Balkrishna Chaube1, Parmanand Malvi1, Shivendra Vikram Singh1, Naoshad Mohammad1, Avtar Singh Meena1,2 e Manoj Kumar Bhat1
1 National Centre for Cell Science, Savitribai Phule Pune University Campus, Ganeshkhind, Pune, India
2 Indirizzo attuale: Dipartimento di Fisiologia, University of Tennessee Health Science Center, Memphis, USA
Corrispondenza: Manoj Kumar Bhat, email: [email protected]
Ricevuto: 16 giugno 2015
Accettato: 23 settembre 2015
Pubblicato: 15 ottobre 2015
Abstract
Il melanoma è una neoplasia cutanea in gran parte incurabile a causa dell’eterogeneità molecolare e metabolica sottostante, confusa dallo sviluppo di resistenza. Le cellule tumorali hanno una flessibilità metabolica che le porta a scegliere la fosforilazione ossidativa (OXPHOS) o la glicolisi per la generazione di ATP a seconda della disponibilità di nutrienti nel microambiente tumorale. In questo studio abbiamo analizzato il coinvolgimento del complesso respiratorio I e della lattato deidrogenasi (LDH) nella progressione del melanoma. Abbiamo dimostrato che l’inibizione del complesso I da parte della metformina promuove la crescita del melanoma nei topi attraverso l’aumento dei livelli di lattato e di VEGF. Al contrario, porta all’arresto della crescita in vitro a causa di una maggiore acidificazione extracellulare dovuta all’aumento della glicolisi. L’inibizione della generazione di LDH o di lattato causa una diminuzione della glicolisi con un concomitante arresto della crescita sia in vitro che in vivo. Il blocco della generazione di lattato in cellule di melanoma trattate con metformina determina una diminuzione della proliferazione cellulare e della progressione tumorale nei topi. È interessante notare che l’inibizione della LDH o del complesso I da soli non inducono l’apoptosi, mentre l’inibizione di entrambi insieme provoca una deplezione del pool di ATP cellulare con conseguente catastrofe metabolica indotta dall’apoptosi. Nel complesso, il nostro studio suggerisce che la LDH e il complesso I svolgono ruoli distinti nella regolazione della glicolisi e della proliferazione cellulare. L’inibizione di questi due elementi aumenta la letalità sintetica nel melanoma.
Parole chiave: melanoma; complesso I; LDH; catastrofe metabolica; letalità sintetica
INTRODUZIONE
Il melanoma maligno è una delle forme più aggressive di cancro della pelle, con un elevato potenziale metastatico e resistenza a molti agenti citotossici [1, 2]. Nonostante le numerose ricerche e i parziali successi ottenuti con l’uso dei farmaci attualmente disponibili, non esiste un trattamento efficace contro il melanoma maligno [1-3]. I casi di melanoma aumentano ogni anno e rappresentano circa il 75% dei decessi legati al cancro della pelle in tutto il mondo [2, 3]. La scarsa risposta alle opzioni terapeutiche attualmente disponibili e lo sviluppo della resistenza alla terapia richiedono l’esplorazione di nuove strategie per il trattamento del melanoma.
L’aumento della glicolisi aerobica è una caratteristica di molti tumori [3-5]. È stato riportato che le cellule di melanoma, a causa della mutazione BRAF, dipendono principalmente dalla glicolisi per la generazione di ATP e presentano una fosforilazione ossidativa disfunzionale [6, 7]. Le cellule tumorali ricavano ATP, intermedi biosintetici ed equivalenti riducenti impegnandosi in modo insolito in vie biochimiche come la glicolisi, la glutaminolisi e la via del pentoso fosfato [5]. Le cellule normali (non cancerose) ricavano ATP principalmente attraverso l’OXPHOS mitocondriale, mentre le cellule tumorali si affidano principalmente alla glicolisi aerobica per generare ATP e intermedi glicolitici che facilitano una crescita rapida [4, 5]. L’aumento della generazione di lattato è stato correlato all’aggressività del cancro. Numerosi studi hanno identificato la lattato deidrogenasi (LDH), che catalizza la conversione del piruvato in lattato, come il marcatore più coerente dei tumori aggressivi e in rapida crescita [8-11]. La LDH svolge un ruolo importante nella regolazione della glicolisi, nel mantenimento dello stato redox cellulare, nella fisiologia mitocondriale e nel mantenimento del tumore [12]. Un metabolismo alterato delle cellule tumorali può essere associato a una disfunzione mitocondriale che comporta l’inibizione dell’OXPHOS, l’aumento delle specie reattive dell’ossigeno (ROS) e la promozione di una crescita incontrollata, che a sua volta favorisce ulteriormente lo sviluppo di un fenotipo metastatico [13,14]. Il complesso respiratorio I è l’enzima più grande e complesso che catalizza l’ossidazione del NADH nella catena di trasporto degli elettroni [15]. Il complesso I svolge un ruolo importante nella generazione di ATP nelle cellule normali ed è uno dei principali siti di generazione di ROS, tuttavia il suo ruolo nella tumorigenesi è in gran parte poco chiaro. La maggior parte dei rapporti suggerisce che l’attività del complesso I è soppressa nelle cellule tumorali e la sua inibizione promuove la proliferazione e le metastasi [16-18]. Al contrario, è stato anche riportato che le cellule di melanoma mostrano un aumento della funzione OXPHOS che causa resistenza ai farmaci [19-21]. Grazie alla flessibilità metabolica nella scelta di varie vie metaboliche, in particolare il metabolismo della glutammina [22], le cellule di melanoma possono sfuggire alla perturbazione di una molecola in una via metabolica [23]. Pertanto, per ottenere un risultato efficace e curativo, è necessario colpire più molecole di diverse vie metaboliche.
Le biguanidi, come la metformina e la fenformina, sono note per la loro capacità di inibire il complesso respiratorio I. La metformina, terapia di prima linea per i pazienti affetti da diabete mellito di tipo 2 (T2DM), è un membro delle biguanidi e recentemente è stata scoperta la sua attività antitumorigenica [24, 25]. A livello fisiologico, la metformina esercita la sua attività biologica diminuendo la glucogenesi epatica, aumentando la sensibilità all’insulina, elevando la captazione periferica del glucosio e riducendo l’assorbimento del glucosio dal tratto gastrointestinale [26]. A livello cellulare, la metformina agisce principalmente inibendo il complesso I mitocondriale, che ostacola la fosforilazione ossidativa con conseguente diminuzione del livello di ATP che porta all’attivazione dell’AMPK [27-30]. Inoltre, è stato dimostrato che la metformina diminuisce la respirazione mitocondriale accoppiata alla generazione di ATP, causando così un aumento della glicolisi [31, 32]. L’azione antitumorale della metformina è stata dimostrata in vari tipi di cancro con meccanismi distinti [33, 34]. Tuttavia, il ruolo della metformina nel melanoma non è molto chiaro. A differenza di altri tipi di cancro, è stato dimostrato che la metformina ha sia un’attività antitumorale che promotrice del tumore nel melanoma [35-37]. Inoltre, è stato dimostrato che la metformina, in combinazione con gli inibitori di BRAF, sopprime la crescita del melanoma [38].
Nel presente studio, con l’ipotesi che l’interruzione delle vie di generazione dell’ATP attraverso l’inibizione del complesso I e della LDH possa essere sinteticamente letale per le cellule di melanoma, abbiamo utilizzato la metformina o la fenformina e l’ossamato/dicloro acetato (DCA) per inibire questi due enzimi utilizzando sia modelli cellulari che animali. Abbiamo dimostrato che l’inibizione del complesso I e dell’attività della LDH ha un impatto distinto sulla crescita e sulla proliferazione cellulare. È interessante notare che l’inibizione del complesso I da parte della metformina promuove ulteriormente la glicolisi aerobica, con conseguente aumento della crescita tumorale nei topi. L’ablazione della generazione di lattato indotta dalla metformina mediante l’uso di ossamato o DCA porta alla citostasi e/o all’apoptosi nelle cellule di melanoma.
RISULTATI
Lametformina esercita azioni distinte in vitro e in vivo sulla crescita del melanoma
La metformina sopprime la crescita tumorale inibendo il complesso I che è influenzato dal glucosio [30]. Inoltre, è noto che il glucosio altera l’attività degli enzimi respiratori [39]. Pertanto, per esplorare le conseguenze dell’inibizione del complesso I e l’influenza del glucosio sull’azione della metformina sulla progressione del melanoma, abbiamo monitorato la progressione dell’isotrapianto/xenotrapianto in topi iperglicemici indotti da streptozotocina (STZ). Abbiamo notato che la metformina promuoveva la progressione dell’isotrapianto derivato da B16F10 nei topi iperglicemici rispetto al controllo non trattato (Figura 1A, 1B e 1C). Inoltre, la metformina ha influenzato positivamente la progressione del tumore nei topi C57BL/6J normoglicemici (Figura 1D, 1E e 1F). Analogamente, la somministrazione orale di metformina ha promosso la crescita dello xenotrapianto A375 in topi NOD/SCID iperglicemici e normoglicemici rispetto al controllo non trattato (Figure 1G, 1H e 1I).
