Балкришна Чаубе1, Пармананд Малви1, Шивендра Викрам Сингх1, Наошад Мохаммад1, Автар Сингх Мина1,2 и Манодж Кумар Бхат1
1 Национальный центр клеточной науки, кампус Университета Савитрибай Фуле Пуна, Ганешхинд, Пуна, Индия
Текущий адрес: Кафедра физиологии, Научный центр здоровья Университета Теннесси, Мемфис, США
Корреспонденция: Манодж Кумар Бхат, e-mail: [email protected]
Получено: 16 июня 2015 г.
Принято: 23 сентября 2015 г.
Опубликовано: 15 октября 2015 г
Аннотация
Меланома является в основном неизлечимой злокачественной опухолью кожи из-за лежащей в ее основе молекулярной и метаболической гетерогенности, осложненной развитием резистентности. Раковые клетки обладают метаболической гибкостью, выбирая окислительное фосфорилирование (OXPHOS) или гликолиз для выработки АТФ в зависимости от наличия питательных веществ в микроокружении опухоли. В данном исследовании мы изучили участие дыхательного комплекса I и лактатдегидрогеназы (ЛДГ) в прогрессировании меланомы. Мы показали, что ингибирование комплекса I метформином способствует росту меланомы у мышей через повышение уровня лактата и VEGF. В отличие от этого, оно приводит к остановке роста in vitro из-за усиленного внеклеточного закисления в результате усиленного гликолиза. Ингибирование выработки LDH или лактата вызывает снижение гликолиза с сопутствующей остановкой роста как in vitro, так и in vivo. Блокирование выработки лактата в клетках меланомы, обработанных метформином, приводит к снижению пролиферации клеток и прогрессированию опухоли у мышей. Интересно, что ингибирование LDH или комплекса I по отдельности не вызывает апоптоза, в то время как ингибирование обоих вместе вызывает истощение клеточного пула АТФ, что приводит к метаболической катастрофе, вызывающей апоптоз. В целом, наше исследование показывает, что LDH и комплекс I играют разные роли в регуляции гликолиза и клеточной пролиферации. Их ингибирование усиливает синтетическую летальность при меланоме.
Ключевые слова: меланома; комплекс I; LDH; метаболическая катастрофа; синтетическая летальность
ВВЕДЕНИЕ
Злокачественная меланома является одной из наиболее агрессивных форм рака кожи с высоким метастатическим потенциалом и устойчивостью ко многим цитотоксическим агентам [1, 2]. Несмотря на обширные исследования и частичные успехи, достигнутые при использовании доступных в настоящее время препаратов, эффективного лечения злокачественной меланомы не существует [1-3]. Число случаев заболевания меланомой растет с каждым годом и составляет около 75% смертей от рака кожи во всем мире [2, 3]. Плохой ответ на доступные в настоящее время терапевтические методы и развитие резистентности к терапии требуют поиска новых стратегий лечения меланомы.
Усиленный аэробный гликолиз является характерной особенностью многих видов рака [3-5]. Сообщается, что клетки меланомы, вследствие мутации BRAF, в основном зависят от гликолиза для производства АТФ и демонстрируют дисфункциональное окислительное фосфорилирование [6, 7]. Раковые клетки получают АТФ, промежуточные продукты биосинтеза и восстановительные эквиваленты путем необычного участия в таких биохимических путях, как гликолиз, глутаминолиз и пентозофосфатный путь [5]. Нормальные (нераковые) клетки получают АТФ в основном через митохондриальный OXPHOS, в то время как раковые клетки полагаются в основном на аэробный гликолиз для получения АТФ и гликолитических промежуточных продуктов, способствующих быстрому росту [4, 5]. Повышенное образование лактата коррелирует с агрессивностью рака. Многочисленные исследования выявили лактатдегидрогеназу (ЛДГ), катализирующую превращение пирувата в лактат, как наиболее устойчивый маркер агрессивных и быстрорастущих раковых опухолей [8-11]. LDH играет важную роль в регуляции гликолиза, поддержании клеточного окислительно-восстановительного состояния, физиологии митохондрий и поддержании опухоли [12]. Измененный метаболизм раковой клетки может быть связан с дисфункцией митохондрий, которая включает ингибирование OXPHOS, увеличение реактивных форм кислорода (ROS) и способствует неконтролируемому росту, что в свою очередь способствует развитию метастатического фенотипа [13,14]. Дыхательный комплекс I является самым крупным и сложным ферментом, катализирующим окисление NADH в электронно-транспортной цепи [15]. Комплекс I играет важную роль в генерации АТФ в нормальных клетках и является основным местом генерации ROS, однако его роль в опухолевом генезе остается неясной. Большинство сообщений свидетельствует о том, что активность комплекса I подавлена в раковых клетках, а его ингибирование способствует пролиферации и метастазированию [16-18]. И наоборот, сообщалось, что клетки меланомы демонстрируют повышенную функцию OXPHOS, что вызывает устойчивость к лекарствам [19-21]. Благодаря метаболической гибкости в выборе различных метаболических путей, особенно метаболизма глутамина [22], клетки меланомы могут спастись от возмущения молекулы в метаболическом пути [23]. Поэтому для достижения эффективного и лечебного результата необходимо воздействовать на более чем одну молекулу различных метаболических путей.
Известно, что бигуаниды, такие как метформин и фенформин, ингибируют дыхательный комплекс I. Метформин, являющийся препаратом первой линии терапии для пациентов с сахарным диабетом 2 типа (СД2), относится к бигуанидам, и недавно было обнаружено, что он обладает противоопухолевой активностью [24, 25]. На физиологическом уровне метформин проявляет свою биологическую активность, снижая печеночный глюкогенез, повышая чувствительность к инсулину, повышая периферическое поглощение глюкозы и снижая абсорбцию глюкозы из желудочно-кишечного тракта [26]. На клеточном уровне метформин действует главным образом путем ингибирования митохондриального комплекса I, что препятствует окислительному фосфорилированию, приводящему к снижению уровня АТФ и активации AMPK [27-30]. Более того, было показано, что метформин снижает митохондриальное дыхание в сочетании с выработкой АТФ, тем самым вызывая увеличение гликолиза [31, 32]. Противораковое действие метформина было показано при различных видах рака с различными механизмами [33, 34]. Однако роль метформина при меланоме не совсем ясна. В отличие от других видов рака, метформин обладает как противоопухолевой, так и противоопухолевой активностью при меланоме [35-37]. Также было показано, что метформин в комбинации с ингибиторами BRAF подавляет рост меланомы [38].
В настоящем исследовании, предполагая, что нарушение путей генерации АТФ путем ингибирования комплекса I и LDH вместе может быть синтетически смертельным для клеток меланомы, мы использовали метформин или фенформин и оксамат/дихлорацетат (DCA) для ингибирования этих двух ферментов на клеточных и животных моделях. Мы продемонстрировали, что ингибирование комплекса I и активности LDH оказывает различное влияние на рост и пролиферацию клеток. Интересно, что ингибирование комплекса I метформином способствует дальнейшему развитию аэробного гликолиза, что приводит к усилению роста опухоли у мышей. Подавление вызванного метформином образования лактата с помощью оксамата или DCA приводит к цитостазу и/или апоптозу в клетках меланомы.
