliferación con metformina y la progresión del xenoinjerto derivado de A375 eran fenotípicamente distintos en comparación con el tumor de control. Esto sugiere un avance de grado del tumor primario, evidente por la morfología alargada de los núcleos en comparación con la morfología redondeada en las secciones de los tumores de control (información no publicada). Se observó que la tinción inmunohistoquímica de la proteína reguladora del ciclo celular ciclina D1 (Figura 2C) era mayor en la sección tumoral de los ratones administrados con metformina. Además, validamos el mayor crecimiento tumoral comprobando el estado de las proteínas reguladoras del ciclo celular mediante inmunotransferencia de lisados tumorales. Observamos que los niveles de las moléculas ciclina D1, CDK4, E2F1 y PCNA aumentaban significativamente en los lisados tumorales de los ratones administrados con metformina en comparación con el control, mientras que el nivel de p21 disminuía (Figura 2D). Estos resultados indican que la metformina, independientemente del estado glucémico de los ratones, promueve el crecimiento del melanoma mediante la modulación de las proteínas reguladoras del ciclo celular. Además, el análisis inmunohistoquímico de las secciones tumorales reforzó esta observación, porque el tratamiento con metformina aumentó los niveles proteicos de CD31, un marcador endotelial (Figura 2E y 2F), y aumentó el nivel sérico de VEGF (Figura 2G), lo que sugiere que la metformina promueve la angiogénesis en los tumores de melanoma.
A continuación, comprobamos el efecto de la metformina sobre el crecimiento y la proliferación de células de melanoma in vitro. En contraste con nuestros hallazgos in vivo, el tratamiento con metformina produjo una supresión del crecimiento de las células de melanoma in vit ro (Figura suplementaria S1A, S1B, S1C y S1D). Posteriormente, investigamos el impacto de la inhibición del complejo I mediante metformina y fenformina en el crecimiento de las células de melanoma, ya que ambas inhibían la actividad del complejo I (Figura suplementaria S2). Descubrimos que la metformina y la fenformina provocaban la detención del crecimiento en las células de melanoma cultivadas en presencia de glucosa alta. Sin embargo, en presencia de glucosa baja, el tratamiento con estos agentes provocó la muerte celular (Figura suplementaria S3A, S3B, S3C y S3D). Es probable que la detención del crecimiento mediada por metformina se deba a la reducción del nivel de glucosa y a la acidificación extracelular del medio (Figuras suplementarias S4A y S4B). Curiosamente, la sustitución del medio cada 12 h por medio fresco con 25 mM de glucosa aumentó la supervivencia clonogénica tras el tratamiento con metformina. Como las células tratadas con metformina utilizaron la glucosa muy rápidamente en comparación con el control, la reposición del medio es esencialmente necesaria para mantener el nivel de glucosa y el pH y, por tanto, la supervivencia de las células en presencia de metformina (Figuras suplementarias S4C y S4D). Estos resultados sugieren que la concentración de glucosa disponible en el medio de cultivo influye en la acción de la metformina.
Lainhibición de la actividad del complejo respiratorio I promueve la glucólisis aeróbica <p>Para reforzar estos hallazgos, exploramos más a fondo el patrón de expresión y la actividad de algunas de las proteínas y enzimas implicadas en la regulación de la glucólisis. Encontramos que la metformina aumentaba los niveles proteicos de las moléculas glucolíticas clave GLUT1, LDHA y ChREBP de forma dependiente de la concentración (Figura 3C). Estos resultados sugieren que la inhibición de la actividad del complejo I por metformina podría forzar a las células cancerosas a adoptar la ruta glucolítica para la generación de ATP, causando un aumento de la progresión tumoral en ratones <br><span id=»2″ class=»referencess blue-text»>2</span> Demierre MF. Epidemiología y prevención del melanoma cutáneo. Curr Treat Options Oncol. 2006; 7:181-186. <br><span id=»3″ class=»referencess blue-text»>3</span> Hersey P, Watts RN, Zhang XD, Hackett J. Metabolic approaches to treatment of melanoma. Clin Cancer Res. 2009; 15:6490-6494. <br><span id=»4″ class=»referencess blue-text»>4</span> Warburg O. On respiratory impairment in cancer cells. Science. 1956; 124:269-270. <br><span id=»5″ class=»referencess blue-text»>5</span> Vander Heiden MG, Cantley LC, Thompson CB. Comprensión del efecto Warburg: los requisitos metabólicos de la proliferación celular. Science. 2009; 324:1029-1033. <br><span id=»6″ class=»referencess blue-text»>6</span> Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C. Dysfunctional oxidative phosphorylation makes malignant melanoma cells addicted to glycolysis driven by the (V600E)BRAF oncogene. Oncotarget. 2013; 4:584-599. <br><span id=»7″ class=»referencess blue-text»>7</span> Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013; 23:302-315. <br><span id=»8″ class=»referencess blue-text»>8</span> Weide B, Elsässer M, Büttner P, Pflugfelder A, Leiter U, Eigentler TK, Bauer J, Witte M, Meier F, Garbe C. Serum markers lactate dehydrogenase and S100B predict independently disease outcome in melanoma patients with distant metastasis. Br J Cancer. 2012; 107:422-428. <br><span id=»9″ class=»referencess blue-text»>9</span> Deichmann M, Benner A, Bock M, Jäckel A, Uhl K, Waldmann V, Näher H. S100-Beta, melanoma-inhibiting activity, and lactate dehydrogenase discriminate progressive from nonprogressive American Joint Committee on Cancer stage IV melanoma. J Clin Oncol. 1999; 17:1891-1896. <br><span id=»10″ class=»referencess blue-text»>10</span> Giatromanolaki A, Sivridis E, Gatter KC, Turley H, Harris AL, Koukourakis MI; Tumor and Angiogenesis Research Group. Lactate dehydrogenase 5 (LDH-5) expression in endometrial cancer relates to the activated VEGF/VEGFR2(KDR) pathway and prognosis. Gynecol Oncol. 2006; 103:912-918. <br><span id=»11″ class=»referencess blue-text»>11</span> Koukourakis MI, Giatromanolaki A, Sivridis E. Lactate dehydrogenase isoenzymes 1 and 5: differential expression by neoplastic and stromal cells in non-small cell lung cancer and other epithelial malignant tumors. Tumour Biol. 2003; 24:199-202. <br><span id=»12″ class=»referencess blue-text»>12</span> Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006; 9:425-434. <br><span id=»13″ class=»referencess blue-text»>13</span> Brandon M, Baldi P, Wallace DC. Mutaciones mitocondriales en el cáncer. Oncogene. 2006; 25:4647-4662. <br><span id=»14″ class=»referencess blue-text»>14</span> Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008; 320:661-664. <br><span id=»15″ class=»referencess blue-text»>15</span> Wallace DC. Un paradigma mitocondrial de las enfermedades metabólicas y degenerativas, el envejecimiento y el cáncer: un amanecer para la medicina evolutiva. Annu Rev Genet. 2005; 39:359-407. <br><span id=»16″ class=»referencess blue-text»>16</span> Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013; 123:1068-1081. <br><span id=»17″ class=»referencess blue-text»>17</span> He X, Zhou A, Lu H, Chen Y, Huang G, Yue X, Zhao P, Wu Y. La supresión del complejo mitocondrial I influye en las propiedades metastásicas de las células. PLoS One. 2013; 8:e61677. <br><span id=»18″ class=»referencess blue-text»>18</span> Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y. Mitochondrial respiratory complex I dysfunction promotes tumorigenesis through ROS alteration and AKT activation. Hum Mol Genet. 2011; 20:4605-4616. <br><span id=»19″ class=»referencess blue-text»>19</span> Gopal YN, Rizos H, Chen G, Deng W, Frederick DT, Cooper ZA, Scolyer RA, Pupo G, Komurov K, Sehgal V, Zhang J, Patel L, Pereira CG et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1α and oxidative phosphorylation in melanoma. Cancer Res. 2014; 74:7037-7047. <br><span id=»20″ class=»referencess blue-text»>20</span> Barbi de Moura M, Vincent G, Fayewicz SL, Bateman NW, Hood BL, Sun M, Suhan J, Duensing S, Yin Y, Sander C, Kirkwood JM, Becker D, Conrads TP, et al. Mitochondrial respiration–an important therapeutic target in melanoma. PLoS One. 2012; 7:e40690. <br><span id=»21″ class=»referencess blue-text»>21</span> Blackman RK, Cheung-Ong K, Gebbia M, Proia DA, He S, Kepros J, Jonneaux A, Marchetti P, Kluza J, Rao PE, Wada Y, Giaever G, Nislow C. Mitochondrial electron transport is the cellular target of the oncology drug elesclomol. PLoS One. 2012; 7:e29798. <br><span id=»22″ class=»referencess blue-text»>22</span> Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Reverse TCA cycle flux through isocitrate dehydrogenases 1 and 2 is required for lipogenesis in hypoxic melanoma cells. Pigment Cell Melanoma Res. 2012; 25:375-383. <br><span id=»23″ class=»referencess blue-text»>23</span> Lim JH, Luo C, Vazquez F, Puigserver P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014; 74:3535-3545. <br><span id=»24″ class=»referencess blue-text»>24</span> Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformina y reducción del riesgo de cáncer en pacientes diabéticos. British Medical Journal. 