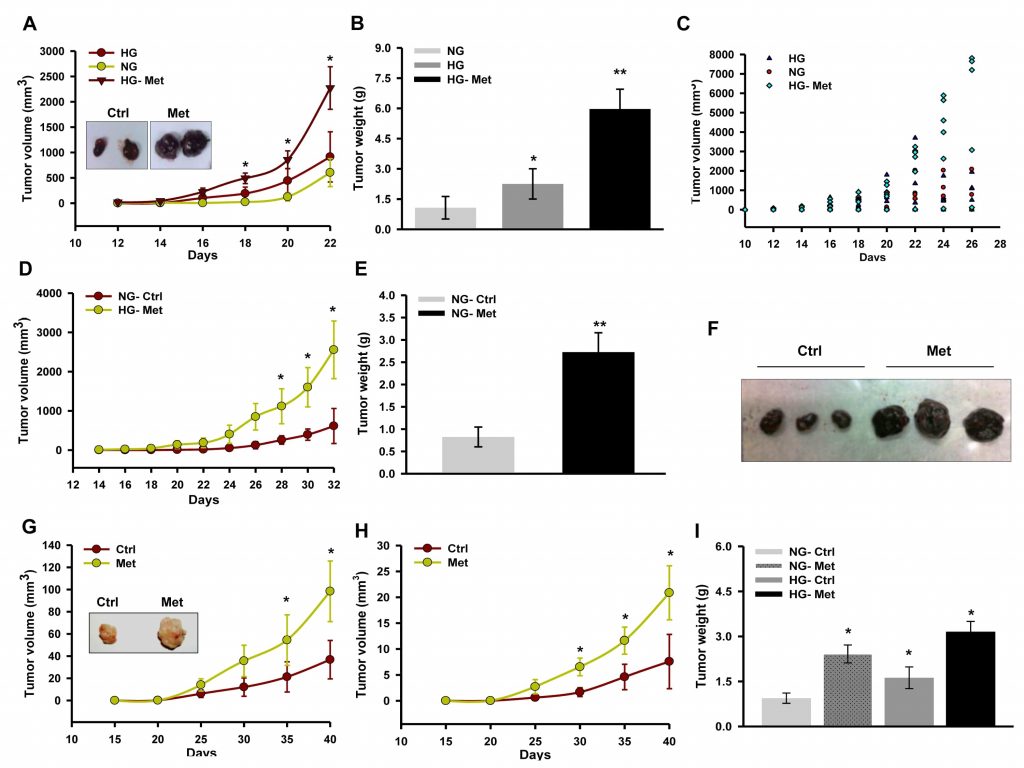
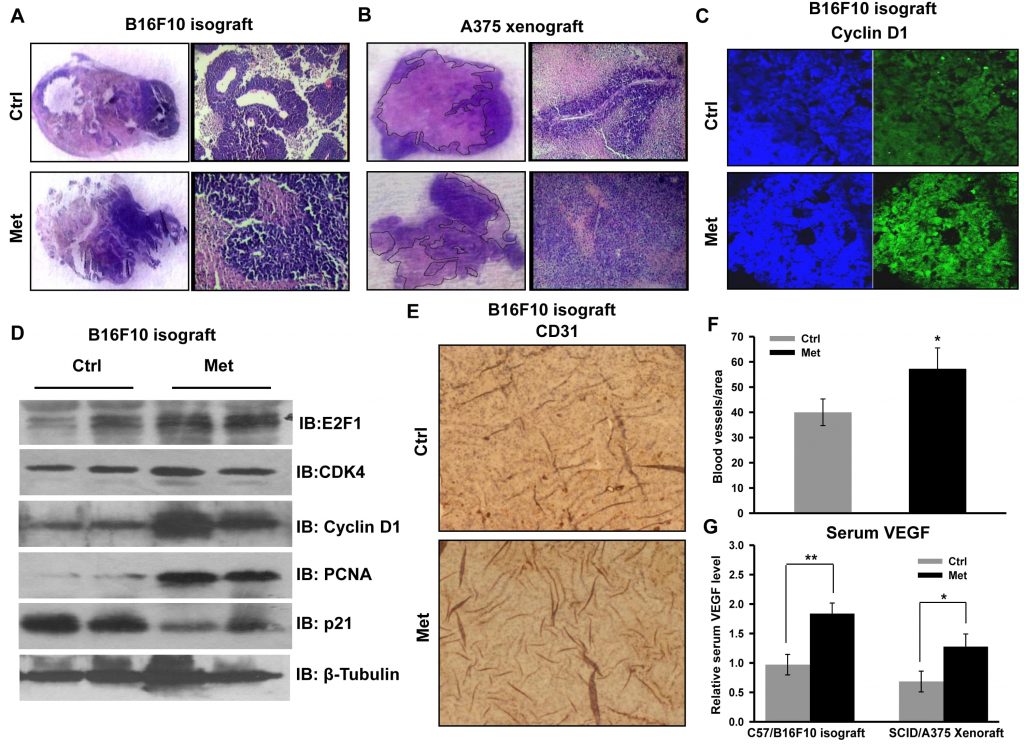
Per verificare gli eventi cellulari e molecolari associati all’aumento della progressione tumorale, sono state esaminate sezioni tumorali per l’analisi istopatologica. L’alta densità cellulare e la ridotta necrosi erano chiaramente visibili nelle sezioni di entrambi i tipi di tumore (isotrapianto derivato da B16F10 e xenotrapianto derivato da A375) dei topi cui era stata somministrata la metformina (Figura 2A e 2B). Abbiamo notato che la proliferazione e la progressione dello xenotrapianto derivato da A375 è stata fenotipicamente distinta rispetto al tumore di controllo. Ciò è suggestivo di un avanzamento di grado del tumore primario, evidente dalla morfologia allungata dei nuclei rispetto alla morfologia arrotondata delle sezioni dei tumori di controllo (informazioni non pubblicate). La colorazione immunoistochimica della proteina regolatrice del ciclo cellulare ciclina D1 (Figura 2C) è risultata più elevata nelle sezioni tumorali dei topi somministrati con metformina. Inoltre, abbiamo convalidato l’aumento della crescita tumorale controllando lo stato delle proteine regolatrici del ciclo cellulare mediante immunoblotting dei lisati tumorali. Abbiamo scoperto che i livelli delle molecole ciclina D1, CDK4, E2F1 e PCNA sono aumentati significativamente nei lisati tumorali dei topi somministrati con metformina rispetto al controllo, mentre il livello di p21 è diminuito (Figura 2D). Questi risultati indicano che la metformina, indipendentemente dallo stato glicemico dei topi, promuove la crescita del melanoma modulando le proteine regolatrici del ciclo cellulare. Inoltre, l’analisi immunoistochimica delle sezioni tumorali ha rafforzato questa osservazione, poiché il trattamento con metformina ha aumentato i livelli proteici di CD31, un marcatore endoteliale (Figura 2E e 2F), e ha aumentato il livello sierico di VEGF (Figura 2G), suggerendo che la metformina promuove l’angiogenesi nei tumori del melanoma.
Successivamente, abbiamo verificato l’effetto della metformina sulla crescita e sulla proliferazione delle cellule di melanoma in vitro. In contrasto con i risultati ottenuti in vivo, il trattamento con metformina ha provocato una soppressione della crescita delle cellule di melanoma in vitro (Figure supplementari S1A, S1B, S1C e S1D). Successivamente, abbiamo analizzato l’impatto dell’inibizione del complesso I utilizzando metformina e fenformina sulla crescita delle cellule di melanoma, poiché entrambe inibiscono l’attività del complesso I (Figura supplementare S2). Abbiamo riscontrato che la metformina e la fenformina hanno causato un arresto della crescita nelle cellule di melanoma coltivate in presenza di glucosio elevato. Tuttavia, in presenza di glucosio basso, il trattamento con questi agenti ha provocato la morte cellulare (Figura S3A, S3B, S3C e S3D). È probabile che l’arresto della crescita mediato dalla metformina sia dovuto alla riduzione del livello di glucosio e all’acidificazione extracellulare del terreno di coltura (Figura S4A e S4B supplementari). È interessante notare che la sostituzione del terreno ogni 12 ore con terreno fresco contenente 25 mM di glucosio ha aumentato la sopravvivenza clonogenica dopo il trattamento con metformina. Poiché le cellule trattate con metformina utilizzano il glucosio molto rapidamente rispetto al controllo, è necessario reintegrare il terreno per mantenere il livello di glucosio e il pH e quindi la sopravvivenza delle cellule in presenza di metformina (Figura S4C e S4D). Questi risultati suggeriscono che la concentrazione di glucosio disponibile nel terreno di coltura influenza l’azione della metformina.
L’inibizione dell’attività del complesso respiratorio I promuove la glicolisi aerobica
La discrepanza tra l’esito del trattamento con metformina in vivo e in vitro ci ha spinto a esplorarne la causa. È noto che la metformina esercita la sua azione principalmente inibendo il complesso I dell’enzima OXPHOS mitocondriale <a href=”#[“> [</a><a href=”https://www.oncotarget.com/article/6134/text/#bib27″>27</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib29″>29</a><a href=”#]”>]</a></sup> e l’inibizione del complesso I determina un aumento della crescita e della proliferazione tumorale<sup><a href=”#14″> [</a><a href=”https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href=”#]”>]</a></sup>. Poiché abbiamo osservato che il trattamento con metformina causava l’acidificazione del terreno di coltura a causa dell’aumento della glicolisi aerobica (Figura 4A e 4B supplementari), abbiamo ipotizzato che questo potesse essere il motivo dell’arresto della crescita cellulare. Abbiamo quindi misurato il livello di glucosio e di lattato nel terreno di coltura esausto raccolto dalle cellule di controllo e da quelle trattate con metformina. Nelle cellule trattate con metformina è stata rilevata una maggiore utilizzazione del glucosio (evidente dalla presenza di meno glucosio residuo nel terreno) e un livello di lattato più elevato rispetto al controllo (Figura 3A e 3B)
<p>Per rafforzare questi risultati, abbiamo esplorato ulteriormente il modello di espressione e l’attività di alcune proteine ed enzimi coinvolti nella regolazione della glicolisi. Abbiamo scoperto che la metformina aumenta i livelli proteici delle molecole glicolitiche chiave GLUT1, LDHA e ChREBP in modo dipendente dalla concentrazione (Figura 3C). Questi risultati suggeriscono che l’inibizione dell’attività del complesso I da parte della metformina potrebbe spingere le cellule tumorali ad adottare la via glicolitica per la generazione di ATP, causando un aumento della progressione tumorale nei topi.</p>
<p>Per valutare se il fenotipo glicolitico possa essere indotto anche <em>in vivo</em> con l’inibizione del complesso I, abbiamo innanzitutto controllato il livello di glucosio nel siero raccolto da topi portatori di tumore insieme a topi normoglicemici trattati o non trattati con metformina. È stata osservata una significativa diminuzione del livello di glucosio sierico nei topi normoglicemici portatori di tumore somministrati con metformina (104,3 ± 13,7 mg/dl nei C57BL/6J e 134,0 ± 7,2 mg/dl nei NOD/SCID) rispetto al controllo non trattato (139,7 ± 16,3 mg/dl nei C57BL/6J e 168,0 ± 8,5 mg/dl nei NOD/SCID) (Figura 3D e 3E). Analogamente, il trattamento con metformina ha causato una riduzione del glucosio sierico nei topi iperglicemici portatori di tumore (Met, 237,3 ± 30,0 mg/dl in C57BL/6J e 227,3 ± 12,3 mg/dl in NOD/SCID) rispetto al controllo (388 ± 31,5 mg/dl in C57BL/6J e 409,3 ± 18,6 mg/dl in NOD/SCID) (Figura 3D e 3E). La secrezione di lattato è un segno distintivo dei tumori glicolitici. Pertanto, abbiamo controllato il livello di lattato, che è risultato molto elevato nel siero di topi normoglicemici e iperglicemici affetti da tumore e somministrati con metformina (Figura 3F e 3G). Un’elevata LDH sierica è associata a tumori aggressivi di alto grado e a una prognosi sfavorevole in caso di melanoma metastatico <sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib8″>8</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=”#]”>]</a></sup>. È stata quindi monitorata l’attività della LDH nel siero, che è risultata molto elevata nel siero raccolto da topi iperglicemici e normoglicemici somministrati con metformina rispetto al controllo (Figura<a href=”https://www.oncotarget.com/article/6134/text/#F3″> </a>3H e 3I). Inoltre, è stato rilevato un aumento dell’attività LDH nei tessuti tumorali dei topi somministrati con metformina rispetto al controllo non trattato (Figura 3J).</p>
<p><strong>L’inibizione del complesso I insieme alla generazione di LDH o di lattato è sinteticamente letale per le cellule di melanoma<br></strong>Per esplorare la possibilità che la metformina promuova la glicolisi o che l’eccesso di generazione di lattato sia la causa principale dell’aumento della crescita e della proliferazione dei tumori di melanoma, la generazione di lattato è stata inibita utilizzando l’inibitore della LDH, l’oxamato, e l’inibitore della PDK1, il DCA. Abbiamo riscontrato che l’oxamato e il DCA da soli hanno un effetto inibitorio sulla crescita delle cellule di melanoma (Figura 4A, 4B e 4C; Figura supplementare S5). È stata osservata una riduzione della sopravvivenza cellulare in seguito al trattamento con ossamato o DCA insieme alla metformina (Figura 4A, 4B e 4C). Analogamente, anche altri noti inibitori glicolitici come la floretina (inibitore di GLUT1), il 2-deossi-D-glucosio (2DG) e lo iodoacetato hanno limitato la proliferazione delle cellule di melanoma trattate con metformina (Figura S6A e S6B supplementari). L’aumento dell’assorbimento del glucosio attraverso GLUT1 e l’aumento della glicolisi aerobica attraverso LDHA e PDK1 sono i segni distintivi delle cellule tumorali. Pertanto, abbiamo scelto l’ossamato e il DCA rispetto ad altri inibitori glicolitici.</p>
<p>Poi, per verificare la sopravvivenza a lungo termine delle cellule di melanoma in presenza di metformina e oxamato/DCA da soli o in combinazione, abbiamo eseguito un saggio di sopravvivenza clonogenica. Non sono state rilevate colonie sopravvissute nelle cellule di melanoma trattate con oxamato e metformina insieme (Figura<a href=”https://www.oncotarget.com/article/6134/text/#F4″> </a>4D, 4E e 4F). Risultati simili sono stati ottenuti anche nelle cellule trattate con metformina e DCA insieme (Figura 4D, 4E e 4F). Successivamente, abbiamo verificato se l’inibizione di questi due enzimi inducesse la morte delle cellule di melanoma, come evidenziato dalla colorazione con Annexin V e PI. Tutti questi agenti da soli hanno causato un arresto della crescita, ma non hanno indotto l’apoptosi. Tuttavia, è stato rilevato un aumento dell’apoptosi nelle cellule A375 e B16F10 trattate con oxamato/DCA insieme alla metformina rispetto al solo agente (Figura 5A e 5B). Questa osservazione è stata ulteriormente confermata dall’immunoblotting per i marcatori apoptotici PARP1, Bax e Bcl-2 (Figura 5C e 5D). Nelle cellule trattate con metformina e oxamato/DCA insieme, rispetto al singolo agente, è stato rilevato un aumento della scissione di PARP e del livello di Bax, mentre è diminuito il livello della proteina antiapoptotica Bcl-2 (Figura 5C e 5D). Questi risultati indicano che l’inibizione del complesso I e della LDH insieme possono promuovere l’induzione dell’apoptosi.</p>