РЕЗУЛЬТАТЫ
Метформин оказывает различное действие in vitro и in vivo на рост меланомы
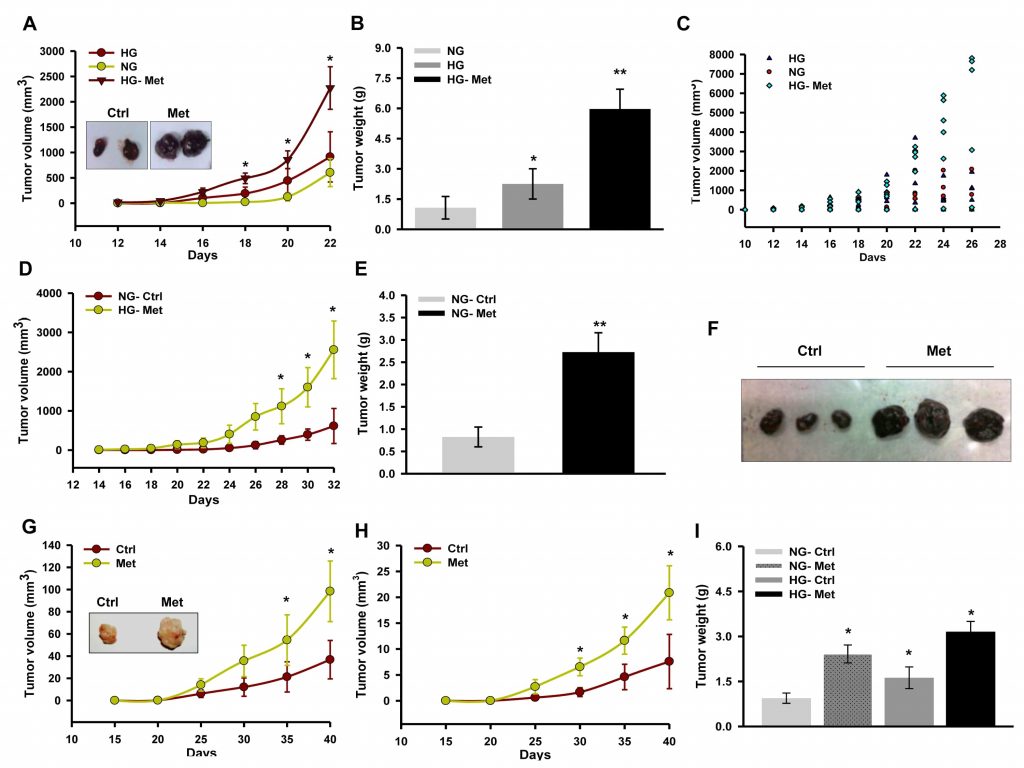
Метформин подавляет рост опухоли путем ингибирования комплекса I, который находится под влиянием глюкозы [30]. Более того, известно, что глюкоза изменяет активность дыхательных ферментов [39]. Поэтому для изучения последствий ингибирования комплекса I и влияния глюкозы на действие метформина на прогрессию меланомы мы наблюдали за развитием изотрансплантата/ксенотрансплантата у мышей с гипергликемией, вызванной стрептозотоцином (STZ). Мы отметили, что метформин способствовал прогрессированию изотрансплантата B16F10 у мышей с гипергликемией по сравнению с контролем без лечения (Рисунок 1A, 1B и 1C). Кроме того, метформин положительно влиял на прогрессирование опухоли у нормогликемических мышей C57BL/6J (Рисунок 1D, 1E и 1F). Аналогично, пероральное введение метформина способствовало росту ксенотрансплантата A375 у гипергликемических, а также у нормогликемических мышей NOD/SCID по сравнению с контролем без лечения (Рисунок 1G, 1H и 1I).
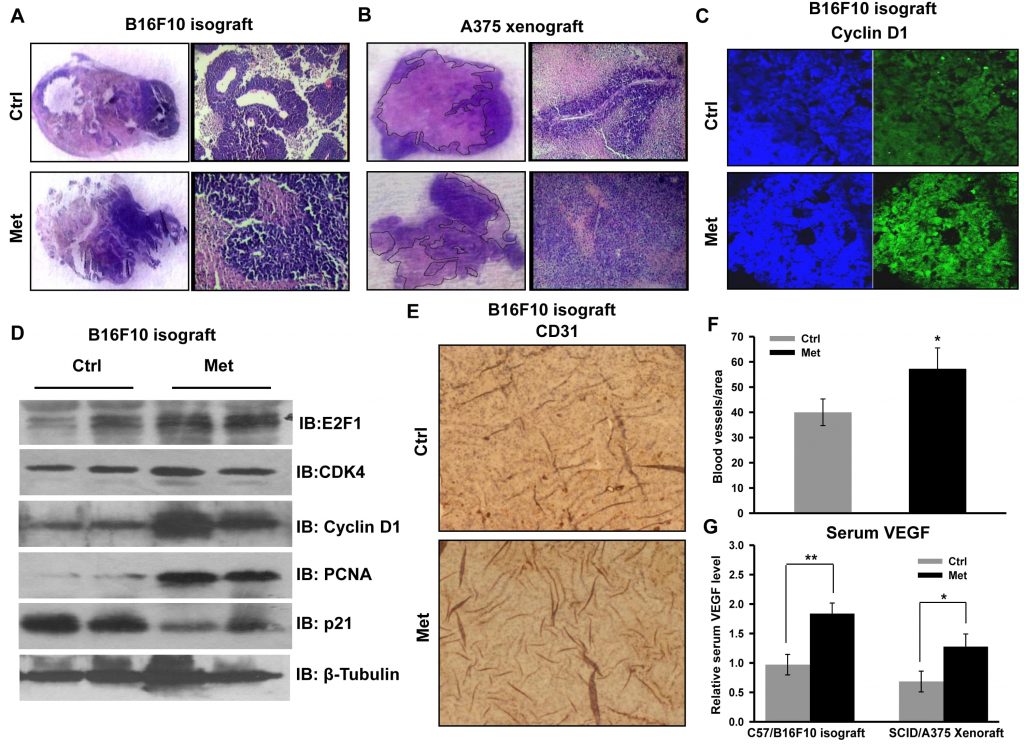
Для проверки клеточных и молекулярных событий, связанных с усилением опухолевой прогрессии, срезы опухоли были подвергнуты гистопатологическому анализу. Высокая плотность клеток и уменьшение некроза были хорошо видны на срезах обоих типов опухолей (изотрансплантат B16F10 и ксенотрансплантат A375), полученных от мышей, получавших метформин (Рисунок 2A и 2B). Мы отметили, что усиленная метформином пролиферация и прогрессия ксенотрансплантата A375 была фенотипически отличной по сравнению с контрольной опухолью. Это свидетельствует о повышении класса первичной опухоли, о чем свидетельствует удлиненная морфология ядер по сравнению с округлой морфологией в контрольных опухолях (неопубликованная информация). Иммуногистохимическое окрашивание регуляторного белка клеточного цикла циклина D1 (рис. 2C) было обнаружено в срезах опухолей, полученных от мышей, получавших метформин. Далее мы подтвердили усиленный рост опухоли, проверив состояние регуляторных белков клеточного цикла с помощью иммуноблотинга лизатов опухоли. Мы обнаружили, что уровни молекул циклина D1, CDK4, E2F1 и PCNA были значительно повышены в лизатах опухолей мышей, получавших метформин, по сравнению с контролем, в то время как уровень p21 был снижен (Рисунок 2D). Эти результаты показывают, что метформин, независимо от гликемического статуса мышей, способствует росту меланомы путем модуляции регуляторных белков клеточного цикла. Более того, иммуногистохимический анализ срезов опухоли подтвердил это наблюдение, поскольку лечение метформином повысило уровень белка CD31, эндотелиального маркера (Рисунок
2Eи 2F), и увеличило сывороточный уровень VEGF (Рисунок 2G), предполагая, что метформин способствует ангиогенезу в опухолях меланомы.