2005; 330:1304-1305. <br><span id=»25″ class=»referencess blue-text «25</span> Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila). 2010; 3:1451-1461. <br><span id=»26″ class=»referencess blue-text»>25</span> Kirpichnikov D, McFarlane SI, Sowers JR. Metformina: una actualización. Ann Intern Med. 2002; 137:25-33. <br><span id=»27″ class=»referencess blue-text»>27</span> Pollak MN. Investigación de la metformina para la prevención y el tratamiento del cáncer: el final del principio. Cancer Discov. 2012; 2:778-790. <br><span id=»28″ class=»referencess blue-text»>28</span> El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000; 275:223-228. <br><span id=»29″ class=»referencess blue-text»>29</span> Owen MR, Doran E, Halestrap AP. Evidencia de que la metformina ejerce sus efectos antidiabéticos a través de la inhibición del complejo 1 de la cadena respiratoria mitocondrial. Biochem J. 2000; 348:607-614. <br><span id=»30″ class=»referencess blue-text»>30</span> Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. Elife. 2014; 3:e02242. <br><span id=»31″ class=»referencess blue-text»>31</span> Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. La metformina actúa directamente sobre las mitocondrias para alterar la bioenergética celular. Cancer Metab. 2014; 2:12. <br><span id=»32″ class=»referencess blue-text»>32</span> Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarié A, Tyler BM, Brem H, Toulas C, Cohen-Jonathan Moyal E, Sarry JE, Skuli N. La metformina inhibe el crecimiento de células de glioblastoma humano y mejora la respuesta terapéutica. PLoS One. 2015; 10:e0123721. <br><span id=»33″ class=»referencess blue-text»>33</span> Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008; 27:3576-3586. <br><span id=»34″ class=»referencess blue-text»>34</span> Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase dependent growth inhibitor for breast cancer cells. Cancer Res. 2006; 66:10269-10273. <br><span id=»35″ class=»referencess blue-text»>35</span> Martin MJ, Hayward R, Viros A, Marais R. Metformin accelerates the growth of BRAF V600E-driven melanoma by upregulating VEGF-A. Cancer Discov. 2012, 2:344-355. <br><span id=»36″ class=»referencess blue-text»>36</span> Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A, Gounon P, Allegra M, Bertolotto C, Bereder JM, Tartare-Deckert S, Bahadoran P, Auberger P, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011; 2:e199. <br><span id=»37″ class=»referencess blue-text»>37</span> Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P, Bertolotto C, Tartare-Deckert S, Ballotti R, Rocchi S. Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol Cancer Ther. 2013; 12:1605-1615. <br><span id=»38″ class=»referencess blue-text»>38</span> Yuan P, Ito K, Perez-Lorenzo R, Del Guzzo C, Lee JH, Shen CH, Bosenberg MW, McMahon M, Cantley LC, Zheng B. Phenformin enhances the therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc Natl Acad Sci USA. 2013; 110:18226-18231. <br><span id=»39″ class=»referencess blue-text»>39</span> Cannino G, El-Khoury R, Pirinen M, Hutz B, Rustin P, Jacobs HT, Dufour E. Glucose modulates respiratory complex I activity in response to acute mitochondrial dysfunction. J Biol Chem. 2012; 287:38729-38740. <br><span id=»40″ class=»referencess blue-text»>40</span> Xu RH, PelicanoH, Zhou Y, CarewJS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005; 65:613-621. <br><span id=»41″ class=»referencess blue-text»>41</span> Pan JG, Mak TW. Metabolic targeting as an anticancer strategy: dawn of a new era? Sci STKE. 2007; 2007:pe14. <br><span id=»42″ class=»referencess blue-text»>42</span> Dalva-Aydemir S, Bajpai R, Martinez M, Adekola KU, Kandela I, Wei C, Singhal S, Koblinski JE, Raje NS, Rosen ST, Shanmugam M. Targeting the Metabolic Plasticity of Multiple Myeloma with FDA-Approved Ritonavir and Metformin. Clin Cancer Res. 2015; 21:1161-1171. <br><span id=»43″ class=»referencess blue-text»>43</span> Menéndez JA, Oliveras-Ferraros C, Cufí S, Corominas-Faja B, Joven J, Martín-Castillo B, Vázquez-Martín A. La metformina es sintéticamente letal con la retirada de glucosa en células cancerosas. Cell Cycle. 2012; 11:2782-2792. <br><span id=»44″ class=»referencess blue-text»>44</span> Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, Nanni P, Bergamini C, Nicoletti G, Giovanni CD, Ghelli A, Giorgio V, Caratozzolo MF, et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013; 1:11. <br><span id=»45″ class=»referencess blue-text»>45</span> Semenza GL. Metabolismo tumoral: las células cancerosas dan y toman lactato. J Clin Invest. 2008; 118:3835-3837. <br><span id=»46″ class=»referencess blue-text»>46</span> Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Bernard Gallez B, Wahl ML, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008; 118:3930-3942. <br><span id=»47″ class=»referencess blue-text»>47</span> Phoenix KN, Vumbaca F, Claffey KP. La activación terapéutica de metformina/AMPK promueve el fenotipo angiogénico en el modelo de cáncer de mama MDA-MB-435 ERalpha negativo. Breast Cancer Res Treat. 2009; 113:101-111. <br><span id=»48″ class=»referencess blue-text»>48</span> Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011; 71:2550-2560. <br><span id=»49″ class=»referencess blue-text»>49</span> Sonveaux P, Copetti T, De Saedeleer CJ, Vegran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frérart F, Gallez B, Ribeiro A, Michiels C, et al. Targeting the lactate transporter MCT1 in endothelial cells inhibits lactate-induced HIF-1 activation and tumor angiogenesis. PLoS ONE. 2012; 7:e33418. <br><span id=»50″ class=»referencess blue-text»>50</span> Wu M, Seto E, Zhang J. E2F1 mejora la glucólisis mediante la supresión de la transcripción de Sirt6 en células cancerosas. Oncotarget. 2015; 6:11252-11263. <br><span id=»51″ class=»referencess blue-text»>51</span> Hernlund E, Strandberg Ihrlund L, Khan O, Ates YO, Linder S, Panaretakis T, Shoshan MC. Potentiation of chemotherapeutic drugs by energy metabolism inhibitors 2-deoxyglucose and etomoxir. Int J Cancer. 2008; 123:476-483. <br><span id=»53″ class=»referencess blue-text»>53</span> Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-target in therapy for cancer. Br J Cancer. 2008; 99:989-994. <br><span id=»54″ class=»referencess blue-text»>54</span> Walenta S, Mueller-Kliese WF. Lactato: espejo y motor de la malignidad tumoral. Semin Radiat Oncol. 2004; 14:267-274. <br><span id=»56″ class=»referencess blue-text»>56</span> Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Inhibition of lactate dehydrogenase A induces oxidative stress and inhibits tumor progression. Proc Natl Acad Sci USA. 2010; 107:2037-2042. <br><span id=»57″ class=»referencess blue-text»>57</span> Zhao YH, Zhou M, Liu H, Ding Y, Khong HT, Yu D, Fodstad O, Tan M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009; 28:3689-3701. <br><span id=»58″ class=»referencess blue-text»>58</span> Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, et al. Inactivation of the HIF-1α/PDK3 signaling axis drives melanoma towards mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012; 72:5035-5047. <br><span id=»59″ class=»referencess blue-text»>59</span> Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cáncer Discov. 2014; 4:423-433. <br><span id=»60″ class=»referencess blue-text»>60</span> Miskimins WK, Ahn HJ, Kim JY, Ryu S, Jung YS, Choi JY. Efecto anticancerígeno sinérgico de la fenformina y el oxamato. PLoS One. 2014; 9:e85576. <br><span id=»61″ class=»referencess blue-text»>61</span> Jin S, DiPaola RS, Mathew R, White E. Metabolic catastrophe as a means to cancer cell death. J Cell Sci. 2007; 120:379-383. <br><span id=»62″ class=»referencess blue-text»>62</span> McLornan DP, List A, Mufti GJ. Aplicación de la letalidad sintética para el tratamiento selectivo del cáncer. N Engl J Med. 2014; 371:1725-1735. <br><span id=»63″ class=»referencess blue-text»>63</span> Kaelin WG Jr. El concepto de letalidad sintética en el contexto de la terapia contra el cáncer. Nat Rev Cancer. 