Balkrishna Chaube1, Parmanand Malvi1, Shivendra Vikram Singh1, Naoshad Mohammad1, Avtar Singh Meena1,2 und Manoj Kumar Bhat1
1 Nationales Zentrum für Zellwissenschaften, Savitribai Phule Pune University Campus, Ganeshkhind, Pune, Indien2
Aktuelle Adresse: Abteilung für Physiologie, University of Tennessee Health Science Center, Memphis, USA
Korrespondenz: Manoj Kumar Bhat, E-Mail: [email protected]
Eingereicht am: 16. Juni
2015Angenommen: 23. September 2015Veröffentlicht
: 15. Oktober 2015
Zusammenfassung
Das Melanom ist eine weitgehend unheilbare bösartige Hauterkrankung, die auf die zugrunde liegende molekulare und metabolische Heterogenität zurückzuführen ist, die durch die Entwicklung von Resistenzen erschwert wird. Krebszellen verfügen über eine metabolische Flexibilität, indem sie je nach Nährstoffverfügbarkeit in der Mikroumgebung des Tumors entweder die oxidative Phosphorylierung (OXPHOS) oder die Glykolyse zur ATP-Erzeugung nutzen. In dieser Studie untersuchten wir die Beteiligung des Atmungskomplexes I und der Laktatdehydrogenase (LDH) an der Progression des Melanoms. Wir zeigen, dass die Hemmung des Komplexes I durch Metformin das Melanomwachstum in Mäusen durch eine Erhöhung der Laktat- und VEGF-Spiegel fördert. Im Gegensatz dazu führt die Hemmung in vitro zu einem Wachstumsstillstand aufgrund einer verstärkten extrazellulären Ansäuerung als Folge einer erhöhten Glykolyse. Die Hemmung der LDH- oder Laktatbildung führt zu einem Rückgang der Glykolyse mit gleichzeitigem Wachstumsstillstand sowohl in vitro als auch in vivo. Die Blockierung der Laktatbildung in mit Metformin behandelten Melanomzellen führt zu einer verminderten Zellproliferation und Tumorprogression in Mäusen. Interessanterweise löst die Hemmung der LDH oder des Komplexes I allein keine Apoptose aus, während die Hemmung beider zusammen zu einer Erschöpfung des zellulären ATP-Pools und damit zu einer durch eine metabolische Katastrophe ausgelösten Apoptose führt. Insgesamt legt unsere Studie nahe, dass LDH und Komplex I bei der Regulierung der Glykolyse und der Zellproliferation unterschiedliche Rollen spielen. Die Hemmung dieser beiden erhöht die synthetische Letalität beim Melanom.
Schlüsselwörter: Melanom; Komplex I; LDH; metabolische Katastrophe; synthetische Letalität
EINLEITUNG
Das maligne Melanom ist eine der aggressivsten Formen von Hautkrebs mit hohem Metastasierungspotenzial und Resistenz gegenüber vielen zytotoxischen Wirkstoffen [1, 2]. Trotz umfangreicher Forschung und Teilerfolgen durch den Einsatz derzeit verfügbarer Medikamente gibt es keine wirksame Behandlung des malignen Melanoms [1-3]. Die Zahl der Melanomfälle nimmt jedes Jahr zu und ist weltweit für etwa 75 % der hautkrebsbedingten Todesfälle verantwortlich [2, 3]. Das schlechte Ansprechen auf die derzeit verfügbaren therapeutischen Optionen und die Entwicklung von Therapieresistenzen rechtfertigen die Erforschung neuer Strategien zur Behandlung des Melanoms.
Eine verstärkte aerobe Glykolyse ist ein charakteristisches Merkmal vieler Krebsarten [3-5]. Es wurde berichtet, dass Melanomzellen aufgrund der BRAF-Mutation hauptsächlich von der Glykolyse zur ATP-Erzeugung abhängen und eine gestörte oxidative Phosphorylierung aufweisen [6, 7]. Krebszellen gewinnen ATP, biosynthetische Zwischenprodukte und reduzierende Äquivalente durch eine ungewöhnliche Beteiligung an biochemischen Stoffwechselwegen wie der Glykolyse, der Glutaminolyse und dem Pentosephosphat-Weg [5]. Normale (nicht krebsartige) Zellen gewinnen ATP hauptsächlich durch mitochondriale OXPHOS, während Krebszellen hauptsächlich auf aerobe Glykolyse angewiesen sind, um ATP und glykolytische Zwischenprodukte zu erzeugen, die ein schnelles Wachstum ermöglichen [4, 5]. Eine verstärkte Laktatbildung wurde mit der Aggressivität von Krebs in Verbindung gebracht. In zahlreichen Studien wurde die Laktatdehydrogenase (LDH), die die Umwandlung von Pyruvat in Laktat katalysiert, als der konsequenteste Marker für aggressive und schnell wachsende Krebsarten identifiziert [8-11]. LDH spielt eine wichtige Rolle bei der Regulierung der Glykolyse, der Aufrechterhaltung des zellulären Redoxzustands, der mitochondrialen Physiologie und der Tumorerhaltung [12]. Ein veränderter Stoffwechsel von Krebszellen kann mit einer mitochondrialen Dysfunktion einhergehen, die eine Hemmung der OXPHOS, eine Zunahme reaktiver Sauerstoffspezies (ROS) und ein unkontrolliertes Wachstum zur Folge hat, was wiederum die Entwicklung eines metastatischen Phänotyps fördert[13,14]. Der Atmungskomplex I ist das größte und komplexeste Enzym, das die Oxidation von NADH in der Elektronentransportkette katalysiert [15]. Komplex I spielt eine wichtige Rolle bei der ATP-Erzeugung in normalen Zellen und ist ein Hauptort der ROS-Erzeugung, seine Rolle bei der Tumorentstehung ist jedoch weitgehend unklar. Die meisten Berichte deuten darauf hin, dass die Aktivität von Komplex I in Krebszellen unterdrückt wird und seine Hemmung die Proliferation und Metastasierung fördert [16-18]. Umgekehrt wurde auch berichtet, dass Melanomzellen eine erhöhte OXPHOS-Funktion aufweisen, die eine Arzneimittelresistenz verursacht [19-21]. Aufgrund der metabolischen Flexibilität bei der Wahl verschiedener Stoffwechselwege, insbesondere des Glutaminstoffwechsels [22]können Melanomzellen einer Störung eines Moleküls in einem Stoffwechselweg entgehen [23]. Daher müssen mehrere Moleküle verschiedener Stoffwechselwege angegangen werden, um ein wirksames und kuratives Ergebnis zu erzielen.
Biguanide wie Metformin und Phenformin sind dafür bekannt, dass sie den Atmungskomplex I hemmen. Metformin, eine Erstlinientherapie für Patienten mit Typ-2-Diabetes mellitus (T2DM), gehört zu den Biguaniden und es wurde kürzlich entdeckt, dass es eine antitumorale Wirkung hat [24, 25]. Auf physiologischer Ebene übt Metformin seine biologische Aktivität aus, indem es die hepatische Glukogenese verringert, die Insulinsensitivität erhöht, die periphere Glukoseaufnahme steigert und die Absorption von Glukose aus dem Gastrointestinaltrakt verringert [26]. Auf zellulärer Ebene wirkt Metformin in erster Linie durch die Hemmung des mitochondrialen Komplexes I, der die oxidative Phosphorylierung behindert, was zu einem verringerten ATP-Spiegel und damit zur Aktivierung der AMPK führt [27-30]. Darüber hinaus hat sich gezeigt, dass Metformin die mitochondriale Atmung in Verbindung mit der ATP-Erzeugung verringert und dadurch eine Zunahme der Glykolyse bewirkt [31, 32]. Die krebshemmende Wirkung von Metformin ist bei verschiedenen Krebsarten mit unterschiedlichen Mechanismen nachgewiesen worden [33, 34]. Die Rolle von Metformin beim Melanom ist jedoch nicht ganz klar. Im Gegensatz zu anderen Krebsarten hat Metformin beim Melanom nachweislich sowohl eine tumorfördernde als auch eine antitumorale Wirkung [35-37]. Außerdem hat sich gezeigt, dass Metformin in Kombination mit BRAF-Inhibitoren das Melanomwachstum unterdrückt [38].
In der vorliegenden Studie wurde spekuliert, dass die Unterbrechung der ATP-Erzeugungswege durch die gleichzeitige Hemmung von Komplex I und LDH für Melanomzellen synthetisch tödlich sein könnte. Wir verwendeten Metformin oder Phenformin und Oxamat/Dichloracetat (DCA), um diese beiden Enzyme sowohl im Zell- als auch im Tiermodell zu hemmen. Wir konnten zeigen, dass die Hemmung der Komplex I- und LDH-Aktivität unterschiedliche Auswirkungen auf das Zellwachstum und die Zellproliferation hat. Interessanterweise fördert die Hemmung von Komplex I durch Metformin die aerobe Glykolyse weiter, was zu einem verstärkten Tumorwachstum bei Mäusen führt. Die Ausschaltung der Metformin-induzierten Laktatbildung durch Oxamat oder DCA führt zu Zytostase und/oder Apoptose in Melanomzellen.
ERGEBNISSE
Metformin zeigt in vitro und in vivo unterschiedliche Wirkungen auf das Wachstum von Melanomen
Metformin unterdrückt das Tumorwachstum durch Hemmung des Komplexes I, der von Glukose beeinflusst wird [30]. Außerdem ist bekannt, dass Glukose die Aktivität von Atmungsenzymen verändert [39]. Um die Folgen der Komplex-I-Hemmung und den Einfluss von Glukose auf die Wirkung von Metformin auf das Wachstum von Melanomen zu untersuchen, haben wir die Entwicklung von Isotransplantaten und Xenotransplantaten in Mäusen mit Streptozotocin (STZ) und Hyperglykämie beobachtet. Wir stellten fest, dass Metformin das Fortschreiten von B16F10-Isotransplantaten in hyperglykämischen Mäusen im Vergleich zur unbehandelten Kontrolle förderte (Abbildung 1A, 1B und 1C). Auch bei normoglykämischen C57BL/6J-Mäusen beeinflusste Metformin das Fortschreiten des Tumors positiv (Abbildung 1D, 1E und 1F). In ähnlicher Weise förderte die orale Verabreichung von Metformin das Wachstum des A375-Xenotransplantats sowohl bei hyperglykämischen als auch bei normoglykämischen NOD/SCID-Mäusen im Vergleich zur unbehandelten Kontrolle (Abbildung 1G, 1H und 1I).