<p>In seguito, abbiamo verificato l’impatto della metformina e dell’oxamato/DCA, da soli o in combinazione, sulla sopravvivenza delle cellule non cancerose. Abbiamo utilizzato tre diverse linee cellulari non cancerose: AML12 (epatociti di topo), L6 (cellule muscolari di ratto) e MEF (fibroblasti embrionali di topo) per verificare l’effetto del trattamento combinato. Sebbene la combinazione di metformina e oxamato/DCA abbia rallentato la proliferazione di queste cellule (Figura supplementare S7A, S7B e S7C), non ha influito sulla vitalità cellulare (Figura supplementare S7D), indicando che questa combinazione è efficace nell’uccidere selettivamente le cellule tumorali.</p>
<h3></h3>
<p><strong>Bloccare il complesso I e la generazione di lattato impone una catastrofe metabolica<br></strong>Per verificare se il co-trattamento con oxamato e metformina influisce sul metabolismo delle cellule di melanoma, abbiamo misurato i parametri metabolici in presenza di questi inibitori. Il trattamento con metformina ha aumentato la glicolisi, mentre il trattamento con oxamato ha determinato una diminuzione dell’utilizzo del glucosio e della secrezione di lattato (Figura 6A e 6B). È interessante notare che l’utilizzo del glucosio e la secrezione di lattato sono stati inibiti in modo significativo quando le cellule sono state trattate con oxamato e metformina insieme (Figura 6A e 6B). Inoltre, sia l’ossamato che la metformina hanno ridotto i livelli di ATP inibendo rispettivamente la fosforilazione del substrato e l’OXPHOS (Figura 6C). Inoltre, è stata rilevata una diminuzione significativa del livello di ATP nelle cellule trattate con oxamato e metformina insieme (Figura 6C). Inoltre, per confermare il coinvolgimento della LDH nella crescita e nella proliferazione delle cellule di melanoma, è stato eseguito il knock down della LDHA (LDHA-KD) utilizzando un siRNA specifico (Figura 6D). È stato osservato un aumento della morte cellulare nelle cellule LDHA-KD trattate con metformina o fenformina (Figura 6E). Nelle cellule LDHA-KD è stata osservata una riduzione significativa della glicolisi (evidente dalla diminuzione dell’utilizzo del glucosio e della secrezione di lattato) e del livello di ATP rispetto al controllo (Figure 6F, 6G e 6H). Inoltre, nelle cellule LDHA-KD trattate con metformina o fenformina è stata osservata una diminuzione del tasso glicolitico e un netto calo del livello di ATP (Figura 6F, 6G e 6H). Questi risultati indicano che l’inibizione simultanea del complesso I e della via di generazione della LDH o del lattato induce una catastrofe metabolica dovuta all’interruzione del pool di ATP cellulare, portando infine all’arresto della crescita e alla morte cellulare.</p>
<p><strong>L’inibizione simultanea del complesso I e della LDH da parte di metformina e ossamato ritarda la progressione tumorale nei topi<br></strong>Per verificare se la combinazione di ossamato e metformina sia efficace anche nel ridurre la progressione tumorale, sono stati sviluppati tumori iniettando cellule B16F10 in topi C57BL/6J. Dopo che i tumori hanno raggiunto le dimensioni di 50 mm<sup>3</sup>, i topi sono stati randomizzati in quattro gruppi: (a) controllo, (b) metformina, (c) oxamato e (d) topi trattati con metformina e oxamato. Abbiamo notato che i topi somministrati con metformina hanno sviluppato un tumore più grande rispetto al controllo non trattato (Figura 7A, 7B e 7C). Mentre nei topi somministrati con oxamato, i tumori sono progrediti a un ritmo più lento rispetto al controllo non trattato e ai topi somministrati con metformina (Figura 7A). È interessante notare che l’oxamato ha ritardato drasticamente la progressione del tumore quando è stato somministrato insieme alla metformina, come dimostra la riduzione del volume e del peso del tumore (Figura 7A, 7B e 7C)
<p>Inoltre, per confermare se l’abrogazione della progressione tumorale indotta dalla metformina da parte dell’oxamato sia una conseguenza della riduzione della glicolisi aerobica, abbiamo misurato i parametri metabolici <em>in vivo</em>. I topi somministrati con metformina presentavano bassi livelli di glucosio sierico e livelli più elevati di lattato e LDH nel siero rispetto al controllo, mentre nei topi trattati con oxamato da solo o metformina insieme a oxamato sono stati rilevati livelli relativamente più elevati di glucosio sierico e bassi di lattato (Figura 7D e 7E). L’attività dell’enzima LDH è risultata significativamente ridotta nei tumori escissi da topi trattati con oxamato da solo o metformina insieme a oxamato (Figura 7F). Un livello relativamente molto basso di ATP è stato rilevato nei tumori dei topi somministrati con oxamato e metformina insieme rispetto al solo agente. Tuttavia, non abbiamo osservato cambiamenti significativi nei livelli proteici delle molecole che regolano la glicolisi (Figura 7I). È importante notare che non è stata rilevata alcuna variazione significativa del peso corporeo nei topi sottoposti al trattamento combinato e che all’esame visivo non sono stati osservati possibili effetti collaterali negativi o sintomi grossolani di tossicità generalizzata (Figura S8A supplementare). Inoltre, non sono state osservate anomalie patologiche negli organi vitali come polmoni, fegato, reni e cuore di questi topi (Figura S8B supplementare). Questi risultati suggeriscono che questa combinazione non esercita una tossicità generalizzata ed è efficace nel ritardare la progressione tumorale nei topi.</p>
<h2>DISCUSSIONE</h2>
<p>Le cellule tumorali, comprese quelle del melanoma, dirottano un’enorme quantità di flusso di carbonio verso la glicolisi <sup><a href=”#5″>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib5″>5</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href=”#]”>]</a></sup>. Ciò aiuta le cellule a generare ATP e altri elementi costitutivi per una rapida crescita tumorale <sup><a href=”#5″>[5]</a></sup>. Diversi rapporti suggeriscono che il bersaglio delle vie metaboliche riduce la progressione del tumore e la crescita cellulare <em>in vitro</em> <sup><a href=”#40″>[40-</a><a href=”#42″>42]</a></sup>. Nel presente studio dimostriamo che la metformina promuove la crescita del melanoma aumentando la glicolisi a causa dell’inibizione della funzione del complesso I, mentre l’inibizione della LDH causa l’arresto della crescita delle cellule. L’inibizione della generazione di lattato nelle cellule di melanoma trattate con metformina influisce sulla sopravvivenza delle cellule, causando una riduzione della progressione del tumore (Figura 8).</p>
<p>Le conseguenze dell’inibizione del complesso I sulla crescita delle cellule tumorali non sono chiaramente comprese, anche se alcuni rapporti suggeriscono che l’inibizione del complesso I porta alla trasformazione cellulare e aumenta la crescita delle cellule<a href=”#[“> <sup>[</sup></a><sup><a href=”https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href=”#]”>]</a></sup>. Al contrario, è stato riportato che l’inibizione del complesso I ritarda la progressione tumorale nei topi <sup><a href=”#30″>[30]</a></sup>. Questi diversi risultati dipendono probabilmente dalla disponibilità di nutrienti nel microambiente tumorale. È stato riportato che quando il glucosio è disponibile in abbondanza, l’inibizione del complesso I promuove la glicolisi e la rapida proliferazione delle cellule, mentre induce la morte cellulare in condizioni di stress metabolico<sup><a href=”#43″> [43]</a></sup>. In questo studio abbiamo osservato che l’inibizione del complesso I da parte della metformina promuove la progressione del tumore del melanoma nei topi.</p>
<p>Mentre è stato dimostrato che la metformina inibisce la proliferazione di molte cellule tumorali e limita anche la progressione tumorale di xenotrapianti nei topi <sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib33″>33</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib34″>34</a><a href=”#]”>]</a></sup>, è noto che esercita effetti di inibizione della crescita e di promozione della crescita nel melanoma<sup> <a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib35″>35</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib37″>37</a><a href=”#]”>]</a></sup>. È interessante notare che i tumori derivati da B16F10 e A375 sono cresciuti rapidamente nei topi a cui è stata somministrata la metformina e il tasso di crescita variava in base allo stato glicemico dei topi (Figura 2D e 2E). Questa osservazione concorda con un recente rapporto che dimostra che la metformina facilita la progressione tumorale delle cellule di melanoma BRAF mutanti<sup><a href=”http://335″> [35]</a></sup>.</p>
<p>La disparità nell’azione della metformina sulla crescita delle cellule di melanoma <em>in vitro</em> e <em>in vivo</em> è probabilmente influenzata dalla disponibilità di glucosio e dalla concentrazione di lattato che potrebbero influenzare la sua azione. La metformina inibisce la crescita cellulare e induce l’apoptosi in condizioni di limitazione del glucosio <sup><a href=”#43″>[43]</a></sup>, mentre abbiamo notato che in presenza di glucosio elevato provoca un arresto della crescita senza influenzare la morte cellulare. Per questa discrepanza abbiamo ipotizzato che le cellule tumorali ricavino ATP <em>tramite</em> OXPHOS in condizioni di stress metabolico, mentre quando il glucosio è abbondante l’ATP viene ricavato principalmente attraverso la glicolisi. L’inibizione del complesso I in condizioni di glucosio elevato favorisce ulteriormente la glicolisi con conseguente generazione di lattato in eccesso, fenomeno già segnalato in precedenza<sup><a href=”#44″> [44]</a></sup>. Nel complesso, questi risultati suggeriscono che l’arresto della crescita indotto dalla metformina nelle cellule di melanoma <em>in vitro</em> è una conseguenza dell’acidificazione del terreno di coltura dovuta all’accumulo di acido lattico in eccesso a causa del rapido utilizzo del glucosio disponibile. In particolare, l’arresto di crescita indotto dalla metformina può essere prevenuto reintegrando frequentemente il terreno di coltura per mantenere le condizioni di crescita ottimali, poiché le cellule trattate con metformina richiedono più glucosio per proliferare a causa dell’aumento della glicolisi aerobica. Questo imita le condizioni <em>in vivo</em>, dove probabilmente la circolazione rapida e costante dell’acido lattico di accesso consente alle cellule tumorali di utilizzare più glucosio, favorendo una rapida proliferazione. Inoltre, il lattato può essere utilizzato dalle cellule tumorali nei <sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib45″>45</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib46″>46</a></sup><a href=”#]”><sup>]</sup></a>. Inoltre, è stato riportato che la metformina induce l’angiogenesi <em>tramite</em> l’aumento del livello di VEGF<sup><a href=”#[“> [</a><a href=”https://www.oncotarget.com/article/6134/text/#bib35″>35</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib47″>47</a><a href=”#]”>]</a></sup>. Abbiamo riportato che l’inibizione del complesso I aumenta la produzione di lattato ed è stato precedentemente suggerito che l’interruttore glicolitico migliora l’angiogenesi<sup> <a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib48″>48</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib49″>49</a><a href=”#]”>]</a></sup>. I nostri risultati mostrano una correlazione positiva tra il lattato sierico e il livello di VEGF nei topi somministrati con metformina. Pertanto, è probabile che la crescita tumorale favorita dalla metformina sia facilitata dall’angiogenesi indotta dal lattato, che può essere mediata dal VEGF <sup><a href=”#48″>[48]</a></sup>. Inoltre, abbiamo osservato un aumento del livello di E2F1, un’importante molecola regolatrice del ciclo cellulare, nei tumori escissi da topi a cui è stata somministrata la metformina. Oltre a promuovere la proliferazione cellulare, E2F1 regola anche molti geni coinvolti nella glicolisi<sup><a href=”#50″> [50]</a></sup>, essenziale per la rapida crescita delle cellule tumorali. Pertanto, è probabile che E2F1 possa svolgere un ruolo importante nel mediare la rapida progressione tumorale indotta dalla metformina probabilmente <em>tramite</em> la regolazione della glicolisi aerobica.</p>