2005; 5:689-698. <br><span id=»64″ class=»referencess blue-text»>64</span> Iaquinta PJ, Lees JA. Decisiones de vida o muerte por los factores de transcripción E2F. Curr Opin Cell Biol. 2007; 19:649-657. <br><span id=»65″ class=»referencess blue-text»>65</span> Houben R, Hesbacher S, Schmid CP, Kauczok CS, Flohr U, Haferkamp S, Müller CS, Schrama D, Wischhusen J, Becker JC. La expresión de alto nivel de p53 de tipo salvaje en células de melanoma se asocia frecuentemente con la inactividad en ensayos de genes reporteros de p53. PLoS One. 2011; 6:e22096. <br><span id=»66″ class=»referencess blue-text»>66</span> Avery-Kiejda KA, Bowden NA, Croft AJ, Scurr LL, Kairupan CF, Ashton KA, Talseth-Palmer BA, Rizos H, Zhang XD, Scott RJ, Hersey P. P53 in human melanoma fails to regulate target genes associated with apoptosis and the cell cycle and may contribute to proliferation. BMC Cancer. 2011; 11:203. <br><span id=»67″ class=»referencess blue-text»>67</span> Lu M, Breyssens H, Salter V, Zhong S, Hu Y, Baer C, Ratnayaka I, Sullivan A, Brown NR, Endicott J, Knapp S, Kessler BM, Middleton MR, et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell. 2013; 23:618-633. <br><span id=»68″ class=»referencess blue-text»>68</span> Pandey V, Vijayakumar MV, Ajay AK, Malvi P, Bhat MK. Diet-induced obesity increases melanoma progression: involvement of Cav-1 and FASN. Int J Cancer. 2012; 130:497-508. <br><span id=»69″ class=»referencess blue-text»>69</span> Leontieva OV, Blagosklonny MV. M(o)TOR del estado pseudohipóxico en el envejecimiento: rapamicina al rescate. Cell Cycle. 2014; 13:509-515. <br><span id=»70″ class=»referencess blue-text»>70</span> Leontieva OV, Blagosklonny MV. Yeast-like chronological senescence in mammalian cells: phenomenon, mechanism and pharmacological suppression. Envejecimiento (Albany NY). 2011; 3:1078-1091. <br><span id=»71″ class=»referencess blue-text»>71</span> Vijayakumar MV, Singh S, Chhipa RR, Bhat MK. The hypoglycaemic activity of fenugreek seed extract is mediated through the stimulation of an insulin signalling pathway. Br J Pharmacol. 2005; 146:41-48. <br><span id=»72″ class=»referencess blue-text»>72</span> Malvi P, Chaube B, Pandey V, Vijayakumar MV, Boreddy PR, Mohammad N, Singh SV, Bhat MK. Obesity induced rapid melanoma progression is reversed by orlistat treatment and dietary intervention: role of adipokines. Mol Oncol. 2015; 9:689-703. <br><span id=»73″ class=»referencess blue-text»>73</span> Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Evaluación de las actividades enzimáticas de la cadena respiratoria mitocondrial en tejidos y células cultivadas. Nat Protoc. 2012; 7: 1235-1246.
La discrepancia en el resultado del tratamiento con metformina in vivo e in vitro nos llevó a explorar la causa de la misma. Se sabe que la metformina ejerce su acción principalmente inhibiendo el complejo I de la enzima OXPHOS mitocondrial <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib27″>27</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib29″>29</a><a href=»#]»>]</a></sup> y la inhibición del complejo I provoca un aumento del crecimiento y la proliferación tumoral</a><a href=»#14″>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href=»#]»>]</a></sup>. Como observamos que el tratamiento con metformina provocaba la acidificación del medio de cultivo debido al aumento de la glucólisis aeróbica (Figuras suplementarias 4A y 4B), especulamos que ésta podría ser la razón de la detención del crecimiento celular. Así pues, medimos el nivel de glucosa y de lactato en el medio de cultivo usado recogido de las células de control y de las tratadas con metformina. Se detectó una mayor utilización de glucosa (evidente por la presencia de menos glucosa residual en el medio) y un mayor nivel de lactato en las células tratadas con metformina en comparación con el control (Figura 3A y 3B)
<p>A continuación, hemos comprobado el impacto de la metformina y el oxamato/DCA solos o en combinación sobre la supervivencia de células no cancerosas. Hemos utilizado tres líneas celulares no cancerosas diferentes: AML12 (hepatocitos de ratón), L6 (células musculares de rata) y MEF (fibroblastos embrionarios de ratón) para comprobar el efecto del tratamiento combinado. Aunque, la combinación de metformina y oxamato/DCA aflojó la proliferación de estas células (Figura Suplementaria S7A, S7B y S7C), no afectó a la viabilidad celular (Figura Suplementaria S7D), indicando que esta combinación es eficaz para matar selectivamente las células cancerosas.</p> <p><strong>Metformina y oxamato/DCA</strong>
<h3></h3>
<p><strong>El bloqueo del complejo I y de la generación de lactato impone una catástrofe metabólica<br></strong>Para asegurar si el co-tratamiento con oxamato y metformina afecta al metabolismo de las células de melanoma, medimos los parámetros metabólicos en presencia de estos inhibidores. El tratamiento con metformina elevó la glucólisis, mientras que el tratamiento con oxamato provocó una disminución de la utilización de glucosa y de la secreción de lactato (Figura 6A y 6B). Curiosamente, la utilización de glucosa y la secreción de lactato se inhibieron significativamente al tratar las células con oxamato y metformina conjuntamente (Figura 6A y 6B). Además, tanto el oxamato como la metformina redujeron los niveles de ATP al inhibir la fosforilación a nivel de sustrato y la OXPHOS respectivamente (Figura 6C). También se detectó una disminución significativa del nivel de ATP en las células tratadas con oxamato y metformina conjuntamente (Figura 6C). Además, para confirmar la implicación de la LDH en el crecimiento y la proliferación de las células de melanoma, se eliminó la LDHA (LDHA-KD) mediante un siARN específico (Figura 6D). Se observó un aumento de la muerte celular en las células LDHA-KD tratadas con metformina o fenformina (Figura 6E). En las células LDHA-KD se observó una reducción significativa de la glucólisis (evidente por la disminución de la utilización de glucosa y la secreción de lactato) y del nivel de ATP en comparación con el control (figuras 6F, 6G y 6H). Además, en las células LDHA-KD tratadas con metformina o fenformina se observó una disminución de la tasa glucolítica y un fuerte descenso del nivel de ATP (figuras 6F, 6G y 6H). Estos resultados indican que la inhibición simultánea tanto del complejo I como de la LDH o de la vía de generación de lactato induce una catástrofe metabólica debido a la alteración de la reserva celular de ATP, lo que finalmente conduce a la detención del crecimiento y a la muerte celular.</p> <p> </p> <p><strong>Células con LDHA-KD</strong>
<figure class=»wp-block-image size-large»><img src=»https://www.dcaguide.org/wp-content/uploads/2021/09/Fig-6-10-1024×768.jpg» alt=»» class=»wp-image-4174″/><figcaption><strong>Figura 6: </strong>La inhibición del complejo I y la generación de lactato inducen conjuntamente una catástrofe metabólica.<strong>A.</strong> y <strong>B.</strong> Las células A375 y B16F10 se trataron con oxamato (25 y 50 mM) solo o con metformina 2 mM durante 36 h. El nivel de glucosa y lactato en el medio de cultivo se midió como se menciona en la sección del método. <strong>C.</strong> Niveles relativos de ATP en células A375 y B16F10 cultivadas en las condiciones mencionadas en <strong>A.</strong>. <strong>D.</strong> Las células de melanoma humano (A375 y SKMel28) se cultivaron en placas de 96 pocillos (60% de confluencia) y se transfectaron con siRNA de control y específico de LDHA como se describe en la sección de métodos. Estas células se trataron con metformina (2 mM) o fenformina (100 µM) durante 48 h adicionales. La inhibición de la expresión de LDHA se confirmó mediante inmunotransferencia. <strong>E.</strong> Supervivencia de A375 y SKMel28 tras la anulación de LDHA cultivados en presencia o ausencia de metformina y fenformina. <strong>F.</strong>-<strong>H.</strong> Las células A375 y SKMel28 se cultivaron en placas de cultivo de 35 mm durante 24 h. Estas células se transfectaron con ARNsi de control o específico de LDHA y se dejaron crecer durante 24 h más. Las células se trataron con 2 mM de metformina o 100 µM de fenformina durante 48 h adicionales. La utilización de glucosa <strong>F.</strong>, la secreción de lactato <strong>G.</strong> y el nivel celular total de ATP <strong>H.</strong> se determinaron en células de melanoma como se describe en la sección del método. Los valores se representan como media ± DE. Los valores *<em>p</em> < 0,05, **<em>p</em> < 0,01, ***<em>p</em> < 0,001 denotan diferencias significativas entre los grupos (<em>n</em> > 3 como mínimo). (Ctrl- control, Met- metformina, Oxa- oxamato, Phen- fenformina).</figcaption></figure>