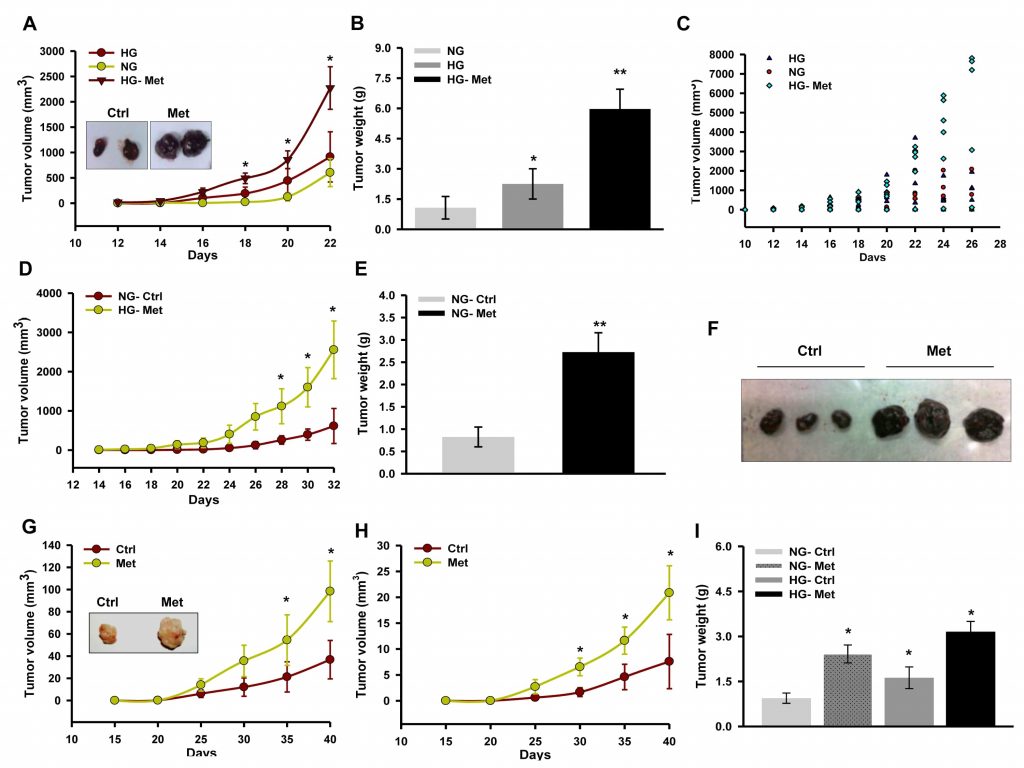
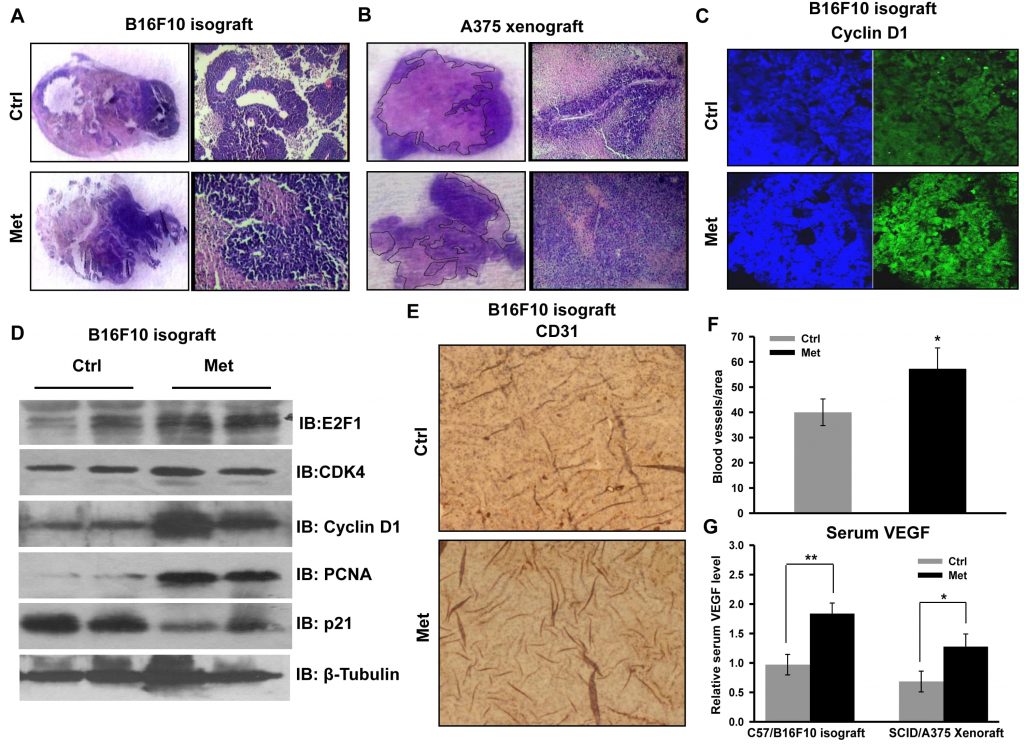
Zur Überprüfung der zellulären und molekularen Vorgänge, die mit einer verstärkten Tumorprogression einhergehen, wurden Tumorschnitte für die histopathologische Analyse untersucht. In den Schnitten beider Tumorarten (B16F10-Isotransplantat und A375-Xenotransplantat) von mit Metformin behandelten Mäusen waren eine hohe Zelldichte und eine geringere Nekrose deutlich zu erkennen (Abbildung 2A und 2B). Wir stellten fest, dass die durch Metformin verstärkte Proliferation und Progression des A375-Xenotransplantats im Vergleich zum Kontrolltumor phänotypisch unterschiedlich war. Dies deutet auf ein Fortschreiten des Primärtumors hin, was an der länglichen Morphologie der Zellkerne im Vergleich zur runden Morphologie in den Kontrolltumorabschnitten zu erkennen ist (unveröffentlichte Informationen). Die immunhistochemische Färbung des zellzyklusregulierenden Proteins Cyclin D1 (Abbildung 2C) war in den Tumorabschnitten der mit Metformin behandelten Mäuse erhöht. Zur Bestätigung des verstärkten Tumorwachstums überprüften wir den Status der zellzyklusregulierenden Proteine durch Immunoblotting von Tumorlysaten. Wir stellten fest, dass die Spiegel der Moleküle Cyclin D1, CDK4, E2F1 und PCNA in den Tumorlysaten der mit Metformin behandelten Mäuse im Vergleich zur Kontrolle deutlich erhöht waren, während der Spiegel von p21 vermindert war (Abbildung 2D). Diese Ergebnisse deuten darauf hin, dass Metformin, unabhängig vom glykämischen Status der Mäuse, das Melanomwachstum durch die Modulation zellzyklusregulierender Proteine fördert. Darüber hinaus untermauerte die immunhistochemische Analyse von Tumorschnitten diese Beobachtung, da die Behandlung mit Metformin die Proteinkonzentration von CD31, einem Endothelmarker (Abbildung 2E und 2F), erhöhte und den Serumspiegel von VEGF (Abbildung 2G) ansteigen ließ, was darauf schließen lässt, dass Metformin die Angiogenese in Melanomtumoren fördert.
Als nächstes überprüften wir die Wirkung von Metformin auf das Wachstum und die Proliferation von Melanomzellen in vitro. Im Gegensatz zu unseren In-vivo-Ergebnissen führte die Behandlung mit Metformin in vitro zu einer Unterdrückung des Wachstums von Melanomzellen (ergänzende Abbildungen S1A, S1B, S1C und S1D). Anschließend untersuchten wir die Auswirkungen der Komplex-I-Hemmung mit Metformin und Phenformin auf das Wachstum der Melanomzellen, da beide die Komplex-I-Aktivität hemmen (ergänzende Abbildung S2). Wir stellten fest, dass Metformin und Phenformin bei Melanomzellen, die unter hohem Glukosegehalt wuchsen, einen Wachstumsstopp verursachten. In Gegenwart von niedrigem Glukosegehalt führte die Behandlung mit diesen Wirkstoffen jedoch zum Zelltod (ergänzende Abbildungen S3A, S3B, S3C und S3D). Es ist wahrscheinlich, dass der durch Metformin vermittelte Wachstumsstillstand auf die Senkung des Glukosespiegels und die extrazelluläre Ansäuerung des Mediums zurückzuführen ist (ergänzende Abbildungen S4A und S4B). Interessanterweise erhöhte der Austausch des Mediums nach jeweils 12 Stunden durch frisches Medium mit 25 mM Glukose das klonogene Überleben nach der Metformin-Behandlung. Da die mit Metformin behandelten Zellen die Glukose im Vergleich zur Kontrolle sehr schnell verbrauchten, ist eine Auffüllung des Mediums im Wesentlichen erforderlich, um den Glukosespiegel und den pH-Wert aufrechtzuerhalten und damit das Überleben der Zellen in Gegenwart von Metformin zu sichern (ergänzende Abbildungen S4C und S4D). Diese Ergebnisse deuten darauf hin, dass die Konzentration der im Kulturmedium verfügbaren Glukose die Wirkung von Metformin beeinflusst.
DieHemmung der Aktivität des Atmungskomplexes I fördert die aerobe Glykolyse
Die Diskrepanz zwischen den Ergebnissen der Metformin-Behandlung in vivo und in vitro veranlasste uns, die Ursache hierfür zu untersuchen. Es ist bekannt, dass Metformin seine Wirkung hauptsächlich durch die Hemmung des mitochondrialen OXPHOS-Enzyms Komplex I<ahref=“#[„> [</a><a href=“https://www.oncotarget.com/article/6134/text/#bib27″>27</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib29″>29</a><a href=“#]“>]</a></sup> und die Hemmung von Komplex I führt zu erhöhtem Tumorwachstum und Proliferation<sup><a href=“#14″> [</a><a href=“https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href=“#]“>]</a></sup>. Da wir beobachteten, dass die Metformin-Behandlung aufgrund der verstärkten aeroben Glykolyse zu einer Ansäuerung des Kulturmediums führte (ergänzende Abbildungen 4A und 4B), vermuteten wir, dass dies der Grund für den Wachstumsstillstand der Zellen sein könnte. Daher haben wir den Glukose- und Laktatgehalt im verbrauchten Kulturmedium von Kontroll- und metforminbehandelten Zellen gemessen. In den mit Metformin behandelten Zellen wurde eine erhöhte Glukoseverwertung (erkennbar an der geringeren Restglukose im Medium) und ein höherer Laktatspiegel im Vergleich zur Kontrolle festgestellt (Abbildung 3A und 3B).</p>
<p>Um diese Ergebnisse zu untermauern, untersuchten wir weiter das Expressionsmuster und die Aktivität einiger Proteine und Enzyme, die an der Regulierung der Glykolyse beteiligt sind. Wir stellten fest, dass Metformin die Proteinkonzentration der wichtigsten glykolytischen Moleküle GLUT1, LDHA und ChREBP in Abhängigkeit von der Konzentration erhöhte (Abbildung 3C). Diese Ergebnisse deuten darauf hin, dass die Hemmung der Komplex-I-Aktivität durch Metformin Krebszellen möglicherweise dazu veranlasst, den glykolytischen Weg zur ATP-Erzeugung einzuschlagen, was die Tumorprogression in Mäusen beschleunigt.</p>
<p>Um herauszufinden, ob der glykolytische Phänotyp auch <em>in vivo</em> durch die Hemmung von Komplex I induziert werden kann, haben wir zunächst den Glukosespiegel im Serum von Mäusen mit Tumor und von normoglykämischen Mäusen, die mit Metformin behandelt oder nicht behandelt wurden, untersucht. Bei normoglykämischen, tumortragenden Mäusen, denen Metformin verabreicht wurde, wurde ein signifikanter Rückgang des Serumglukosespiegels (104,3 ± 13,7 mg/dl bei C57BL/6J und 134,0 ± 7,2 mg/dl bei NOD/SCID) im Vergleich zur unbehandelten Kontrolle (139,7 ± 16,3 mg/dl bei C57BL/6J und 168,0 ± 8,5 mg/dl bei NOD/SCID) beobachtet (Abbildung 3D und 3E). In ähnlicher Weise führte die Behandlung mit Metformin zu einer Verringerung des Serumglukosespiegels in den tumortragenden hyperglykämischen Mäusen (Met, 237,3 ± 30,0 mg/dl in C57BL/6J und 227,3 ± 12,3 mg/dl in NOD/SCID) im Vergleich zur Kontrolle (388 ± 31,5 mg/dl in C57BL/6J und 409,3 ± 18,6 mg/dl in NOD/SCID-Mäusen) (Abbildung 3D und 3E). Die Sekretion von Laktat ist ein Kennzeichen glykolytischer Tumoren. Daher überprüften wir den Laktatspiegel, der sowohl im Serum von normoglykämischen als auch von hyperglykämischen Mäusen, die mit Metformin behandelt wurden, sehr hoch war (Abbildung 3F und 3G). Erhöhte LDH-Werte im Serum werden mit hochgradig aggressiven Tumoren und einer schlechten Prognose bei metastasierten Melanomen in Verbindung gebracht <sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib8″>8</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=“#]“>]</a></sup>. So wurde die LDH-Aktivität im Serum von hyperglykämischen und normoglykämischen Mäusen, denen Metformin verabreicht wurde, im Vergleich zur Kontrolle als sehr hoch eingestuft (Abbildung<a href=“https://www.oncotarget.com/article/6134/text/#F3″> </a>3H und 3I). Außerdem wurde eine erhöhte LDH-Aktivität im Tumorgewebe der mit Metformin behandelten Mäuse im Vergleich zur unbehandelten Kontrollgruppe festgestellt (Abbildung 3J).</p>
<p><strong>Die Hemmung von Komplex I zusammen mit LDH oder Laktatbildung ist synthetisch tödlich für Melanomzellen<br></strong>Um zu untersuchen, ob Metformin die Glykolyse fördert oder eine übermäßige Laktatbildung die Hauptursache für das verstärkte Wachstum und die Proliferation von Melanomtumoren ist, wurde die Laktatbildung mit dem LDH-Hemmer Oxamat und dem PDK1-Hemmer DCA gehemmt. Wir stellten fest, dass Oxamat und DCA allein eine hemmende Wirkung auf das Melanomzellwachstum haben (Abbildung 4A, 4B und 4C; ergänzende Abbildung S5). Bei der Behandlung mit Oxamat oder DCA zusammen mit Metformin wurde eine verringerte Zellüberlebensrate beobachtet (Abbildung 4A, 4B und 4C). Auch andere bekannte glykolytische Inhibitoren wie Phloretin (GLUT1-Inhibitor), 2-Desoxy-D-Glucose (2DG) und Iodacetat schränkten die Proliferation von Melanomzellen ein, die mit Metformin behandelt wurden (ergänzende Abbildungen S6A und S6B). Eine gesteigerte Glukoseaufnahme durch GLUT1 und eine erhöhte aerobe Glykolyse durch LDHA und PDK1 sind das Markenzeichen von Krebszellen. Daher haben wir Oxamat und DCA gegenüber anderen glykolytischen Inhibitoren bevorzugt.</p>
<p>Um die langfristige Überlebensfähigkeit von Melanomzellen in Anwesenheit von Metformin und Oxamat/DCA allein oder in Kombination zu überprüfen, führten wir als nächstes einen klonogenen Überlebenstest durch. In Melanomzellen, die mit Oxamat und Metformin zusammen behandelt wurden, wurden keine überlebenden Kolonien festgestellt (Abbildung<a href=“https://www.oncotarget.com/article/6134/text/#F4″> </a>4D, 4E und 4F). Ähnliche Ergebnisse wurden auch bei Zellen erzielt, die mit Metformin und DCA zusammen behandelt wurden (Abbildung 4D, 4E und 4F). Anschließend überprüften wir, ob die Hemmung dieser beiden Enzyme den Tod der Melanomzellen auslöste, was durch Annexin V und PI-Färbung nachgewiesen werden konnte. Alle diese Wirkstoffe allein bewirkten einen Wachstumsstopp, lösten aber keine Apoptose aus. Bei A375- und B16F10-Zellen, die mit Oxamat/DCA zusammen mit Metformin behandelt wurden, wurde jedoch eine erhöhte Apoptose festgestellt, verglichen mit dem Einzelwirkstoff allein (Abbildung 5A und 5B). Diese Beobachtung wurde durch Immunoblotting für die Apoptosemarker PARP1, Bax und Bcl-2 bestätigt (Abbildung 5C und 5D). In den Zellen, die mit Metformin und Oxamat/DCA zusammen behandelt wurden, wurde eine erhöhte PARP-Spaltung und ein erhöhter Bax-Spiegel festgestellt, während der Spiegel des antiapoptotischen Proteins Bcl-2 im Vergleich zu den Zellen, die nur mit einem Wirkstoff behandelt wurden, abnahm (Abbildung 5C und 5D). Diese Ergebnisse deuten darauf hin, dass die Hemmung von Komplex I und LDH zusammen die Induktion der Apoptose fördern kann.</p>