<p>Inibitori glicolitici come il 2-deossi glucosio, la lonimide, il 3-bromopiruvato, il DCA e farmaci che interferiscono con le vie metaboliche hanno mostrato risultati promettenti nella soppressione della crescita tumorale<sup> <a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib40″>40</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib42″>42</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib51″>51</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href=”#]”>]</a></sup>. Inoltre, colpire la via di generazione del lattato è interessante soprattutto nei tumori glicolitici <sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib54″>54</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib55″>55</a><a href=”#]”>]</a></sup>. Le cellule di melanoma sono metabolicamente glicolitiche e fanno quindi affidamento principalmente sull’elevata attività della LDH per la generazione di ATP <sup><a href=”#6″>[6]</a></sup>. Le cellule di melanoma sovraesprimono la LDH e l’elevato livello sierico è spesso correlato a una prognosi sfavorevole e alla sopravvivenza dei pazienti<sup> <a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib8″>8</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=”#]”>]</a></sup>. I nostri risultati indicano che l’interruzione della conversione del piruvato in lattato influisce profondamente sulla proliferazione delle cellule di melanoma. Ciò è in accordo con quanto riportato che i tumori di tipi di cellule glicolitiche sono più sensibili all’inibizione della LDH-A con FX11<sup><a href=”#56″> [56]</a></sup>.</p>
<p>LaLDH svolge un ruolo importante nell’omeostasi metabolica e nel mantenimento dei tumori<sup> <a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib12″>12</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib57″>57</a><a href=”#]”>]</a></sup>. Abbiamo utilizzato l’ossamato e il DCA per inibire la generazione di lattato nelle cellule di melanoma. Il DCA è una piccola molecola somministrabile per via orale che è stata utilizzata per il trattamento dell’acidosi lattica e il ripristino della funzionalità dell’OXPHOS da parte del DCA ha dimostrato di indurre la morte cellulare<sup><a href=”#[“> [</a><a href=”https://www.oncotarget.com/article/6134/text/#bib52″>52</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href=”#]”>]</a></sup>. Inoltre, è stato precedentemente dimostrato che il DCA riduce la produzione di lattato e innesca l’apoptosi nelle cellule di melanoma <sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib58″>58</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib59″>59</a><a href=”#]”>]</a></sup>. Anche l’ossamato, un analogo del piruvato, viene utilizzato per inibire la LDH. Un recente studio di Miskimins et al. suggerisce che colpire il complesso I e la LDH insieme può essere una strategia promettente per arrestare la crescita di tumori aggressivi <sup><a href=”#60″>[60]</a></sup>. In accordo con ciò, abbiamo anche notato che impedendo la generazione di lattato e inibendo contemporaneamente il complesso I con oxamato/DCA o con siRNA specifici per la LDH e metformina, si ottiene una diminuzione del pool di ATP cellulare. La diminuzione del pool di ATP cellulare evoca una catastrofe metabolica che porta all’apoptosi nelle cellule di melanoma. La morte cellulare indotta dalla catastrofe metabolica è considerata una strategia promettente per arrestare la progressione del cancro<sup><a href=”#61″> [61]</a></sup>. Inoltre, il nostro studio evidenzia il ruolo distinto del complesso I e della LDH nella proliferazione cellulare, e questi due costituiscono la via di generazione dell’ATP attraverso l’OXPHOS o la fosforilazione a livello del substrato rispettivamente. Il blocco di uno dei due enzimi da solo non induce la morte cellulare, poiché le cellule possono passare a una via alternativa per la generazione di ATP. Invece, l’inibizione simultanea di entrambi induce l’apoptosi. Ciò indica chiaramente che le cellule dipendono dalla funzione di questi due enzimi che formano una coppia sinteticamente letale, un fenomeno promettente che può essere impiegato per colpire selettivamente le cellule di melanoma<a href=”#[“> </a><sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib62″>62</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib63″>63</a><a href=”#]”>]</a></sup>. Dato che E2F1 è coinvolto nella regolazione dell’apoptosi <sup><a href=”#64″>[64]</a></sup>, è probabile che l’apoptosi indotta dalla combinazione di metformina e oxamato/DCA possa coinvolgere E2F1. La metformina promuove l’apoptosi nelle cellule tumorali <em>tramite</em> l’attivazione di p53 <sup><a href=”#37″>[37]</a></sup>. Tuttavia, nel caso del melanoma, il ruolo di p53 non è chiaro. Come suggerisce la letteratura, la p53 non è funzionale nel melanoma e i suoi livelli sono paradossalmente elevati nei gradi avanzati di melanoma <sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib65″>65</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib67″>67</a><a href=”#]”>]</a></sup>. In precedenza, abbiamo riportato che un livello elevato di p53 è associato a un aumento della crescita tumorale<sup><a href=”#68″> [68]</a></sup>. Pertanto, è improbabile che p53 sia coinvolta nell’apoptosi indotta dal trattamento combinato e, quindi, sono necessari ulteriori studi per valutare la funzionalità di E2F1 e p53 nel melanoma.</p>
<p>L’uso di combinazioni di farmaci diversi che possono promuovere sinergicamente la morte delle cellule tumorali, può anche aumentare la tossicità per le cellule normali. Pertanto, è fondamentale valutare l’impatto del trattamento combinato sulle cellule normali. I nostri dati suggeriscono che questa combinazione è efficace nell’uccidere le cellule tumorali sia <em>in vitro</em> che <em>in vivo</em>, e ha un impatto minimo sulla sopravvivenza delle cellule normali. La diversa sensibilità tra melanoma e cellule normali dovuta alla combinazione di metformina e oxamato/DCA può essere attribuita al fatto che le cellule di melanoma sono altamente glicolitiche e sovraesprimono molecole coinvolte nella generazione e nella secrezione di lattato rispetto alle cellule normali <sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib6″>6</a>-<a href=”https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=”#]”>]</a></sup>. È interessante notare che, rispetto alle cellule normali, le cellule tumorali mostrano una maggiore respirazione mitocondriale disaccoppiata<sup><a href=”#31″> [31]</a></sup>. Le cellule tumorali, in seguito al trattamento con metformina, mostrano una maggiore elevazione compensativa della glicolisi rispetto alle cellule normali <sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib31″>31</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib32″>32</a><a href=”#]”>]</a></sup>, appesantendo così la suscettibilità metabolica delle cellule tumorali. Pertanto, l’inibizione della generazione/secrezione di LDH o di lattato sopprime la crescita delle cellule tumorali, mentre le cellule normali sono meno colpite in quanto è ancora possibile produrre una quantità sufficiente di ATP dall’OXPHOS, poiché la metformina è un debole inibitore del complesso I. Poiché l’attività OXPHOS è più elevata nelle cellule normali rispetto a quelle del melanoma, è probabile che la combinazione di metformina e oxamato/DCA abbia un impatto negativo minimo sulla respirazione delle cellule normali.</p>
<p>Il nostro studio apre nuove strade per colpire il metabolismo delle cellule tumorali e può essere ulteriormente coinvolto nella sperimentazione di altri farmaci clinicamente approvati noti per inibire la glicolisi insieme alla metformina. Il presente studio suggerisce che qualsiasi farmaco/inibitore che blocchi la generazione di lattato può essere utilizzato in combinazione con la metformina per una migliore gestione e prevenzione della crescita tumorale. Ad esempio, la rapamicina, un farmaco clinicamente approvato, ha dimostrato di prevenire la generazione di lattato<sup> <a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib69″>69</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib70″>70</a><a href=”#]”>]</a></sup>. Analogamente, è stato dimostrato che l’inibizione di BRAF comporta la soppressione della glicolisi <sup><a href=”#[“>[</a><a href=”https://www.oncotarget.com/article/6134/text/#bib6″>6</a>, <a href=”https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href=”#]”>]</a></sup>. Pertanto, la rapamicina e gli inibitori di BRAF possono essere utilizzati insieme alla metformina per migliorare i risultati terapeutici con effetti collaterali ridotti. Nel complesso, i nostri risultati indicano che la LDH o altri meccanismi che controllano la generazione o la secrezione di lattato sono fondamentali per la rapida progressione del melanoma in condizioni di compromissione dell’OXPHOS e possono essere sfruttati come bersaglio terapeutico adatto per controllare la crescita delle cellule tumorali glicolitiche. Sono necessari studi più approfonditi per valutare la funzionalità della LDH e del complesso I in altri modelli di cancro e, successivamente, la loro implicazione nella terapia del cancro in generale.</p>
<h2>MATERIALI E METODI</h2>
