Balkrishna Chaube1, Parmanand Malvi1, Shivendra Vikram Singh1, Naoshad Mohammad1, Avtar Singh Meena1,2 et Manoj Kumar Bhat1
1 National Centre for Cell Science, Savitribai Phule Pune University Campus, Ganeshkhind, Pune, Inde
2 Adresse actuelle : Département de physiologie, University of Tennessee Health Science Center, Memphis, États-Unis
Correspondance : Manoj Kumar Bhat, courriel : [email protected]
Reçue : 16 juin 2015
Accepté : 23 septembre 2015
Publié : 15 octobre 2015
Résumé
Le mélanome est une malignité cutanée largement incurable en raison de l’hétérogénéité moléculaire et métabolique sous-jacente confondue avec le développement de la résistance. Les cellules cancéreuses ont une flexibilité métabolique en choisissant la phosphorylation oxydative (OXPHOS) ou la glycolyse pour la génération d’ATP en fonction de la disponibilité des nutriments dans le microenvironnement tumoral. Dans cette étude, nous avons examiné l’implication du complexe respiratoire I et de la lactate déshydrogénase (LDH) dans la progression du mélanome. Nous montrons que l’inhibition du complexe I par la metformine favorise la croissance du mélanome chez la souris via l’augmentation des niveaux de lactate et de VEGF. En revanche, elle entraîne l’arrêt de la croissance in vitro en raison d’une acidification extracellulaire accrue résultant d’une augmentation de la glycolyse. L’inhibition de la LDH ou de la production de lactate entraîne une diminution de la glycolyse et un arrêt de la croissance concomitant, tant in vitro qu’in vivo. Le blocage de la production de lactate dans les cellules de mélanome traitées à la metformine entraîne une diminution de la prolifération cellulaire et de la progression tumorale chez la souris. Il est intéressant de noter que l’inhibition de la LDH ou du complexe I seul n’induit pas d’apoptose, alors que l’inhibition conjointe des deux entraîne une déplétion du pool d’ATP cellulaire, ce qui provoque une apoptose induite par une catastrophe métabolique. Globalement, notre étude suggère que la LDH et le complexe I jouent des rôles distincts dans la régulation de la glycolyse et de la prolifération cellulaire. L’inhibition de ces deux éléments augmente la létalité synthétique dans le mélanome.
Mots clés : mélanome ; complexe I ; LDH ; catastrophe métabolique ; létalité synthétique
INTRODUCTION
Le mélanome malin est l’une des formes les plus agressives de cancer de la peau, avec un potentiel métastatique élevé et une résistance à de nombreux agents cytotoxiques [1, 2]. Malgré des recherches approfondies et des succès partiels obtenus par l’utilisation des médicaments actuellement disponibles, il n’existe aucun traitement efficace contre le mélanome malin [1-3]. Les cas de mélanome augmentent chaque année et représentent environ 75 % des décès liés au cancer de la peau dans le monde [2, 3]. La faible réponse aux options thérapeutiques actuellement disponibles et le développement d’une résistance à la thérapie justifient l’exploration de nouvelles stratégies pour traiter le mélanome.
L’augmentation de la glycolyse aérobie est une caractéristique de nombreux cancers [3-5]. Il a été signalé que les cellules de mélanome, en raison de la mutation BRAF, dépendent principalement de la glycolyse pour la production d’ATP et présentent un dysfonctionnement de la phosphorylation oxydative [6, 7]. Les cellules cancéreuses obtiennent de l’ATP, des intermédiaires biosynthétiques et des équivalents réducteurs en s’engageant de manière inhabituelle dans des voies biochimiques telles que la glycolyse, la glutaminolyse et la voie des pentoses phosphates [5]. Les cellules normales (non cancéreuses) obtiennent de l’ATP principalement par l’OXPHOS mitochondriale, tandis que les cellules cancéreuses s’appuient principalement sur la glycolyse aérobie pour générer de l’ATP et des intermédiaires glycolytiques qui facilitent leur croissance rapide [4, 5]. La production accrue de lactate a été corrélée à l’agressivité du cancer. De nombreuses études ont identifié la lactate déshydrogénase (LDH), qui catalyse la conversion du pyruvate en lactate, comme le marqueur le plus constant des cancers agressifs et à croissance rapide [8-11]. La LDH joue un rôle important dans la régulation de la glycolyse, le maintien de l’état d’oxydoréduction cellulaire, la physiologie mitochondriale et l’entretien des tumeurs [12]. L’altération du métabolisme des cellules cancéreuses peut être associée à un dysfonctionnement mitochondrial qui implique l’inhibition de l’OXPHOS, l’augmentation des espèces réactives de l’oxygène (ROS) et la promotion d’une croissance incontrôlée, qui à son tour favorise le développement d’un phénotype métastatique [13,14]. Le complexe respiratoire I est l’enzyme la plus grande et la plus complexe qui catalyse l’oxydation du NADH dans la chaîne de transport des électrons [15]. Le complexe I joue un rôle important dans la génération d’ATP dans les cellules normales et est un site principal de génération de ROS, cependant, son rôle dans la tumorigenèse n’est pas clair. La plupart des rapports suggèrent que l’activité du complexe I est supprimée dans les cellules cancéreuses et que son inhibition favorise la prolifération et les métastases [16-18]. À l’inverse, il a également été signalé que les cellules de mélanome présentent une fonction OXPHOS accrue qui entraîne une résistance aux médicaments [19-21]. En raison de la flexibilité métabolique dans le choix de diverses voies métaboliques, en particulier le métabolisme de la glutamine [22], les cellules de mélanome peuvent échapper à la perturbation d’une molécule dans une voie métabolique [23]. Par conséquent, plusieurs molécules de différentes voies métaboliques doivent être ciblées pour obtenir un résultat efficace et curatif.
Les biguanides comme la metformine et la phenformine sont connus pour inhiber le complexe respiratoire I. La metformine, un traitement de première intention pour les patients atteints de diabète de type 2 (DT2), est un membre des biguanides et on a récemment découvert qu’elle avait une activité anti-tumorigène [24, 25]. Au niveau physiologique, la metformine exerce son activité biologique en diminuant la glucogenèse hépatique, en augmentant la sensibilité à l’insuline, en augmentant l’absorption périphérique du glucose et en réduisant l’absorption du glucose dans le tractus gastro-intestinal [26]. Au niveau cellulaire, la metformine agit principalement en inhibant le complexe I mitochondrial, ce qui entrave la phosphorylation oxydative et entraîne une diminution du niveau d’ATP, ce qui conduit à l’activation de l’AMPK [27-30]. De plus, il a été démontré que la metformine diminue la respiration mitochondriale couplée à la génération d’ATP, provoquant ainsi une augmentation de la glycolyse [31, 32]. L’action anticancéreuse de la metformine a été démontrée dans divers cancers avec des mécanismes distincts [33, 34]. Cependant, le rôle de la metformine dans le mélanome n’est pas très clair. Contrairement à d’autres types de cancer, il a été démontré que la metformine a une activité à la fois promotrice et antitumorale dans le mélanome [35-37]. De plus, il a été démontré que la metformine, en association avec les inhibiteurs de BRAF, supprime la croissance du mélanome [38].
Dans la présente étude, en supposant que la perturbation des voies de production d’ATP par l’inhibition simultanée du complexe I et de la LDH pourrait être synthétiquement létale pour les cellules de mélanome, nous avons utilisé la metformine ou la phenformine et l’oxamate/acétate de dichloro (DCA) pour inhiber ces deux enzymes en utilisant des modèles cellulaires et animaux. Nous démontrons que l’inhibition de l’activité du complexe I et de la LDH a un impact distinct sur la croissance et la prolifération des cellules. Il est intéressant de noter que l’inhibition du complexe I par la metformine favorise davantage la glycolyse aérobie, ce qui entraîne une augmentation de la croissance tumorale chez la souris. L’ablation de la génération de lactate induite par la metformine en utilisant l’oxamate ou le DCA conduit à la cytostase et/ou à l’apoptose dans les cellules de mélanome.