<p>Als Nächstes haben wir die Auswirkungen von Metformin und Oxamat/DCA entweder allein oder in Kombination auf das Überleben von Nicht-Krebszellen untersucht. Wir haben drei verschiedene nicht-krebsartige Zelllinien verwendet, nämlich AML12 (Maushepatozyten), L6 (Rattenmuskelzellen) und MEF (embryonale Mausfibroblasten), um die Wirkung der Kombinationsbehandlung zu testen. Obwohl die Kombination von Metformin und Oxamat/DCA die Proliferation dieser Zellen verringerte (ergänzende Abbildungen S7A, S7B und S7C), beeinträchtigte sie nicht die Lebensfähigkeit der Zellen (ergänzende Abbildung S7D), was darauf hindeutet, dass diese Kombination Krebszellen selektiv abtöten kann.</p>
<h3></h3>
<p><strong>Die Blockierung von Komplex I und der Laktatbildung führt zu einer metabolischen Katastrophe<br></strong>Um sicherzustellen, ob die gleichzeitige Behandlung mit Oxamat und Metformin den Stoffwechsel von Melanomzellen beeinflusst, haben wir die metabolischen Parameter in Gegenwart dieser Inhibitoren gemessen. Die Behandlung mit Metformin erhöhte die Glykolyse, während die Behandlung mit Oxamat zu einem Rückgang der Glukoseverwertung und der Laktatausscheidung führte (Abbildung 6A und 6B). Interessanterweise wurden die Glukoseverwertung und die Laktatsekretion deutlich gehemmt, wenn die Zellen mit Oxamat und Metformin zusammen behandelt wurden (Abbildung 6A und 6B). Darüber hinaus verringerten sowohl Oxamat als auch Metformin den ATP-Spiegel durch Hemmung der Substratphosphorylierung bzw. der OXPHOS (Abbildung 6C). Außerdem wurde ein signifikanter Rückgang des ATP-Spiegels in Zellen festgestellt, die mit Oxamat und Metformin zusammen behandelt wurden (Abbildung 6C). Um die Beteiligung von LDH am Wachstum und an der Proliferation von Melanomzellen zu bestätigen, wurde LDHA (LDHA-KD) mit spezifischer siRNA ausgeschaltet (Abbildung 6D). Bei LDHA-KD-Zellen, die mit Metformin oder Phenformin behandelt wurden, wurde ein erhöhter Zelltod beobachtet (Abbildung 6E). Eine signifikante Verringerung der Glykolyse (erkennbar an der verringerten Glukoseverwertung und Laktatsekretion) und des ATP-Spiegels wurde in LDHA-KD-Zellen im Vergleich zur Kontrolle beobachtet (Abbildung 6F, 6G und 6H). Darüber hinaus wurde in LDHA-KD-Zellen, die entweder mit Metformin oder Phenformin behandelt wurden, eine Abnahme der glykolytischen Rate sowie ein starker Rückgang des ATP-Spiegels beobachtet (Abbildung 6F, 6G und 6H). Diese Ergebnisse deuten darauf hin, dass die gleichzeitige Hemmung sowohl des Komplexes I als auch der LDH oder des Laktatbildungsweges eine metabolische Katastrophe aufgrund der Störung des zellulären ATP-Pools auslöst, die schließlich zum Wachstumsstillstand und Zelltod führt.</p>
<p><strong>Die gleichzeitige Hemmung von Komplex I und LDH durch Metformin und Oxamat verzögert die Tumorprogression bei Mäusen<br></strong>Um zu überprüfen, ob die Kombination von Oxamat und Metformin auch bei der Verringerung der Tumorprogression wirksam ist, wurden Tumore durch Injektion von B16F10-Zellen in C57BL/6J-Mäuse entwickelt. Nachdem die Tumore eine Größe von bis zu 50 mm<sup>3</sup> erreicht hatten, wurden die Mäuse nach dem Zufallsprinzip in vier Gruppen eingeteilt: (a) Kontrolle, (b) Metformin, (c) Oxamat und (d) mit Metformin und Oxamat behandelte Mäuse. Wir stellten fest, dass Mäuse, denen Metformin verabreicht wurde, im Vergleich zur unbehandelten Kontrollgruppe größere Tumore entwickelten (Abbildung 7A, 7B und 7C). Bei Mäusen, denen Oxamat verabreicht wurde, entwickelten sich die Tumore langsamer als bei der unbehandelten Kontrolle und bei Mäusen, denen Metformin verabreicht wurde (Abbildung 7A). Interessanterweise beobachteten wir, dass Oxamat die Tumorprogression drastisch verlangsamte, wenn es zusammen mit Metformin verabreicht wurde, was sich in einer Verringerung des Tumorvolumens und des Tumorgewichts zeigte (Abbildung 7A, 7B und 7C).</p>
<p>Um zu bestätigen, ob die Aufhebung der Metformin-induzierten Tumorprogression durch Oxamat eine Folge der Verringerung der aeroben Glykolyse ist, haben wir außerdem <em>in vivo</em> metabolische Parameter gemessen. Die mit Metformin behandelten Mäuse wiesen im Vergleich zur Kontrollgruppe niedrige Serumglukose-, höhere Laktat- und LDH-Werte im Serum auf, während bei den mit Oxamat allein oder mit Metformin zusammen mit Oxamat behandelten Mäusen relativ höhere Serumglukose- und niedrige Laktatwerte festgestellt wurden (Abbildung 7D und 7E). Die Aktivität des Enzyms LDH war in den Tumoren der Mäuse, die mit Oxamat allein oder mit Metformin zusammen mit Oxamat behandelt wurden, signifikant vermindert (Abbildung 7F). In den Tumoren von Mäusen, die sowohl mit Oxamat als auch mit Metformin behandelt wurden, wurde ein relativ niedriger ATP-Gehalt festgestellt, verglichen mit den Tumoren von Mäusen, die nur einen Wirkstoff erhielten. Wir konnten jedoch keine signifikanten Veränderungen in den Proteinkonzentrationen der Moleküle beobachten, die die Glykolyse regulieren (Abbildung 7I). Wichtig ist, dass bei den Mäusen, die eine Kombinationsbehandlung erhielten, keine signifikante Veränderung des Körpergewichts festgestellt wurde, und dass bei der visuellen Inspektion keine möglichen unerwünschten Nebenwirkungen oder grobe Symptome einer allgemeinen Toxizität beobachtet wurden (ergänzende Abbildung S8A). Außerdem wurden scheinbar keine pathologischen Anomalien in den lebenswichtigen Organen wie Lunge, Leber, Niere und Herz dieser Mäuse beobachtet (ergänzende Abbildung S8B). Diese Ergebnisse deuten darauf hin, dass diese Kombination keine allgemeine Toxizität ausübt und das Fortschreiten des Tumors bei Mäusen wirksam verzögert.</p>
<h2>DISKUSSION</h2>
<p>Krebszellen, einschließlich Melanomzellen, leiten große Mengen an Kohlenstoff in die Glykolyse <sup><a href=“#5″>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib5″>5</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href=“#]“>]</a></sup>. Dies hilft den Zellen, ATP und andere Bausteine für ein schnelles Tumorwachstum zu erzeugen <sup><a href=“#5″>[5]</a></sup>. Mehrere Berichte deuten darauf hin, dass die Beeinflussung von Stoffwechselwegen die Tumorprogression und das Zellwachstum <em>in vitro</em> <sup><a href=“#40″>[40-</a><a href=“#42″>42]</a></sup> reduziert. In der vorliegenden Studie zeigen wir, dass Metformin das Melanomwachstum durch eine Erhöhung der Glykolyse aufgrund der Hemmung der Komplex-I-Funktion fördert, während die Hemmung der LDH einen Wachstumsstopp in den Zellen bewirkt. Die Hemmung der Laktatbildung in Melanomzellen, die mit Metformin behandelt wurden, wirkt sich auf das Überleben der Zellen aus und führt so zu einer Verringerung der Tumorprogression (Abbildung 8).</p>
<p>Die Folgen einer Hemmung des Komplexes I auf das Wachstum von Krebszellen sind nicht eindeutig geklärt. Berichte deuten jedoch darauf hin, dass eine Hemmung des Komplexes I zu einer Zelltransformation führt und das Zellwachstum<a href=“#[„><sup>[</sup></a><sup><a href=“https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href=“#]“>]</a></sup> fördert. Umgekehrt wurde auch berichtet, dass die Hemmung von Komplex I das Fortschreiten von Tumoren bei Mäusen verzögert <sup><a href=“#30″>[30]</a></sup>. Diese unterschiedlichen Ergebnisse hängen wahrscheinlich von der Verfügbarkeit von Nährstoffen in der Mikroumgebung des Tumors ab. Es wurde berichtet, dass, wenn Glukose im Überfluss vorhanden ist, die Hemmung von Komplex I die Glykolyse und die schnelle Proliferation von Zellen fördert, während sie unter metabolischen Stressbedingungen den Zelltod auslöst<sup><a href=“#43″>[43]</a></sup>. In dieser Studie haben wir beobachtet, dass die Hemmung von Komplex I durch Metformin das Fortschreiten des Melanomtumors bei Mäusen fördert.</p>
<p>Während Metformin nachweislich die Proliferation vieler Krebszellen hemmt und auch die Tumorprogression von Xenotransplantaten in Mäusen einschränkt <sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib33″>33</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib34″>34</a><a href=“#]“>]</a></sup>, ist bekannt, dass es sowohl wachstumshemmende als auch wachstumsfördernde Effekte bei Melanomen<sup> <a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib35″>35</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib37″>37</a><a href=“#]“>]</a></sup> ausübt. Interessanterweise stellten wir fest, dass B16F10- und A375-Tumore in Mäusen, denen Metformin verabreicht wurde, schnell wuchsen, und die Wachstumsrate variierte in Abhängigkeit vom glykämischen Status der Mäuse (Abbildung 2D und 2E). Diese Beobachtung stimmt mit dem jüngsten Bericht überein, der zeigt, dass Metformin die Tumorprogression von BRAF-mutierten Melanomzellen fördert<sup><a href=“http://335″>[35]</a></sup>.</p>