Далее мы проверили влияние метформина на рост и пролиферацию клеток меланомы in vitro. В отличие от результатов, полученных нами in vivo, лечение метформином привело к подавлению роста клеток меланомы in vitro (дополнительные рисунки S1A, S1B, S1C и S1D). Затем мы исследовали влияние ингибирования комплекса I с помощью метформина и фенформина на рост клеток меланомы, поскольку оба они подавляли активность комплекса I (Дополнительный рисунок S2). Мы обнаружили, что метформин и фенформин вызывали остановку роста клеток меланомы, выращенных в условиях высокого содержания глюкозы. Однако в присутствии низкой глюкозы лечение этими агентами приводило к гибели клеток (Дополнительный рисунок S3A, S3B, S3C и S3D). Вероятно, что метформин-опосредованная остановка роста обусловлена снижением уровня глюкозы и внеклеточным подкислением среды (Дополнительный рисунок S4A и S4B). Интересно, что замена среды через каждые 12 ч свежей средой, содержащей 25 мМ глюкозы, увеличивала выживаемость клоногенных клеток при лечении метформином. Поскольку клетки, обработанные метформином, очень быстро расходуют глюкозу по сравнению с контролем, поэтому пополнение среды необходимо для поддержания уровня глюкозы и рН и, следовательно, выживания клеток в присутствии метформина (Дополнительный рисунок S4C и S4D). Эти результаты позволяют предположить, что концентрация глюкозы в культуральной среде влияет на действие метформина.
Ингибирование активности дыхательного комплекса I способствует аэробному гликолизу <p>Чтобы усилить эти выводы, мы дополнительно изучили характер экспрессии и активность некоторых белков и ферментов, участвующих в регуляции гликолиза. Мы обнаружили, что метформин повышает уровень белка ключевых гликолитических молекул GLUT1, LDHA и ChREBP в зависимости от концентрации (Рисунок 3C). Эти результаты позволяют предположить, что ингибирование активности комплекса I метформином может заставить раковые клетки перейти на гликолитический путь выработки АТФ, что приводит к усилению опухолевой прогрессии у мышей.</p> <p>Следующим этапом мы проверили влияние метформина и оксамата/DCA по отдельности или в комбинации на выживаемость нераковых клеток. Мы использовали три различные линии нераковых клеток: AML12 (гепатоциты мыши), L6 (мышечные клетки крысы) и MEF (эмбриональные фибробласты мыши) для проверки эффекта комбинированного лечения. Хотя комбинация метформина и оксамата/DCA замедлила пролиферацию этих клеток (Дополнительные рисунки S7A, S7B и S7C), она не повлияла на жизнеспособность клеток (Дополнительный рисунок S7D), что указывает на то, что эта комбинация эффективна в избирательном уничтожении раковых клеток.</p> <p><strong>Блокирование комплекса I и выработки лактата приводит к метаболической катастрофе<br></strong>Чтобы убедиться, влияет ли совместное лечение оксаматом и метформином на метаболизм клеток меланомы, мы измерили метаболические параметры в присутствии этих ингибиторов. Лечение метформином повышало гликолиз, в то время как лечение оксаматом приводило к снижению утилизации глюкозы и секреции лактата (рис. 6A и 6B). Интересно, что утилизация глюкозы и секреция лактата значительно подавлялись при обработке клеток оксаматом и метформином вместе (Рисунок 6A и 6B). Кроме того, как оксамат, так и метформин снижали уровень АТФ путем ингибирования фосфорилирования на уровне субстратов и OXPHOS соответственно (Рисунок 6C). Также значительное снижение уровня АТФ было обнаружено в клетках, обработанных оксаматом и метформином вместе (Рисунок 6C). Далее, чтобы подтвердить участие LDH в росте и пролиферации клеток меланомы, было проведено нокаутирование LDHA (LDHA-KD) с помощью специфической siRNA (Рисунок 6D). Повышенная гибель клеток наблюдалась в клетках LDHA-KD, обработанных метформином или фенформином (Рисунок 6E). Значительное снижение гликолиза (о чем свидетельствует уменьшение утилизации глюкозы и выделения лактата) и уровня АТФ наблюдалось в клетках LDHA-KD по сравнению с контролем (Рисунок 6F, 6G и 6H). Более того, снижение гликолитической скорости, а также резкое снижение уровня АТФ наблюдалось в клетках LDHA-KD, обработанных метформином или фенформином (Рисунок 6F, 6G и 6H). Эти результаты показывают, что одновременное ингибирование комплекса I и ЛДГ или пути выработки лактата вызывает метаболическую катастрофу из-за нарушения клеточного пула АТФ, что в конечном итоге приводит к остановке роста и гибели клеток.</p> <br><span id=»2″ class=»referencess blue-text»>2</span> Demierre MF. Эпидемиология и профилактика кожной меланомы. Curr Treat Options Oncol. 2006; 7:181-186. <br><span id=»3″ class=»referencess blue-text»>3</span> Hersey P, Watts RN, Zhang XD, Hackett J. Metabolic approaches to treatment of melanoma. Clin Cancer Res. 2009; 15:6490-6494. <br><span id=»4″ class=»referencess blue-text»>4</span> Варбург О. О нарушении дыхания в раковых клетках. Science. 1956; 124:269-270. <br><span id=»5″ class=»referencess blue-text»>5</span> Vander Heiden MG, Cantley LC, Thompson CB. Понимание эффекта Варбурга: метаболические требования клеточной пролиферации. Science. 2009; 324:1029-1033. <br><span id=»6″ class=»referencess blue-text»>6</span> Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C. Дисфункциональное окислительное фосфорилирование делает клетки злокачественной меланомы зависимыми от гликолиза, управляемого онкогеном (V600E)BRAF. Oncotarget. 2013; 4:584-599. <br><span id=»7″ class=»referencess blue-text»>7</span> Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. 2013; 23:302-315. <br><span id=»8″ class=»referencess blue-text»>8</span> Weide B, Elsässer M, Büttner P, Pflugfelder A, Leiter U, Eigentler TK, Bauer J, Witte M, Meier F, Garbe C. Serum markers lactate dehydrogenase and S100B predict independently disease outcome in melanoma patients with distant metastasis. Br J Cancer. 2012; 107:422-428. <br><span id=»9″ class=»referencess blue-text»>9</span> Deichmann M, Benner A, Bock M, Jäckel A, Uhl K, Waldmann V, Näher H. S100-Beta, ингибирующая активность меланомы и лактатдегидрогеназа отличают прогрессирующую меланому IV стадии Американского объединенного комитета по раку от непрогрессирующей. J Clin Oncol. 1999; 17:1891-1896. <br><span id=»10″ class=»referencess blue-text»>10</span> Giatromanolaki A, Sivridis E, Gatter KC, Turley H, Harris AL, Koukourakis MI; Tumor and Angiogenesis Research Group. Экспрессия лактатдегидрогеназы 5 (LDH-5) при раке эндометрия связана с активированным путем VEGF/VEGFR2(KDR) и прогнозом. Gynecol Oncol. 2006; 103:912-918. <br><span id=»11″ class=»referencess blue-text»>11</span> Koukourakis MI, Giatromanolaki A, Sivridis E. Lactate dehydrogenase isoenzymes 1 and 5: differential expression by neoplastic and stromal cells in non-small cell lung cancer and other epithelial malignant tumors. Tumour Biol. 2003; 24:199-202. <br><span id=»12″ class=»referencess blue-text»>12</span> Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006; 9:425-434. <br><span id=»13″ class=»referencess blue-text»>13</span> Brandon M, Baldi P, Wallace DC. Митохондриальные мутации в раке. Oncogene. 