RÉSULTATS
La metformine présente des actions distinctes in vitro et in vivo sur la croissance du mélanome
La metformine supprime la croissance tumorale en inhibant le complexe I qui est influencé par le glucose [30]. De plus, le glucose est connu pour modifier l’activité des enzymes respiratoires [39]. Par conséquent, pour explorer la ou les conséquences de l’inhibition du complexe I et l’influence du glucose sur l’action de la metformine sur la progression du mélanome, nous avons suivi la progression des isogreffes/xénogreffes chez des souris hyperglycémiques induites par la streptozotocine (STZ). Nous avons constaté que la metformine favorisait la progression des isogreffes dérivées de B16F10 chez les souris hyperglycémiques par rapport au contrôle non traité (Figure 1A, 1B et 1C). De même, la metformine a influencé positivement la progression de la tumeur chez les souris C57BL/6J normoglycémiques (Figure 1D, 1E et 1F). De même, l’administration orale de metformine a favorisé la croissance de la xénogreffe A375 chez les souris NOD/SCID hyperglycémiques et normoglycémiques par rapport au contrôle non traité (Figure 1G, 1H et 1I).
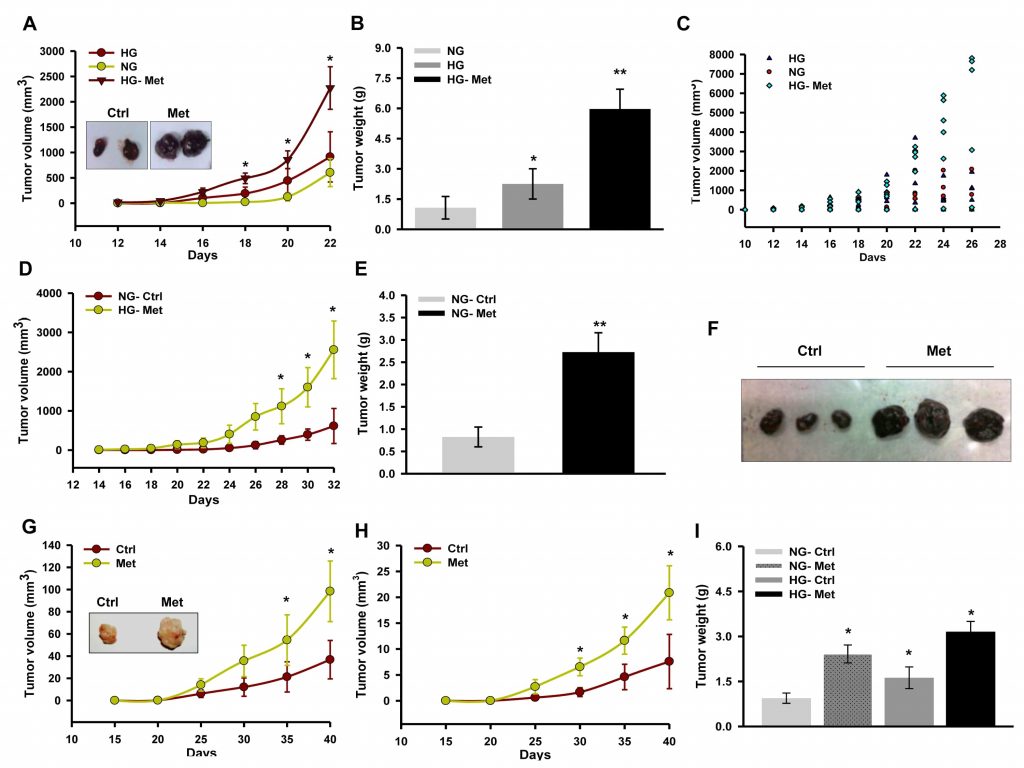
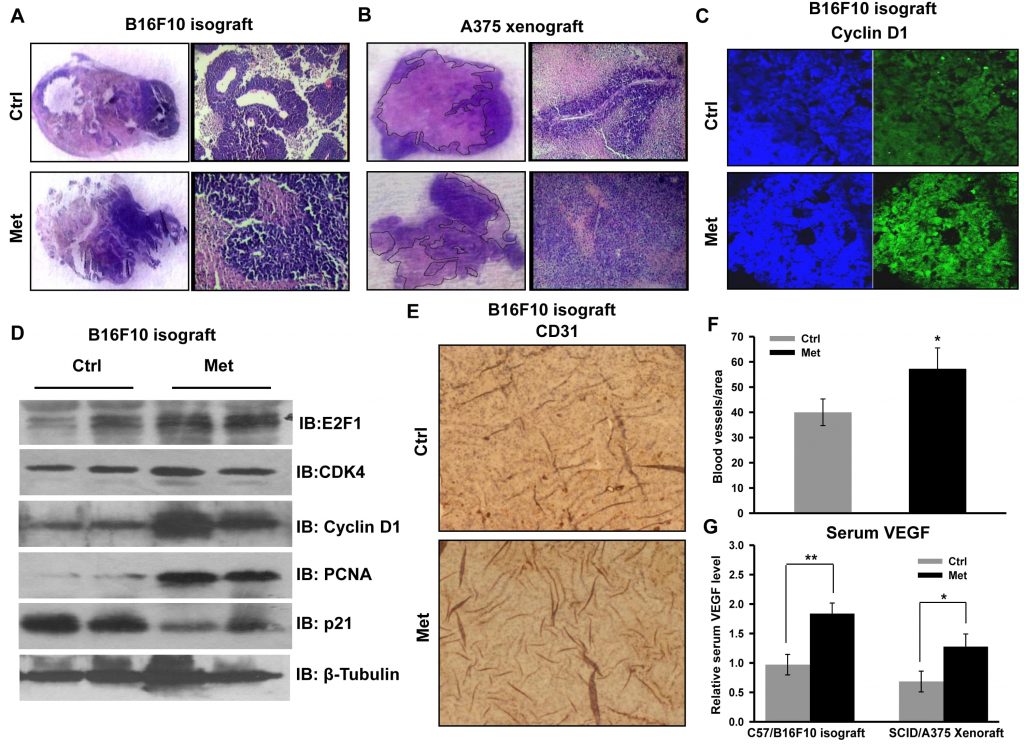
Pour vérifier les événements cellulaires et moléculaires associés à une progression tumorale accrue, des sections de tumeurs ont été examinées pour une analyse histopathologique. Une densité cellulaire élevée et une nécrose réduite étaient clairement visibles dans les sections des deux types de tumeurs (isogreffe dérivée de B16F10 et xénogreffe dérivée de A375) chez les souris ayant reçu de la metformine (Figure 2A et 2B). Nous avons noté que la prolifération et la progression de la xénogreffe dérivée de l’A375 étaient phénotypiquement distinctes par rapport à la tumeur témoin. Cela suggère un avancement de grade de la tumeur primaire, mis en évidence par la morphologie allongée des noyaux par rapport à la morphologie arrondie dans les sections de tumeurs de contrôle (informations non publiées). La coloration immunohistochimique de la protéine régulatrice du cycle cellulaire, la cycline D1 (Figure 2C), s’est avérée plus élevée dans les sections de tumeurs des souris ayant reçu de la metformine. De plus, nous avons validé la croissance tumorale accrue en vérifiant le statut des protéines régulatrices du cycle cellulaire par immunoblotting des lysats tumoraux. Nous avons constaté que les niveaux des molécules cycline D1, CDK4, E2F1 et PCNA étaient significativement augmentés dans les lysats tumoraux des souris ayant reçu la metformine par rapport au contrôle, tandis que le niveau de p21 était diminué (Figure 2D). Ces résultats indiquent que la metformine, indépendamment du statut glycémique des souris, favorise la croissance du mélanome en modulant les protéines régulatrices du cycle cellulaire. De plus, l’analyse immunohistochimique des coupes de tumeurs a renforcé cette observation, car le traitement à la metformine a augmenté les niveaux de protéines de CD31, un marqueur endothélial (Figure 2E et 2F), et a augmenté le niveau sérique de VEGF (Figure 2G), suggérant que la metformine favorise l’angiogenèse dans les tumeurs de mélanome.