<p><strong>Esperimenti sugli animali<br></strong>I topi sono stati procurati dalla Experimental Animal Facility (EAF) del National Centre for Cell Science (NCCS), Pune, India. L’iperglicemia nei topi è stata indotta utilizzando STZ come precedentemente descritto<sup><a href=”#71″> [71]</a></sup> con lievi modifiche. In breve, i topi maschi C57BL/6J e NOD/SCID sono stati digiunati per 6 ore prima dell’iniezione intraperitoneale di STZ (50 mg/kg) in tampone citrato 0,01 M (pH 4,4) per tre giorni consecutivi. La misurazione della glicemia è stata eseguita tagliando la coda e applicando il sangue su un analizzatore di glucosio (Accu-Chek Active, Roche, Germania). I topi con valori di glucosio nel sangue superiori a 200 mg/dl sono stati considerati iperglicemici. Il tumore è stato indotto iniettando cellule B16F10 (2 × 10<sup>5</sup>) o A375 (1 × 10<sup>6</sup>) in 100 µl di PBS sterile per via sottocutanea sul fianco destro di topi C57BL/6J e NOD/SCID rispettivamente. La progressione del tumore è stata monitorata regolarmente misurandone le dimensioni con un calibro digitale (Sigma, USA) dopo la comparsa di tumori palpabili. La somministrazione orale di metformina a giorni alterni è iniziata una settimana prima del challenge tumorale nei topi iperglicemici. Altrimenti, la metformina è stata somministrata solo dopo che il tumore ha raggiunto una dimensione ottimale e il trattamento è stato continuato fino al completamento dell’esperimento. Al termine dell’esperimento, i topi sono stati sacrificati; i tumori sono stati escissi e conservati a -80<sup>°</sup>C o in soluzione di formalina al 10% per ulteriori studi. In un’altra serie di esperimenti, i topi (background C57BL/6J, maschi) sono stati divisi a caso in 4 gruppi dopo la comparsa di un tumore palpabile. Per esaminare l’esito del trattamento con metformina in combinazione con l’inibitore LDH oxamato, ai topi è stata somministrata per via orale metformina (200 mg/kg) o oxamato da solo (500 mg/kg) o entrambi insieme a giorni alterni. La progressione del tumore è stata regolarmente monitorata con un calibro. Al termine dell’esperimento, i tessuti tumorali, muscolari ed epatici sono stati prelevati, pesati e conservati a -80<sup>°</sup>C per ulteriori analisi. Tutti gli esperimenti sugli animali sono stati eseguiti secondo le linee guida del Comitato per il controllo e la supervisione degli esperimenti sugli animali (CPCSEA), Governo dell’India, e dopo aver ottenuto l’autorizzazione del Comitato etico per gli animali dell’Istituto (IAEC).</p>
<p><strong>Linee cellulari e condizioni di coltura<br></strong>Il melanoma murino B16F10, le cellule di melanoma umano A375 e SKMel28 sono state acquistate dall’ATCC (VA, USA) e mantenute nel nostro archivio interno. Le cellule non cancerose, AML12 (epatociti di topo), L6 (cellule muscolari di ratto) e MEF (fibroblasti embrionali di topo) sono state utilizzate in parallelo con le cellule tumorali come controllo. Tutte le cellule sono state coltivate nei rispettivi terreni di coltura contenenti 1 mM o 25 mM di glucosio a seconda degli esperimenti e integrate con il 10% di siero fetale bovino inattivato al calore (Hyclone, UT, USA), penicillina (100 U/ml) e streptomicina 100 µg/ml (Life Technologies, NY, USA), a 37<sup>°</sup>C in presenza del 5%<sub> </sub>CO<sub>2</sub>.</p>
<p><strong>Farmaci e reagenti<br></strong>Metformina, fenformina, oligomicina, rotenone, decilubiquinone, streptozotocina (STZ), [3-(4, 5-dimetiltiazol-2-il)-2, 5difenilterazolio bromuro] (MTT), NAD+, NADH, ATP, AMP, D-glucosio, sodio ossamato, iodoacetato, 2-deossi-D-glucosio, dicloroacetato (DCA), diaminobenzidina (DAB) e floretina sono stati acquistati da Sigma (MO, USA). Gli anticorpi per ChREBP (1:1000), GLUT1 (1:1000), LDHA (1:1000), Ciclina D1 (1:1000), PCNA (1:1000), CDK4 (1:1000), p21 (1:1000), E2F1 (1:1000) CD31 (1:100), PARP-1 (1.:1000), Bcl-2 (1:1000), Bax (1:1000), β-tubulina (1:1000), GAPDH (1:1000), HSP60 (1:1000) e VEGF (1:50) sono stati prodotti da Santa Cruz Biotechnology (CA, USA).</p>
<p><strong>Preparazione del lisato cellulare e immunoblotting<br></strong>Le cellule sono state lavate tre volte con soluzione fisiologica tamponata con fosfato (PBS) e lisate in un tampone di lisi contenente 50 mM di Tris-Cl (pH 7.5), 120 mM NaCl, 10 mM fluoruro di sodio, 10 mM pirofosfato di sodio, 2 mM EDTA, 1 mM ortovanadato di sodio, 1 mM fenilmetilsulfonil fluoruro, 1% NP-40 e cocktail di inibitori delle proteasi (Roche, Germania). I lisati tumorali sono stati preparati tagliando i tessuti tumorali in pezzi sottili, lavati cinque volte con soluzione salina allo 0,85% contenente cocktail di inibitori delle proteasi e lisati in tampone di lisi mediante omogeneizzazione con un omogeneizzatore di tessuti (Sigma, USA) seguita da sonicazione. I lisati sono stati chiarificati mediante centrifugazione a 15.000 rpm<em> </em>per 30 minuti. I lisati cellulari sono stati preparati in condizioni di refrigerazione. Circa 50-100 µg di proteine da lisati cellulari interi sono stati risolti su un gel di SDS-poliacrilammide all’8-12%, successivamente trasferito su membrana PVDF (Millipore, Germania). La membrana è stata sondata con gli anticorpi primari desiderati, seguiti da anticorpi secondari coniugati con HRP. Gli immunoblots sono stati rilevati con il reagente luminale (Santa Cruz Biotechnology). Quando necessario, i blot sono stati strippati incubando la membrana a 50<sup>°</sup>C per 15 minuti in un tampone di strippaggio (62,5 mM Tris-Cl, pH 6,8, 100 mM mercaptoetanolo e 2% SDS) con agitazione intermittente. Le membrane sono state lavate accuratamente con soluzione salina tamponata Tris (TBS) e sono state riprodotte con gli anticorpi desiderati.</p>
<p><strong>Immunofluorescenza o microscopia confocale<br></strong>Le cellule sono state poste su vetrini a camera Labtek (Nunc, USA) e lasciate crescere per 24 h. Il trattamento con metformina e altri inibitori è stato somministrato per i periodi e le concentrazioni desiderate. Le cellule sono state lavate con PBS sterile raffreddato e fissate con una soluzione di paraformaldeide al 3,7% a temperatura ambiente per 10 minuti. Sono state quindi permeabilizzate con Triton X-100 allo 0,025% in PBS per 10 minuti e successivamente bloccate con BSA al 5% in PBS per 1 ora a 37<sup>°</sup>C. Le cellule sono state incubate con diluizioni 1:100 di anticorpi primari nella soluzione bloccante per 2 ore a temperatura ambiente e lavate con TBST (TBS contenente lo 0,05% di Tween-20) almeno cinque volte prima di essere incubate con anticorpi secondari marcati appropriati (1:200) nella soluzione bloccante per un’altra ora a temperatura ambiente. Dopo cinque lavaggi con TBST, i campioni sono stati stratificati con terreno di montaggio contenente DAPI (Santa Cruz Biotechnology, USA). I vetrini sono stati sigillati, esaminati con un microscopio confocale a scansione laser (LSM510 Carl Zeiss, Germania) e le immagini sono state acquisite. Le immagini sono state successivamente elaborate dal software di analisi delle immagini LSM.</p>
<p><strong>Immunoistochimica e istopatologia<br></strong>L’immunoistochimica e l’istopatologia sono state eseguite secondo le indicazioni di Malvi et al. <sup><a href=”#72″>[72]</a></sup>. In breve, sezioni sottili del tumore e di altri organi sono state realizzate con microtomo, fissate su vetrini e parafinizzate. Per l’immunoistochimica, i vetrini sono stati deparaffinati in soluzione di xilene due volte per 10 minuti e successivamente lavati tre volte con etanolo al 100%, 95%, 70% e 50%. I vetrini sono stati nuovamente lavati con acqua distillata e poi con PBS per 5 minuti. Per il recupero dell’antigene, i vetrini sono stati bolliti in tampone di sodio citrato (0,01 M, pH 4,5) in forno a microonde per 10 minuti e lasciati raffreddare a temperatura ambiente per altri 20 minuti. La BSA o il siero di capra normale (2%) sono stati utilizzati per il blocco in camera umidificata o in cella frigorifera per 1 ora. I vetrini sono stati testati con gli anticorpi desiderati (diluizione 1:100) in PBST (PBS contenente lo 0,025% di Tween-20) contenente lo 0,01% di BSA per 2 ore a temperatura ambiente o durante la notte a 4<sup>°</sup>C. I vetrini sono stati lavati con PBST 4 volte per 5 minuti e sono stati testati con anticorpi secondari compatibili coniugati con HRP per 1 ora. Sono stati nuovamente lavati e colorati con diaminobenzidina (DAB) per 10 minuti, quindi lavati e controcolorati con ematossilina. I vetrini sono stati ulteriormente lavati con acqua, disidratati con alcool assoluto e poi stratificati con terreno di montaggio, quindi analizzati per l’espressione delle molecole desiderate. Per l’istopatologia, i vetrini deparaffinati sono stati colorati con ematossilina ed eosina, e l’analisi microscopica della densità cellulare, della morfologia cellulare, delle metastasi, della citotossicità e della necrosi è stata eseguita da patologi in cieco presso il KEM Hospital di Pune, India.</p>
<p><strong>Saggio di citotossicità cellulare e sopravvivenza</strong><br>Circa 5 × 10<sup>3</sup> (B16F10) e 10 × 10<sup>3</sup> (A375 e SKMel28) sono state seminate in ogni pozzetto di piastre di coltura tissutale da 96 pozzetti e lasciate aderire per 24 ore a 37<sup>°</sup>C. Le cellule sono state trattate secondo le esigenze sperimentali e la proliferazione o la vitalità sono state valutate mediante il saggio MTT. La MTT (50 µl, 1 mg/ml in DMEM senza rosso fenolo) è stata aggiunta a ciascun pozzetto e incubata per 4 h a 37<sup>°</sup>C. I cristalli di formazan sono stati solubilizzati<sup> </sup>in 100 µl di isopropanolo incubando con agitazione a temperatura ambiente per 5 min. L’assorbanza è stata rilevata a 570 nm utilizzando 630 nm come filtro di riferimento. Le cellule non trattate sono state considerate come controllo (100% di sopravvivenza cellulare).</p>
<p><strong>Rilevazione dell’apoptosi mediante colorazione con annexina V<br></strong>Le cellule sono state seminate a una densità di circa 3 × 10<sup>5</sup> cellule in piastre da 35 mm e lasciate crescere per 24 h. Le cellule sono state trattate con o senza ossamato e DCA da soli o con metformina per 48 h o secondo le esigenze sperimentali. Le cellule sono state raccolte mediante tripsinizzazione e trattate per la citometria a flusso. L’apoptosi è stata rilevata mediante doppia colorazione di Annexina V e PI utilizzando il kit per il dosaggio dell’apoptosi (BD Bioscience, USA) secondo le istruzioni del produttore.</p>
<p><strong>Analisi del ciclo cellulare<br></strong>Le cellule sono state seminate a una densità di circa 3 × 10<sup>5</sup> cellule in piastre da 35 mm e lasciate crescere per 24 h. Le cellule sono state trattate con o senza metformina, oxamato e altri inibitori glicolitici da soli o insieme, come indicato, per 48 h o secondo le esigenze sperimentali. Le cellule sono state raccolte mediante tripsinizzazione e trattate per la citometria a flusso. In breve, le cellule sono state lavate con PBS freddo e fissate in etanolo al 70% in ghiaccio per 30 minuti. Dopo il trattamento con RNasi (200 μg/ml) per 30 minuti a 37°C, 50 μg/ml di PI sono stati aggiunti al pellet cellulare e incubati al buio per 30 minuti in ghiaccio. La fluorescenza di PI è stata registrata attraverso un filtro da 585 nm in un citometro a flusso (FACS Calibur, Becton Dickinson, California, USA). I dati sono stati analizzati utilizzando il software Cell Quest Pro per 10.000 cellule.</p>