<p>Die unterschiedliche Wirkung von Metformin auf das Wachstum von Melanomzellen <em>in vitro</em> und <em>in vivo</em> wird wahrscheinlich durch die Verfügbarkeit von Glukose und die Laktatkonzentration beeinflusst, die seine Wirkung beeinflussen könnten. Metformin hemmt das Zellwachstum und löst unter glukoselimitierenden Bedingungen <sup><a href=“#43″>[43]</a></sup> Apoptose aus, während wir feststellten, dass es in Gegenwart von hoher Glukose einen Wachstumsstillstand bewirkt, ohne den Zelltod zu beeinflussen. Wir begründeten diese Diskrepanz damit, dass Krebszellen unter metabolischem Stress ATP <em>über</em> OXPHOS gewinnen, während ATP bei reichlich Glukose hauptsächlich durch Glykolyse gewonnen wird. Die Hemmung von Komplex I unter hohen Glukosebedingungen fördert die Glykolyse weiter, was zu einer übermäßigen Laktatbildung führt, und über dieses Phänomen wurde bereits früher berichtet<sup><a href=“#44″>[44]</a></sup>. Insgesamt deuten diese Ergebnisse darauf hin, dass der durch Metformin induzierte Wachstumsstillstand bei Melanomzellen <em>in vitro</em> eine Folge der Ansäuerung des Mediums ist, die auf eine übermäßige Milchsäurebildung aufgrund der schnellen Verwertung der verfügbaren Glukose zurückzuführen ist. Der durch Metformin ausgelöste Wachstumsstillstand kann verhindert werden, indem das Medium häufig aufgefüllt wird, um optimale Wachstumsbedingungen aufrechtzuerhalten, da die mit Metformin behandelten Zellen aufgrund der verstärkten aeroben Glykolyse mehr Glukose benötigen, um sich zu vermehren. Dies ahmt die <em>in vivo</em>-Bedingungen nach, bei denen die Krebszellen aufgrund der wahrscheinlich schnellen und konstanten Zirkulation der zugeführten Milchsäure mehr Glukose verbrauchen können, was zu einer schnellen Vermehrung beiträgt. Darüber hinaus kann Laktat auch von Krebszellen in Tumoren <sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib45″>45</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib46″>46</a></sup><a href=“#]“><sup>]</sup></a> verwertet werden. Außerdem wurde berichtet, dass Metformin die Angiogenese <em>über</em> eine Erhöhung des VEGF-Spiegels<sup><a href=“#[„> [</a><a href=“https://www.oncotarget.com/article/6134/text/#bib35″>35</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib47″>47</a><a href=“#]“>]</a></sup> fördert. Wir berichten, dass die Hemmung von Komplex I die Laktatproduktion erhöht, und es wurde bereits früher vermutet, dass der glykolytische Schalter die Angiogenese fördert<sup> <a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib48″>48</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib49″>49</a><a href=“#]“>]</a></sup>. Unsere Ergebnisse zeigen eine positive Korrelation zwischen Serumlaktat und VEGF-Spiegel bei Mäusen, denen Metformin verabreicht wurde. Daher ist es wahrscheinlich, dass das durch Metformin verstärkte Tumorwachstum durch eine laktatinduzierte Angiogenese begünstigt wird, die durch VEGF <sup><a href=“#48″>[48]</a></sup> vermittelt werden kann. Darüber hinaus beobachteten wir in den Tumoren von Mäusen, denen Metformin verabreicht wurde, eine erhöhte Konzentration von E2F1, einem wichtigen Molekül zur Regulierung des Zellzyklus. E2F1 fördert nicht nur die Zellproliferation, sondern reguliert auch viele Gene, die an der Glykolyse<sup><a href=“#50″>[50]</a></sup> beteiligt sind, die für das schnelle Wachstum von Krebszellen unerlässlich ist. Daher ist es wahrscheinlich, dass E2F1 eine wichtige Rolle bei der Vermittlung der Metformin-induzierten schnellen Tumorprogression spielt, wahrscheinlich <em>über</em> die Regulierung der aeroben Glykolyse.</p>
<p>Glykolytische Inhibitoren wie 2-Desoxyglukose, Lonimid, 3-Bromopyruvat, DCA und Medikamente, die in Stoffwechselwege eingreifen, haben vielversprechende Ergebnisse bei der Unterdrückung des Tumorwachstums gezeigt<sup> <a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib40″>40</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib42″>42</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib51″>51</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href=“#]“>]</a></sup>. Darüber hinaus ist die Beeinflussung der Laktatbildung besonders bei glykolytischen Krebsarten interessant <sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib54″>54</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib55″>55</a><a href=“#]“>]</a></sup>. Melanomzellen haben einen glykolytischen Stoffwechsel und sind daher in erster Linie auf die hohe Aktivität von LDH zur ATP-Erzeugung angewiesen <sup><a href=“#6″>[6]</a></sup>. Melanomzellen überexprimieren LDH und ein hoher Serumspiegel korreliert häufig mit einer schlechten Prognose und dem Überleben der Patienten<sup> <a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib8″>8</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=“#]“>]</a></sup>. Unsere Ergebnisse zeigen, dass die Unterbrechung der Umwandlung von Pyruvat in Laktat die Proliferation von Melanomzellen stark beeinträchtigt. Dies stimmt mit Berichten überein, wonach Tumoren glykolytischer Zelltypen empfindlicher auf die Hemmung von LDH-A durch FX11<sup><a href=“#56″>[56]</a></sup> reagieren.</p>
<p>LDH spielt eine wichtige Rolle bei der metabolischen Homöostase und der Tumorerhaltung<sup> <a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib12″>12</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib57″>57</a><a href=“#]“>]</a></sup>. Wir haben Oxamat und DCA verwendet, um die Laktatbildung in Melanomzellen zu hemmen. DCA ist ein oral verabreichbares kleines Molekül, das zur Behandlung von Laktatazidose eingesetzt wird, und die Wiederherstellung der OXPHOS-Funktionalität durch DCA induziert nachweislich den Zelltod<sup><a href=“#[„> [</a><a href=“https://www.oncotarget.com/article/6134/text/#bib52″>52</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href=“#]“>]</a></sup>. Außerdem hat sich gezeigt, dass DCA die Laktatproduktion verringert und die Apoptose in Melanomzellen auslöst <sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib58″>58</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib59″>59</a><a href=“#]“>]</a></sup>. Oxamat, ein Analogon von Pyruvat, wird ebenfalls zur Hemmung der LDH eingesetzt. Eine kürzlich von Miskimins et al. durchgeführte Studie legt nahe, dass die gemeinsame Bekämpfung von Komplex I und LDH eine vielversprechende Strategie sein kann, um das Wachstum von aggressiven Krebsarten zu stoppen <sup><a href=“#60″>[60]</a></sup>. In Übereinstimmung damit haben wir auch festgestellt, dass die Verhinderung der Laktatbildung und die gleichzeitige Hemmung von Komplex I durch Oxamat/DCA oder durch LDH-spezifische siRNA bzw. Metformin zu einer Verarmung des zellulären ATP-Pools führt. Der Rückgang des zellulären ATP-Pools löst eine metabolische Katastrophe aus, die zur Apoptose in Melanomzellen führt. Der durch die Stoffwechselkatastrophe ausgelöste Zelltod gilt als vielversprechende Strategie, um das Fortschreiten von Krebs zu stoppen. Darüber hinaus weist unsere Studie auf die unterschiedliche Rolle von Komplex I und LDH bei der Zellproliferation hin, wobei diese beiden Enzyme entweder durch OXPHOS oder durch Phosphorylierung auf Substratebene ATP erzeugen. Die Blockierung eines der beiden Enzyme allein führt nicht zum Zelltod, da die Zellen auf einen alternativen Weg zur ATP-Erzeugung umschalten können. Die gleichzeitige Hemmung beider Enzyme hingegen führt zur Apoptose. Dies deutet eindeutig darauf hin, dass die Zellen von der Funktion dieser beiden Enzyme abhängen, die ein synthetisch tödliches Paar bilden, was ein vielversprechendes Phänomen ist, das für die selektive Bekämpfung von Melanomzellen genutzt werden kann<a href=“#[„> </a><sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib62″>62</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib63″>63</a><a href=“#]“>]</a></sup>. Da E2F1 an der Regulierung der Apoptose <sup><a href=“#64″>[64]</a></sup> beteiligt ist, ist es wahrscheinlich, dass an der durch die Kombination von Metformin und Oxamat/DCA ausgelösten Apoptose E2F1 beteiligt sein könnte. Metformin fördert die Apoptose in Krebszellen <em>über</em> die Aktivierung von p53 <sup><a href=“#37″>[37]</a></sup>. Im Falle des Melanoms ist die Rolle von p53 jedoch unklar. Wie aus der Literatur hervorgeht, ist p53 beim Melanom nicht funktionsfähig, und seine Werte sind paradoxerweise in fortgeschrittenen Melanomstadien erhöht <sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib65″>65</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib67″>67</a><a href=“#]“>]</a></sup>. Zuvor haben wir berichtet, dass ein erhöhter p53-Spiegel mit einer Zunahme des Tumorwachstums verbunden ist<sup><a href=“#68″>[68]</a></sup>. Daher ist es unwahrscheinlich, dass p53 an der durch die Kombinationsbehandlung induzierten Apoptose beteiligt ist. Daher sind weitere Studien erforderlich, um die Funktionalität von E2F1 und p53 beim Melanom zu bewerten.</p>
<p>Bei der Verwendung von Kombinationen verschiedener Medikamente, die synergistisch den Tod von Krebszellen fördern können, kann dies auch die Toxizität für normale Zellen erhöhen. Daher ist es von entscheidender Bedeutung, die Auswirkungen der Kombinationsbehandlung auf normale Zellen zu untersuchen. Unsere Daten deuten darauf hin, dass diese Kombination Krebszellen sowohl <em>in vitro</em> als auch <em>in vivo</em> wirksam abtötet und die geringsten Auswirkungen auf das Überleben normaler Zellen hat. Die unterschiedliche Empfindlichkeit von Melanom- und normalen Zellen gegenüber der Kombination aus Metformin und Oxamat/DCA lässt sich darauf zurückführen, dass Melanomzellen stark glykolytisch sind und im Vergleich zu normalen Zellen <sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib6″>6</a>-<a href=“https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=“#]“>]</a></sup> Moleküle überexprimieren, die an der Laktatbildung und -sekretion beteiligt sind. Interessanterweise zeigen Krebszellen im Vergleich zu normalen Zellen eine höhere ungekoppelte mitochondriale Atmung<sup><a href=“#31″>[31]</a></sup>. Krebszellen zeigen bei Behandlung mit Metformin eine stärkere kompensatorische Erhöhung der Glykolyse als normale Zellen<sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib31″>31</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib32″>32</a><a href=“#]“>]</a></sup>, wodurch die metabolische Anfälligkeit von Krebszellen erhöht wird. Daher unterdrückt die Hemmung der LDH- oder Laktatbildung/-ausscheidung das Wachstum von Krebszellen, während normale Zellen am wenigsten betroffen sind, da immer noch genügend ATP aus der OXPHOS produziert werden kann, da Metformin ein schwacher Komplex-I-Inhibitor ist. Da die OXPHOS-Aktivität in normalen Zellen höher ist als in Melanomzellen, hat die Kombination aus Metformin und Oxamat/DCA wahrscheinlich die geringsten negativen Auswirkungen auf die Atmung normaler Zellen.</p>