<p><strong>La inhibición simultánea del complejo I y de la LDH por metformina y oxamato retrasa la progresión tumoral en ratones<br></strong>Para verificar si la combinación de oxamato y metformina es también eficaz para disminuir la progresión tumoral, se desarrollaron tumores inyectando células B16F10 en ratones C57BL/6J. Una vez que los tumores alcanzaron un tamaño de 50 mm<sup>3</sup>, los ratones fueron distribuidos aleatoriamente en cuatro grupos: (a) control, (b) metformina, (c) oxamato, y (d) ratones tratados conjuntamente con metformina y oxamato. Observamos que los ratones administrados con metformina desarrollaron tumores de mayor tamaño en comparación con el control no tratado (Figura 7A, 7B y 7C). Mientras que en los ratones administrados con oxamato, los tumores progresaron a un ritmo más lento que en el control no tratado, así como en los ratones administrados con metformina (Figura 7A). Curiosamente, observamos que el oxamato retrasaba drásticamente la progresión tumoral cuando se administraba junto con metformina, como evidenciaba la reducción del volumen tumoral y del peso tumoral (Figura 7A, 7B y 7C).</p> <p>
<p>Además, para confirmar si la anulación de la progresión tumoral inducida por metformina mediante oxamato es consecuencia de la reducción de la glucólisis aeróbica, medimos parámetros metabólicos <em>in vivo</em>. Los ratones administrados con metformina presentaban niveles bajos de glucosa sérica y niveles más altos de lactato y LDH en suero en comparación con el control, mientras que en los ratones tratados con oxamato solo o metformina junto con oxamato se detectaron niveles relativamente más altos de glucosa sérica y bajos de lactato (Figura 7D y 7E). La actividad de la enzima LDH disminuyó significativamente en los tumores extirpados de los ratones tratados con oxamato solo o metformina junto con oxamato (Figura 7F). Se detectó un nivel relativamente bajo de ATP en los tumores de los ratones administrados conjuntamente con oxamato y metformina, en comparación con el agente único. Sin embargo, no observamos cambios significativos en los niveles proteicos de las moléculas que regulan la glucólisis (Figura 7I). Es importante destacar que no se detectaron cambios significativos en el peso corporal de los ratones que recibieron el tratamiento combinado y que, tras una inspección visual, no se observaron posibles efectos secundarios adversos ni síntomas macroscópicos de toxicidad generalizada (Figura suplementaria S8A). Además, aparentemente no se observaron anomalías patológicas en los órganos vitales como pulmón, hígado, riñón y corazón de estos ratones (Figura suplementaria S8B). Estos resultados sugieren que esta combinación no ejerce toxicidad generalizada y es eficaz para retardar la progresión tumoral en ratones.</p> <p> </p> <p><strong>Consecuencias adversas</strong>
<figure class=»wp-block-image size-large»><img src=»https://www.dcaguide.org/wp-content/uploads/2021/09/Fig-7-4-1024×768.jpg» alt=»» class=»wp-image-4177″/><figcaption><strong>Figura 7: </strong>La inhibición de la actividad LDH restringe la progresión tumoral del melanoma tras la inhibición del complejo I</strong>. A.</strong> Progresión del isoinjerto B16F10 en ratones C57BL/6J administrados con metformina (200 mg/kg, por vía oral) y oxamato (500 mg/kg, por vía oral) solos o juntos (<em>n</em> = 5). Los valores se representan como media ± DE. <strong>B.</strong> y <strong>C.</strong> Peso tumoral e imagen representativa de tumores extirpados de ratones de los grupos indicados. <strong>D.</strong> y <strong>E.</strong> nivel de glucosa y lactato en el suero recogido de los ratones portadores de tumores de los grupos de tratamiento indicados (<em>n</em> = 4) <strong>F.</strong> Actividad enzimática de LDH en el lisado tumoral <strong>G.</strong> Actividad de LDH en el suero de los ratones de los grupos de tratamiento indicados (<em>n</em> = 3). <strong>H.</strong> Niveles relativos de ATP en los tumores de los grupos de tratamiento indicados (<em>n</em> = 3). <strong>I.</strong> Inmunoblots que muestran los niveles de proteínas de las moléculas indicadas en los lisados tumorales completos. Los valores se representan como media ± DE. Los valores *<em>p</em> < 0,05, **<em>p</em> < 0,01 denotan diferencias significativas entre los grupos. (Ctrl- control, Met- metformina, Oxa- oxamato).<br></figcaption></figure>
<h2>DISCUSIÓN</h2>
<p>Las células cancerosas, incluidas las del melanoma, desvían una enorme cantidad de flujo de carbono a la glucólisis <sup><a href=»#5″>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib5″>5</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href=»#]»>]</a></sup>. Esto ayuda a las células a generar ATP y otros componentes básicos para el rápido crecimiento tumoral <sup><a href=»#5″>[5]</a></sup>. Varios informes sugieren que atacar las vías metabólicas reduce la progresión tumoral y el crecimiento celular <em>in vitro</em> <sup><a href=»#40″>[40-</a><a href=»#42″>42]</a></sup>. En el presente estudio, demostramos que la metformina promueve el crecimiento del melanoma al elevar la glucólisis debido a la inhibición de la función del complejo I, mientras que la inhibición de la LDH provoca la detención del crecimiento en las células. La inhibición de la generación de lactato en células de melanoma tratadas con metformina afecta a la supervivencia celular causando así la reducción de la progresión tumoral (Figura 8).</p> <p>
<p>Las consecuencias de la inhibición del complejo I en el crecimiento de las células cancerosas no se conocen con claridad, aunque los informes sugieren que la inhibición del complejo I conduce a la transformación celular y aumenta el crecimiento celular<a href=»#[«> <sup>[</sup></a><sup><a href=»https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href=»#]»>]</a></sup>. Por el contrario, también se ha observado que la inhibición del complejo I retrasa la progresión tumoral en ratones <sup><a href=»#30″>[30]</a></sup>. Es probable que estos diversos resultados dependan de la disponibilidad de nutrientes en el microambiente tumoral. Se ha descrito que cuando la glucosa está disponible en abundancia, la inhibición del complejo I promueve la glucólisis y la proliferación rápida de las células, mientras que induce la muerte celular en condiciones de estrés metabólico<sup><a href=»#43″>[43]</a></sup>. En este estudio, observamos que la inhibición del complejo I por metformina promueve la progresión tumoral del melanoma en ratones.</p> <p>
<p>Aunque se ha demostrado que la metformina inhibe la proliferación de muchas células cancerosas y también restringe la progresión tumoral del xenoinjerto en ratones <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib33″>33</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib34″>34</a><a href=»#]»>]</a></sup>, se sabe que ejerce efectos inhibidores del crecimiento así como promotores del crecimiento en melanoma<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib35″>35</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib37″>37</a><a href=»#]»>]</a></sup>. Curiosamente, detectamos que los tumores derivados de B16F10 y A375 crecían rápidamente en ratones administrados con metformina y la tasa de crecimiento variaba en función del estado glucémico de los ratones (Figura 2D y 2E). Esta observación concuerda con un informe reciente que demuestra que la metformina facilita la progresión tumoral de células de melanoma mutantes de BRAF<sup><a href=»http://335″>[35]</a></sup>.</p> <p><a href=»http://335″>[35]</a></sup>
<p>Es probable que la disparidad en la acción de la metformina sobre el crecimiento de células de melanoma <em>in vitro</em> e <em>in vivo</em> esté influida por la disponibilidad de glucosa y la concentración de lactato, que podrían influir en su acción. La metformina inhibe el crecimiento celular e induce la apoptosis en condiciones de glucosa limitada <sup><a href=»#43″>[43]</a></sup>, mientras que observamos que en presencia de glucosa elevada provoca la detención del crecimiento sin afectar a la muerte celular. Razonamos esta discrepancia en que las células cancerosas obtienen ATP <em>vía</em> OXPHOS bajo estrés metabólico, mientras que cuando la glucosa es abundante el ATP se obtiene principalmente a través de la glucólisis. La inhibición del complejo I en condiciones de glucosa elevada fomenta aún más la glucólisis, lo que da lugar a una generación excesiva de lactato, y este fenómeno ya se había descrito anteriormente<sup><a href=»#44″>[44]</a></sup>. En conjunto, estos hallazgos sugieren que la detención del crecimiento inducida por metformina en células de melanoma <em>in vitro</em> es consecuencia de la acidificación del medio debido a la acumulación excesiva de ácido láctico por la rápida utilización de la glucosa disponible. En particular, la detención del crecimiento inducida por la metformina