<p><strong>Saggio di sopravvivenza cellulare a lungo termine<br></strong>Le cellule (5 × 10<sup>2</sup>) sono state poste in piastre da 12 pozzetti per 24 h. Le cellule sono state trattate senza o con 25 mM o 50 mM ossamato e 10 o 20 mM DCA insieme alla metformina e hanno continuato per altre 48 h. Successivamente, il terreno è stato sostituito con terreno fresco privo di farmaci. Le piastre sono state incubate per altri 10-15 giorni a 37°C in incubatore a CO<sub>2</sub> con cambio di terreno ogni 2-3 giorni. Le cellule sono state quindi fissate (paraformaldeide al 3% e glutaraldeide allo 0,02% in PBS) e colorate con cristalvioletto allo 0,05%
<p><strong>Saggio di utilizzo del glucosio<br></strong>Le cellule (3 × 10<sup>5</sup>) sono state coltivate in DMEM contenente 25 mM di glucosio. Dopo 24 ore, il terreno è stato sostituito con il rispettivo terreno contenente concentrazioni crescenti di metformina (0,1 mM, 0,5 mM, 1 mM e 2 mM) o rotenone per 24 ore e il glucosio residuo presente nel terreno esaurito è stato monitorato utilizzando il kit per il dosaggio del glucosio basato su GOD-POD (Spinreact, Spagna) secondo le istruzioni del produttore. Per misurare l’utilizzo del glucosio in presenza di inibitori glicolitici, le cellule sono state trattate con 50 mM di ossamato, 100 µM di floretina e 20 mM di DCA, da soli o insieme a 2 mM di metformina o 100 µM di fenformina. Il glucosio consumato è stato stimato sottraendo il glucosio rimanente nel terreno di coltura dalla concentrazione iniziale nel terreno di controllo (450 mg/dl). Gli esperimenti sono stati eseguiti almeno in triplo e i valori sono stati normalizzati al numero totale di cellule.</p>
<p><strong>Saggio di stima del lattato<br></strong>La concentrazione di lattato nel terreno di coltura raccolto dalle cellule trattate con o senza metformina, oxamato e altri inibitori glicolitici, è stata determinata utilizzando un kit di stima del lattato disponibile in commercio (Spinreact, Spagna) secondo le istruzioni del produttore. In breve, il terreno di coltura o il siero sono stati diluiti in soluzione fisiologica allo 0,9% fino a 10 volte e 10 μl di campione sono stati aggiunti a ciascun pozzetto della piastra ELISA a 96 pozzetti. 150 µl del reagente fornito con il kit sono stati aggiunti a ciascun pozzetto contenente il campione, mantenendo il terreno non diluito e il reagente da solo come bianco e l’assorbanza è stata registrata a 405 nm utilizzando uno spettrofotometro (Thermo-Scientific, USA). Gli esperimenti sono stati eseguiti almeno in triplo e i valori finali sono stati normalizzati con il numero totale di cellule.</p>
<p><strong>Trasfezione di siRNA<br></strong>Il siRNA specifico contro LDHA è stato acquistato da Santa Cruz Biotechnology (USA). La trasfezione è stata effettuata utilizzando Lipofectamina 2000 (Life Technologies, USA) secondo le istruzioni del produttore. In breve, le cellule sono state piastrate a circa il 60% di confluenza. Il giorno successivo, il terreno è stato sostituito con OptiMEM (Life Technologies, USA) e mantenuto per 3 ore. I siRNA desiderati sono stati disciolti nei tamponi forniti insieme a loro. Lipofectamina2000 e siRNA sono stati diluiti separatamente in OptiMEM e incubati per 5 minuti. Successivamente, i reagenti diluiti sono stati mescolati e ulteriormente incubati per 30 minuti a temperatura ambiente. Il precipitato risultante è stato lasciato sulle cellule per 6 ore, dopodiché è stato aggiunto DMEM fresco integrato con FBS al 10% e incubato per altre 24-36 ore. L’efficienza di trasfezione è stata valutata trasfettando contemporaneamente il plasmide pEGFPN1. L’immunoblotting è stato eseguito per garantire l’inibizione della rispettiva espressione genica.</p>
<p><strong>Preparazione della frazione ricca di mitocondri da cellule e tessuti<br></strong>La frazione ricca di mitocondri è stata preparata da cellule coltivate e da tumori o tessuti normali come precedentemente riportato<sup><a href=”#73″> [73]</a></sup>. Brevemente, le cellule (1 × 10<sup>6</sup>) sono state piastrate in piastre da 10 cm e sono state tripsinizzate dopo il trattamento con gli inibitori desiderati per 24 ore. La sospensione cellulare è stata sottoposta a tre cicli di congelamento-scongelamento in tampone ipotonico (20 mM di fosfato di potassio). Questa sospensione è stata centrifugata a 50.000 rpm per 1 h per ottenere un surnatante ricco di mitocondri. Allo stesso modo, per isolare i mitocondri dai tessuti, i tessuti congelati sono stati tagliati in pezzi fini e omogeneizzati in tampone di omogeneizzazione (tampone Tris 0,5 M pH 7,5, 100 mM EGTA e 250 mM saccarosio), seguito da una procedura ciclica di congelamento/scongelamento e centrifugato a 50.000 rpm per 1 h. Il surnatante è stato raccolto come frazione mitocondriale e utilizzato per la determinazione della funzione mitocondriale e dell’attività dell’enzima OXPHOS.</p>
<p><strong>Saggi enzimatici<br></strong>Le cellule sono state omogeneizzate in tampone ipotonico (20 mM) di fosfato di potassio (pH 7,5) contenente cocktail di inibitori delle proteasi (Roche, Germania), vortexate e lisate con tre cicli di congelamento e scongelamento. Per preparare l’estratto dai tessuti, i campioni tumorali sono stati tagliati in pezzi sottili e omogeneizzati in tampone di omogeneizzazione (0,1 M Tris, 0,1 M KCl, 350 mM EDTA e 1 M saccarosio, pH 7,5) usando un omogeneizzatore di tessuti. Gli omogenati sono stati chiarificati centrifugando a 10.000 rpm per 10 minuti a 4<sup>°</sup>C e conservati in ghiaccio fino all’esecuzione dei saggi.</p>
<p><strong><em>Saggio dell’attività LDH:</em> </strong>L’attività della lattato deidrogenasi nei lisati cellulari o nell’estratto tumorale e nel siero è stata determinata utilizzando il kit per il dosaggio dell’attività LDH (Spinreact, Spagna) secondo le istruzioni del produttore.</p>
<p><em><strong>Saggio dell’attività del complesso I:</strong></em> Il saggio dell’enzima complesso I dell’OXPHOS mitocondriale è stato eseguito come descritto altrove<sup><a href=”#73″> [73]</a></sup>. In breve, la frazione ricca di mitocondri dell’omogenato cellulare o tissutale (20-50 µg di proteine dall’omogenato tissutale o 10-20 µg di frazione ricca di mitocondri) è stata aggiunta a 700 µl di acqua distillata prelevata in una cuvetta da 1 ml, seguita dall’aggiunta di 100 µl di tampone fosfato di potassio (0,5 M, pH 7,5), 60 µl di BSA priva di acidi grassi (50 mg/ml), 30 µl di KCN (10 mM) e 10 µl di NADH (10 mM). Il volume finale è stato regolato a 994 µl con acqua distillata, mescolato capovolgendo le cuvette e la lettura della linea di base è stata acquisita a 340 nm per 2 minuti. La reazione è stata avviata con l’aggiunta di 6 µl di decilubiquinone (10 mM, Sigma, USA) mescolati adeguatamente capovolgendo le cuvette. La diminuzione dell’assorbanza a 340 nm è stata seguita per 10 minuti. Una procedura simile è stata seguita per il calcolo dell’attività del complesso I in presenza di metformina 2 mM e fenformina 100 µM. Il rotenone (10 µM) è stato utilizzato come controllo positivo. I valori finali sono stati normalizzati con il contenuto totale di proteine cellulari.</p>
<p><strong>Misurazione dell’ATP cellulare totale<br></strong>Il livello di ATP è stato misurato utilizzando il kit di bioluminescenza dell’ATP disponibile in commercio (Roche, Germania) secondo le istruzioni del produttore. In breve, le cellule sono state coltivate in presenza dei farmaci indicati. Le cellule sono state raccolte e lisate in tampone Tris-EDTA 100 mM contenente NP-40 allo 0,01% e bollite per 1 minuto, seguite da un ciclo di congelamento/scongelamento. La luminescenza è stata registrata sia per il bianco che per i campioni. Per misurare l’ATP generato esclusivamente attraverso l’OXPHOS e la glicolisi, le cellule sono state trattate con oligomicina (10 µM) e ossamato o DCA rispettivamente con o senza metformina. Gli esperimenti sono stati eseguiti in triplo e ripetuti almeno una volta, e i valori finali sono stati normalizzati con il contenuto di proteine totali.</p>
<p><strong>Statistica</strong><br> L’analisi statistica è stata eseguita utilizzando Sigma Plot 12.0 (Systat Software Inc., CA, USA). La maggior parte degli esperimenti è stata ripetuta almeno una volta, con un minimo di triplicati, se non diversamente indicato. I dati sono stati rappresentati come media ± SD, tranne che per gli esperimenti indicati. Laddove richiesto, sia in coppia che non, per calcolare il valore p è stato utilizzato il test <em>t</em> di Student a due code, ipotizzando una varianza disuguale, se non diversamente indicato. I valori *<em>p</em> < 0,05, **<em>p</em> < 0,01, ***<em>p</em> < 0,001 indicano differenze significative tra i gruppi (<em>n</em> > 3 almeno).</p>
<h2>ACCETTAZIONI</h2>
<p>Gli autori ringraziano il Dr. S.C. Mande, Direttore, NCCS, Pune, India, e il Dr. G.C. Mishra, ex Direttore, NCCS, Pune, India per averci sostenuto e incoraggiato a portare a termine questo lavoro. Ringraziamo il Dr. Benoit violet (INSERM, Francia) per aver fornito le MEF, il Dr. Mahesh J Kulkarni (National Chemical Laboratory, India) per le cellule L6 e il Dr. Rakesh K Tyagi (Jawaharlal Nehru University, India) per aver fornito le cellule AML12. Si ringrazia anche il Dr. Vijayakumar MV per l’aiuto negli esperimenti sugli animali e per la lettura critica del manoscritto. BC e NM ringraziano il Council of Scientific and Industrial Research (CSIR) India; PM e SVS ringraziano la University Grant Commission (UGC), India, per aver fornito una borsa di studio. Si ringrazia il supporto degli altri membri del laboratorio e dei colleghi dell’NCCS, nonché i membri dello staff della Experimental Animal Facility, del FACS, del Confocal e della spettrometria di massa.</p>
<h2>SOSTEGNO FINANZIARIO</h2>
<p>Questo lavoro è stato supportato da una borsa di studio intramurale del NCCS finanziata dal Dipartimento di Biotecnologia del governo indiano.</p>
<h2>CONFLITTI DI INTERESSE</h2>
<p>Gli autori dichiarano di non avere potenziali conflitti di interesse</p>
<h3>NOTA</h3>
<p>Questo lavoro è stato svolto in parziale adempimento di una tesi di dottorato (di laurea) da presentare all’Università Savitribai Phule Pune, Pune, India.</p>
<p></p>
<h2>REFERENZE</h2>
<br><span id=”1″ class=”referencess blue-text”>1</span> Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007; 445:851-857.