2006; 25:4647-4662. <br><span id=»14″ class=»referencess blue-text»>14</span> Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, Nakada K, Honma Y, Hayashi J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008; 320:661-664. <br><span id=»15″ class=»referencess blue-text»>15</span> Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005; 39:359-407. <br><span id=»16″ class=»referencess blue-text»>16</span> Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013; 123:1068-1081. <br><span id=»17″ class=»referencess blue-text»>17</span> He X, Zhou A, Lu H, Chen Y, Huang G, Yue X, Zhao P, Wu Y. Подавление митохондриального комплекса I влияет на метастатические свойства клеток. PLoS One. 2013; 8:e61677. <br><span id=»18″ class=»referencess blue-text»>18</span> Sharma LK, Fang H, Liu J, Vartak R, Deng J, Bai Y. Дисфункция митохондриального дыхательного комплекса I способствует развитию опухолевого генеза через изменение ROS и активацию AKT. Hum Mol Genet. 2011; 20:4605-4616. <br><span id=»19″ class=»referencess blue-text»>19</span> Gopal YN, Rizos H, Chen G, Deng W, Frederick DT, Cooper ZA, Scolyer RA, Pupo G, Komurov K, Sehgal V, Zhang J, Patel L, Pereira CG et al. Inhibition of mTORC1/2 overcomes resistance to MAPK pathway inhibitors mediated by PGC1α and oxidative phosphorylation in melanoma. Cancer Res. 2014; 74:7037-7047. <br><span id=»20″ class=»referencess blue-text»>20</span> Barbi de Moura M, Vincent G, Fayewicz SL, Bateman NW, Hood BL, Sun M, Suhan J, Duensing S, Yin Y, Sander C, Kirkwood JM, Becker D, Conrads TP, et al. Mitochondrial respiration—an important therapeutic target in melanoma. PLoS One. 2012; 7:e40690. <br><span id=»21″ class=»referencess blue-text»>21</span> Blackman RK, Cheung-Ong K, Gebbia M, Proia DA, He S, Kepros J, Jonneaux A, Marchetti P, Kluza J, Rao PE, Wada Y, Giaever G, Nislow C. Митохондриальный электронный транспорт — клеточная мишень онкологического препарата элоскломол. PLoS One. 2012; 7:e29798. <br><span id=»22″ class=»referencess blue-text»>22</span> Filipp FV, Scott DA, Ronai ZA, Osterman AL, Smith JW. Обратный поток цикла ТСА через изоцитратдегидрогеназы 1 и 2 необходим для липогенеза в гипоксических клетках меланомы. Pigment Cell Melanoma Res. 2012; 25:375-383. <br><span id=»23″ class=»referencess blue-text»>23</span> Lim JH, Luo C, Vazquez F, Puigserver P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. 2014; 74:3535-3545. <br><span id=»24″ class=»referencess blue-text»>24</span> Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Метформин и снижение риска развития рака у пациентов с сахарным диабетом. Британский медицинский журнал. 2005; 330:1304-1305. <br><span id=»25″ class=»referencess blue-text «25</span> Decensi A, Puntoni M, Goodwin P, Cazzaniga M, Gennari A, Bonanni B, Gandini S. Metformin and cancer risk in diabetic patients: a systematic review and meta-analysis. Cancer Prev Res (Phila). 2010; 3:1451-1461. <br><span id=»26″ class=»referencess blue-text»>25</span> Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med. 2002; 137:25-33. <br><span id=»27″ class=»referencess blue-text»>27</span> Pollak MN. Исследование метформина для профилактики и лечения рака: конец начала. Cancer Discov. 2012; 2:778-790. <br><span id=»28″ class=»referencess blue-text»>28</span> El-Mir MY, Nogueira V, Fontaine E, Averet N, Rigoulet M, Leverve X. Dimethylbiguanide inhibits cell respiration through a indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000; 275:223-228. <br><span id=»29″ class=»referencess blue-text»>29</span> Owen MR, Doran E, Halestrap AP. Доказательства того, что метформин оказывает свое антидиабетическое действие через ингибирование комплекса 1 митохондриальной дыхательной цепи. Biochem J. 2000; 348:607-614. <br><span id=»30″ class=»referencess blue-text»>30</span> Wheaton WW, Weinberg SE, Hamanaka RB, Soberanes S, Sullivan LB, Anso E, Glasauer A, Dufour E, Mutlu GM, Budigner GS, Chandel NS. Метформин ингибирует митохондриальный комплекс I раковых клеток для уменьшения туморигенеза. Elife. 2014; 3:e02242. <br><span id=»31″ class=»referencess blue-text»>31</span> Andrzejewski S, Gravel SP, Pollak M, St-Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014; 2:12. <br><span id=»32″ class=»referencess blue-text»>32</span> Sesen J, Dahan P, Scotland SJ, Saland E, Dang VT, Lemarié A, Tyler BM, Brem H, Toulas C, Cohen-Jonathan Moyal E, Sarry JE, Skuli N. Metformin inhibits growth of human glioblastoma cells and enhances therapeutic response. PLoS One. 2015; 10:e0123721. <br><span id=»33″ class=»referencess blue-text»>33</span> Ben Sahra I, Laurent K, Loubat A, Giorgetti-Peraldi S, Colosetti P, Auberger P, Tanti JF, Le Marchand-Brustel Y, Bost F. Противодиабетический препарат метформин оказывает противоопухолевое действие in vitro и in vivo через снижение уровня циклина D1. Oncogene. 2008; 27:3576-3586. <br><span id=»34″ class=»referencess blue-text»>34</span> Zakikhani M, Dowling R, Fantus IG, Sonenberg N, Pollak M. Metformin is an AMP kinase dependent growth inhibitor for breast cancer cells. Cancer Res. 2006; 66:10269-10273. <br><span id=»35″ class=»referencess blue-text»>35</span> Martin MJ, Hayward R, Viros A, Marais R. Metformin accelerates the growth of BRAF V600E-driven melanoma by upregulating VEGF-A. Cancer Discov. 2012, 2:344-355. <br><span id=»36″ class=»referencess blue-text»>36</span> Tomic T, Botton T, Cerezo M, Robert G, Luciano F, Puissant A, Gounon P, Allegra M, Bertolotto C, Bereder JM, Tartare-Deckert S, Bahadoran P, Auberger P, et al. Metformin inhibits melanoma development through autophagy and apoptosis mechanisms. Cell Death Dis. 2011; 2:e199. <br><span id=»37″ class=»referencess blue-text»>37</span> Cerezo M, Tichet M, Abbe P, Ohanna M, Lehraiki A, Rouaud F, Allegra M, Giacchero D, Bahadoran P, Bertolotto C, Tartare-Deckert S, Ballotti R, Rocchi S. Metformin blocks melanoma invasion and metastasis development in AMPK/p53-dependent manner. Mol Cancer Ther. 2013; 12:1605-1615. <br><span id=»38″ class=»referencess blue-text»>38</span> Yuan P, Ito K, Perez-Lorenzo R, Del Guzzo C, Lee JH, Shen CH, Bosenberg MW, McMahon M, Cantley LC, Zheng B. Phenformin enhances therapeutic benefit of BRAF(V600E) inhibition in melanoma. Proc Natl Acad Sci USA. 2013; 110:18226-18231. <br><span id=»39″ class=»referencess blue-text»>39</span> Cannino G, El-Khoury R, Pirinen M, Hutz B, Rustin P, Jacobs HT, Dufour E. Glucose modulates respiratory complex I activity in response to acute mitochondrial dysfunction. J Biol Chem. 2012; 287:38729-38740. <br><span id=»40″ class=»referencess blue-text»>40</span> Xu RH, PelicanoH, Zhou Y, CarewJS, Feng L, Bhalla KN, Keating MJ, Huang P. Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia. Cancer Res. 2005; 65:613-621. <br><span id=»41″ class=»referencess blue-text»>41</span> Pan JG, Mak TW. Метаболический таргетинг как противораковая стратегия: рассвет новой эры? Sci STKE. 2007; 2007:pe14. <br><span id=»42″ class=»referencess blue-text»>42</span> Dalva-Aydemir S, Bajpai R, Martinez M, Adekola KU, Kandela I, Wei C, Singhal S, Koblinski JE, Raje NS, Rosen ST, Shanmugam M. Targeting the Metabolic Plasticity of Multiple Myeloma with FDA-Approved Ritonavir and Metformin. Clin Cancer Res. 2015; 21:1161-1171. <br><span id=»43″ class=»referencess blue-text»>43</span> Menendez JA, Oliveras-Ferraros C, Cufí S, Corominas-Faja B, Joven J, Martin-Castillo B, Vazquez-Martin A. Metformin is synthetically lethal with glucose withdrawal in cancer cells. Cell Cycle. 