puede evitarse reponiendo el medio con frecuencia para mantener unas condiciones de crecimiento óptimas, ya que las células tratadas con metformina necesitan más glucosa para proliferar debido al aumento de la glucólisis aeróbica. Esto imita las condiciones <em>in vivo</em>, en las que probablemente la circulación rápida y constante de ácido láctico de acceso permite a las células cancerosas utilizar más glucosa, lo que contribuye a una rápida proliferación. Además, el lactato también puede ser utilizado por las células cancerosas en el tumor <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib45″>45</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib46″>46</a></sup><a href=»#]»><sup>]</sup></a>. Además, se ha informado de que la metformina induce la angiogénesis <em>vía</em> elevando el nivel de VEGF<sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib35″>35</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib47″>47</a><a href=»#]»>]</a></sup>. Informamos de que la inhibición del complejo I aumenta la producción de lactato y se ha sugerido previamente que el cambio glucolítico mejora la angiogénesis<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib48″>48</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib49″>49</a><a href=»#]»>]</a></sup>. Nuestros resultados muestran una correlación positiva entre el lactato sérico y el nivel de VEGF en ratones administrados con metformina. Por lo tanto, es probable que el crecimiento tumoral potenciado por metformina se vea facilitado por la angiogénesis inducida por lactato que puede estar mediada por VEGF <sup><a href=»#48″>[48]</a></sup>. Además, observamos un aumento del nivel de E2F1, una importante molécula reguladora del ciclo celular, en los tumores extirpados de ratones administrados con metformina. Además de promover la proliferación celular, E2F1 también regula muchos genes implicados en la glucólisis<sup><a href=»#50″> [50]</a></sup> que es esencial para el crecimiento rápido de las células cancerosas. Por lo tanto, es probable que, E2F1 podría desempeñar un papel importante en la mediación de la progresión tumoral rápida inducida por metformina probablemente <em>vía</em> la regulación de la glucólisis aeróbica.</p> <p>
<p>Los inhibidores glucolíticos como la 2-deoxiglucosa, la lonimida, el 3-bromopiruvato, el DCA y los fármacos que interfieren en las vías metabólicas han mostrado resultados prometedores en la supresión del crecimiento tumoral<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib40″>40</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib42″>42</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib51″>51</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href=»#]»>]</a></sup>. Además, dirigirse a la vía de generación de lactato resulta especialmente atractivo en cánceres glucolíticos <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib54″>54</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib55″>55</a><a href=»#]»>]</a></sup>. Las células de melanoma son metabólicamente glucolíticas, por lo que dependen principalmente de la elevada actividad de la LDH para la generación de ATP <sup><a href=»#6″>[6]</a></sup>. Las células de melanoma sobreexpresan LDH y un nivel elevado en suero suele correlacionarse con un mal pronóstico y la supervivencia de los pacientes<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib8″>8</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=»#]»>]</a></sup>. Nuestros resultados indican que la interrupción de la conversión de piruvato en lactato afecta profundamente a la proliferación de células de melanoma. Esto concuerda con los informes que indican que los tumores de tipos celulares glucolíticos son más susceptibles a la inhibición de LDH-A con FX11<sup><a href=»#56″>[56]</a></sup>.</p> <p><a href=»#56″>[56]</a></sup>
<p>LDH desempeña un papel importante en la homeostasis metabólica y el mantenimiento tumoral<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib12″>12</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib57″>57</a><a href=»#]»>]</a></sup>. Utilizamos oxamato y DCA para inhibir la generación de lactato en células de melanoma. El DCA es una pequeña molécula de administración oral que se ha utilizado para el tratamiento de la acidosis láctica, y se ha demostrado que la restauración de la funcionalidad de la OXPHOS mediante DCA induce la muerte celular<sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib52″>52</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href=»#]»>]</a></sup>. Además, se ha demostrado anteriormente que el DCA reduce la producción de lactato y desencadena la apoptosis en células de melanoma <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib58″>58</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib59″>59</a><a href=»#]»>]</a></sup>. El oxamato, un análogo del piruvato, también se utiliza para inhibir la LDH. Un estudio reciente de Miskimins et al. sugiere que atacar conjuntamente el complejo I y la LDH puede ser una estrategia prometedora para detener el crecimiento de cánceres agresivos <sup><a href=»#60″>[60]</a></sup>. En concordancia con esto, también observamos que la prevención de la generación de lactato y la inhibición simultánea del complejo I mediante oxamato/DCA o mediante siRNA específico de LDH y metformina, respectivamente, resulta en el agotamiento de la reserva celular de ATP. La disminución del ATP celular provoca una catástrofe metabólica que conduce a la apoptosis de las células de melanoma. La muerte celular inducida por catástrofe metabólica se considera una estrategia prometedora para detener la progresión del cáncer. Además, nuestro estudio pone de manifiesto el papel diferenciado del complejo I y de la LDH en la proliferación celular, y estos dos constituyen la ruta de generación de ATP a través de la OXPHOS o de la fosforilación a nivel de sustrato, respectivamente. El bloqueo de cualquiera de las enzimas por sí sola no induce la muerte celular, ya que las células pueden cambiar a una vía alternativa para la generación de ATP. Sin embargo, la inhibición simultánea de ambas induce la apoptosis. Esto indica claramente que las células dependen de la función de estas dos enzimas que forman un par sintéticamente letal, lo cual es un fenómeno prometedor que puede emplearse para la selección de células de melanoma<a href=»#[«> </a><sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib62″>62</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib63″>63</a><a href=»#]»>]</a></sup>. Dado que E2F1 está implicado en la regulación de la apoptosis <sup><a href=»#64″>[64]</a></sup>, es probable que la apoptosis inducida por la combinación de metformina y oxamato/DCA pueda implicar a E2F1. La metformina promueve la apoptosis en células cancerosas <em>vía</em> la activación de p53 <sup><a href=»#37″>[37]</a></sup>. Sin embargo, en el caso del melanoma, el papel de p53 no está claro. Como sugiere la literatura, p53 no es funcional en el melanoma y sus niveles están paradójicamente elevados en grados avanzados de melanoma <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib65″>65</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib67″>67</a><a href=»#]»>]</a></sup>. Anteriormente, hemos informado de que un nivel elevado de p53 se asocia con un aumento del crecimiento tumoral<sup><a href=»#68″>[68]</a></sup>. Por lo tanto, es poco probable que p53 esté implicado en la apoptosis inducida por el tratamiento combinado y, por lo tanto, se necesitan más estudios para evaluar la funcionalidad de E2F1 y p53 en el melanoma.</p> <p><a href=»#68″>[68]</a></sup>
<p>Al utilizar combinaciones de diferentes fármacos que pueden promover sinérgicamente la muerte de las células cancerosas, esto también puede aumentar la toxicidad para las células normales. Por lo tanto, es crucial acceder al impacto del tratamiento combinado en las células normales. Es importante destacar que nuestros datos sugieren que esta combinación es eficaz para destruir las células cancerosas tanto <em>in vitro</em> como <em>in vivo</em>, y tiene un impacto mínimo en la supervivencia de las células normales. La sensibilidad diferencial entre células de melanoma y normales debida a la combinación de metformina y oxamato/DCA puede atribuirse al hecho de que las células de melanoma son altamente glucolíticas y sobreexpresan moléculas implicadas en la generación y secreción de lactato en comparación con las células normales <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib6″>6</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=»#]»>]</a></sup>. Curiosamente, en comparación con las células normales, las células cancerosas muestran una mayor respiración mitocondrial desacoplada<sup><a href=»#31″>[31]</a></sup>. Las células cancerosas tras el tratamiento con metformina muestran una mayor elevación compensatoria de la glucólisis que las células normales <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib31″>31</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib32″>32</a><a href=»#]»>]</a></sup>, ponderando así la susceptibilidad metabólica de las células cancerosas. Por lo tanto, la inhibición de la LDH o de la generación/secreción de lactato suprime el crecimiento de las células cancerosas, mientras que las células normales se ven menos afectadas, ya que se puede seguir produciendo suficiente ATP a partir de la OXPHOS porque la metformina es un inhibidor débil del complejo I. Como la actividad OXPHOS es mayor en las células normales que en las células de melanoma, es probable que la combinación de metformina y oxamato/DCA tenga el menor impacto adverso sobre la respiración de las células normales.</p> <p>