<br><span id=”2″ class=”referencess blue-text”>2</span> Demierre MF. Epidemiologia e prevenzione del melanoma cutaneo. Curr Treat Options Oncol. 2006; 7:181-186.
<br><span id=”3″ class=”referencess blue-text”>3</span> Hersey P, Watts RN, Zhang XD, Hackett J. Metabolic approaches to treatment of melanoma. Clin Cancer Res. 2009; 15:6490-6494.
<br><span id=”4″ class=”referencess blue-text”>4</span> Warburg O. On respiratory impairment in cancer cells. Science. 1956; 124:269-270.
<br><span id=”5″ class=”referencess blue-text”>5</span> Vander Heiden MG, Cantley LC, Thompson CB. Capire l’effetto Warburg: i requisiti metabolici della proliferazione cellulare. Science. 2009; 324:1029-1033.
<br><span id=”6″ class=”referencess blue-text”>6</span> Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C. La fosforilazione ossidativa disfunzionale rende le cellule di melanoma maligno dipendenti dalla glicolisi guidata dall’oncogene (V600E)BRAF. Oncotarget. 2013; 4:584-599.
<br><span id=”7″ class=”referencess blue-text”>7</span> Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013; 23:302-315.
<br><span id=”8″ class=”referencess blue-text”>8</span> Weide B, Elsässer M, Büttner P, Pflugfelder A, Leiter U, Eigentler TK, Bauer J, Witte M, Meier F, Garbe C. I marcatori sierici lattato deidrogenasi e S100B predicono in modo indipendente l’esito della malattia nei pazienti con melanoma con metastasi a distanza. Br J Cancer. 2012; 107:422-428.
<br><span id=”9″ class=”referencess blue-text”>9</span> Deichmann M, Benner A, Bock M, Jäckel A, Uhl K, Waldmann V, Näher H. S100-Beta, attività inibitoria del melanoma e lattato deidrogenasi discriminano il melanoma progressivo da quello non progressivo di stadio IV dell’American Joint Committee on Cancer. J Clin Oncol. 1999; 17:1891-1896.
<br><span id=”10″ class=”referencess blue-text”>10</span> Giatromanolaki A, Sivridis E, Gatter KC, Turley H, Harris AL, Koukourakis MI; Tumor and Angiogenesis Research Group. L’espressione della lattato deidrogenasi 5 (LDH-5) nel tumore dell’endometrio è correlata al percorso VEGF/VEGFR2(KDR) attivato e alla prognosi. Gynecol Oncol. 2006; 103:912-918.
<br><span id=”11″ class=”referencess blue-text”>11</span> Koukourakis MI, Giatromanolaki A, Sivridis E. Lactate dehydrogenase isoenzymes 1 and 5: differential expression by neoplastic and stromal cells in non-small cell lung cancer and other epithelial malignant tumors. Tumour Biol. 2003; 24:199-202.
<br><span id=”12″ class=”referencess blue-text”>12</span> Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncoversal link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006; 9:425-434.
<br><span id=”13″ class=”referencess blue-text”>13</span> Brandon M, Baldi P, Wallace DC. Mutazioni mitocondriali nel cancro. Oncogene. 2006; 25:4647-4662.
<br><span id=”14″ class=”referencess blue-text”>14</span> Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. Le mutazioni del DNA mitocondriale che generano ROS possono regolare la metastasi delle cellule tumorali. Science. 2008; 320:661-664.
<br><span id=”15″ class=”referencess blue-text”>15</span> Wallace DC. Un paradigma mitocondriale delle malattie metaboliche e degenerative, dell’invecchiamento e del cancro: un’alba per la medicina evolutiva. Annu Rev Genet. 2005; 39:359-407.
<br><span id=”16″ class=”referencess blue-text”>16</span> Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013; 123:1068-1081.
<br><span id=”17″ class=”referencess blue-text”>17</span> He X, Zhou A, Lu H, Chen Y, Huang G, Yue X, Zhao P, Wu Y. La soppressione del complesso mitocondriale I influenza le proprietà metastatiche delle cellule. PLoS One. 2013; 8:e61677.
<br><span id=”18″ class=”referencess blue-text”>18</span> Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y. La disfunzione del complesso respiratorio I mitocondriale promuove la tumorigenesi attraverso l’alterazione dei ROS e l’attivazione di AKT. Hum Mol Genet. 2011; 20:4605-4616.
<br><span id=”19″ class=”referencess blue-text”>19</span> Gopal YN, Rizos H, Chen G, Deng W, Frederick DT, Cooper ZA, Scolyer RA, Pupo G, Komurov K, Sehgal V, Zhang J, Patel L, Pereira CG et al. L’inibizione di mTORC1/2 supera la resistenza agli inibitori della via MAPK mediata da PGC1α e dalla fosforilazione ossidativa nel melanoma. Cancer Res. 2014; 74:7037-7047.
<br><span id=”20″ class=”referencess blue-text”>20</span> Barbi de Moura M, Vincent G, Fayewicz SL, Bateman NW, Hood BL, Sun M, Suhan J, Duensing S, Yin Y, Sander C, Kirkwood JM, Becker D, Conrads TP, et al. Mitochondrial respiration–an important therapeutic target in melanoma. PLoS One. 2012; 7:e40690.
<br><span id=”21″ class=”referencess blue-text”>21</span> Blackman RK, Cheung-Ong K, Gebbia M, Proia DA, He S, Kepros J, Jonneaux A, Marchetti P, Kluza J, Rao PE, Wada Y, Giaever G, Nislow C. Il trasporto di elettroni mitocondriali è il bersaglio cellulare del farmaco oncologico elesclomol. PLoS One. 2012; 7:e29798.
<br><span id=”22″ class=”referencess blue-text”>22</span> Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Il flusso inverso del ciclo TCA attraverso le isocitrato deidrogenasi 1 e 2 è necessario per la lipogenesi nelle cellule di melanoma ipossiche. Pigment Cell Melanoma Res. 2012; 25:375-383.
<br><span id=”23″ class=”referencess blue-text”>23</span> Lim JH, Luo C, Vazquez F, Puigserver P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014; 74:3535-3545.
<br><span id=”24″ class=”referencess blue-text”>24</span> Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformina e riduzione del rischio di cancro nei pazienti diabetici. British Medical Journal. 2005; 330:1304-1305.
<br><span id=”25″ class=”referencess blue-text “25</span> Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformina e rischio di cancro nei pazienti diabetici: una revisione sistematica e una meta-analisi. Cancer Prev Res (Phila). 2010; 3:1451-1461.
<br><span id=”26″ class=”referencess blue-text”>25</span> Kirpichnikov D, McFarlane SI, Sowers JR. Metformina: un aggiornamento. Ann Intern Med. 2002; 137:25-33.
<br><span id=”27″ class=”referencess blue-text”>27</span> Pollak MN. Lo studio della metformina per la prevenzione e il trattamento del cancro: la fine dell’inizio. Cancer Discov. 2012; 2:778-790.
<br><span id=”28″ class=”referencess blue-text”>28</span> El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inibisce la respirazione cellulare attraverso un effetto indiretto mirato al complesso I della catena respiratoria. J Biol Chem. 2000; 275:223-228.
<br><span id=”29″ class=”referencess blue-text”>29</span> Owen MR, Doran E, Halestrap AP. Prove che la metformina esercita i suoi effetti antidiabetici attraverso l’inibizione del complesso 1 della catena respiratoria mitocondriale. Biochem J. 2000; 348:607-614.
<br><span id=”30″ class=”referencess blue-text”>30</span> Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. La metformina inibisce il complesso mitocondriale I delle cellule tumorali per ridurre la tumorigenesi. Elife. 2014; 3:e02242.
<br><span id=”31″ class=”referencess blue-text”>31</span> Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. La metformina agisce direttamente sui mitocondri per alterare la bioenergetica cellulare. Cancer Metab. 2014; 2:12.
<br><span id=”32″ class=”referencess blue-text”>32</span> Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarié A, Tyler BM, Brem H, Toulas C, Cohen-Jonathan Moyal E, Sarry JE, Skuli N. La metformina inibisce la crescita delle cellule di glioblastoma umano e migliora la risposta terapeutica. PLoS One. 2015; 10:e0123721.
<br><span id=”33″ class=”referencess blue-text”>33</span> Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. Il farmaco antidiabetico metformina esercita un effetto antitumorale in vitro e in vivo attraverso una diminuzione del livello di ciclina D1. Oncogene. 2008; 27:3576-3586.
<br><span id=”34″ class=”referencess blue-text”>34</span> Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase dependent growth inhibitor for breast cancer cells. Cancer Res. 2006; 66:10269-10273.
<br><span id=”35″ class=”referencess blue-text”>35</span> Martin MJ, Hayward R, Viros A, Marais R. La metformina accelera la crescita del melanoma BRAF V600E-driven attraverso l’upregolazione di VEGF-A. Cancer Discov. 2012, 2:344-355.
<br><span id=”36″ class=”referencess blue-text”>36</span> Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A, Gounon P, Allegra M, Bertolotto C, Bereder JM, Tartare-Deckert S, Bahadoran P, Auberger P, et al. La metformina inibisce lo sviluppo del melanoma attraverso meccanismi di autofagia e apoptosi. Cell Death Dis. 2011; 2:e199.