2012; 11:2782-2792. <br><span id=»44″ class=»referencess blue-text»>44</span> Calabrese C, Iommarini L, Kurelac I, Calvaruso MA, Capristo M, Lollini PL, Nanni P, Bergamini C, Nicoletti G, Giovanni CD, Ghelli A, Giorgio V, Caratozzolo MF, et al. Respiratory complex I is essential to induce a Warburg profile in mitochondria-defective tumor cells. Cancer Metab. 2013; 1:11. <br><span id=»45″ class=»referencess blue-text»>45</span> Semenza GL. Опухолевый метаболизм: раковые клетки отдают и принимают лактат. J Clin Invest. 2008; 118:3835-3837. <br><span id=»46″ class=»referencess blue-text»>46</span> Sonveaux P, Vegran F, Schroeder T, Wergin MC, Verrax J, Rabbani ZN, De Saedeleer CJ, Kennedy KM, Diepart C, Jordan BF, Kelley MJ, Bernard Gallez B, Wahl ML, et al. Targeting lactate-fueled respiration selectively kills hypoxic tumor cells in mice. J Clin Invest. 2008; 118:3930-3942. <br><span id=»47″ class=»referencess blue-text»>47</span> Phoenix KN, Vumbaca F, Claffey KP. Терапевтическая активация метформина/AMPK способствует развитию ангиогенного фенотипа в модели ERalpha-негативного рака молочной железы MDA-MB-435. Breast Cancer Res Treat. 2009; 113:101-111. <br><span id=»48″ class=»referencess blue-text»>48</span> Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res. 2011; 71:2550-2560. <br><span id=»49″ class=»referencess blue-text»>49</span> Sonveaux P, Copetti T, De Saedeleer CJ, Vegran F, Verrax J, Kennedy KM, Moon EJ, Dhup S, Danhier P, Frérart F, Gallez B, Ribeiro A, Michiels C, et al. Таргетинг транспортера лактата MCT1 в эндотелиальных клетках подавляет индуцированную лактатом активацию HIF-1 и опухолевый ангиогенез. PLoS ONE. 2012; 7:e33418. <br><span id=»50″ class=»referencess blue-text»>50</span> Wu M, Seto E, Zhang J. E2F1 усиливает гликолиз через подавление транскрипции Sirt6 в раковых клетках. Oncotarget. 2015; 6:11252-11263. <br><span id=»51″ class=»referencess blue-text»>51</span> Hernlund E, Strandberg Ihrlund L, Khan O, Ates YO, Linder S, Panaretakis T, Shoshan MC. Потенцирование химиотерапевтических препаратов ингибиторами энергетического метаболизма 2-дезоксиглюкозой и этомоксиром. Int J Cancer. 2008; 123:476-483. <br><span id=»53″ class=»referencess blue-text»>53</span> Michelakis ED, Webster L, Mackey JR. Дихлорацетат (DCA) как потенциальная метаболическая мишень в терапии рака. Br J Cancer. 2008; 99:989-994. <br><span id=»54″ class=»referencess blue-text»>54</span> Walenta S, Mueller-Kliese WF. Лактат: зеркало и двигатель злокачественности опухоли. Semin Radiat Oncol. 2004; 14:267-274. <br><span id=»56″ class=»referencess blue-text»>56</span> Le A, Cooper CR, Gouw AM, Dinavahi R, Maitra A, Deck LM, Royer RE, Vander Jagt DL, Semenza GL, Dang CV. Ингибирование лактатдегидрогеназы А вызывает окислительный стресс и тормозит прогрессию опухоли. Proc Natl Acad Sci USA. 2010; 107:2037-2042. <br><span id=»57″ class=»referencess blue-text»>57</span> Zhao YH, Zhou M, Liu H, Ding Y, Khong HT, Yu D, Fodstad O, Tan M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene. 2009; 28:3689-3701. <br><span id=»58″ class=»referencess blue-text»>58</span> Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, et al. Инактивация сигнальной оси HIF-1α/PDK3 стимулирует меланому к митохондриальному окислительному метаболизму и потенцирует терапевтическую активность про-оксидантов. Cancer Res. 2012; 72:5035-5047. <br><span id=»59″ class=»referencess blue-text»>59</span> Parmenter TJ, Kleinschmidt M, Kinross KM, Bond ST, Li J, Kaadige MR, Rao A, Sheppard KE, Hugo W, Pupo GM, Pearson RB, McGee SL, Long GV, et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014; 4:423-433. <br><span id=»60″ class=»referencess blue-text»>60</span> Miskimins WK, Ahn HJ, Kim JY, Ryu S, Jung YS, Choi JY. Синергетический противораковый эффект фенформина и оксамата. PLoS One. 2014; 9:e85576. <br><span id=»61″ class=»referencess blue-text»>61</span> Jin S, DiPaola RS, Mathew R, White E. Metabolic catastrophe as a means to cancer cell death. J Cell Sci. 2007; 120:379-383. <br><span id=»62″ class=»referencess blue-text»>62</span> McLornan DP, List A, Mufti GJ. Применение синтетической летальности для избирательного воздействия на рак. N Engl J Med. 2014; 371:1725-1735. <br><span id=»63″ class=»referencess blue-text»>63</span> Kaelin WG Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005; 5:689-698. <br><span id=»64″ class=»referencess blue-text»>64</span> Iaquinta PJ, Lees JA. Решения о жизни и смерти, принимаемые транскрипционными факторами E2F. Curr Opin Cell Biol. 2007; 19:649-657. <br><span id=»65″ class=»referencess blue-text»>65</span> Houben R, Hesbacher S, Schmid CP, Kauczok CS, Flohr U, Haferkamp S, Müller CS, Schrama D, Wischhusen J, Becker JC. Высокий уровень экспрессии дикого типа р53 в клетках меланомы часто ассоциируется с неактивностью в анализах репортерных генов р53. PLoS One. 2011; 6:e22096. <br><span id=»66″ class=»referencess blue-text»>66</span> Avery-Kiejda KA, Bowden NA, Croft AJ, Scurr LL, Kairupan CF, Ashton KA, Talseth-Palmer BA, Rizos H, Zhang XD, Scott RJ, Hersey P. P53 в меланоме человека не регулирует целевые гены, связанные с апоптозом и клеточным циклом, и может способствовать пролиферации. BMC Cancer. 2011; 11:203. <br><span id=»67″ class=»referencess blue-text»>67</span> Lu M, Breyssens H, Salter V, Zhong S, Hu Y, Baer C, Ratnayaka I, Sullivan A, Brown NR, Endicott J, Knapp S, Kessler BM, Middleton MR, et al. Restoring p53 function in human melanoma cells by inhibiting MDM2 and cyclin B1/CDK1-phosphorylated nuclear iASPP. Cancer Cell. 2013; 23:618-633. <br><span id=»68″ class=»referencess blue-text»>68</span> Pandey V, Vijayakumar MV, Ajay AK, Malvi P, Bhat MK. Ожирение, вызванное диетой, увеличивает прогрессию меланомы: вовлечение Cav-1 и FASN. Int J Cancer. 2012; 130:497-508. <br><span id=»69″ class=»referencess blue-text»>69</span> Leontieva OV, Blagosklonny MV. M(o)TOR псевдогипоксического состояния при старении: рапамицин на помощь. Cell Cycle. 2014; 13:509-515. <br><span id=»70″ class=»referencess blue-text»>70</span> Leontieva OV, Blagosklonny MV. Дрожжеподобная хронологическая старость в клетках млекопитающих: феномен, механизм и фармакологическое подавление. Aging (Albany NY). 2011; 3:1078-1091. <br><span id=»71″ class=»referencess blue-text»>71</span> Vijayakumar MV, Singh S, Chhipa RR, Bhat MK. Гипогликемическая активность экстракта семян пажитника опосредована через стимуляцию инсулинового сигнального пути. Br J Pharmacol. 2005; 146:41-48. <br><span id=»72″ class=»referencess blue-text»>72</span> Malvi P, Chaube B, Pandey V, Vijayakumar MV, Boreddy PR, Mohammad N, Singh SV, Bhat MK. Быстрая прогрессия меланомы, вызванная ожирением, обращается вспять при лечении орлистатом и диетическом вмешательстве: роль адипокинов. Mol Oncol. 2015; 9:689-703. <br><span id=»73″ class=»referencess blue-text»>73</span> Spinazzi M, Casarin A, Pertegato V, Salviati L, Angelini C. Оценка ферментативной активности митохондриальной дыхательной цепи на тканях и культивируемых клетках. Nat Protoc. 2012; 7: 1235-1246.