Ensuite, nous avons vérifié l’effet de la metformine sur la croissance et la prolifération des cellules de mélanome in vitro. Contrairement à nos résultats in vivo, le traitement à la metformine a entraîné une suppression de la croissance des cellules de mélanome in vitro (figures supplémentaires S1A, S1B, S1C et S1D). Nous avons ensuite étudié l’impact de l’inhibition du complexe I en utilisant la metformine et la phenformine sur la croissance des cellules de mélanome, car ces deux substances inhibent l’activité du complexe I (figure supplémentaire S2). Nous avons constaté que la metformine et la phenformine provoquaient un arrêt de la croissance des cellules de mélanome cultivées en présence de glucose élevé. Cependant, en présence d’un faible taux de glucose, le traitement avec ces agents a entraîné la mort des cellules (figures supplémentaires S3A, S3B, S3C et S3D). Il est probable que l’arrêt de croissance médié par la metformine soit dû à la réduction du taux de glucose et à l’acidification extracellulaire du milieu (figures supplémentaires S4A et S4B). Il est intéressant de noter que le remplacement du milieu toutes les 12 heures par un milieu frais contenant 25 mM de glucose a augmenté la survie clonogénique lors du traitement à la metformine. Comme les cellules traitées à la metformine ont utilisé le glucose très rapidement par rapport au témoin, le renouvellement du milieu est essentiellement nécessaire pour maintenir le niveau de glucose et le pH, et donc la survie des cellules en présence de metformine (Figure supplémentaire S4C et S4D). Ces résultats suggèrent que la concentration de glucose disponible dans le milieu de culture influence l’action de la metformine.
L’inhibition de l’activité du complexe respiratoire I favorise la glycolyse aérobie <p><strong>Dossiers enzymatiques<br></strong>Les cellules ont été homogénéisées dans un tampon phosphate de potassium hypotonique (20 mM) (pH 7,5) contenant un cocktail d’inhibiteurs de protéases (Roche, Allemagne), vortexées et lysées par trois cycles de procédure de congélation et décongélation. Pour préparer l’extrait de tissus, les échantillons de tumeurs ont été coupés en fins morceaux et homogénéisés dans un tampon d’homogénéisation (Tris 0,1 M, KCl 0,1 M, EDTA 350 mM et saccharose 1 M, pH 7,5) en utilisant un homogénéisateur de tissus. Les homogénats ont été clarifiés par centrifugation à 10 000 rpm pendant 10 min à 4<sup>°</sup>C et conservés sur la glace jusqu’à la réalisation des essais.</p> <p><strong><em>Dosage de l’activité LDH:</em> </strong>L’activité de la lactate déshydrogénase dans les lysats cellulaires ou l’extrait de tumeur et le sérum a été déterminée à l’aide du kit de dosage de l’activité LDH (Spinreact, Espagne) selon les instructions du fabricant.</p> <p> <p>Les auteurs remercient le Dr S.C. Mande, directeur du NCCS, Pune, Inde, et le Dr G.C. Mishra, ancien directeur du NCCS, Pune, Inde, pour avoir été d’un grand soutien et avoir donné tous les encouragements nécessaires à la réalisation de ce travail. Nous remercions le Dr Benoit Violet (INSERM, France) pour avoir fourni les MEF, le Dr Mahesh J Kulkarni (National Chemical Laboratory, Inde) pour les cellules L6 et le Dr Rakesh K Tyagi (Jawaharlal Nehru University, Inde) pour avoir fourni les cellules AML12. Nous remercions également le Dr Vijayakumar MV pour son aide dans les expériences sur les animaux et pour sa lecture critique du manuscrit. BC et NM remercient le Council of Scientific and Industrial Research (CSIR), Inde ; PM et SVS remercient la University Grant Commission (UGC), Inde pour l’octroi d’une bourse. Le soutien des autres membres du laboratoire et des collègues du NCCS, ainsi que des membres du personnel de l’animalerie expérimentale, de l’installation FACS, confocale et de spectrométrie de masse est dûment reconnu.</p> <p>Les auteurs déclarent qu’ils n’ont pas de conflits d’intérêts potentiels</p> <p>Ce travail a été réalisé dans le cadre de l’accomplissement partiel d’une thèse de doctorat (de B.C.) qui sera soumise à l’Université Savitribai Phule Pune, Pune, Inde.</p>
La divergence des résultats du traitement à la metformine in vivo et in vitro nous a incités à en rechercher la cause. Il est connu que la metformine exerce son action principalement en inhibant le complexe I de l’enzyme OXPHOS mitochondriale <a href= »#[« > [</a><a href= »https://www.oncotarget.com/article/6134/text/#bib27″>27</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib29″>29</a><a href= »#] »>]</a></sup> et l’inhibition du complexe I entraîne une augmentation de la croissance et de la prolifération tumorales<sup><a href= »#14″> [</a><a href= »https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href= »#] »>]</a></sup>. Comme nous avons observé que le traitement à la metformine provoquait une acidification du milieu de culture en raison de l’augmentation de la glycolyse aérobie (Figures supplémentaires 4A et 4B), nous avons donc supposé que cela pouvait être la raison de l’arrêt de la croissance cellulaire. Nous avons donc mesuré le taux de glucose et de lactate dans le milieu de culture usé provenant des cellules témoins et des cellules traitées à la metformine. Une utilisation accrue du glucose (comme en témoigne la présence de moins de glucose résiduel dans le milieu) et un niveau de lactate plus élevé ont été détectés dans les cellules traitées à la metformine par rapport au contrôle (Figure 3A et 3B).</p>
</p> <p>Pour renforcer ces résultats, nous avons exploré plus avant le schéma d’expression et l’activité de certaines des protéines et enzymes impliquées dans la régulation de la glycolyse. Nous avons constaté que la metformine augmentait les niveaux de protéines des molécules glycolytiques clés GLUT1, LDHA et ChREBP d’une manière dépendante de la concentration (Figure 3C). Ces résultats suggèrent que l’inhibition de l’activité du complexe I par la metformine pourrait éventuellement contraindre les cellules cancéreuses à adopter la voie glycolytique pour la génération d’ATP provoquant une amélioration de la progression tumorale chez la souris.</p>
</p> <p>Pour évaluer si le phénotype glycolytique peut également être induit <em>in vivo</em> lors de l’inhibition du complexe I, nous avons d’abord vérifié le niveau de glucose dans le sérum collecté chez des souris porteuses de tumeurs en même temps que des souris normoglycémiques traitées ou non par la metformine. Une baisse significative du taux de glucose sérique a été observée chez les souris normoglycémiques porteuses de tumeurs et traitées par la metformine (104,3 ± 13,7 mg/dl chez les C57BL/6J et 134,0 ± 7,2 mg/dl chez les NOD/SCID) par rapport au contrôle non traité (139,7 ± 16,3 mg/dl chez les C57BL/6J et 168,0 ± 8,5 mg/dl chez les NOD/SCID) (Figures 3D et 3E). De même, le traitement à la metformine a entraîné une réduction du glucose sérique chez les souris hyperglycémiques porteuses de tumeurs (Met, 237,3 ± 30,0 mg/dl chez les C57BL/6J, et 227,3 ± 12,3 mg/dl chez les NOD/SCID) par rapport au contrôle (388 ± 31,5 mg/dl chez les C57BL/6J, et 409,3 ± 18,6 mg/dl chez les NOD/SCID) (Figures 3D et 3E). La sécrétion de lactate est une caractéristique des tumeurs glycolytiques. Nous avons donc vérifié le taux de lactate, qui s’est avéré très élevé dans le sérum des souris normoglycémiques et hyperglycémiques porteuses de tumeurs et traitées à la metformine (Figure 3F et 3G). Une LDH sérique élevée est associée à des tumeurs agressives de haut grade et à un mauvais pronostic en cas de mélanome métastatique <sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib8″>8</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href= »#] »>]</a></sup>. Ainsi, l’activité LDH sérique a été surveillée et s’est avérée très élevée dans le sérum recueilli chez les souris hyperglycémiques et normoglycémiques administrées avec la metformine par rapport au contrôle (Figure<a href= »https://www.oncotarget.com/article/6134/text/#F3″> </a>3H et 3I). En outre, une augmentation de l’activité LDH a été détectée dans les tissus tumoraux des souris administrées avec de la metformine par rapport au contrôle non traité (Figure 3J).</p>