<br><span id=”37″ class=”referencess blue-text”>37</span> Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P, Bertolotto C, Tartare-Deckert S, Ballotti R, Rocchi S. La metformina blocca l’invasione del melanoma e lo sviluppo di metastasi in modo AMPK/p53-dipendente. Mol Cancer Ther. 2013; 12:1605-1615.
<br><span id=”38″ class=”referencess blue-text”>38</span> Yuan P, Ito K, Perez-Lorenzo R, Del Guzzo C, Lee JH, Shen CH, Bosenberg MW, McMahon M, Cantley LC, Zheng B. La fenformina aumenta il beneficio terapeutico dell’inibizione di BRAF(V600E) nel melanoma. Proc Natl Acad Sci USA. 2013; 110:18226-18231.
<br><span id=”39″ class=”referencess blue-text”>39</span> Cannino G, El-Khoury R, Pirinen M, Hutz B, Rustin P, Jacobs HT, Dufour E. Il glucosio modula l’attività del complesso respiratorio I in risposta alla disfunzione mitocondriale acuta. J Biol Chem. 2012; 287:38729-38740.
<br><span id=”40″ class=”referencess blue-text”>40</span> Xu RH, PelicanoH, Zhou Y, CarewJS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005; 65:613-621.
<br><span id=”41″ class=”referencess blue-text”>41</span> Pan JG, Mak TW. Il targeting metabolico come strategia antitumorale: l’alba di una nuova era? Sci STKE. 2007; 2007:pe14.
<br><span id=”42″ class=”referencess blue-text”>42</span> Dalva-Aydemir S, Bajpai R, Martinez M, Adekola KU, Kandela I, Wei C, Singhal S, Koblinski JE, Raje NS, Rosen ST, Shanmugam M. Targeting the Metabolic Plasticity of Multiple Myeloma with FDA-Approved Ritonavir and Metformin. Clin Cancer Res. 2015; 21:1161-1171.
<br><span id=”43″ class=”referencess blue-text”>43</span> Menendez JA, Oliveras-Ferraros C, Cufí S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. La metformina è sinteticamente letale con la sospensione del glucosio nelle cellule tumorali. Cell Cycle. 2012; 11:2782-2792.
<br><span id=”44″ class=”referencess blue-text”>44</span> Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, Nanni P, Bergamini C, Nicoletti G, Giovanni CD, Ghelli A, Giorgio V, Caratozzolo MF, et al. Il complesso respiratorio I è essenziale per indurre un profilo di Warburg in cellule tumorali con difetti dei mitocondri. Cancer Metab. 2013; 1:11.
<br><span id=”45″ class=”referencess blue-text”>45</span> Semenza GL. Metabolismo tumorale: le cellule tumorali danno e prendono lattato. J Clin Invest. 2008; 118:3835-3837.
<br><span id=”46″ class=”referencess blue-text”>46</span> Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Bernard Gallez B, Wahl ML, et al. Il bersaglio della respirazione alimentata dal lattato uccide selettivamente le cellule tumorali ipossiche nei topi. J Clin Invest. 2008; 118:3930-3942.
<br><span id=”47″ class=”referencess blue-text”>47</span> Phoenix KN, Vumbaca F, Claffey KP. L’attivazione terapeutica di metformina/AMPK promuove il fenotipo angiogenico nel modello di carcinoma mammario MDA-MB-435 ERalfa negativo. Breast Cancer Res Treat. 2009; 113:101-111.
<br><span id=”48″ class=”referencess blue-text”>48</span> Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. L’afflusso di lattato attraverso il trasportatore monocarbossilato delle cellule endoteliali MCT1 supporta un percorso NF-κB/IL-8 che guida l’angiogenesi tumorale. Cancer Res. 2011; 71:2550-2560.
<br><span id=”49″ class=”referencess blue-text”>49</span> Sonveaux P, Copetti T, De Saedeleer CJ, Vegran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frérart F, Gallez B, Ribeiro A, Michiels C, et al. Il target del trasportatore del lattato MCT1 nelle cellule endoteliali inibisce l’attivazione di HIF-1 indotta dal lattato e l’angiogenesi tumorale. PLoS ONE. 2012; 7:e33418.
<br><span id=”50″ class=”referencess blue-text”>50</span> Wu M, Seto E, Zhang J. E2F1 aumenta la glicolisi attraverso la soppressione della trascrizione di Sirt6 nelle cellule tumorali. Oncotarget. 2015; 6:11252-11263.
<br><span id=”51″ class=”referencess blue-text”>51</span> Hernlund E, Strandberg Ihrlund L, Khan O, Ates YO, Linder S, Panaretakis T, Shoshan MC. Potenziamento dei farmaci chemioterapici da parte degli inibitori del metabolismo energetico 2-deossiglucosio ed etomoxir. Int J Cancer. 2008; 123:476-483.
<br><span id=”52 “class=”referencess blue-text”>52</span>. Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Il dicloroacetato induce l’apoptosi nelle cellule del cancro endometriale. Gynecol Oncol. 2008; 109:402.
<br><span id=”53″ class=”referencess blue-text”>53</span> Michelakis ED, Webster L, Mackey JR. Il dicloroacetato (DCA) come potenziale bersaglio metabolico nella terapia del cancro. Br J Cancer. 2008; 99:989-994.
<br><span id=”54″ class=”referencess blue-text”>54</span> Walenta S, Mueller-Kliese WF. Il lattato: specchio e motore della malignità tumorale. Semin Radiat Oncol. 2004; 14:267-274.
<br><span id=”55″ class=”referencess blue-text”>55</span> Doherty JR, Cleveland JL. Prendere di mira il metabolismo del lattato per la terapia del cancro. J Clin Invest. 2013; 123:3685-3692.
<br><span id=”56″ class=”referencess blue-text”>56</span> Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. L’inibizione della lattato deidrogenasi A induce stress ossidativo e inibisce la progressione tumorale. Proc Natl Acad Sci USA. 2010; 107:2037-2042.
<br><span id=”57″ class=”referencess blue-text”>57</span> Zhao YH, Zhou M, Liu H, Ding Y, Khong HT, Yu D, Fodstad O, Tan M. L’up-regolazione della lattato deidrogenasi A da parte di ErbB2 attraverso il fattore di shock termico 1 promuove la glicolisi e la crescita delle cellule di cancro al seno. Oncogene. 2009; 28:3689-3701.
<br><span id=”58″ class=”referencess blue-text”>58</span> Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, et al. L’inattivazione dell’asse di segnalazione HIF-1α/PDK3 spinge il melanoma verso il metabolismo ossidativo mitocondriale e potenzia l’attività terapeutica dei pro-ossidanti. Cancer Res. 2012; 72:5035-5047.
<br><span id=”59″ class=”referencess blue-text”>59</span> Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, et al. La risposta del melanoma BRAF-mutante all’inibizione di BRAF è mediata da una rete di regolatori trascrizionali della glicolisi. Cancer Discov. 2014; 4:423-433.
<br><span id=”60″ class=”referencess blue-text”>60</span> Miskimins WK, Ahn HJ, Kim JY, Ryu S, Jung YS, Choi JY. Effetto antitumorale sinergico di fenformina e ossamato. PLoS One. 2014; 9:e85576.
<br><span id=”61″ class=”referencess blue-text”>61</span> Jin S, DiPaola RS, Mathew R, White E. Metabolic catastrophe as a means to cancer cell death. J Cell Sci. 2007; 120:379-383.
<br><span id=”62″ class=”referencess blue-text”>62</span> McLornan DP, List A, Mufti GJ. Applicazione della letalità sintetica per la lotta selettiva contro il cancro. N Engl J Med. 2014; 371:1725-1735.
<br><span id=”63″ class=”referencess blue-text”>63</span> Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005; 5:689-698.
<br><span id=”64″ class=”referencess blue-text”>64</span> Iaquinta PJ, Lees JA. Decisioni di vita e di morte da parte dei fattori di trascrizione E2F. Curr Opin Cell Biol. 2007; 19:649-657.
<br><span id=”65″ class=”referencess blue-text”>65</span> Houben R, Hesbacher S, Schmid CP, Kauczok CS, Flohr U, Haferkamp S, Müller CS, Schrama D, Wischhusen J, Becker JC. L’espressione ad alto livello di p53 wild-type nelle cellule di melanoma è spesso associata all’inattività nei test del gene reporter p53. PLoS One. 2011; 6:e22096.
<br><span id=”66″ class=”referencess blue-text”>66</span> Avery-Kiejda KA, Bowden NA, Croft AJ, Scurr LL, Kairupan CF, Ashton KA, Talseth-Palmer BA, Rizos H, Zhang XD, Scott RJ, Hersey P. P53 nel melanoma umano non riesce a regolare geni bersaglio associati all’apoptosi e al ciclo cellulare e può contribuire alla proliferazione. BMC Cancer. 2011; 11:203.
<br><span id=”67″ class=”referencess blue-text”>67</span> Lu M, Breyssens H, Salter V, Zhong S, Hu Y, Baer C, Ratnayaka I, Sullivan A, Brown NR, Endicott J, Knapp S, Kessler BM, Middleton MR, et al. Ripristino della funzione di p53 nelle cellule di melanoma umano attraverso l’inibizione di MDM2 e della ciclina B1/CDK1-fosforilata iASPP nucleare. Cancer Cell. 2013; 23:618-633.
<br><span id=”68″ class=”referencess blue-text”>68</span> Pandey V, Vijayakumar MV, Ajay AK, Malvi P, Bhat MK. L’obesità indotta dalla dieta aumenta la progressione del melanoma: coinvolgimento di Cav-1 e FASN. Int J Cancer. 2012; 130:497-508.
<br><span id=”69″ class=”referencess blue-text”>69</span> Leontieva OV, Blagosklonny MV. M(o)TOR dello stato pseudo-ipossico nell’invecchiamento: la rapamicina in soccorso. Cell Cycle. 2014; 13:509-515.
<br><span id=”70″ class=”referencess blue-text”>70</span> Leontieva OV, Blagosklonny MV. Senescenza cronologica simile a quella del lievito in cellule di mammifero: fenomeno, meccanismo e soppressione farmacologica. Aging (Albany NY). 2011; 3:1078-1091.
<br><span id=”71″ class=”referencess blue-text”>71</span> Vijayakumar MV, Singh S, Chhipa RR, Bhat MK. L’attività ipoglicemizzante dell’estratto di semi di fieno greco è mediata dalla stimolazione di una via di segnalazione insulinica. Br J Pharmacol. 2005; 146:41-48.
<br><span id=”72″ class=”referencess blue-text”>72</span> Malvi P, Chaube B, Pandey V, Vijayakumar MV, Boreddy PR, Mohammad N, Singh SV, Bhat MK. La rapida progressione del melanoma indotta dall’obesità è invertita dal trattamento con orlistat e dall’intervento dietetico: ruolo delle adipochine. Mol Oncol. 2015; 9:689-703.
<br><span id=”73″ class=”referencess blue-text”>73</span> Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Valutazione delle attività enzimatiche della catena respiratoria mitocondriale su tessuti e cellule in coltura. Nat Protoc. 2012; 7: 1235-1246.
<p></p>