Несоответствие результатов лечения метформином in vivo и in vitro побудило нас изучить причину этого. Известно, что метформин оказывает свое действие главным образом путем ингибирования митохондриального OXPHOS фермента комплекса I <a href=»#[«> [</a><a href=»https://www.oncotarget.com/article/6134/text/#bib27″>27</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib29″>29</a><a href=»#]»>]</a></sup> и ингибирование комплекса I приводит к увеличению роста и пролиферации опухоли<sup><a href=»#14″> [</a><a href=»https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href=»#]»>]</a></sup>. Поскольку мы наблюдали, что лечение метформином вызывало закисление культуральной среды вследствие усиления аэробного гликолиза (Дополнительный рисунок 4A и 4B), мы предположили, что это может быть причиной остановки роста клеток. Таким образом, мы измерили уровень глюкозы, а также лактата в отработанной культуральной среде, собранной из контрольных и обработанных метформином клеток. В клетках, обработанных метформином, было обнаружено повышенное использование глюкозы (о чем свидетельствует наличие меньшего количества остаточной глюкозы в среде) и более высокий уровень лактата по сравнению с контролем (Рисунок 3A и 3B).</p>
<h2>ДИСКУССИЯ</h2>
<p>Раковые клетки, включая клетки меланомы, направляют огромное количество углеродного потока на гликолиз <sup><a href=»#5″>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib5″>5</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href=»#]»>]</a></sup>. Это помогает клеткам вырабатывать АТФ и другие строительные блоки для быстрого роста опухоли <sup><a href=»#5″>[5]</a></sup>. По некоторым данным, воздействие на метаболические пути уменьшает прогрессию опухоли и рост клеток <em>in vitro</em> <sup><a href=»#40″>[40-</a><a href=»#42″>42]</a></sup>. В настоящем исследовании мы продемонстрировали, что метформин способствует росту меланомы, повышая гликолиз вследствие ингибирования функции комплекса I, в то время как ингибирование LDH вызывает остановку роста клеток. Ингибирование выработки лактата в клетках меланомы, обработанных метформином, влияет на выживаемость клеток, тем самым вызывая снижение прогрессии опухоли (Рисунок 8).</p>
<p>Последствия ингибирования комплекса I на рост раковых клеток четко не изучены, однако, согласно сообщениям, ингибирование комплекса I приводит к трансформации клеток и усиливает их рост<a href=»#[«> <sup>[</sup></a><sup><a href=»https://www.oncotarget.com/article/6134/text/#bib14″>14</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib18″>18</a><a href=»#]»>]</a></sup>. И наоборот, сообщалось, что ингибирование комплекса I замедляет развитие опухоли у мышей <sup><a href=»#30″>[30]</a></sup>. Эти различные результаты, вероятно, зависят от доступности питательных веществ в микросреде опухоли. Сообщалось, что когда глюкоза доступна в изобилии, ингибирование комплекса I способствует гликолизу и быстрой пролиферации клеток, в то время как в условиях метаболического стресса оно вызывает гибель клеток<sup><a href=»#43″>[43]</a></sup>. В данном исследовании мы наблюдали, что ингибирование комплекса I метформином способствует прогрессированию опухоли меланомы у мышей.</p>
<p>Хотя было показано, что метформин подавляет пролиферацию многих раковых клеток, а также ограничивает опухолевую прогрессию ксенотрансплантата у мышей <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib33″>33</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib34″>34</a><a href=»#]»>]</a></sup>, известно, что он оказывает как ингибирующее, так и стимулирующее рост действие в меланоме<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib35″>35</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib37″>37</a><a href=»#]»>]</a></sup>. Интересно, что мы обнаружили, что опухоли, полученные из B16F10 и A375, быстро росли у мышей, получавших метформин, и скорость роста варьировала в зависимости от гликемического статуса мышей (Рисунок 2D и 2E). Это наблюдение согласуется с недавним сообщением о том, что метформин способствует опухолевой прогрессии BRAF-мутантных клеток меланомы<sup><a href=»http://335″> [35]</a></sup>.</p>
<p>Различия в действии метформина на рост клеток меланомы <em>in vitro</em> и <em>in vivo</em>, вероятно, зависят от наличия глюкозы и концентрации лактата, которые могут влиять на его действие. Метформин подавляет рост клеток и индуцирует апоптоз в условиях ограничения глюкозы <sup><a href=»#43″>[43]</a></sup>, в то время как мы заметили, что в присутствии высокой глюкозы он вызывает остановку роста, не влияя на гибель клеток. Мы объяснили это несоответствие тем, что раковые клетки получают АТФ <em>через</em> OXPHOS при метаболическом стрессе, тогда как при изобилии глюкозы АТФ получается в основном через гликолиз. Ингибирование комплекса I в условиях высокого содержания глюкозы еще больше способствует гликолизу, приводящему к избыточному образованию лактата, о чем сообщалось ранее<sup><a href=»#44″> [44]</a></sup>. В совокупности эти данные позволяют предположить, что остановка роста, индуцированная метформином в клетках меланомы <em>in vitro</em>, является следствием закисления среды из-за избыточного накопления молочной кислоты вследствие быстрого использования доступной глюкозы. Примечательно, что остановка роста, вызванная метформином, может быть предотвращена путем частого пополнения среды для поддержания оптимальных условий роста, поскольку клетки, обработанные метформином, требуют больше глюкозы для пролиферации из-за усиленного аэробного гликолиза. Это имитирует условия <em>in vivo</em>, в которых, вероятно, быстрая, постоянная циркуляция молочной кислоты позволяет раковым клеткам использовать больше глюкозы, что способствует быстрой пролиферации. Более того, лактат также может быть использован раковыми клетками в опухоли <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib45″>45</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib46″>46</a></sup><a href=»#]»><sup>]</sup></a>. Кроме того, сообщалось, что метформин индуцирует ангиогенез <em>через</em> повышение уровня VEGF<sup><a href=»#[«> [</a><a href=»https://www.oncotarget.com/article/6134/text/#bib35″>35</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib47″>47</a><a href=»#]»>]</a></sup>. Мы сообщаем, что ингибирование комплекса I увеличивает производство лактата, а ранее было высказано предположение, что гликолитический переключатель усиливает ангиогенез<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib48″>48</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib49″>49</a><a href=»#]»>]</a></sup>. Наши результаты показывают положительную корреляцию между лактатом сыворотки и уровнем VEGF у мышей, получавших метформин. Поэтому вполне вероятно, что усиленный метформином рост опухоли способствует ангиогенезу, индуцированному лактатом, который может быть опосредован VEGF <sup><a href=»#48″>[48]</a></sup>. Кроме того, мы наблюдали повышенный уровень E2F1, важной молекулы регуляции клеточного цикла, в опухолях, вырезанных у мышей, получавших метформин. Помимо содействия клеточной пролиферации, E2F1 также регулирует многие гены, участвующие в гликолизе<sup><a href=»#50″> [50]</a></sup>, который необходим для быстрого роста раковых клеток. Поэтому вполне вероятно, что E2F1 может играть важную роль в опосредовании вызванной метформином быстрой опухолевой прогрессии, вероятно <em>видя</em> регуляцию аэробного гликолиза.</p>