<p><strong>L’inhibition du complexe I conjointement avec la LDH ou la génération de lactate est synthétiquement létale pour les cellules de mélanome<br></strong>Pour explorer la possibilité que la metformine favorise la glycolyse ou que la génération excessive de lactate soit la cause principale de la croissance et de la prolifération accrues des tumeurs de mélanome, la génération de lactate a été inhibée en utilisant l’oxamate, un inhibiteur de la LDH, et le DCA, un inhibiteur de la PDK1. Nous avons constaté que l’oxamate et le DCA seuls ont un effet inhibiteur sur la croissance des cellules de mélanome (Figure 4A, 4B et 4C ; Figure supplémentaire S5). Une réduction de la survie des cellules a été observée lors du traitement par l’oxamate ou le DCA avec la metformine (Figure 4A, 4B et 4C). De même, d’autres inhibiteurs glycolytiques bien connus comme la phlorétine (inhibiteur du GLUT1), le 2-désoxy-D-glucose (2DG) et l’iodoacétate ont également limité la prolifération des cellules de mélanome traitées par la metformine (figures supplémentaires S6A et S6B). L’augmentation de l’absorption du glucose par GLUT1 et de la glycolyse aérobie par LDHA et PDK1 est la caractéristique des cellules cancéreuses. Par conséquent, nous avons choisi l’oxamate et le DCA plutôt que d’autres inhibiteurs glycolytiques.</p>
<p>Puis, pour vérifier la capacité de survie à long terme des cellules de mélanome en présence de metformine et d’oxamate/DCA seuls ou en combinaison ensemble, nous avons effectué un test de survie clonogénique. Aucune colonie survivante n’a été détectée dans les cellules de mélanome traitées par l’oxamate et la metformine ensemble (Figure<a href= »https://www.oncotarget.com/article/6134/text/#F4″> </a>4D, 4E et 4F). Des résultats similaires ont également été obtenus dans les cellules traitées par la metformine et le DCA ensemble (Figure 4D, 4E et 4F). Par la suite, nous avons vérifié si l’inhibition de ces deux enzymes induisait la mort des cellules de mélanome, comme le montrent les colorations à l’Annexin V et au PI. Tous ces agents ont provoqué à eux seuls un arrêt de la croissance, mais n’ont pas induit d’apoptose. Cependant, une augmentation de l’apoptose a été détectée dans les cellules A375 et B16F10 traitées avec l’oxamate/DCA et la metformine, par rapport à l’agent unique (figures 5A et 5B). Cette observation a été confirmée par immunoblotting pour les marqueurs apoptotiques PARP1, Bax et Bcl-2 (figures 5C et 5D). Une augmentation du clivage de PARP et du niveau de Bax, ainsi qu’une diminution du niveau de la protéine antiapoptotique Bcl-2 ont été détectées dans les cellules traitées par la metformine et l’oxamate/DCA ensemble, par rapport à l’agent unique (Figure 5C et 5D). Ces résultats indiquent que l’inhibition du complexe I et de la LDH ensemble peut favoriser l’induction de l’apoptose.</p>
<p>Puis, nous avons vérifié l’impact de la metformine et de l’oxamate/DCA, seuls ou en association, sur la survie des cellules non cancéreuses. Nous avons utilisé trois différentes lignées cellulaires non cancéreuses à savoir AML12 (hépatocytes de souris), L6 (cellules musculaires de rat) et MEF (fibroblastes embryonnaires de souris) pour tester l’effet du traitement combiné. Bien que, la combinaison de la metformine et de l’oxamate/DCA ait ralenti la prolifération de ces cellules (Figure supplémentaire S7A, S7B et S7C), elle n’a pas affecté la viabilité cellulaire (Figure supplémentaire S7D), ce qui indique que cette combinaison est efficace pour tuer sélectivement les cellules cancéreuses.</p>
<h3></h3>
<p><strong>Le blocage du complexe I et de la génération de lactate impose une catastrophe métabolique<br></strong>Pour s’assurer que le co-traitement par oxamate et metformine affecte le métabolisme des cellules de mélanome, nous avons mesuré les paramètres métaboliques en présence de ces inhibiteurs. Le traitement par la metformine a augmenté la glycolyse, tandis que le traitement par l’oxamate a entraîné une diminution de l’utilisation du glucose et de la sécrétion de lactate (Figure 6A et 6B). Il est intéressant de noter que l’utilisation du glucose et la sécrétion de lactate ont été inhibées de manière significative lors du traitement des cellules avec l’oxamate et la metformine ensemble (Figure 6A et 6B). De plus, l’oxamate ainsi que la metformine ont réduit les niveaux d’ATP en inhibant la phosphorylation du substrat et l’OXPHOS respectivement (Figure 6C). En outre, une baisse significative du niveau d’ATP a été détectée dans les cellules traitées par l’oxamate et la metformine ensemble (Figure 6C). En outre, pour confirmer l’implication de la LDH dans la croissance et la prolifération des cellules de mélanome, un knock down de la LDHA (LDHA-KD) a été réalisé en utilisant un siRNA spécifique (Figure 6D). Une augmentation de la mort cellulaire a été observée dans les cellules LDHA-KD traitées avec de la metformine ou de la phenformine (Figure 6E). Une réduction significative de la glycolyse (mise en évidence par une diminution de l’utilisation du glucose et de la sécrétion de lactate) et du niveau d’ATP a été observée dans les cellules LDHA-KD par rapport au contrôle (Figure 6F, 6G et 6H). De plus, une diminution du taux glycolytique ainsi qu’une forte baisse du niveau d’ATP ont été observées dans les cellules LDHA-KD traitées avec de la metformine ou de la phenformine (Figure 6F, 6G et 6H). Ces résultats indiquent que l’inhibition simultanée du complexe I et de la voie de génération de LDH ou de lactate induit une catastrophe métabolique en raison de la perturbation du pool d’ATP cellulaire, conduisant finalement à l’arrêt de la croissance et à la mort cellulaire.</p>
<p><strong>L’inhibition simultanée du complexe I et de la LDH par la metformine et l’oxamate retarde la progression tumorale chez la souris<br></strong>Pour vérifier si la combinaison de l’oxamate et de la metformine est également efficace pour diminuer la progression tumorale, des tumeurs ont été développées en injectant des cellules B16F10 à des souris C57BL/6J. Une fois que les tumeurs ont atteint une taille de 50 mm<sup>3</sup>, les souris ont été réparties au hasard en quatre groupes : (a) contrôle, (b) metformine, (c) oxamate, et (d) souris traitées conjointement par la metformine et l’oxamate. Nous avons remarqué que les souris traitées à la metformine ont développé des tumeurs plus importantes que les souris témoins non traitées (Figure 7A, 7B et 7C). Alors que chez les souris traitées à l’oxamate, les tumeurs ont progressé plus lentement que chez le témoin non traité ainsi que chez les souris traitées à la metformine (Figure 7A). De manière intéressante, nous avons observé que l’oxamate retardait considérablement la progression des tumeurs lorsqu’il était administré en même temps que la metformine, comme en témoigne la réduction du volume et du poids des tumeurs (Figure 7A, 7B et 7C).</p>
</p> <p>En outre, pour confirmer si l’abrogation de la progression tumorale induite par la metformine par l’oxamate est une conséquence de la réduction de la glycolyse aérobie, nous avons mesuré les paramètres métaboliques <em>in vivo</em>. Les souris administrées par la metformine présentaient un faible taux de glucose sérique, des taux de lactate et de LDH plus élevés dans le sérum par rapport au contrôle, alors qu’un taux de glucose sérique relativement plus élevé et un faible taux de lactate ont été détectés chez les souris traitées par l’oxamate seul ou par la metformine en même temps que l’oxamate (Figure 7D et 7E). L’activité de l’enzyme LDH était significativement diminuée dans les tumeurs excisées chez les souris traitées par l’oxamate seul ou par la metformine en association avec l’oxamate (Figure 7F). Un niveau relativement très faible d’ATP a été détecté dans les tumeurs des souris ayant reçu à la fois l’oxamate et la metformine, par rapport à l’agent unique. Cependant, nous n’avons pas observé de changements significatifs dans les niveaux de protéines des molécules régulant la glycolyse (Figure 7I). Il est important de noter qu’aucun changement significatif du poids corporel n’a été détecté chez les souris recevant le traitement combiné et qu’aucun effet secondaire indésirable possible ou symptôme brut de toxicité généralisée n’a été observé lors de l’inspection visuelle (figure supplémentaire S8A). De plus, aucune anomalie pathologique n’a été observée dans les organes vitaux comme les poumons, le foie, les reins et le cœur de ces souris (figure supplémentaire S8B). Ces résultats suggèrent que cette combinaison n’exerce pas de toxicité généralisée et qu’elle est efficace pour retarder la progression tumorale chez les souris.</p>