<p>Nuestro estudio abre nuevas vías en la lucha contra el metabolismo de las células cancerosas y puede implicarse aún más en el ensayo de otros fármacos clínicamente aprobados que se sabe que inhiben la glucólisis junto con la metformina. El presente estudio sugiere que cualquier fármaco/inhibidor que bloquee la generación de lactato puede utilizarse en combinación con la metformina para mejorar el tratamiento y prevenir el crecimiento tumoral. Por ejemplo, se ha demostrado que la rapamicina, un fármaco clínicamente aprobado, previene la generación de lactato<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib69″>69</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib70″>70</a><a href=»#]»>]</a></sup>. Del mismo modo, se ha demostrado que la inhibición de BRAF provoca la supresión de la glucólisis <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib6″>6</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href=»#]»>]</a></sup>. Por lo tanto, tanto la rapamicina como los inhibidores de BRAF pueden utilizarse junto con la metformina para mejorar el resultado terapéutico con efectos secundarios reducidos. En conjunto, nuestros resultados indican que la LDH u otro mecanismo que controle la generación o secreción de lactato es crítico para la rápida progresión del melanoma en condiciones de OXPHOS comprometida, y esto puede ser explotado como un objetivo terapéutico adecuado para controlar el crecimiento de las células cancerosas glicolíticas. Se necesitan estudios más amplios para evaluar la funcionalidad de la LDH y el complejo I en otros modelos de cáncer, y posteriormente su implicación en la terapia del cáncer en general.</p> <p>
<figure class=»wp-block-image size-large»><img src=»https://www.dcaguide.org/wp-content/uploads/2021/09/Fig-8-1-1024×730.jpg» alt=»» class=»wp-image-4179″/><figcaption><strong>Figura 8: </strong>Representación esquemática de la letalidad sintética inducida por la combinación del tratamiento con metformina y oxamato en melanoma.<strong> A.</strong> La inhibición del complejo respiratorio I por metformina o fenformina solas da lugar a un aumento de la glucólisis debido a la activación de la lactato deshidrogenasa (LDH). La generación de bloques de construcción y ATP exclusivamente a través de la glucólisis contribuye al rápido crecimiento tumoral. <strong>B.</strong> Si se ataca la LDH o la generación de lactato mediante oxamato y DCA o mediante ARNsi, se produce la activación de la OXPHOS y la consiguiente generación de ATP. Sin embargo, esto conduce a la supresión de la proliferación celular sin inducir la muerte celular. <strong>C.</strong> El bloqueo conjunto de ambas enzimas induce una catástrofe metabólica debido al agotamiento de la reserva de ATP celular. Esto conduce al inicio de la apoptosis y a la supresión del crecimiento tumoral. El complejo I y el LDHA forman una pareja sintéticamente letal en las células de melanoma
<h2>MATERIALES Y MÉTODOS</h2>
<p><strong>Experimentos con animales</strong></strong>Los ratones se obtuvieron del Experimental Animal Facility (EAF) del National Centre for Cell Science (NCCS), Pune, India. La hiperglucemia en ratones se indujo utilizando STZ como se ha descrito previamente<sup><a href=»#71″> [71]</a></sup> con ligeras modificaciones. Brevemente, ratones machos C57BL/6J y NOD/SCID fueron ayunados durante 6 h antes de la inyección intraperitoneal de STZ (50 mg/kg) en tampón citrato 0,01 M (pH 4,4) durante tres días consecutivos. La medición de la glucosa en sangre se realizó pellizcando la cola y aplicando sangre en un analizador de glucosa (Accu-Chek Active, Roche, Alemania). Los ratones con valores de glucosa en sangre superiores a 200 mg/dl se consideraron hiperglucémicos. El tumor se indujo inyectando células B16F10 (2 × 10<sup>5</sup>) o A375 (1 × 10<sup>6</sup>) en 100 µl de PBS estéril por vía subcutánea en el flanco derecho de ratones C57BL/6J y NOD/SCID respectivamente. La progresión tumoral se controló periódicamente midiendo su tamaño con un calibre digital (Sigma, EE.UU.) tras la aparición de tumores palpables. La administración oral de metformina en días alternos se inició una semana antes de la provocación tumoral en los ratones hiperglucémicos. De lo contrario, la metformina se administró sólo después de que el tumor alcanzara un tamaño óptimo y el tratamiento se continuó hasta la finalización del experimento. Al final del experimento, se sacrificaron los ratones; se extirparon los tumores y se almacenaron a -80<sup>°</sup>C o en una solución de formol al 10% para estudios posteriores. En otra serie de experimentos, los ratones (C57BL/6J, machos) se dividieron aleatoriamente en 4 grupos tras la aparición del tumor palpable. Para examinar el resultado del tratamiento con metformina en combinación con el inhibidor de la LDH oxamato, se administró a los ratones por vía oral metformina (200 mg/kg) u oxamato solo (500 mg/kg) o ambos juntos en días alternos. La progresión tumoral se controló periódicamente con un calibrador. Al final del experimento, se extirparon los tejidos tumorales, musculares y hepáticos, se pesaron y se almacenaron a -80<sup>°</sup>C para su posterior análisis. Todos los experimentos con animales se han realizado según las directrices del comité con fines de control y supervisión de experimentos con animales (CPCSEA), Gobierno de la India, y tras obtener el permiso del Comité de Ética Animal del Instituto (IAEC).</p> <p>
<p><strong>Líneas celulares y condiciones de cultivo</strong></strong>Las células de melanoma murino B16F10 y las células de melanoma humano A375 y SKMel28 se adquirieron a ATCC (VA, EE.UU.) y se mantuvieron en nuestro depósito interno. Se utilizaron células no cancerosas, AML12 (hepatocitos de ratón), L6 (células musculares de rata) y MEF (fibroblastos embrionarios de ratón) en paralelo con células cancerosas como control. Todas las células se cultivaron en sus respectivos medios que contenían 1 mM o 25 mM de glucosa en función de los experimentos y se complementaron con un 10% de suero fetal bovino inactivado por calor (Hyclone, UT, EE.UU.), penicilina (100 U/ml) y estreptomicina 100 µg/ml (Life Technologies, NY, EE.UU.), a 37<sup>°</sup>C en presencia de un 5%<sub> </sub>CO<sub>2</sub>.</p> <p>
<p><strong>Productos químicos y reactivos</strong></strong>Metformina, fenformina, oligomicina, rotenona, decilubiquinona, estreptozotocina (STZ), [bromuro de 3-(4, 5-dimetiltiazol-2-il)-2, 5-difenilterazolio] (MTT), NAD+, NADH, ATP, AMP, D-glucosa, oxamato sódico, yodoacetato, 2-deoxi-D-glucosa, dicloroacetato (DCA), Diaminobenzidina (DAB) y phloretina se adquirieron a Sigma (MO, EE.UU.). Los anticuerpos para ChREBP (1:1000), GLUT1 (1:1000), LDHA (1:1000), Ciclina D1 (1:1000), PCNA (1:1000), CDK4 (1:1000), p21 (1:1000), E2F1 (1:1000) CD31 (1:100), PARP-1 (11000), Bcl-2 (1:1000), Bax (1:1000), β-tubulina (1:1000), GAPDH (1:1000), HSP60 (1:1000) y VEGF (1:50) procedían de Santa Cruz Biotechnology (CA, EE.UU.).</p>