<p>Unsere Studie eröffnet neue Wege für die gezielte Beeinflussung des Stoffwechsels von Krebszellen und kann bei der Erprobung anderer klinisch zugelassener Medikamente, von denen bekannt ist, dass sie zusammen mit Metformin die Glykolyse hemmen, weiter berücksichtigt werden. Die vorliegende Studie deutet darauf hin, dass jedes Medikament/jeder Hemmstoff, der die Laktatbildung blockiert, in Kombination mit Metformin eingesetzt werden kann, um die Behandlung zu verbessern und das Tumorwachstum zu verhindern. So hat sich beispielsweise gezeigt, dass Rapamycin, ein klinisch zugelassenes Medikament, die Laktatbildung<sup> <a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib69″>69</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib70″>70</a><a href=“#]“>]</a></sup> verhindert. Ebenso hat sich gezeigt, dass die Hemmung von BRAF zu einer Unterdrückung der Glykolyse führt <sup><a href=“#[„>[</a><a href=“https://www.oncotarget.com/article/6134/text/#bib6″>6</a>, <a href=“https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href=“#]“>]</a></sup>. Daher können sowohl Rapamycin als auch BRAF-Inhibitoren zusammen mit Metformin eingesetzt werden, um das therapeutische Ergebnis bei geringeren Nebenwirkungen zu verbessern. Insgesamt deuten unsere Ergebnisse darauf hin, dass LDH oder andere Mechanismen, die die Laktatbildung oder -sekretion kontrollieren, für ein schnelles Fortschreiten des Melanoms unter OXPHOS-gestörten Bedingungen entscheidend sind, und dass dies als geeignetes therapeutisches Ziel für die Kontrolle des Wachstums glykolytischer Krebszellen genutzt werden kann. Es sind weitere Studien erforderlich, um die Funktionalität von LDH und Komplex I in anderen Krebsmodellen zu bewerten und anschließend ihre Bedeutung für die Krebstherapie im Allgemeinen zu untersuchen.</p>
<h2>MATERIALIEN UND METHODEN</h2>
<p><strong>Tierversuche<br></strong>Mäuse wurden von der Experimental Animal Facility (EAF) am National Centre for Cell Science (NCCS), Pune, Indien, beschafft. Hyperglykämie bei Mäusen wurde mit STZ induziert, wie zuvor beschrieben<sup><a href=“#71″>[71]</a></sup> mit leichten Änderungen. Kurz gesagt, männliche C57BL/6J- und NOD/SCID-Mäuse wurden vor der intraperitonealen Injektion von STZ (50 mg/kg) in 0,01 M Citratpuffer (pH 4,4) an drei aufeinanderfolgenden Tagen 6 Stunden lang gefastet. Die Blutzuckermessung erfolgte durch Abklemmen des Schwanzes und Auftragen des Blutes auf ein Glukoseanalysegerät (Accu-Chek Active, Roche, Deutschland). Mäuse mit einem Blutzuckerwert von über 200 mg/dl wurden als hyperglykämisch eingestuft. Der Tumor wurde durch Injektion von B16F10 (2 × 10<sup>5</sup>) oder A375 (1 × 10<sup>6</sup>) Zellen in 100 µl sterilem PBS subkutan in die rechte Flanke von C57BL/6J- bzw. NOD/SCID-Mäusen induziert. Das Fortschreiten des Tumors wurde regelmäßig überwacht, indem seine Größe mit einer digitalen Schieblehre (Sigma, USA) nach dem Auftreten von tastbaren Tumoren gemessen wurde. Die orale Verabreichung von Metformin an abwechselnden Tagen wurde bei hyperglykämischen Mäusen eine Woche vor der Tumorprovokation begonnen. Ansonsten wurde Metformin erst verabreicht, nachdem der Tumor eine optimale Größe erreicht hatte, und die Behandlung wurde bis zum Abschluss des Versuchs fortgesetzt. Am Ende des Versuchs wurden die Mäuse getötet; die Tumore wurden entfernt und entweder bei -80<sup>°</sup>C oder in 10%iger Formalinlösung für weitere Untersuchungen aufbewahrt. In einer weiteren Versuchsreihe wurden Mäuse (C57BL/6J-Hintergrund, männlich) nach dem Auftreten eines tastbaren Tumors zufällig in 4 Gruppen aufgeteilt. Um das Ergebnis der Metformin-Behandlung in Kombination mit dem LDH-Hemmer Oxamat zu untersuchen, wurde den Mäusen an jedem zweiten Tag entweder Metformin (200 mg/kg) oder Oxamat allein (500 mg/kg) oder beides zusammen oral verabreicht. Das Wachstum des Tumors wurde regelmäßig mit einer Schieblehre überwacht. Am Ende des Versuchs wurden Tumor-, Muskel- und Lebergewebe entnommen, gewogen und zur weiteren Analyse bei -80<sup>°</sup>C gelagert. Alle Tierversuche wurden gemäß den Richtlinien des Komitees zur Kontrolle und Überwachung von Tierversuchen (CPCSEA) der indischen Regierung und nach Genehmigung durch das Ethikkomitee des Instituts für Tiere (IAEC) durchgeführt.</p>
<p><strong>Zelllinien und Kulturbedingungen<br></strong>B16F10-Mausmelanom-, A375- und SKMel28-Humanmelanomzellen wurden von ATCC (VA, USA) erworben und in unserem hauseigenen Repository aufbewahrt. Die Nicht-Krebszellen AML12 (Maushepatozyten), L6 (Rattenmuskelzellen) und MEF (embryonale Mausfibroblasten) wurden parallel zu den Krebszellen als Kontrolle verwendet. Alle Zellen wurden in ihrem jeweiligen Medium gezüchtet, das je nach Experiment entweder 1 mM oder 25 mM Glukose enthielt und mit 10 % hitzeinaktiviertem fötalen Rinderserum (Hyclone, UT, USA), Penicillin (100 U/ml) und Streptomycin 100 µg/ml (Life Technologies, NY, USA) bei 37<sup>°</sup>C in Gegenwart von 5 %<sub> </sub>CO<sub>2</sub> ergänzt wurde.</p>
<p><strong>Chemikalien und Reagenzien<br></strong>Metformin, Phenformin, Oligomycin, Rotenon, Decylubiquinon, Streptozotocin (STZ), [3-(4, 5-Dimethylthiazol-2-yl)-2, 5-Diphenylterazoliumbromid] (MTT), NAD+, NADH, ATP, AMP, D-Glucose, Natriumoxamat, Iodacetat, 2-Desoxy-D-Glucose, Dichloracetat (DCA), Diaminobenzidin (DAB) und Phloretin wurden von Sigma (MO, USA) bezogen. Antikörper für ChREBP (1:1000), GLUT1 (1:1000), LDHA (1:1000), Cyclin D1 (1:1000), PCNA (1:1000), CDK4 (1:1000), p21 (1:1000), E2F1 (1:1000) CD31 (1:100), PARP-1 (1:1000), Bcl-2 (1:1000), Bax (1:1000), β-Tubulin (1:1000), GAPDH (1:1000), HSP60 (1:1000) und VEGF (1:50) waren von Santa Cruz Biotechnology (CA, USA).</p>
<p><strong>Zelllysatvorbereitung und Immunoblotting<br></strong>Die Zellen wurden dreimal mit phosphatgepufferter Kochsalzlösung (PBS) gewaschen und in Lysispuffer mit 50 mM Tris-Cl (pH 7.5), 120 mM NaCl, 10 mM Natriumfluorid, 10 mM Natriumpyrophosphat, 2 mM EDTA, 1 mM Natriumorthovanadat, 1 mM Phenylmethylsulfonylfluorid, 1% NP-40 und Proteaseinhibitor-Cocktail (Roche, Deutschland). Zur Herstellung von Tumorlysaten wurde das Tumorgewebe in feine Stücke geschnitten, fünfmal mit 0,85%iger Kochsalzlösung, die einen Proteaseinhibitor-Cocktail enthielt, gewaschen und in Lysispuffer durch Homogenisierung mit einem Gewebehomogenisator (Sigma, USA) und anschließende Beschallung lysiert. Die Lysate wurden durch Zentrifugation bei 15.000 rpm<em> </em>für 30 Minuten geklärt. Die Zelllysate wurden unter gekühlten Bedingungen hergestellt. Etwa 50-100 µg Protein aus den Ganzzell-Lysaten wurden auf einem 8-12%igen SDS-Polyacrylamid-Gel aufgelöst, das anschließend auf eine PVDF-Membran (Millipore, Deutschland) übertragen wurde. Die Membran wurde mit den gewünschten primären Antikörpern und anschließend mit HRP-konjugierten sekundären Antikörpern beschichtet. Die Immunblots wurden mit Luminalreagenz (Santa Cruz Biotechnology) nachgewiesen. Wenn erforderlich, wurden die Blots durch 15-minütige Inkubation der Membran bei 50<sup>°</sup>C in Stripping-Puffer (62,5 mM Tris-Cl, pH 6,8, 100 mM Mercaptoethanol und 2 % SDS) unter intermittierendem Schütteln gestrippt. Die Membranen wurden gründlich mit Tris-gepufferter Kochsalzlösung (TBS) gewaschen und mit den gewünschten Antikörpern rebediert.</p>
<p><strong>Immunfluoreszenz oder konfokale Mikroskopie<br></strong>Die Zellen wurden in Labtek-Kammer-Objektträger (Nunc, USA) plattiert und 24 Stunden lang wachsen gelassen. Die Behandlung mit Metformin und anderen Inhibitoren erfolgte für die gewünschten Zeiträume und Konzentrationen. Die Zellen wurden mit gekühltem, sterilem PBS gewaschen und mit 3,7%iger Paraformaldehydlösung bei Raumtemperatur 10 Minuten lang fixiert. Anschließend wurden sie mit 0,025 % Triton X-100 in PBS 10 Minuten lang permeabilisiert und anschließend mit 5 % BSA in PBS 1 Stunde lang bei 37<sup>°</sup>C blockiert. Die Zellen wurden mit 1:100-Verdünnungen der primären Antikörper in der Blockierungslösung 2 Stunden lang bei Raumtemperatur inkubiert und mindestens fünfmal mit TBST (TBS mit 0,05 % Tween-20) gewaschen, bevor sie mit entsprechend markierten sekundären Antikörpern (1:200) in Blockierungslösung weitere 1 Stunde lang bei Raumtemperatur inkubiert wurden. Nach fünfmaligem Waschen mit TBST wurden die Proben mit DAPI-haltigem Eindeckmittel (Santa Cruz Biotechnology, USA) beschichtet. Die Objektträger wurden versiegelt, unter einem konfokalen Laser-Scanning-Mikroskop (LSM510 Carl Zeiss, Deutschland) untersucht und die Bilder aufgenommen. Die Bilder wurden anschließend mit der LSM-Bildanalysesoftware bearbeitet.</p>