<h2>DISCUSSION</h2>
<p>Les cellules cancéreuses, y compris les cellules de mélanome, détournent une énorme quantité de flux de carbone vers la glycolyse <sup><a href= »#5″>[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib5″>5</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href= »#] »>]</a></sup>. Cela aide les cellules à générer de l’ATP et d’autres éléments constitutifs pour une croissance tumorale rapide <sup><a href= »#5″>[5]</a></sup>. Plusieurs rapports suggèrent que le ciblage des voies métaboliques réduit la progression tumorale et la croissance cellulaire <em>in vitro</em> <sup><a href= »#40″>[40-</a><a href= »#42″>42]</a></sup>. Dans la présente étude, nous démontrons que la metformine favorise la croissance du mélanome en augmentant la glycolyse en raison de l’inhibition de la fonction du complexe I, tandis que l’inhibition de la LDH entraîne un arrêt de la croissance des cellules. L’inhibition de la génération de lactate dans les cellules de mélanome traitées par la metformine affecte la survie cellulaire provoquant ainsi une réduction de la progression tumorale (Figure 8).</p>
</p> <p>Les conséquences de l’inhibition du complexe I sur la croissance des cellules cancéreuses ne sont pas clairement comprises bien que des rapports suggèrent que l’inhibition du complexe I entraîne une transformation cellulaire et augmente la croissance cellulaire<a href= »#[« > <sup>[</sup></a><sup><a href= »https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href= »#] »>]</a></sup>. Inversement, l’inhibition du complexe I a également été signalée pour retarder la progression tumorale chez les souris <sup><a href= »#30″>[30]</a></sup>. Ces divers résultats sont susceptibles de dépendre de la disponibilité des nutriments dans le microenvironnement tumoral. Il a été rapporté que lorsque le glucose est disponible en abondance, l’inhibition du complexe I favorise la glycolyse et la prolifération rapide des cellules, alors qu’elle induit la mort cellulaire dans des conditions de stress métabolique<sup><a href= »#43″> [43]</a></sup>. Dans cette étude, nous avons observé que l’inhibition du complexe I par la metformine favorise la progression tumorale du mélanome chez la souris.</p>
</p> <p>Alors qu’il a été démontré que la metformine inhibe la prolifération de nombreuses cellules cancéreuses et limite également la progression tumorale des xénogreffes chez la souris <sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib33″>33</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib34″>34</a><a href= »#] »>]</a></sup>, il est connu pour exercer des effets inhibiteurs de croissance ainsi que des effets promoteurs de croissance dans le mélanome<sup> </a><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib35″>35</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib37″>37</a><a href= »#] »>]</a></sup>. De manière intéressante, nous avons détecté que les tumeurs dérivées de B16F10 et A375 se développaient rapidement chez les souris administrées avec de la metformine et que le taux de croissance variait en fonction du statut glycémique des souris (Figure 2D et 2E). Cette observation concorde avec un rapport récent démontrant que la metformine facilite la progression tumorale des cellules de mélanome mutant BRAF<sup><a href= »http://335″> [35]</a></sup>.</p>
<p>La disparité de l’action de la metformine sur la croissance des cellules de mélanome <em>in vitro</em> et <em>in vivo</em> est susceptible d’être influencée par la disponibilité du glucose et la concentration en lactate qui pourraient influencer son action. La metformine inhibe la croissance cellulaire et induit l’apoptose dans des conditions limitant le glucose <sup><a href= »#43″>[43]</a></sup>, alors que nous avons remarqué qu’en présence d’un glucose élevé, elle provoque un arrêt de croissance sans affecter la mort cellulaire. Nous avons expliqué cette divergence par le fait que les cellules cancéreuses dérivent l’ATP <em>via</em> l’OXPHOS sous stress métabolique, alors que lorsque le glucose est abondant, l’ATP est dérivé principalement par la glycolyse. L’inhibition du complexe I dans des conditions de glucose élevé favorise davantage la glycolyse, ce qui entraîne une production excessive de lactate, et l’occurrence de ce phénomène a été signalée précédemment<sup><a href= »#44″> [44]</a></sup>. Collectivement, ces résultats suggèrent que l’arrêt de croissance induit par la metformine dans les cellules de mélanome <em>in vitro</em> est une conséquence de l’acidification du milieu due à l’accumulation excessive d’acide lactique en raison de l’utilisation rapide du glucose disponible. Notamment, l’arrêt de croissance induit par la metformine peut être évité en renouvelant fréquemment le milieu pour maintenir des conditions de croissance optimales, car les cellules traitées par la metformine ont besoin de plus de glucose pour proliférer en raison d’une glycolyse aérobie accrue. Cela imite les conditions <em>in vivo</em> dans lesquelles la circulation probablement rapide et constante de l’acide lactique d’accès permet aux cellules cancéreuses d’utiliser plus de glucose, ce qui favorise une prolifération rapide. De plus, le lactate peut également être utilisé par les cellules cancéreuses dans les <sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib45″>45</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib46″>46</a></sup><a href= »#] »><sup>]</sup></a>. De plus, il a été rapporté que la metformine induit l’angiogenèse <em>via</em> l’augmentation du niveau de VEGF<sup><a href= »#[« > [</a><a href= »https://www.oncotarget.com/article/6134/text/#bib35″>35</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib47″>47</a><a href= »#] »>]</a></sup>. Nous rapportons que l’inhibition du complexe I augmente la production de lactate et il a été précédemment suggéré que le switch glycolytique améliore l’angiogenèse<sup> <a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib48″>48</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib49″>49</a><a href= »#] »>]</a></sup>. Nos résultats montrent une corrélation positive entre le lactate sérique et le niveau de VEGF chez les souris administrées avec la metformine. Par conséquent, il est probable que la croissance tumorale favorisée par la metformine soit facilitée par l’angiogenèse induite par le lactate qui peut être médiée par le VEGF <sup><a href= »#48″>[48]</a></sup>. De plus, nous avons observé une augmentation du niveau de E2F1, une importante molécule régulatrice du cycle cellulaire, dans les tumeurs excisées des souris administrées avec la metformine. En plus de favoriser la prolifération cellulaire, E2F1 régule également de nombreux gènes impliqués dans la glycolyse<sup><a href= »#50″> [50]</a></sup> qui est essentielle pour la croissance rapide des cellules cancéreuses. Par conséquent, il est probable que, E2F1 pourrait jouer un rôle important dans la médiation de la progression tumorale rapide induite par la metformine probablement <em>via</em> la régulation de la glycolyse aérobie.</p>
</p> <p>Les inhibiteurs glycolytiques tels que le 2-désoxy glucose, le lonimide, le 3-bromopyruvate, le DCA, et les médicaments interférant avec les voies métaboliques ont montré des résultats prometteurs dans la suppression de la croissance tumorale<sup> <a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib40″>40</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib42″>42</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib51″>51</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href= »#] »>]</a></sup>. De plus, cibler la voie de génération du lactate est attrayant notamment dans les cancers glycolytiques <sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib54″>54</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib55″>55</a><a href= »#] »>]</a></sup>. Les cellules de mélanome sont métaboliquement glycolytiques, et dépendent donc principalement de la forte activité de la LDH pour la génération d’ATP <sup><a href= »#6″>[6]</a></sup>. Les cellules de mélanome surexpriment la LDH et un niveau sérique élevé est souvent corrélé avec un mauvais pronostic et la survie des patients<sup> <a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib8″>8</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href= »#] »>]</a></sup>. Nos résultats indiquent que la perturbation de la conversion du pyruvate en lactate affecte profondément la prolifération des cellules de mélanome. Ceci est en accord avec les rapports selon lesquels les tumeurs des types de cellules glycolytiques sont plus sensibles à l’inhibition de la LDH-A avec FX11<sup><a href= »#56″> [56]</a></sup>.</p>