<p>Ингибиторы гликолиза, такие как 2-дезоксиглюкоза, лонимид, 3-бромпируват, DCA, и препараты, вмешивающиеся в метаболические пути, показали многообещающий результат в подавлении роста опухоли<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib40″>40</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib42″>42</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib51″>51</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href=»#]»>]</a></sup>. Кроме того, воздействие на путь образования лактата является привлекательным, особенно в гликолитических раковых опухолях <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib54″>54</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib55″>55</a><a href=»#]»>]</a></sup>. Клетки меланомы являются метаболически гликолитическими, поэтому в первую очередь полагаются на высокую активность LDH для выработки АТФ <sup><a href=»#6″>[6]</a></sup>. Клетки меланомы сверхэкспрессируют LDH, а высокий сывороточный уровень часто коррелирует с плохим прогнозом и выживаемостью пациентов<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib8″>8</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=»#]»>]</a></sup>. Наши результаты показывают, что нарушение превращения пирувата в лактат значительно влияет на пролиферацию клеток меланомы. Это согласуется с данными о том, что опухоли из гликолитических типов клеток более восприимчивы к ингибированию LDH-A с помощью FX11<sup><a href=»#56″> [56]</a></sup>.</p>
<p>ЛДГ играет важную роль в метаболическом гомеостазе и поддержании опухолей<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib12″>12</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib57″>57</a><a href=»#]»>]</a></sup>. Мы использовали оксамат и DCA для подавления выработки лактата в клетках меланомы. DCA является перорально доставляемой малой молекулой, которая используется для лечения молочнокислого ацидоза, а восстановление функциональности OXPHOS с помощью DCA, как было показано, вызывает гибель клеток<sup><a href=»#[«> [</a><a href=»https://www.oncotarget.com/article/6134/text/#bib52″>52</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib53″>53</a><a href=»#]»>]</a></sup>. Более того, ранее было показано, что DCA снижает выработку лактата и запускает апоптоз в клетках меланомы <sup><a href=»#[«>[</a><a><a href=»https://www.oncotarget.com/article/6134/text/#bib58″>58</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib59″>59</a><a href=»#]»>]</a></sup>. Оксамат, аналог пирувата, также используется для ингибирования LDH. Недавнее исследование Miskimins et al. предполагает, что совместное воздействие на комплекс I и LDH может быть многообещающей стратегией для остановки роста агрессивных раковых опухолей <sup><a href=»#60″>[60]</a></sup>. В соответствии с этим, мы также заметили, что предотвращение выработки лактата и одновременное ингибирование комплекса I оксаматом/DCA или LDH-специфической siRNA и метформином, соответственно, приводит к истощению клеточного пула АТФ. Снижение клеточного пула АТФ вызывает метаболическую катастрофу, которая приводит к апоптозу в клетках меланомы. Вызванная метаболической катастрофой гибель клеток рассматривается как перспективная стратегия для остановки прогрессии рака<sup><a href=»#61″> [61]</a></sup>. Более того, наше исследование свидетельствует о различной роли комплекса I и LDH в клеточной пролиферации, причем эти два фермента генерируют АТФ либо через OXPHOS, либо через фосфорилирование на уровне субстрата соответственно. Блокирование одного из ферментов в отдельности не вызывает гибели клеток, так как клетки могут переключиться на альтернативный путь генерации АТФ. В то время как одновременное ингибирование обоих ферментов вызывает апоптоз. Это ясно указывает на то, что клетки зависят от функции этих двух ферментов, образующих синтетически летальную пару, что является перспективным явлением, которое может быть использовано для избирательного воздействия на клетки меланомы<a href=»#[«> </a><sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib62″>62</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib63″>63</a><a href=»#]»>]</a></sup>. Учитывая, что E2F1 участвует в регуляции апоптоза <sup><a href=»#64″>[64]</a></sup>, вполне вероятно, что апоптоз, индуцированный комбинацией метформина и оксамата/DCA, может быть связан с E2F1. Метформин способствует апоптозу в раковых клетках <em>через</em> активацию p53 <sup><a href=»#37″>[37]</a></sup>. Однако в случае меланомы роль р53 не ясна. Согласно литературным данным, р53 в меланоме нефункционален, а его уровень парадоксально повышен в поздних стадиях меланомы <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib65″>65</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib67″>67</a><a href=»#]»>]</a></sup>. Ранее мы сообщали, что повышенный уровень р53 связан с увеличением роста опухоли<sup><a href=»#68″> [68]</a></sup>. Поэтому маловероятно, что p53 участвует в апоптозе, индуцированном комбинированным лечением, и, таким образом, необходимы дополнительные исследования для оценки функциональности E2F1 и p53 в меланоме.</p>
<p>При использовании комбинаций различных препаратов, которые могут синергетически способствовать гибели раковых клеток, это может также усилить токсичность для нормальных клеток. Поэтому очень важно получить доступ к воздействию комбинированного лечения на нормальные клетки. Важно отметить, что, по нашим данным, эта комбинация эффективно убивает раковые клетки как <em>in vitro</em>, так и <em>in vivo</em>, и оказывает наименьшее влияние на выживание нормальных клеток. Различная чувствительность меланомы и нормальных клеток к комбинации метформина и оксамата/ДКА может быть объяснена тем, что клетки меланомы являются высоко гликолитическими и сверхэкспрессируют молекулы, участвующие в выработке и секреции лактата, по сравнению с нормальными клетками <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib3″>3</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib6″>6</a>-<a href=»https://www.oncotarget.com/article/6134/text/#bib10″>10</a><a href=»#]»>]</a></sup>. Интересно, что по сравнению с нормальными клетками, раковые клетки демонстрируют более высокий уровень несвязанного митохондриального дыхания<sup><a href=»#31″> [31]</a></sup>. Раковые клетки при лечении метформином демонстрируют большее компенсаторное повышение гликолиза, чем нормальные клетки <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib31″>31</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib32″>32</a><a href=»#]»>]</a></sup>, что утяжеляет метаболическую восприимчивость раковых клеток. Поэтому ингибирование выработки/секреции LDH или лактата подавляет рост раковых клеток, в то время как нормальные клетки подвергаются наименьшему воздействию, поскольку достаточное количество АТФ все еще может вырабатываться из OXPHOS, так как метформин является слабым ингибитором комплекса I. Поскольку активность OXPHOS в нормальных клетках выше по сравнению с клетками меланомы, комбинация метформина и оксамата/DCA, вероятно, будет иметь наименьшее негативное воздействие на дыхание нормальных клеток.</p>