<p><strong>Immunhistochemie und Histopathologie<br></strong>Die immunhistochemische und histopathologische Untersuchung wurde nach dem Verfahren von Malvi et al. <sup><a href=“#72″>[72]</a></sup> durchgeführt. Kurz gesagt, wurden feine Schnitte von Tumoren und anderen Organen mit dem Mikrotom angefertigt, auf Glasobjektträgern fixiert und paraffiniert. Für die Immunhistochemie wurden die Objektträger zweimal für 10 Minuten in Xylol-Lösung entparaffiniert und anschließend dreimal mit 100%, 95%, 70% und 50% Ethanol gewaschen. Die Objektträger wurden erneut mit destilliertem Wasser gewaschen und anschließend 5 Minuten lang mit PBS gespült. Zum Antigen-Retrieval wurden die Objektträger 10 Minuten lang in Natriumcitratpuffer (0,01 M, pH 4,5) in der Mikrowelle gekocht und anschließend 20 Minuten lang bei Raumtemperatur abgekühlt. Die Objektträger wurden mit den gewünschten Antikörpern (1:100 Verdünnung) in PBST (PBS mit 0,025 % Tween-20) mit 0,01 % BSA für 2 Stunden bei Raumtemperatur oder über Nacht bei 4<sup>°</sup>C sondiert. Die Objektträger wurden 4 Mal 5 Minuten lang mit PBST gewaschen und 1 Stunde lang mit kompatiblen HRP-konjugierten sekundären Antikörpern sondiert. Die Objektträger wurden erneut gewaschen und 10 Minuten lang mit Diaminobenzidin (DAB) gefärbt, anschließend gewaschen und mit Hämatoxylin gegengefärbt. Die Objektträger wurden anschließend mit Wasser gewaschen, mit absolutem Alkohol dehydriert und mit Einbettungsmedium beschichtet, bevor sie auf die Expression der gewünschten Moleküle untersucht wurden. Für die Histopathologie wurden die entparaffinierten Objektträger mit Hämatoxylin und Eosin angefärbt, und die mikroskopische Analyse auf Zelldichte, Zellmorphologie, Metastasierung, Zytotoxizität und Nekrose wurde von Pathologen im KEM-Krankenhaus in Pune, Indien, verblindet durchgeführt.</p>
<p><strong>Zellulärer Zytotoxizitäts- und Überlebenstest</strong><br>Ungefähr 5 × 10<sup>3</sup> (B16F10) und 10 × 10<sup>3</sup> (A375 und SKMel28) Zellen wurden in jede Vertiefung von 96-Well-Gewebekulturplatten gesät und für 24 h bei 37<sup>°</sup>C anhaften gelassen. Die Zellen wurden entsprechend den experimentellen Anforderungen behandelt, und die Proliferation bzw. Lebensfähigkeit wurde mittels MTT-Assay bewertet. MTT (50 µl, 1 mg/ml in DMEM ohne Phenolrot) wurde in jede Vertiefung gegeben und 4 Stunden lang bei 37<sup>°</sup>C inkubiert. Die Formazankristalle wurden in 100 µl Isopropanol durch 5-minütiges Schütteln bei Raumtemperatur aufgelöst. Die Absorption wurde bei 570 nm mit 630 nm als Referenzfilter gemessen. Unbehandelte Zellen wurden als Kontrolle betrachtet (100% Zellüberleben).</p>
<p><strong>Nachweis der Apoptose durch Annexin V-Färbung<br></strong>Die Zellen wurden in einer Dichte von etwa 3 × 10<sup>5</sup> Zellen in 35-mm-Platten ausgesät und für 24 h wachsen gelassen. Die Zellen wurden mit oder ohne Oxamat und DCA entweder allein oder mit Metformin für 48 h oder nach experimenteller Anforderung behandelt. Die Zellen wurden durch Trypsinierung geerntet und für die Durchflusszytometrie aufbereitet. Apoptose wurde durch zweifache Färbung mit Annexin V und PI unter Verwendung eines Apoptose-Assay-Kits (BD Bioscience, USA) gemäß den Anweisungen des Herstellers nachgewiesen.</p>
<p><strong>Zellzyklusanalyse<br></strong>Die Zellen wurden in einer Dichte von etwa 3 × 10<sup>5</sup> Zellen in 35-mm-Platten ausgesät und 24 h lang wachsen gelassen. Die Zellen wurden mit oder ohne Metformin, Oxamat und anderen glykolytischen Inhibitoren allein oder zusammen, wie angegeben, 48 h lang oder nach experimenteller Anforderung behandelt. Die Zellen wurden durch Trypsinierung geerntet und für die Durchflusszytometrie aufbereitet. Die Zellen wurden mit gekühltem PBS gewaschen und 30 Minuten lang in 70%igem Ethanol auf Eis fixiert. Nach einer 30-minütigen RNase-Behandlung (200 μg/ml) bei 37 °C wurden 50 μg/ml PI zum Zellpellet gegeben und 30 Minuten im Dunkeln auf Eis inkubiert. Die Fluoreszenz von PI wurde durch einen 585-nm-Filter in einem Durchflusszytometer (FACS Calibur, Becton Dickinson, Kalifornien, USA) aufgezeichnet. Die Daten wurden mit der Software Cell Quest Pro für 10.000 Zellen analysiert.</p>
<p><strong>Langzeit-Zellüberlebenstest<br></strong>Zellen (5 × 10<sup>2</sup>) wurden für 24 h in 12-Well-Platten plattiert. Die Zellen wurden ohne oder mit 25 mM oder 50 mM Oxamat und 10 oder 20 mM DCA zusammen mit Metformin behandelt und für weitere 48 h beibehalten. Anschließend wurde das Medium durch frisches, wirkstofffreies Medium ersetzt. Die Platten wurden weitere 10-15 Tage bei 37°C im CO<sub>2</sub>-Inkubator bebrütet, wobei das Medium alle 2-3 Tage gewechselt wurde. Die Zellen wurden dann fixiert (3% Paraformaldehyd und 0,02% Glutaraldehyd in PBS) und mit 0,05% Kristallviolett gefärbt.</p>
<p><strong>Glukoseverwertungstest<br></strong>Zellen (3 × 10<sup>5</sup>) wurden in DMEM mit 25 mM Glukose kultiviert. Nach 24 Stunden wurde das Medium durch ein entsprechendes Medium ersetzt, das 24 Stunden lang eine steigende Konzentration von Metformin (0,1 mM, 0,5 mM, 1 mM und 2 mM) oder Rotenon enthielt, und die im verbrauchten Medium vorhandene Restglukose wurde mit einem Glukose-Assay-Kit auf GOD-POD-Basis (Spinreact, Spanien) gemäß den Anweisungen des Herstellers gemessen. Zur Messung der Glukoseverwertung in Gegenwart glykolytischer Inhibitoren wurden die Zellen mit 50 mM Oxamat, 100 µM Phloretin und 20 mM DCA behandelt, entweder allein oder zusammen mit 2 mM Metformin oder 100 µM Phenformin. Die verbrauchte Glukose wurde durch Subtraktion der verbleibenden Glukose im Medium von der Anfangskonzentration im Kontrollmedium (450 mg/dl) geschätzt. Die Experimente wurden mindestens in dreifacher Ausführung durchgeführt und die Werte wurden auf die Gesamtzahl der Zellen normiert.</p>
<p><strong>Laktatbestimmung<br></strong>Die Laktatkonzentration im verbrauchten Medium der Zellen, die mit oder ohne Metformin, Oxamat und anderen glykolytischen Inhibitoren behandelt wurden, wurde mit einem handelsüblichen Laktatbestimmungs-Kit (Spinreact, Spanien) gemäß den Anweisungen des Herstellers bestimmt. Kurz gesagt, Kulturmedium oder Serum wurde bis zum 10-fachen in 0,9%iger Kochsalzlösung verdünnt und 10 μl der Probe wurden in jede Vertiefung der 96-Well-ELISA-Platte gegeben. 150 µl des mit dem Kit gelieferten Reagens wurden in jede Probe enthaltende Vertiefung gegeben, wobei das nicht verbrauchte Medium und das Reagens allein als Leerwert beibehalten wurden, und die Absorption wurde bei 405 nm mit einem Spektrophotometer (Thermo-Scientific, USA) aufgezeichnet. Die Experimente wurden mindestens in dreifacher Ausführung durchgeführt und die Endwerte wurden mit der Gesamtzahl der Zellen normalisiert.</p>
<p><strong>siRNA-Transfektion<br></strong>Spezifische siRNA gegen LDHA wurde von Santa Cruz Biotechnology (USA) erworben. Die Transfektion erfolgte mit Lipofectamine 2000 (Life Technologies, USA) nach den Anweisungen des Herstellers. Die Zellen wurden bei einer Konfluenz von etwa 60 % ausgeplattet. Am nächsten Tag wurde das Medium durch OptiMEM (Life Technologies, USA) ersetzt und für 3 h belassen. Die gewünschten siRNAs wurden in den mitgelieferten Puffern gelöst. Lipofectamine2000 und siRNA wurden getrennt in OptiMEM verdünnt und 5 Minuten lang inkubiert. Danach wurden die verdünnten Reagenzien gemischt und weitere 30 Minuten bei Raumtemperatur inkubiert. Das entstandene Präzipitat wurde 6 Stunden lang auf den Zellen belassen. Danach wurde frisches DMEM mit 10 % FBS hinzugefügt und weitere 24-36 Stunden lang inkubiert. Die Transfektionseffizienz wurde durch gleichzeitige Transfektion des pEGFPN1-Plasmids bewertet. Immunoblotting wurde durchgeführt, um die Hemmung der jeweiligen Genexpression sicherzustellen.</p>
<p><strong>Zubereitung der mitochondrienreichen Fraktion aus Zellen und Geweben<br></strong>Die mitochondrienreiche Fraktion wurde aus kultivierten Zellen und Tumoren oder normalem Gewebe hergestellt, wie bereits berichtet<sup><a href=“#73″>[73]</a></sup>. Die Zellen (1 × 10<sup>6</sup>) wurden in 10-cm-Platten ausplattiert und nach 24-stündiger Behandlung mit den gewünschten Inhibitoren trypsiniert. Die Zellsuspension wurde in hypotonischem Puffer (20 mM Kaliumphosphat) dreimal gefroren und aufgetaut. Diese Suspension wurde 1 Stunde lang bei 50.000 U/min zentrifugiert, um einen mitochondrienreichen Überstand zu erhalten. Um Mitochondrien aus Geweben zu isolieren, wurden gefrorene Gewebe in feine Stücke geschnitten und in Homogenisierungspuffer (0,5 M Tris-Puffer pH 7,5, 100 mM EGTA und 250 mM Saccharose) homogenisiert, gefolgt von einem zyklischen Gefrier-Auftau-Verfahren und Zentrifugation bei 50.000 U/min für 1 h. Der Überstand wurde als mitochondriale Fraktion gesammelt und für die Bestimmung der mitochondrialen Funktion und der OXPHOS-Enzymaktivität verwendet.</p>
<p><strong>Enzymassays<br></strong>Zellen wurden in hypotonischem (20 mM) Kaliumphosphatpuffer (pH 7,5) mit Proteaseinhibitorcocktail (Roche, Deutschland) homogenisiert, vortexed und durch drei Zyklen von Einfrieren und Auftauen lysiert. Zur Herstellung des Gewebeextrakts wurden die Tumorproben in feine Stücke geschnitten und in Homogenisierungspuffer (0,1 M Tris, 0,1 M KCl, 350 mM EDTA und 1 M Saccharose, pH 7,5) mit einem Gewebehomogenisator homogenisiert. Die Homogenate wurden durch Zentrifugieren bei 10.000 U/min für 10 Minuten bei 4<sup>°</sup>C geklärt und bis zur Durchführung der Assays auf Eis gelagert.</p>
<p><strong><em>LDH-Aktivitätstest:</em> </strong>Die Laktatdehydrogenase-Aktivität in Zelllysaten oder Tumorextrakt und Serum wurde mit einem LDH-Aktivitäts-Testkit (Spinreact, Spanien) gemäß den Anweisungen des Herstellers bestimmt.</p>
<p><em><strong>Komplex-I-Aktivitätstest:</strong></em> Der Test des mitochondrialen OXPHOS-Komplex-I-Enzyms wurde wie an anderer Stelle beschrieben<sup><a href=“#73″>[73]</a></sup> durchgeführt. Kurz gesagt, mitochondrienreiche Fraktion von Zell- oder Gewebehomogenat (20-50 µg Protein aus Gewebehomogenat oder 10-20 µg mitochondrienreiche Fraktion) wurde zu 700 µl destilliertem Wasser in einer 1-ml-Küvette gegeben, gefolgt von der Zugabe von 100 µl Kaliumphosphatpuffer (0,5 M, pH 7,5), 60 µl fettsäurefreiem BSA (50 mg/ml), 30 µl KCN (10 mM) und 10 µl NADH (10 mM). Das Endvolumen wurde mit destilliertem Wasser auf 994 µl eingestellt, durch Umdrehen der Küvetten gemischt und die Basislinie bei 340 nm für 2 min gemessen. Die Reaktion wurde durch Zugabe von 6 µl Decylubiquinon (10 mM, Sigma, USA) gestartet und durch Umdrehen der Küvetten gut gemischt. Die Abnahme der Absorption bei 340 nm wurde 10 Minuten lang beobachtet. Ein ähnliches Verfahren wurde für die Berechnung der Komplex-I-Aktivität in Gegenwart von 2 mM Metformin und 100 µM Phenformin angewandt. Rotenon (10 µM) wurde als Positivkontrolle verwendet. Die Endwerte wurden mit dem gesamten zellulären Proteingehalt normalisiert.</p>