<p>Le DLH joue un rôle important dans l’homéostasie métabolique et le maintien des tumeurs<sup> <a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib12″>12</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib57″>57</a><a href= »#] »>]</a></sup>. Nous avons utilisé l’oxamate et le DCA pour inhiber la génération de lactate dans les cellules de mélanome. Le DCA est une petite molécule délivrable par voie orale qui a été utilisée pour le traitement de l’acidose lactique, et il a été démontré que la restauration de la fonctionnalité OXPHOS par le DCA induit la mort cellulaire<sup><a href= »#[« > [</a><a href= »https://www.oncotarget.com/article/6134/text/#bib52″>52</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href= »#] »>]</a></sup>. En outre, il a été précédemment démontré que le DCA réduit la production de lactate et déclenche l’apoptose dans les cellules de mélanome <sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib58″>58</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib59″>59</a><a href= »#] »>]</a></sup>. L’oxamate, un analogue du pyruvate, est également utilisé pour inhiber la LDH. Une étude récente de Miskimins et al, suggère que le ciblage conjoint du complexe I et de la LDH peut être une stratégie prometteuse pour arrêter la croissance des cancers agressifs <sup><a href= »#60″>[60]</a></sup>. En concordance avec cela, nous avons également remarqué que la prévention de la génération de lactate et l’inhibition simultanée du complexe I par l’oxamate/DCA ou par le siRNA spécifique de la LDH et la metformine, respectivement, entraîne un appauvrissement du pool d’ATP cellulaire. La diminution du pool d’ATP cellulaire provoque une catastrophe métabolique qui entraîne l’apoptose des cellules de mélanome. La mort cellulaire induite par la catastrophe métabolique est considérée comme une stratégie prometteuse pour arrêter la progression du cancer<sup><a href= »#61″> [61]</a></sup>. De plus, notre étude signifie le rôle distinct du complexe I et de la LDH dans la prolifération cellulaire, et ces deux constituent la voie de génération d’ATP soit par OXPHOS ou par phosphorylation au niveau du substrat respectivement. Le blocage de l’une ou l’autre des enzymes seules n’induit pas la mort cellulaire car les cellules peuvent passer à une autre voie de génération d’ATP. En revanche, l’inhibition simultanée de ces deux enzymes induit l’apoptose. Cela indique clairement que les cellules dépendent de la fonction de ces deux enzymes qui forment une paire synthétiquement létale, ce qui est un phénomène prometteur qui peut être employé pour le ciblage sélectif des cellules de mélanome<a href= »#[« > </a><sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib62″>62</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib63″>63</a><a href= »#] »>]</a></sup>. Étant donné que E2F1 est impliqué dans la régulation de l’apoptose <sup><a href= »#64″>[64]</a></sup>, il est probable que l’apoptose induite par l’association de la metformine et de l’oxamate/DCA puisse impliquer E2F1. La metformine favorise l’apoptose des cellules cancéreuses <em>via</em> l’activation de p53 <sup><a href= »#37″>[37]</a></sup>. Cependant, dans le cas du mélanome, le rôle de p53 n’est pas clair. Comme la littérature le suggère, p53 est non fonctionnel dans le mélanome et ses niveaux sont paradoxalement élevés dans les grades avancés de mélanome <sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib65″>65</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib67″>67</a><a href= »#] »>]</a></sup>. Précédemment, nous avons signalé qu’un niveau élevé de p53 est associé à une augmentation de la croissance tumorale<sup><a href= »#68″> [68]</a></sup>. Par conséquent, il est peu probable que p53 soit impliqué dans l’apoptose induite par le traitement combiné et, ainsi, d’autres études sont nécessaires pour évaluer la fonctionnalité de E2F1 et p53 dans le mélanome.</p>
</p> <p>Lorsque l’on utilise des combinaisons de différents médicaments qui peuvent favoriser de manière synergique la mort des cellules cancéreuses, cela peut également renforcer la toxicité pour les cellules normales. Par conséquent, il est crucial d’accéder à l’impact du traitement combiné sur les cellules normales. Il est important de noter que nos données suggèrent que cette combinaison est efficace pour tuer les cellules cancéreuses aussi bien <em>in vitro</em> que <em>in vivo</em>, et a le moins d’impact sur la survie des cellules normales. La sensibilité différentielle entre les cellules de mélanome et les cellules normales due à la combinaison metformine et oxamate/DCA peut être attribuée au fait que les cellules de mélanome sont hautement glycolytiques et surexpriment des molécules impliquées dans la génération et la sécrétion de lactate par rapport aux cellules normales <sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib6″>6</a>-<a href= »https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href= »#] »>]</a></sup>. Il est intéressant de noter que, par rapport aux cellules normales, les cellules cancéreuses présentent une respiration mitochondriale découplée plus élevée<sup><a href= »#31″> [31]</a></sup>. Lors d’un traitement à la metformine, les cellules cancéreuses présentent une plus grande élévation compensatoire de la glycolyse que les cellules normales <sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib31″>31</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib32″>32</a><a href= »#] »>]</a></sup>, pesant ainsi sur la susceptibilité métabolique des cellules cancéreuses. Par conséquent, l’inhibition de la LDH ou de la génération/sécrétion de lactate supprime la croissance des cellules cancéreuses alors que les cellules normales sont le moins affectées car une quantité suffisante d’ATP peut encore être produite à partir de l’OXPHOS, la metformine étant un inhibiteur faible du complexe I. Comme l’activité OXPHOS est plus élevée dans les cellules normales par rapport aux cellules de mélanome, l’association metformine et oxamate/DCA est susceptible d’avoir le moins d’impact négatif sur la respiration des cellules normales.</p>
</p> <p>Notre étude ouvre de nouvelles voies pour cibler le métabolisme des cellules cancéreuses et peut être davantage impliquée dans le test d’autres médicaments cliniquement approuvés connus pour inhiber la glycolyse avec la metformine. La présente étude suggère que tout médicament/inhibiteur qui bloque la génération de lactate peut être utilisé en association avec la metformine pour une meilleure prise en charge et la prévention de la croissance tumorale. Par exemple, il est démontré que la rapamycine, un médicament cliniquement approuvé, empêche la génération de lactate<sup> <a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib69″>69</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib70″>70</a><a href= »#] »>]</a></sup>. De même, il a été montré que l’inhibition de BRAF entraîne une suppression de la glycolyse <sup><a href= »#[« >[</a><a href= »https://www.oncotarget.com/article/6134/text/#bib6″>6</a>, <a href= »https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href= »#] »>]</a></sup>. Par conséquent, la rapamycine ainsi que les inhibiteurs de BRAF peuvent être utilisés avec la metformine pour améliorer le résultat thérapeutique avec des effets secondaires réduits. Collectivement, nos résultats indiquent que la LDH ou d’autres mécanismes qui contrôlent la génération ou la sécrétion de lactate sont essentiels pour la progression rapide du mélanome dans des conditions où l’OXPHOS est compromise, et cela peut être exploité comme une cible thérapeutique appropriée pour contrôler la croissance des cellules cancéreuses glycolytiques. Des études plus approfondies sont nécessaires pour évaluer la fonctionnalité de la LDH et du complexe I dans d’autres modèles de cancer, et par la suite, leur implication dans la thérapie du cancer en général.</p>
<h2>MATERIELS ET METHODES</h2>