<p><strong>Preparación del lisado celular e inmunotransferencia<br></strong>Las células se lavaron tres veces con tampón fosfato salino (PBS) y se lisaron en tampón de lisis que contenía 50 mM Tris-Cl (pH 7.5), 120 mM NaCl, 10 mM fluoruro sódico, 10 mM pirofosfato sódico, 2 mM EDTA, 1 mM ortovanadato sódico, 1 mM fluoruro de fenilmetilsulfonilo, 1% NP-40 y cóctel inhibidor de proteasas (Roche, Alemania). Los lisados tumorales se prepararon cortando los tejidos tumorales en trozos finos, se lavaron cinco veces con solución salina al 0,85% que contenía cóctel inhibidor de proteasas y se lisaron en tampón de lisis mediante homogeneización con un homogeneizador de tejidos (Sigma, EE.UU.) seguida de sonicación. Los lisados se clarificaron mediante centrifugación a 15.000 rpm<em> </em>durante 30 minutos. Los lisados celulares se prepararon en condiciones de refrigeración. Aproximadamente, 50-100 µg de proteína de lisados de células enteras se resolvieron en un gel de SDS-poliacrilamida al 8-12% que posteriormente se transfirió a una membrana de PVDF (Millipore, Alemania). La membrana se sondó con los anticuerpos primarios deseados seguidos de anticuerpos secundarios conjugados con HRP. Los inmunoblots se detectaron con el reactivo luminal (Santa Cruz Biotechnology). Cuando fue necesario, los blots se decaparon incubando la membrana a 50<sup>°</sup>C durante 15 min en tampón de decapado (62,5 mM Tris-Cl, pH 6,8, 100 mM mercaptoetanol y 2% SDS) con agitación intermitente. Las membranas se lavaron minuciosamente con solución salina tamponada con Tris (TBS), y se volvieron a recubrir con los anticuerpos deseados.</p> <p><strong>Posicionamiento de los anticuerpos</strong>
<p><strong>Inmunofluorescencia o microscopía confocal<br></strong>Las células se colocaron en portaobjetos con cámara Labtek (Nunc, EE.UU.) y se dejaron crecer durante 24 h. Se administraron tratamientos con metformina y otros inhibidores durante los periodos de tiempo y las concentraciones deseados. Las células se lavaron con PBS estéril refrigerado y se fijaron con una solución de paraformaldehído al 3,7% a temperatura ambiente durante 10 min. A continuación se permeabilizaron con Triton X-100 al 0,025% en PBS durante 10 min y posteriormente se bloquearon con BSA al 5% en PBS durante 1 h a 37<sup>°</sup>C. Las células se incubaron con diluciones 1:100 de anticuerpos primarios en la solución de bloqueo durante 2 h a temperatura ambiente y se lavaron con TBST (TBS con 0,05% de Tween-20) al menos cinco veces antes de incubarlas con anticuerpos secundarios marcados apropiados (1:200) en solución de bloqueo durante 1 h más a temperatura ambiente. Tras cinco lavados con TBST, las muestras se cubrieron con medio de montaje que contenía DAPI (Santa Cruz Biotechnology, EE.UU.). Se sellaron los portaobjetos, se examinaron con un microscopio confocal de barrido láser (LSM510 Carl Zeiss, Alemania) y se capturaron imágenes. Posteriormente, las imágenes se procesaron con el software de análisis de imágenes LSM.</p> <p>
<p><strong>Inmunohistoquímica e histopatología<br></strong>La inmunohistoquímica y la histopatología se realizaron de acuerdo con el método de Malvi et al. <sup><a href=»#72″>[72]</a></sup>. Brevemente, se realizaron secciones finas de tumor, y otros órganos con micrótomo fijadas en portaobjetos de vidrio y parafinizadas. Para la inmunohistoquímica, los portaobjetos se desparafinizaron en una solución de xileno dos veces durante 10 minutos, y posteriormente se lavaron tres veces con etanol al 100%, 95%, 70% y 50%. Los portaobjetos se lavaron de nuevo con agua destilada y después con PBS durante 5 minutos. Para la recuperación de antígenos, los portaobjetos se hirvieron en tampón de citrato sódico (0,01 M, pH 4,5) en un horno microondas durante 10 minutos y se dejaron enfriar a temperatura ambiente durante otros 20 minutos. Se utilizó BSA o suero de cabra normal (2%) para el bloqueo en cámara humidificada o en caja fría durante 1 h. Los portaobjetos se sondaron con los anticuerpos deseados (dilución 1:100) en PBST (PBS que contenía 0,025% de Tween-20) con 0,01% de BSA durante 2 h a temperatura ambiente o durante la noche a 4<sup>°</sup>C. Los portaobjetos se lavaron con PBST (PBS que contenía 0,025% de Tween-20) que contenía 0,01% de BSA. Los portaobjetos se lavaron con PBST 4 veces durante 5 minutos y se cubrieron con anticuerpos secundarios compatibles conjugados con HRP durante 1 h. Se lavaron de nuevo y se tiñeron con Diaminobenzidina (DAB) durante 10 minutos, seguidos de lavado y contratinción con hematoxilina. Los portaobjetos se volvieron a lavar con agua, se deshidrataron con alcohol absoluto, se cubrieron con medio de montaje y se analizó la expresión de las moléculas deseadas. Para la histopatología, los portaobjetos deparafinizados se tiñeron con hematoxilina y eosina, y el análisis microscópico de la densidad celular, morfología celular, metástasis, citotoxicidad y necrosis fue realizado por patólogos de forma ciega en el Hospital KEM de Pune, India.</p> <p>
<p><strong>Ensayo de citotoxicidad celular y supervivencia</strong><br>Se sembraron aproximadamente 5 × 10<sup>3</sup> (B16F10) y 10 × 10<sup>3</sup> (A375 y SKMel28) células en cada pocillo de placas de cultivo tisular de 96 pocillos y se dejaron adherir durante 24 h a 37<sup>°</sup>C. Las células se trataron según los requisitos experimentales y la proliferación o viabilidad se evaluó mediante el ensayo MTT. Se añadió MTT (50 µl, 1 mg/ml en DMEM sin rojo de fenol) a cada pocillo y se incubó durante 4 h a 37<sup>°</sup>C. Los cristales de formacán se solubilizaron<sup> </sup>en 100 µl de isopropanol incubando con agitación a temperatura ambiente durante 5 min. La absorbancia se tomó a 570 nm utilizando 630 nm como filtro de referencia. Las células no tratadas se consideraron como control (100% de supervivencia celular)</p> <p>
<p><strong>Detección de apoptosis mediante tinción con anexina V<br></strong>Las células se sembraron a una densidad de aproximadamente 3 × 10<sup>5</sup> células en placas de 35 mm y se dejaron crecer durante 24 h. Las células se trataron con o sin oxamato y DCA solos o con metformina durante 48 h o según el requerimiento experimental. Las células se cosecharon por tripsinización y se procesaron para citometría de flujo. La apoptosis se detectó mediante tinción dual de Annexin V y PI utilizando el kit de ensayo de apoptosis (BD Bioscience, EE.UU.) de acuerdo con las instrucciones del fabricante.</p> <p><strong>Detección de apoptosis</strong>
<p><strong>Análisis del ciclo celular</strong></strong>Las células se sembraron a una densidad de aproximadamente 3 × 10<sup>5</sup> células en placas de 35 mm y se dejaron crecer durante 24 h. Las células se trataron con o sin metformina, oxamato y otros inhibidores glucolíticos solos o juntos según se indica durante 48 h o según el requisito experimental. Las células se cosecharon por tripsinización y se procesaron para citometría de flujo. Brevemente, las células se lavaron con PBS frío y se fijaron en etanol al 70% en hielo durante 30 min. Tras el tratamiento con RNasa (200 μg/ml) durante 30 min a 37 °C, se añadieron 50 μg/ml de PI al sedimento celular y se incubaron en oscuridad durante 30 min en hielo. La fluorescencia de PI se registró a través de un filtro de 585 nm en un citómetro de flujo (FACS Calibur, Becton Dickinson, California, EE.UU.). Los datos se analizaron utilizando el software Cell Quest Pro para 10.000 células.</p> <p>
<p><strong>Ensayo de supervivencia celular a largo plazo</strong></strong>Las células (5 × 10<sup>2</sup>) se sembraron en placas de 12 pocillos durante 24 h. Las células se trataron sin o con 25 mM o 50 mM de oxamato y 10 o 20 mM de DCA junto con metformina y se continuó durante 48 h más. Posteriormente, el medio se sustituyó por medio fresco libre de fármacos. Las placas se incubaron durante otros 10-15 días a 37°C en un incubador de CO<sub>2</sub> con cambio de medio cada 2-3 días. A continuación, las células se fijaron (paraformaldehído al 3% y glutaraldehído al 0,02% en PBS) y se tiñeron con violeta cristal al 0,05%.</p> <p>
<p><strong>Ensayo de utilización de glucosa</strong></strong>Las células (3 × 10<sup>5</sup>) se cultivaron en DMEM que contenía 25 mM de glucosa. Después de 24 h, el medio se sustituyó por otro que contenía concentraciones crecientes de metformina (0,1 mM, 0,5 mM, 1 mM y 2 mM) o rotenona durante 24 h y la glucosa residual presente en el medio utilizado se controló mediante un kit de ensayo de glucosa basado en GOD-POD (Spinreact, España) de acuerdo con las instrucciones del fabricante. Para medir la utilización de glucosa en presencia de inhibidores glucolíticos, las células se trataron con 50 mM de oxamato, 100 µM de floretina y 20 mM de DCA, solos o junto con 2 mM de metformina o 100 µM de fenformina. La glucosa consumida se estimó restando la glucosa restante en el medio de la concentración inicial en el medio de control (450 mg/dl). Los experimentos se realizaron al menos por triplicado y los valores se normalizaron con respecto al número total de células.</p> <p