<p><strong>Gesamtzelluläre ATP-Messung<br></strong>Der ATP-Gehalt wurde mit einem handelsüblichen ATP-Biolumineszenz-Kit (Roche, Deutschland) gemäß den Anweisungen des Herstellers gemessen. Kurz gesagt, die Zellen wurden in Gegenwart der angegebenen Medikamente gezüchtet. Die Zellen wurden geerntet und in 100 mM Tris-EDTA-Puffer mit 0,01% NP-40 lysiert und 1 Minute lang gekocht, gefolgt von einem Gefrier-Auftauzyklus. Die Lumineszenz wurde sowohl für die Leerwerte als auch für die Proben aufgezeichnet. Zur Messung des ausschließlich durch OXPHOS und Glykolyse erzeugten ATP wurden die Zellen mit Oligomycin (10 µM) und Oxamat bzw. DCA mit oder ohne Metformin behandelt. Die Experimente wurden in dreifacher Ausführung durchgeführt und mindestens einmal wiederholt, und die Endwerte wurden mit dem Gesamtproteingehalt normalisiert.</p>
<p><strong>Statistik</strong><br>Die statistische Analyse wurde mit Sigma Plot 12.0 (Systat Software Inc., CA, USA) durchgeführt. Die meisten Experimente wurden mindestens einmal und mindestens in dreifacher Ausführung wiederholt, sofern nicht anders angegeben. Die Daten wurden als Mittelwert ± SD dargestellt, außer bei den angegebenen Experimenten. Wo immer erforderlich, wurde bei den Experimenten, sofern nicht anders angegeben, ein gepaarter oder ungepaarter zweiseitiger Student-<em>t</em>-Test unter der Annahme ungleicher Varianz verwendet, um den p-Wert zu berechnen. Die Werte *<em>p</em> < 0,05, **<em>p</em> < 0,01, ***<em>p</em> < 0,001 kennzeichnen signifikante Unterschiede zwischen den Gruppen (<em>n</em> > 3 mindestens).</p>
<h2>HINWEISE</h2>
<p>Die Autoren danken Dr. S.C. Mande, Direktor des NCCS, Pune, Indien, und Dr. G.C. Mishra, ehemaliger Direktor des NCCS, Pune, Indien, für ihre große Unterstützung und die Ermutigung zur Durchführung dieser Arbeit. Wir danken Dr. Benoit Violet (INSERM, Frankreich) für die Bereitstellung von MEFs, Dr. Mahesh J Kulkarni (National Chemical Laboratory, Indien) für L6-Zellen und Dr. Rakesh K Tyagi (Jawaharlal Nehru University, Indien) für die Bereitstellung von AML12-Zellen. Wir danken auch Dr. Vijayakumar MV für seine Hilfe bei den Tierversuchen und für das kritische Lesen des Manuskripts. BC und NM danken dem Council of Scientific and Industrial Research (CSIR), Indien; PM und SVS danken der University Grant Commission (UGC), Indien, für die Bereitstellung eines Stipendiums. Die Unterstützung durch andere Labormitglieder und Kollegen am NCCS sowie durch Mitarbeiter der Experimental Animal Facility, der FACS-, Konfokal- und Massenspektrometrie-Einrichtung wird gebührend gewürdigt.</p>
<h2>FINANZIELLE UNTERSTÜTZUNG</h2>
<p>Diese Arbeit wurde durch ein intramurales Stipendium des NCCS unterstützt, das vom Department of Biotechnology der indischen Regierung finanziert wurde.</p>
<h2>INTERESSENSKONFLIKTE</h2>
<p>Die Autoren erklären, dass sie keine potenziellen Interessenkonflikte haben</p>
<h3>Hinweis</h3>
<p>Diese Arbeit wurde in teilweiser Erfüllung einer Doktorarbeit (von B.C.) durchgeführt, die an der Savitribai Phule Pune University, Pune, Indien, eingereicht werden soll.</p>
<p></p>
<h2>REFERENZEN</h2>
<br><span id=“1″ class=“referencess blue-text“>1</span> Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007; 445:851-857.
<br><span id=“2″ class=“referencess blue-text“>2</span> Demierre MF. Epidemiologie und Prävention des kutanen Melanoms. Curr Treat Options Oncol. 2006; 7:181-186.
<br><span id=“3″ class=“referencess blue-text“>3</span> Hersey P, Watts RN, Zhang XD, Hackett J. Metabolic approaches to treatment of melanoma. Clin Cancer Res. 2009; 15:6490-6494.
<br><span id=“4″ class=“referencess blue-text“>4</span> Warburg O. On respiratory impairment in cancer cells. Science. 1956; 124:269-270.
<br><span id=“5″ class=“referencess blue-text“>5</span> Vander Heiden MG, Cantley LC, Thompson CB. Den Warburg-Effekt verstehen: die metabolischen Anforderungen der Zellproliferation. Science. 2009; 324:1029-1033.
<br><span id=“6″ class=“referencess blue-text“>6</span> Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C. Dysfunktionale oxidative Phosphorylierung macht maligne Melanomzellen süchtig nach Glykolyse, angetrieben durch das (V600E)BRAF-Onkogen. Oncotarget. 2013; 4:584-599.
<br><span id=“7″ class=“referencess blue-text“>7</span> Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, et al. Oncogenic BRAF reguliert den oxidativen Stoffwechsel über PGC1α und MITF. Cancer Cell. 2013; 23:302-315.
<br><span id=“8″ class=“referencess blue-text“>8</span> Weide B, Elsässer M, Büttner P, Pflugfelder A, Leiter U, Eigentler TK, Bauer J, Witte M, Meier F, Garbe C. Serummarker Laktatdehydrogenase und S100B sagen unabhängig den Krankheitsverlauf bei Melanompatienten mit Fernmetastasen voraus. Br J Cancer. 2012; 107:422-428.
<br><span id=“9″ class=“referencess blue-text“>9</span> Deichmann M, Benner A, Bock M, Jäckel A, Uhl K, Waldmann V, Näher H. S100-Beta, melanomhemmende Aktivität und Laktatdehydrogenase diskriminieren progressives von nicht-progressivem American Joint Committee on Cancer Melanom Stadium IV. J Clin Oncol. 1999; 17:1891-1896.
<br><span id=“10″ class=“referencess blue-text“>10</span> Giatromanolaki A, Sivridis E, Gatter KC, Turley H, Harris AL, Koukourakis MI; Tumor and Angiogenesis Research Group. Die Expression von Laktatdehydrogenase 5 (LDH-5) bei Endometriumkrebs steht in Zusammenhang mit dem aktivierten VEGF/VEGFR2(KDR)-Signalweg und der Prognose. Gynecol Oncol. 2006; 103:912-918.
<br><span id=“11″ class=“referencess blue-text“>11</span> Koukourakis MI, Giatromanolaki A, Sivridis E. Lactate dehydrogenase isoenzymes 1 and 5: differential expression by neoplastic and stromal cells in non-small cell lung cancer and other epithelial malignant tumors. Tumor Biol. 2003; 24:199-202.
<br><span id=“12″ class=“referencess blue-text“>12</span> Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006; 9:425-434.
<br><span id=“13″ class=“referencess blue-text“>13</span> Brandon M, Baldi P, Wallace DC. Mitochondriale Mutationen bei Krebs. Oncogene. 2006; 25:4647-4662.
<br><span id=“14″ class=“referencess blue-text“>14</span> Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-erzeugende mitochondriale DNA-Mutationen können die Metastasierung von Tumorzellen steuern. Wissenschaft. 2008; 320:661-664.
<br><span id=“15″ class=“referencess blue-text“>15</span> Wallace DC. Ein mitochondriales Paradigma für Stoffwechsel- und degenerative Krankheiten, Alterung und Krebs: ein Aufbruch für die evolutionäre Medizin. Annu Rev Genet. 2005; 39:359-407.
<br><span id=“16″ class=“referencess blue-text“>16</span> Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013; 123:1068-1081.
<br><span id=“17″ class=“referencess blue-text“>17</span> He X, Zhou A, Lu H, Chen Y, Huang G, Yue X, Zhao P, Wu Y. Die Unterdrückung des mitochondrialen Komplexes I beeinflusst die metastatischen Eigenschaften von Zellen. PLoS One. 2013; 8:e61677.
<br><span id=“18″ class=“referencess blue-text“>18</span> Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y. Dysfunktion des mitochondrialen Atmungskomplexes I fördert die Tumorentstehung durch ROS-Veränderung und AKT-Aktivierung. Hum Mol Genet. 2011; 20:4605-4616.
<br><span id=“19″ class=“referencess blue-text“>19</span> Gopal YN, Rizos H, Chen G, Deng W, Frederick DT, Cooper ZA, Scolyer RA, Pupo G, Komurov K, Sehgal V, Zhang J, Patel L, Pereira CG et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1α and oxidative phosphorylation in melanoma. Cancer Res. 2014; 74:7037-7047.
<br><span id=“20″ class=“referencess blue-text“>20</span> Barbi de Moura M, Vincent G, Fayewicz SL, Bateman NW, Hood BL, Sun M, Suhan J, Duensing S, Yin Y, Sander C, Kirkwood JM, Becker D, Conrads TP, et al. Mitochondrial respiration–an important therapeutic target in melanoma. PLoS One. 2012; 7:e40690.
<br><span id=“21″ class=“referencess blue-text“>21</span> Blackman RK, Cheung-Ong K, Gebbia M, Proia DA, He S, Kepros J, Jonneaux A, Marchetti P, Kluza J, Rao PE, Wada Y, Giaever G, Nislow C. Mitochondrial electron transport is the cellular target of the oncology drug elesclomol. PLoS One. 2012; 7:e29798.
<br><span id=“22″ class=“referencess blue-text“>22</span> Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Umgekehrter TCA-Zyklus-Fluss durch Isocitratdehydrogenasen 1 und 2 ist für die Lipogenese in hypoxischen Melanomzellen erforderlich. Pigment Cell Melanoma Res. 2012; 25:375-383.
<br><span id=“23″ class=“referencess blue-text“>23</span> Lim JH, Luo C, Vazquez F, Puigserver P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014; 74:3535-3545.
<br><span id=“24″ class=“referencess blue-text“>24</span> Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin und verringertes Krebsrisiko bei Diabetikern. British Medical Journal. 2005; 330:1304-1305.
<br><span id=“25″ class=“referencess blue-text „25</span> Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila). 2010; 3:1451-1461.
<br><span id=“26″ class=“referencess blue-text“>25</span> Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med. 2002; 137:25-33.
<br><span id=“27″ class=“referencess blue-text“>27</span> Pollak MN. Untersuchung von Metformin zur Krebsprävention und -behandlung: das Ende vom Anfang. Cancer Discov. 2012; 2:778-790.
<br><span id=“28″ class=“referencess blue-text“>28</span> El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibiert die Zellatmung über eine indirekte Wirkung, die auf den Atmungskettenkomplex I abzielt. J Biol Chem. 2000; 275:223-228.