<p><strong>Expériences sur les animaux<br></strong>Les souris ont été obtenues auprès de l’Experimental Animal Facility (EAF) du National Centre for Cell Science (NCCS), Pune, Inde. L’hyperglycémie chez les souris a été induite en utilisant le STZ comme décrit précédemment<sup><a href= »#71″> [71]</a></sup> avec de légères modifications. En bref, les souris C57BL/6J et NOD/SCID mâles ont été mises à jeun pendant 6 heures avant l’injection intrapéritonéale de STZ (50 mg/kg) dans un tampon citrate 0,01 M (pH 4,4) pendant trois jours consécutifs. La mesure de la glycémie a été effectuée en pinçant la queue et en appliquant le sang sur un analyseur de glucose (Accu-Chek Active, Roche, Allemagne). Les souris ayant une valeur de glycémie supérieure à 200 mg/dl ont été considérées comme hyperglycémiques. La tumeur a été induite en injectant des cellules B16F10 (2 × 10<sup>5</sup>) ou A375 (1 × 10<sup>6</sup>) dans 100 µl de PBS stérile par voie sous-cutanée au niveau du flanc droit de souris C57BL/6J et NOD/SCID respectivement. La progression de la tumeur a été suivie régulièrement en mesurant sa taille avec un pied à coulisse numérique (Sigma, USA) suite à l’apparition de tumeurs palpables. L’administration orale de metformine un jour sur deux a été initiée une semaine avant le test de provocation de la tumeur chez les souris hyperglycémiques. Sinon, la metformine n’a été administrée qu’après que la tumeur ait atteint une taille optimale et le traitement a été poursuivi jusqu’à la fin de l’expérience. A la fin de l’expérience, les souris ont été sacrifiées ; les tumeurs ont été excisées et stockées soit à -80<sup>°</sup>C soit dans une solution de formol à 10% pour des études ultérieures. Dans une autre série d’expériences, les souris (fond C57BL/6J, mâles) ont été divisées au hasard en 4 groupes après l’apparition de la tumeur palpable. Pour examiner le résultat du traitement par la metformine en association avec l’oxamate, un inhibiteur de la LDH, les souris ont reçu par voie orale soit de la metformine (200 mg/kg), soit de l’oxamate seul (500 mg/kg), soit les deux ensemble, un jour sur deux. La progression de la tumeur a été régulièrement suivie à l’aide d’un pied à coulisse. A la fin de l’expérience, les tissus tumoraux, musculaires et hépatiques ont été excisés, pesés et conservés à -80<sup>°</sup>C pour des analyses ultérieures. Toutes les expériences sur les animaux ont été réalisées conformément aux directives du comité pour le contrôle et la supervision des expériences sur les animaux (CPCSEA), gouvernement de l’Inde, et après avoir obtenu la permission du comité d’éthique animale de l’institut (IAEC).</p>
<p><strong>Lignées cellulaires et conditions de culture<br></strong>Les cellules de mélanome murin B16F10, de mélanome humain A375 et SKMel28 ont été achetées auprès de l’ATCC (VA, USA) et maintenues dans notre dépôt interne. Des cellules non cancéreuses, AML12 (hépatocytes de souris), L6 (cellules musculaires de rat) et MEF (fibroblastes embryonnaires de souris) ont été utilisées en parallèle avec des cellules cancéreuses comme contrôle. Toutes les cellules ont été cultivées dans leur milieu respectif contenant soit 1 mM ou 25 mM de glucose en fonction des expériences et complété par 10% de sérum fœtal bovin inactivé par la chaleur (Hyclone, UT, USA), de la pénicilline (100 U/ml) et de la streptomycine 100 µg/ml (Life Technologies, NY, USA), à 37<sup>°</sup>C en présence de 5%<sub> </sub>CO<sub>2</sub>.</p>
</p><strong>Chimiques et réactifs<br></strong>Metformine, phenformine, oligomycine, roténone, décylubiquinone, streptozotocine (STZ), [bromure de 3-(4, 5-diméthylthiazol-2-yl)-2, 5diphénylterazolium] (MTT), NAD+, NADH, ATP, AMP, D-glucose, oxamate de sodium, iodoacétate, 2-désoxy-D-glucose, dichloroacétate (DCA), Diaminobenzidine (DAB) et phlorétine ont été achetés chez Sigma (MO, USA). Les anticorps pour ChREBP (1:1000), GLUT1 (1:1000), LDHA (1:1000), Cyclin D1 (1:1000), PCNA (1:1000), CDK4 (1:1000), p21 (1:1000), E2F1 (1:1000) CD31 (1:100), PARP-1 (1 :1000), Bcl-2 (1:1000), Bax (1:1000), β-tubuline (1:1000), GAPDH (1:1000), HSP60 (1:1000) et VEGF (1:50) provenaient de Santa Cruz Biotechnology (CA, USA).</p>
</p><strong>Préparation du lysat cellulaire et immunoblotting<br></strong>Les cellules ont été lavées trois fois avec une solution saline tamponnée au phosphate (PBS) et lysées dans un tampon de lyse contenant 50 mM Tris-Cl (pH 7.5), 120 mM de NaCl, 10 mM de fluorure de sodium, 10 mM de pyrophosphate de sodium, 2 mM d’EDTA, 1 mM d’orthovanadate de sodium, 1 mM de fluorure de phénylméthylsulfonyle, 1% de NP-40 et un cocktail d’inhibiteurs de protéase (Roche, Allemagne). Les lysats tumoraux ont été préparés en découpant les tissus tumoraux en fins morceaux, lavés cinq fois avec une solution saline à 0,85 % contenant un cocktail d’inhibiteurs de protéase et lysés dans un tampon de lyse en les homogénéisant avec un homogénéisateur de tissus (Sigma, USA) suivi d’une sonication. Les lysats ont été clarifiés par centrifugation à 15 000 rpm<em> </em>pendant 30 minutes. Les lysats cellulaires ont été préparés dans des conditions réfrigérées. Environ 50 à 100 µg de protéines provenant de lysats de cellules entières ont été résolus sur un gel de SDS-polyacrylamide à 8-12% qui a ensuite été transféré sur une membrane PVDF (Millipore, Allemagne). La membrane a été sondée avec les anticorps primaires souhaités, puis avec des anticorps secondaires conjugués à la HRP. Les immunoblots ont été détectés par le réactif luminal (Santa Cruz Biotechnology). Lorsque cela était nécessaire, les blots ont été strippés en incubant la membrane à 50<sup>°</sup>C pendant 15 min dans un tampon de strippage (62,5 mM Tris-Cl, pH 6,8, 100 mM mercaptoéthanol et 2% SDS) avec une agitation intermittente. Les membranes ont été lavées soigneusement avec une solution saline tamponnée au Tris (TBS), et ont été ré-imprégnées avec les anticorps souhaités.</p>
<p><strong>Immunofluorescence ou microscopie confocale<br></strong>Les cellules ont été placées dans des lames à chambre Labtek (Nunc, USA) et laissées à croître pendant 24 h. Un traitement de metformine et d’autres inhibiteurs a été administré pendant les périodes et les concentrations souhaitées. Les cellules ont été lavées avec du PBS stérile refroidi et fixées avec une solution de paraformaldéhyde à 3,7 % à température ambiante pendant 10 minutes. Elles ont ensuite été perméabilisées avec du Triton X-100 à 0,025 % dans du PBS pendant 10 min, puis bloquées avec du BSA à 5 % dans du PBS pendant 1 h à 37<sup>°</sup>C. Les cellules ont été incubées avec des dilutions 1:100 d’anticorps primaires dans la solution de blocage pendant 2 h à température ambiante et lavées avec du TBST (TBS contenant 0,05% de Tween-20) au moins cinq fois avant d’être incubées avec des anticorps secondaires marqués appropriés (1:200) dans la solution de blocage pendant 1 h supplémentaire à température ambiante. Après cinq lavages avec TBST, les échantillons ont été recouverts de milieu de montage contenant du DAPI (Santa Cruz Biotechnology, USA). Les lames ont été scellées, examinées sous un microscope confocal à balayage laser (LSM510 Carl Zeiss, Allemagne) et les images ont été capturées. Les images ont ensuite été traitées par le logiciel d’analyse d’image LSM.</p>
<p><strong>Immunohistochimie et histopathologie<br></strong>L’immunohistochimie et l’histopathologie ont été réalisées selon la méthode de Malvi et al. <sup><a href= »#72″>[72]</a></sup>. Brièvement, des sections fines de la tumeur, et des autres organes ont été réalisées au microtome fixées sur des lames de verre et paraffinées. Pour l’immunohistochimie, les lames ont été déparaffinées dans une solution de xylène deux fois pendant 10 minutes, puis lavées trois fois avec de l’éthanol à 100%, 95%, 70% et 50%. Les lames ont été lavées à nouveau avec de l’eau distillée, puis avec du PBS pendant 5 minutes. Pour la récupération de l’antigène, les lames ont été bouillies dans un tampon de citrate de sodium (0,01 M, pH 4,5) dans un four à micro-ondes pendant 10 minutes et laissées refroidir à température ambiante pendant 20 minutes supplémentaires. Du BSA ou du sérum de chèvre normal (2%) a été utilisé pour le blocage dans une chambre humidifiée ou dans une boîte froide pendant 1 h. Les lames ont été sondées avec les anticorps souhaités (dilution 1:100) dans du PBST (PBS contenant 0,025% de Tween-20) contenant 0,01% de BSA pendant 2 h à température ambiante ou pendant la nuit à 4<sup>°</sup>C. Les lames ont été lavées avec du PBST 4 fois pendant 5 min, et ont été sondées avec des anticorps secondaires compatibles conjugués à la HRP pendant 1 h. Elles ont été à nouveau lavées et colorées avec de la Diaminobenzidine (DAB) pendant 10 min, puis lavées et contre-colorées avec de l’hématoxyline. Les lames ont ensuite été lavées à l’eau, déshydratées à l’alcool absolu puis recouvertes de milieu de montage, avant d’être analysées pour l’expression des molécules souhaitées. Pour l’histopathologie, les lames déparaffinées ont été colorées avec de l’hématoxyline et de l’éosine, et l’analyse microscopique de la densité cellulaire, de la morphologie cellulaire, des métastases, de la cytotoxicité et de la nécrose a été effectuée par des pathologistes en aveugle à l’hôpital KEM de Pune, en Inde.</p>