<p><strong>Ensayo de estimación de lactato</strong></strong>La concentración de lactato en el medio de cultivo recogido de las células tratadas con o sin metformina, oxamato y otros inhibidores glucolíticos, se determinó utilizando un kit de estimación de lactato disponible en el mercado (Spinreact, España) siguiendo las instrucciones del fabricante. Brevemente, el medio de cultivo o el suero se diluyó en solución salina al 0,9% hasta 10 veces y se añadieron 10 μl de muestra a cada pocillo de la placa ELISA de 96 pocillos. se añadieron 150 µl del reactivo suministrado con el kit a cada pocillo que contenía la muestra, manteniendo el medio no utilizado y el reactivo solo como blanco, y se registró la absorbancia a 405 nm utilizando un espectrofotómetro (Thermo-Scientific, EE.UU.). Los experimentos se realizaron al menos por triplicado y los valores finales se normalizaron con el número total de células.</p> <p
<p><strong>Transfección de ARNsi<br></strong>El ARNsi específico contra LDHA se adquirió en Santa Cruz Biotechnology (EE.UU.). La transfección se realizó con Lipofectamine 2000 (Life Technologies, EE.UU.) siguiendo las instrucciones del fabricante. En resumen, las células se sembraron a una confluencia aproximada del 60%. Al día siguiente, el medio se sustituyó por OptiMEM (Life Technologies, EE.UU.) y se mantuvo durante 3 h. Los siARN deseados se disolvieron en los tampones suministrados junto con ellos. La lipofectamina2000 y el siARN se diluyeron por separado en OptiMEM y se incubaron durante 5 minutos. A continuación, se mezclaron los reactivos diluidos y se incubaron durante 30 minutos a temperatura ambiente. El precipitado resultante se dejó en las células durante 6 h, tras lo cual se añadió DMEM fresco suplementado con un 10% de FBS y se incubó durante otras 24-36 h. La eficacia de la transfección se evaluó transfectando simultáneamente el plásmido pEGFPN1. Se realizó inmunotransferencia para asegurar la inhibición de la expresión génica respectiva
<p><strong>Preparación de la fracción rica en mitocondrias a partir de células y tejidos<br></strong>La fracción rica en mitocondrias se preparó a partir de células cultivadas y tumores o tejidos normales como se informó previamente<sup><a href=»#73″>[73]</a></sup>. Brevemente, se sembraron células (1 × 10<sup>6</sup>) en placas de 10 cm y se tripsinizaron tras el tratamiento con los inhibidores deseados durante 24 h. La suspensión celular se sometió a tres ciclos de congelación-descongelación en tampón hipotónico (20 mM de fosfato potásico). Esta suspensión se centrifugó a 50.000 rpm durante 1 h para obtener sobrenadante rico en mitocondrias. Del mismo modo, para aislar las mitocondrias de los tejidos, los tejidos congelados se cortaron en trozos finos y se homogeneizaron en tampón de homogeneización (tampón Tris 0,5 M pH 7,5, 100 mM EGTA, y 250 mM de sacarosa), seguido de un procedimiento cíclico de congelación-descongelación y se centrifugaron a 50.000 rpm durante 1 h. El sobrenadante se recogió como fracción mitocondrial y se utilizó para la determinación de la función mitocondrial y la actividad enzimática OXPHOS.</p> <p>
<p><strong>Ensayos enzimáticos</strong></strong>Las células se homogeneizaron en tampón fosfato potásico hipotónico (20 mM) (pH 7,5) que contenía un cóctel inhibidor de proteasas (Roche, Alemania), se agitaron en vórtex y se lisaron mediante tres ciclos de congelación y descongelación. Para preparar el extracto de tejidos, las muestras tumorales se cortaron en trozos finos y se homogeneizaron en tampón de homogeneización (0,1 M Tris, 0,1 M KCl, 350 mM EDTA y 1 M sacarosa, pH 7,5) utilizando un homogeneizador de tejidos. Los homogeneizados se clarificaron mediante centrifugación a 10.000 rpm durante 10 min a 4<sup>°</sup>C y se mantuvieron en hielo hasta la realización de los ensayos.</p> <p
<p><strong><em>Ensayo de actividad LDH:</em> </strong>La actividad de la lactato deshidrogenasa en lisados celulares o extracto tumoral y suero se determinó utilizando el kit de ensayo de actividad LDH (Spinreact, España) de acuerdo con las instrucciones del fabricante.</p> <p>
<p><em><strong>Asayo de actividad del complejo I:</strong></em> El ensayo enzimático del complejo I de la OXPHOS mitocondrial se realizó como se describe en otra parte<sup><a href=»#73″>[73]</a></sup>. Brevemente, se añadió fracción rica en mitocondrias de homogeneizado celular o tisular (20-50 µg de proteína de homogeneizado tisular o 10-20 µg de fracción rica en mitocondrias) a 700 µl de agua destilada tomada en una cubeta de 1 ml, seguido de la adición de 100 µl de tampón fosfato potásico (0,5 M, pH 7,5), 60 µl de BSA libre de ácidos grasos (50 mg/ml), 30 µl de KCN (10 mM) y 10 µl de NADH (10 mM). El volumen final se ajustó a 994 µl utilizando agua destilada, se mezcló invirtiendo las cubetas y se adquirió la lectura de línea de base a 340 nm durante 2 min. La reacción se inició añadiendo 6 µl de decilubiquinona (10 mM, Sigma, EE.UU.) mezclados adecuadamente invirtiendo las cubetas. Se siguió la disminución de la absorbancia a 340 nm durante 10 min. Se siguió un procedimiento similar para el cálculo de la actividad del complejo I en presencia de 2 mM de metformina y 100 µM de fenformina. Se utilizó rotenona (10 µM) como control positivo. Los valores finales se normalizaron con el contenido total de proteína celular.</p> <p>
<p><strong>Medición del ATP celular total<br></strong>El nivel de ATP se midió utilizando un kit de bioluminiscencia de ATP disponible en el mercado (Roche, Alemania) siguiendo las instrucciones del fabricante. Brevemente, las células se cultivaron en presencia de los fármacos indicados. Las células se cosecharon y se lisaron en tampón Tris-EDTA 100 mM que contenía 0,01% de NP-40 y se hirvieron durante 1 minuto seguido de un ciclo de congelación-descongelación. Se registró la luminiscencia tanto del blanco como de las muestras. Para medir el ATP generado exclusivamente a través de la OXPHOS y la glucólisis, las células se trataron con oligomicina (10 µM) y oxamato o DCA respectivamente con o sin metformina. Los experimentos se realizaron por triplicado y se repitieron al menos una vez, y los valores finales se normalizaron con el contenido total de proteínas.</p> <p
<p><strong>Estadística</strong><br>El análisis estadístico se realizó con Sigma Plot 12.0 (Systat Software Inc., CA, EE.UU.). La mayoría de los experimentos se repitieron al menos una vez, como mínimo por triplicado, a menos que se indique lo contrario. Los datos se representaron como media ± DE, salvo en los experimentos indicados. Siempre que fue necesario, se empleó la prueba <em>t</em> de Student de dos colas, emparejada o no, para calcular el valor p en los experimentos asumiendo una varianza desigual, a menos que se indique lo contrario. Los valores *<em>p</em> < 0,05, **<em>p</em> < 0,01, ***<em>p</em> < 0,001 denotan diferencias significativas entre los grupos (<em>n</em> > 3 como mínimo).</p>
<h2>AGRADECIMIENTOS</h2>
<p>Los autores agradecen al Dr. S.C. Mande, Director, NCCS, Pune, India, y al Dr. G.C. Mishra, antiguo Director, NCCS, Pune, India, por su gran apoyo y por darnos todo el ánimo para llevar a cabo este trabajo. Damos las gracias al Dr. Benoit Violet (INSERM, Francia) por proporcionar MEFs, al Dr. Mahesh J Kulkarni (National Chemical Laboratory, India) por las células L6 y al Dr. Rakesh K Tyagi (Jawaharlal Nehru University, India) por proporcionar células AML12. También damos las gracias al Dr. Vijayakumar MV por su ayuda en los experimentos con animales y por la lectura crítica del manuscrito. BC y NM agradecen al Consejo de Investigación Científica e Industrial (CSIR) de la India; PM y SVS agradecen a la Comisión de Subvenciones Universitarias (UGC) de la India la concesión de becas. El apoyo de otros miembros del laboratorio y colegas en NCCS, así como los miembros del personal de Experimental Animal Facility, FACS, Confocal y la instalación de espectrometría de masas es debidamente recognized.</p> <p>
<h2>APOYOS FINANCIEROS</h2>
<p>Este trabajo fue apoyado por la beca intramural de NCCS financiado por el Departamento de Biotecnología, Gobierno de la India.</p>
<h2>CONFLICTOS DE INTERÉS</h2>
<p>Los autores declaran que no tienen ningún posible conflicto de intereses</p>
<h3>NOTA</h3>
<p>Este trabajo se realizó en cumplimiento parcial de una tesis doctoral (de B.C.) que se presentará a la Universidad Savitribai Phule Pune, Pune, India.</p>
<p></p>
<h2>REFERENCIAS</h2>
<br><span id=»1″ class=»referencess blue-text»>1</span> Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007; 445:851-857.
<br><span id=»52 «class=»referencess blue-text»>52</span>. Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloroacetate induces apoptosis in endometrial cancer cells. Gynecol Oncol. 2008; 109:402.
<br><span id=»55″ class=»referencess blue-text»>55</span> Doherty JR, Cleveland JL. Targeting lactate metabolism for cancer therapeutics. J Clin Invest. 2013; 123:3685-3692.
<p></p>