<br><span id=“29″ class=“referencess blue-text“>29</span> Owen MR, Doran E, Halestrap AP. Beweise, dass Metformin seine antidiabetischen Wirkungen durch Hemmung von Komplex 1 der mitochondrialen Atmungskette ausübt. Biochem J. 2000; 348:607-614.
<br><span id=“30″ class=“referencess blue-text“>30</span> Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Metformin hemmt den mitochondrialen Komplex I von Krebszellen und reduziert so die Tumorentstehung. Elife. 2014; 3:e02242.
<br><span id=“31″ class=“referencess blue-text“>31</span> Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin wirkt direkt auf Mitochondrien und verändert die zelluläre Bioenergetik. Cancer Metab. 2014; 2:12.
<br><span id=“32″ class=“referencess blue-text“>32</span> Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarié A, Tyler BM, Brem H, Toulas C, Cohen-Jonathan Moyal E, Sarry JE, Skuli N. Metformin hemmt das Wachstum menschlicher Glioblastomzellen und verbessert die therapeutische Reaktion. PLoS One. 2015; 10:e0123721.
<br><span id=“33″ class=“referencess blue-text“>33</span> Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. The antidiabetic drug metformin exerts an antitumoral effect in vitro and in vivo through a decrease of cyclin D1 level. Oncogene. 2008; 27:3576-3586.
<br><span id=“34″ class=“referencess blue-text“>34</span> Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin ist ein AMP-Kinase-abhängiger Wachstumsinhibitor für Brustkrebszellen. Cancer Res. 2006; 66:10269-10273.
<br><span id=“35″ class=“referencess blue-text“>35</span> Martin MJ, Hayward R, Viros A, Marais R. Metformin beschleunigt das Wachstum von BRAF V600E-getriebenen Melanomen durch Hochregulieren von VEGF-A. Cancer Discov. 2012, 2:344-355.
<br><span id=“36″ class=“referencess blue-text“>36</span> Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A, Gounon P, Allegra M, Bertolotto C, Bereder JM, Tartare-Deckert S, Bahadoran P, Auberger P, et al. Metformin hemmt die Melanomentwicklung durch Autophagie- und Apoptosemechanismen. Cell Death Dis. 2011; 2:e199.
<br><span id=“37″ class=“referencess blue-text“>37</span> Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P, Bertolotto C, Tartare-Deckert S, Ballotti R, Rocchi S. Metformin blockiert die Invasion und Metastasenbildung von Melanomen in AMPK/p53-abhängiger Weise. Mol Cancer Ther. 2013; 12:1605-1615.
<br><span id=“38″ class=“referencess blue-text“>38</span> Yuan P, Ito K, Perez-Lorenzo R, Del Guzzo C, Lee JH, Shen CH, Bosenberg MW, McMahon M, Cantley LC, Zheng B. Phenformin steigert den therapeutischen Nutzen der BRAF(V600E)-Hemmung beim Melanom. Proc Natl Acad Sci USA. 2013; 110:18226-18231.
<br><span id=“39″ class=“referencess blue-text“>39</span> Cannino G, El-Khoury R, Pirinen M, Hutz B, Rustin P, Jacobs HT, Dufour E. Glucose modulates respiratory complex I activity in response to acute mitochondrial dysfunction. J Biol Chem. 2012; 287:38729-38740.
<br><span id=“40″ class=“referencess blue-text“>40</span> Xu RH, PelicanoH, Zhou Y, CarewJS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005; 65:613-621.
<br><span id=“41″ class=“referencess blue-text“>41</span> Pan JG, Mak TW. Metabolic Targeting als Krebsbekämpfungsstrategie: Anbruch einer neuen Ära? Sci STKE. 2007; 2007:pe14.
<br><span id=“42″ class=“referencess blue-text“>42</span> Dalva-Aydemir S, Bajpai R, Martinez M, Adekola KU, Kandela I, Wei C, Singhal S, Koblinski JE, Raje NS, Rosen ST, Shanmugam M. Targeting the Metabolic Plasticity of Multiple Myeloma with FDA-Approved Ritonavir and Metformin. Clin Cancer Res. 2015; 21:1161-1171.
<br><span id=“43″ class=“referencess blue-text“>43</span> Menendez JA, Oliveras-Ferraros C, Cufí S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle. 2012; 11:2782-2792.
<br><span id=“44″ class=“referencess blue-text“>44</span> Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, Nanni P, Bergamini C, Nicoletti G, Giovanni CD, Ghelli A, Giorgio V, Caratozzolo MF, et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013; 1:11.
<br><span id=“45″ class=“referencess blue-text“>45</span> Semenza GL. Tumorstoffwechsel: Krebszellen geben und nehmen Laktat. J Clin Invest. 2008; 118:3835-3837.
<br><span id=“46″ class=“referencess blue-text“>46</span> Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Bernard Gallez B, Wahl ML, et al. Targeting lactate-fueled respiration tötet selektiv hypoxische Tumorzellen in Mäusen. J Clin Invest. 2008; 118:3930-3942.
<br><span id=“47″ class=“referencess blue-text“>47</span> Phoenix KN, Vumbaca F, Claffey KP. Therapeutische Metformin/AMPK-Aktivierung fördert den angiogenen Phänotyp im ERalpha negativen MDA-MB-435 Brustkrebsmodell. Breast Cancer Res Treat. 2009; 113:101-111.
<br><span id=“48″ class=“referencess blue-text“>48</span> Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011; 71:2550-2560.
<br><span id=“49″ class=“referencess blue-text“>49</span> Sonveaux P, Copetti T, De Saedeleer CJ, Vegran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frérart F, Gallez B, Ribeiro A, Michiels C, et al. Der Angriff auf den Laktattransporter MCT1 in Endothelzellen hemmt die Laktat-induzierte HIF-1-Aktivierung und die Tumorangiogenese. PLoS ONE. 2012; 7:e33418.
<br><span id=“50″ class=“referencess blue-text“>50</span> Wu M, Seto E, Zhang J. E2F1 enhances glycolysis through suppressing Sirt6 transcription in cancer cells. Oncotarget. 2015; 6:11252-11263.
<br><span id=“51″ class=“referencess blue-text“>51</span> Hernlund E, Strandberg Ihrlund L, Khan O, Ates YO, Linder S, Panaretakis T, Shoshan MC. Potenzierung von Chemotherapeutika durch die Energiestoffwechsel-Hemmer 2-Desoxyglucose und Etomoxir. Int J Cancer. 2008; 123:476-483.
<br><span id=“52 „class=“referencess blue-text“>52</span>. Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Dichloracetat induziert Apoptose in Endometriumkrebszellen. Gynecol Oncol. 2008; 109:402.
<br><span id=“53″ class=“referencess blue-text“>53</span> Michelakis ED, Webster L, Mackey JR. Dichloracetat (DCA) als potenzielles metabolisches Ziel in der Krebstherapie. Br J Cancer. 2008; 99:989-994.
<br><span id=“54″ class=“referencess blue-text“>54</span> Walenta S, Mueller-Kliese WF. Laktat: Spiegel und Motor der Malignität von Tumoren. Semin Radiat Oncol. 2004; 14:267-274.
<br><span id=“55″ class=“referencess blue-text“>55</span> Doherty JR, Cleveland JL. Der Laktatstoffwechsel als Ziel für Krebstherapeutika. J Clin Invest. 2013; 123:3685-3692.
<br><span id=“56″ class=“referencess blue-text“>56</span> Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Die Hemmung der Laktatdehydrogenase A induziert oxidativen Stress und hemmt die Tumorprogression. Proc Natl Acad Sci USA. 2010; 107:2037-2042.
<br><span id=“57″ class=“referencess blue-text“>57</span> Zhao YH, Zhou M, Liu H, Ding Y, Khong HT, Yu D, Fodstad O, Tan M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009; 28:3689-3701.
<br><span id=“58″ class=“referencess blue-text“>58</span> Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, et al. Inactivation of the HIF-1α/PDK3 signaling axis drives melanoma towards mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012; 72:5035-5047.
<br><span id=“59″ class=“referencess blue-text“>59</span> Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014; 4:423-433.
<br><span id=“60″ class=“referencess blue-text“>60</span> Miskimins WK, Ahn HJ, Kim JY, Ryu S, Jung YS, Choi JY. Synergistischer Anti-Krebs-Effekt von Phenformin und Oxamat. PLoS One. 2014; 9:e85576.
<br><span id=“61″ class=“referencess blue-text“>61</span> Jin S, DiPaola RS, Mathew R, White E. Metabolic catastrophe as a means to cancer cell death. J Cell Sci. 2007; 120:379-383.
<br><span id=“62″ class=“referencess blue-text“>62</span> McLornan DP, List A, Mufti GJ. Anwendung der synthetischen Letalität für die selektive Krebsbekämpfung. N Engl J Med. 2014; 371:1725-1735.
<br><span id=“63″ class=“referencess blue-text“>63</span> Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005; 5:689-698.
<br><span id=“64″ class=“referencess blue-text“>64</span> Iaquinta PJ, Lees JA. Entscheidungen über Leben und Tod durch die E2F-Transkriptionsfaktoren. Curr Opin Cell Biol. 2007; 19:649-657.
<br><span id=“65″ class=“referencess blue-text“>65</span> Houben R, Hesbacher S, Schmid CP, Kauczok CS, Flohr U, Haferkamp S, Müller CS, Schrama D, Wischhusen J, Becker JC. Hohe Expression von Wildtyp p53 in Melanomzellen ist häufig mit Inaktivität in p53-Reportergen-Assays verbunden. PLoS One. 2011; 6:e22096.
<br><span id=“66″ class=“referencess blue-text“>66</span> Avery-Kiejda KA, Bowden NA, Croft AJ, Scurr LL, Kairupan CF, Ashton KA, Talseth-Palmer BA, Rizos H, Zhang XD, Scott RJ, Hersey P. P53 in humanen Melanomen reguliert keine Zielgene, die mit Apoptose und dem Zellzyklus in Verbindung stehen, und kann zur Proliferation beitragen. BMC Cancer. 2011; 11:203.
<br><span id=“67″ class=“referencess blue-text“>67</span> Lu M, Breyssens H, Salter V, Zhong S, Hu Y, Baer C, Ratnayaka I, Sullivan A, Brown NR, Endicott J, Knapp S, Kessler BM, Middleton MR, et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell. 2013; 23:618-633.
<br><span id=“68″ class=“referencess blue-text“>68</span> Pandey V, Vijayakumar MV, Ajay AK, Malvi P, Bhat MK. Diät-induzierte Fettleibigkeit erhöht die Melanomprogression: Beteiligung von Cav-1 und FASN. Int J Cancer. 2012; 130:497-508.
<br><span id=“69″ class=“referencess blue-text“>69</span> Leontieva OV, Blagosklonny MV. M(o)TOR des pseudo-hypoxischen Zustands bei der Alterung: Rapamycin zur Rettung. Cell Cycle. 2014; 13:509-515.
<br><span id=“70″ class=“referencess blue-text“>70</span> Leontieva OV, Blagosklonny MV. Hefeähnliche chronologische Seneszenz in Säugetierzellen: Phänomen, Mechanismus und pharmakologische Unterdrückung. Aging (Albany NY). 2011; 3:1078-1091.
<br><span id=“71″ class=“referencess blue-text“>71</span> Vijayakumar MV, Singh S, Chhipa RR, Bhat MK. Die hypoglykämische Aktivität von Bockshornkleesamen-Extrakt wird durch die Stimulierung eines Insulin-Signalweges vermittelt. Br J Pharmacol. 2005; 146:41-48.
<br><span id=“72″ class=“referencess blue-text“>72</span> Malvi P, Chaube B, Pandey V, Vijayakumar MV, Boreddy PR, Mohammad N, Singh SV, Bhat MK. Adipositas-induziertes schnelles Fortschreiten des Melanoms wird durch Orlistat-Behandlung und diätetische Intervention rückgängig gemacht: Rolle der Adipokine. Mol Oncol. 2015; 9:689-703.
<br><span id=“73″ class=“referencess blue-text“>73</span> Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Bewertung der enzymatischen Aktivitäten der mitochondrialen Atmungskette an Geweben und kultivierten Zellen. Nat Protoc. 2012; 7: 1235-1246.
<p></p>