</p><strong>Test de cytotoxicité cellulaire et de survie</strong><br>Approximativement 5 × 10<sup>3</sup> (B16F10) et 10 × 10<sup>3</sup> (A375 et SKMel28) cellules ont été ensemencées dans chaque puits de plaques de culture tissulaire de 96 puits et laissées adhérer pendant 24 h à 37<sup>°</sup>C. Les cellules ont été traitées selon les besoins expérimentaux et la prolifération ou la viabilité a été évaluée par le test MTT. Du MTT (50 µl, 1 mg/ml dans du DMEM sans rouge de phénol) a été ajouté à chaque puits et incubé pendant 4 h à 37<sup>°</sup>C. Les cristaux de formazan ont été solubilisés<sup> </sup>dans 100 µl d’isopropanol en les incubant sous agitation à température ambiante pendant 5 min. L’absorbance a été prise à 570 nm en utilisant 630 nm comme filtre de référence. Les cellules non traitées ont été considérées comme un contrôle (100 % de survie cellulaire).</p>
</p> <p><strong>Détection de l’apoptose par coloration à l’annexine V<br></strong>Les cellules ont été ensemencées à une densité d’environ 3 × 10<sup>5</sup> cellules dans des plaques de 35 mm et laissées à croître pendant 24 h. Les cellules ont été traitées avec ou sans oxamate et DCA soit seuls, soit avec la metformine pendant 48 h ou selon les besoins expérimentaux. Les cellules ont été récoltées par trypsinisation et traitées pour la cytométrie de flux. L’apoptose a été détectée par une double coloration de l’Annexin V et du PI en utilisant le kit de test d’apoptose (BD Bioscience, USA) selon les instructions du fabricant.</p>
<p><strong>Analyse du cycle cellulaire<br></strong>Les cellules ont été ensemencées à une densité d’environ 3 × 10<sup>5</sup> cellules dans des plaques de 35 mm et ont été autorisées à croître pendant 24 h. Les cellules ont été traitées avec ou sans metformine, oxamate et autres inhibiteurs glycolytiques seuls ou ensemble comme indiqué pendant 48 h ou selon les besoins expérimentaux. Les cellules ont été récoltées par trypsinisation et traitées pour la cytométrie de flux. En bref, les cellules ont été lavées avec du PBS refroidi et fixées dans de l’éthanol à 70 % sur de la glace pendant 30 minutes. Après un traitement à la RNase (200 μg/ml) pendant 30 min à 37°C, 50 μg/ml de PI ont été ajoutés au culot cellulaire et incubés dans l’obscurité pendant 30 min sur glace. La fluorescence de PI a été enregistrée à travers un filtre de 585 nm dans un cytomètre en flux (FACS Calibur, Becton Dickinson, Californie, USA). Les données ont été analysées à l’aide du logiciel Cell Quest Pro pour 10 000 cellules.</p>
</p><strong>Dossier de survie cellulaire à long terme<br></strong>Les cellules (5 × 10<sup>2</sup>) ont été placées dans des plaques à 12 puits pendant 24 h. Les cellules ont été traitées sans ou avec 25 mM ou 50 mM d’oxamate et 10 ou 20 mM de DCA en même temps que la metformine et poursuivies pendant 48 h supplémentaires. Ensuite, le milieu a été remplacé par un milieu frais sans médicament. Les plaques ont été incubées pendant 10-15 jours supplémentaires à 37°C dans un incubateur CO<sub>2</sub> avec un changement de milieu tous les 2-3 jours. Les cellules ont ensuite été fixées (paraformaldéhyde à 3 % et glutaraldéhyde à 0,02 % dans du PBS) et colorées avec du cristal violet à 0,05 %.</p>
<p><strong>Dossier d’utilisation du glucose<br></strong>Les cellules (3 × 10<sup>5</sup>) ont été cultivées dans du DMEM contenant 25 mM de glucose. Après 24 h, le milieu a été remplacé par le milieu respectif contenant des concentrations croissantes de metformine (0,1 mM, 0,5 mM, 1 mM et 2 mM) ou de roténone pendant 24 h et le glucose résiduel présent dans le milieu usé a été contrôlé à l’aide d’un kit de dosage du glucose basé sur GOD-POD (Spinreact, Espagne) selon les instructions du fabricant. Pour mesurer l’utilisation du glucose en présence d’inhibiteurs glycolytiques, les cellules ont été traitées avec 50 mM d’oxamate, 100 µM de phlorétine et 20 mM de DCA, seuls ou avec 2 mM de metformine ou 100 µM de phenformine. Le glucose consommé a été estimé en soustrayant le glucose restant dans le milieu de la concentration initiale dans le milieu témoin (450 mg/dl). Les expériences ont été réalisées au moins en triplicata et les valeurs ont été normalisées par rapport au nombre total de cellules.</p>
<p><strong>Dossier d’estimation du lactate<br></strong>La concentration en lactate dans le milieu usé collecté à partir des cellules traitées avec ou sans metformine, oxamate et autres inhibiteurs glycolytiques, a été déterminée à l’aide d’un kit d’estimation du lactate disponible dans le commerce (Spinreact, Espagne) selon les instructions du fabricant. Brièvement, le milieu de culture ou le sérum a été dilué dans une solution saline à 0,9 % jusqu’à 10 fois et 10 μl d’échantillon ont été ajoutés à chaque puits d’une plaque ELISA de 96 puits. 150 µl de réactif fourni avec le kit ont été ajoutés à chaque échantillon contenant des puits en gardant le milieu non dépensé et le réactif seul comme blanc et l’absorbance a été enregistrée à 405 nm en utilisant un spectrophotomètre (Thermo-Scientific, USA). Les expériences ont été faites au moins en triplicata et les valeurs finales ont été normalisées avec le nombre total de cellules.</p>
<p><strong>Transfection de l’ARNsi<br></strong>Les ARNsi spécifiques contre LDHA ont été achetés auprès de Santa Cruz Biotechnology (USA). La transfection a été réalisée à l’aide de la Lipofectamine 2000 (Life Technologies, USA) selon les instructions du fabricant. En bref, les cellules ont été placées à environ 60 % de confluence. Le jour suivant, le milieu a été remplacé par de l’OptiMEM (Life Technologies, USA) et conservé pendant 3 h. Les siRNA souhaités ont été dissous dans les tampons fournis avec eux. La Lipofectamine2000 et le siRNA ont été dilués séparément dans l’OptiMEM et incubés pendant 5 minutes. Ensuite, les réactifs dilués ont été mélangés et incubés pendant 30 minutes à température ambiante. Le précipité résultant a été laissé sur les cellules pendant 6 heures, après quoi du DMEM frais complété par 10% de FBS a été ajouté et incubé pendant 24-36 heures supplémentaires. L’efficacité de la transfection a été évaluée en transfectant simultanément le plasmide PEGFPN1. L’immunoblotting a été effectué pour s’assurer de l’inhibition de l’expression des gènes respectifs.</p>
<p><strong>Préparation de la fraction riche en mitochondries à partir de cellules et de tissus<br></strong>La fraction riche en mitochondries a été préparée à partir de cellules cultivées et de tumeurs ou de tissus normaux comme précédemment rapporté<sup><a href= »#73″> [73]</a></sup>. Brièvement, des cellules (1 × 10<sup>6</sup>) ont été placées dans une plaque de 10 cm et ont été trypsinisées après avoir été traitées avec les inhibiteurs souhaités pendant 24 h. La suspension cellulaire a été soumise à trois cycles de congélation-décongélation dans un tampon hypotonique (phosphate de potassium 20 mM). Cette suspension a été centrifugée à 50 000 rpm pendant 1 h pour obtenir un surnageant riche en mitochondries. De même, pour isoler les mitochondries des tissus, les tissus congelés ont été découpés en fins morceaux et homogénéisés dans un tampon d’homogénéisation (tampon Tris 0,5 M pH 7,5, 100 mM EGTA et 250 mM saccharose), suivi d’une procédure de congélation-décongélation cyclique et centrifugé à 50 000 rpm pendant 1 h. Le surnageant a été recueilli comme fraction mitochondriale et utilisé pour la détermination de la fonction mitochondriale et de l’activité enzymatique OXPHOS.</p>