<p>Наше исследование открывает новые возможности для воздействия на метаболизм раковых клеток и может быть использовано для тестирования других клинически одобренных препаратов, ингибирующих гликолиз, наряду с метформином. Настоящее исследование предполагает, что любой препарат/ингибитор, блокирующий выработку лактата, может быть использован в комбинации с метформином для улучшения лечения и предотвращения роста опухоли. Например, рапамицин, клинически одобренный препарат, предотвращает образование лактата<sup> <a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib69″>69</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib70″>70</a><a href=»#]»>]</a></sup>. Аналогично, было показано, что ингибирование BRAF приводит к подавлению гликолиза <sup><a href=»#[«>[</a><a href=»https://www.oncotarget.com/article/6134/text/#bib6″>6</a>, <a href=»https://www.oncotarget.com/article/6134/text/#bib7″>7</a><a href=»#]»>]</a></sup>. Таким образом, рапамицин, а также ингибиторы BRAF могут использоваться вместе с метформином для улучшения терапевтического результата при уменьшении побочных эффектов. В целом, наши результаты показывают, что LDH или другой механизм, контролирующий выработку или секрецию лактата, имеет решающее значение для быстрой прогрессии меланомы в условиях нарушенного OXPHOS, и это может быть использовано в качестве подходящей терапевтической мишени для контроля роста гликолитических раковых клеток. Необходимы более обширные исследования для оценки функциональности LDH и комплекса I в других моделях рака, а затем их применения в терапии рака в целом.</p>
<figure class=»wp-block-image size-large»><img src=»https://www.dcaguide.org/wp-content/uploads/2021/09/Fig-8-1-1024×730.jpg» alt=»» class=»wp-image-4179″/><figcaption><strong>Рисунок 8: </strong> Схематическое изображение синтетической летальности, индуцированной комбинацией метформина и оксамата при лечении меланомы.<strong>А.</strong> Ингибирование дыхательного комплекса I метформином или фенформином приводит к усилению гликолиза вследствие активации лактатдегидрогеназы (ЛДГ). Генерация строительных блоков и АТФ исключительно за счет гликолиза способствует быстрому росту опухоли. <strong>B.</strong> Таргетинг LDH или выработки лактата с помощью оксамата и DCA или siRNA приводит к активации OXPHOS и последующей генерации АТФ. Однако это приводит к подавлению пролиферации клеток, не вызывая их гибели. <strong>C.</strong> Блокирование обоих ферментов вместе вызывает метаболическую катастрофу из-за истощения клеточного пула АТФ. Это приводит к запуску апоптоза и подавлению роста опухоли. Комплекс I и LDHA образуют синтетически летальную пару в клетках меланомы.<br></figcaption></figure>
<h2>МАТЕРИАЛЫ И МЕТОДЫ</h2>
<p><strong>Эксперименты на животных<br></strong>Мыши были получены из экспериментального животноводческого комплекса (EAF) Национального центра клеточной науки (NCCS), Пуна, Индия. Гипергликемию у мышей вызывали с помощью STZ, как описано ранее<sup><a href=»#71″> [71]</a></sup> с небольшими изменениями. Вкратце, самцы мышей C57BL/6J и NOD/SCID голодали в течение 6 часов перед внутрибрюшинным введением STZ (50 мг/кг) в 0,01 М цитратном буфере (pH 4,4) в течение трех последовательных дней. Измерение уровня глюкозы в крови проводилось путем перерезания хвоста и подачи крови на анализатор глюкозы (Accu-Chek Active, Roche, Германия). Мыши, у которых уровень глюкозы в крови превышал 200 мг/дл, считались гипергликемическими. Опухоль индуцировали путем введения клеток B16F10 (2 × 10<sup>5</sup>) или A375 (1 × 10<sup>6</sup>) в 100 мкл стерильного PBS подкожно в правый фланг мышей C57BL/6J и NOD/SCID соответственно. За развитием опухолей следили регулярно, измеряя их размер цифровым штангенциркулем (Sigma, США) после появления пальпируемых опухолей. Пероральное введение метформина в разные дни было начато за неделю до опухолевого теста у мышей с гипергликемией. В остальных случаях метформин давали только после того, как опухоль достигала оптимального размера, и лечение продолжали до завершения эксперимента. В конце эксперимента мышей приносили в жертву, опухоли вырезали и хранили либо в -80<sup>°</sup>C, либо в 10% растворе формалина для дальнейших исследований. В другом эксперименте мышей (C57BL/6J, самцы) случайным образом разделили на 4 группы после появления пальпируемой опухоли. Для изучения результатов лечения метформином в сочетании с ингибитором LDH оксаматом, мышам перорально давали либо метформин (200 мг/кг), либо только оксамат (500 мг/кг), либо оба препарата вместе каждый день. Прогрессирование опухоли регулярно отслеживалось с помощью штангенциркуля. В конце эксперимента ткани опухоли, мышц и печени вырезали, взвешивали и хранили при -80<sup>°</sup>C для дальнейшего анализа. Все эксперименты на животных проводились в соответствии с рекомендациями комитета по контролю и надзору за экспериментами на животных (CPCSEA), правительство Индии, и после получения разрешения Комитета по этике животных института (IAEC).</p>
<p><strong>Клеточные линии и условия культивирования<br></strong>Клетки мышиной меланомы B16F10, меланомы человека A375 и SKMel28 были приобретены у ATCC (VA, США) и поддерживались в нашем внутреннем хранилище. Нераковые клетки, AML12 (гепатоциты мыши), L6 (мышечные клетки крысы) и MEF (эмбриональные фибробласты мыши) использовались параллельно с раковыми клетками в качестве контроля. Все клетки выращивали в соответствующей среде, содержащей 1 мМ или 25 мМ глюкозы в зависимости от экспериментов и дополненной 10% термоинактивированной фетальной бычьей сывороткой (Hyclone, UT, США), пенициллином (100 Ед/мл) и стрептомицином 100 мкг/мл (Life Technologies, NY, США), при 37<sup>°</sup>C в присутствии 5%<sub> </sub>CO<sub>2</sub>.</p>
<p><strong>Химические вещества и реактивы<br></strong>Метформин, фенформин, олигомицин, ротенон, децилубихинон, стрептозотоцин (STZ), [3-(4, 5-диметилтиазол-2-ил)-2, 5-дифенилтеразолия бромид] (МТТ), NAD+, NADH, АТФ, AMP, D-глюкоза, оксамат натрия, йодоацетат, 2-дезокси-D-глюкоза, дихлорацетат (DCA), диаминобензидин (DAB) и флуретин были приобретены у компании Sigma (MO, США). Антитела к ChREBP (1:1000), GLUT1 (1:1000), LDHA (1:1000), Cyclin D1 (1:1000), PCNA (1:1000), CDK4 (1:1000), p21 (1:1000), E2F1 (1:1000) CD31 (1:100), PARP-1 (1:1000), Bcl-2 (1:1000), Bax (1:1000), β-тубулин (1:1000), GAPDH (1:1000), HSP60 (1:1000) и VEGF (1:50) были получены от Santa Cruz Biotechnology (CA, США).</p>
<p><strong>Подготовка клеточного лизата и иммуноблоттинг<br></strong>Клетки трижды промывали фосфатным забуференным физраствором (PBS) и лизировали в буфере для лизиса, содержащем 50 мМ Tris-Cl (pH 7.5), 120 мМ NaCl, 10 мМ фторида натрия, 10 мМ пирофосфата натрия, 2 мМ ЭДТА, 1 мМ ортованадата натрия, 1 мМ фенилметилсульфонилфторида, 1% NP-40 и коктейль ингибиторов протеаз (Roche, Германия). Лизаты опухоли готовили путем измельчения опухолевых тканей на мелкие кусочки, промывали пять раз 0,85% физиологическим раствором, содержащим коктейль ингибиторов протеаз, и лизировали в буфере для лизиса путем гомогенизации с помощью гомогенизатора тканей (Sigma, США) с последующей сонификацией. Лизаты осветляли центрифугированием при 15 000 об/мин <em> </em>в течение 30 минут. Лизаты клеток готовили в охлажденных условиях. Приблизительно 50-100 мкг белка из лизатов целых клеток растворяли на 8-12% SDS-полиакриламидном геле, который затем переносили на PVDF мембрану (Millipore, Германия). На мембрану наносили нужные первичные антитела, а затем вторичные антитела, конъюгированные с HRP. Иммуноблоты детектировали люминальным реагентом (Santa Cruz Biotechnology). В случае необходимости блоты снимали, инкубируя мембрану при 50<sup>°</sup>C в течение 15 минут в буфере для снятия (62,5 мМ Tris-Cl, pH 6,8, 100 мМ меркаптоэтанол и 2% SDS) при периодическом встряхивании. Мембраны тщательно промывали Tris buffered saline (TBS) и окрашивали желаемыми антителами.</p>
<p><strong>Иммунофлуоресценция или конфокальная микроскопия<br></strong>Клетки помещали в камерные предметные стекла Labtek (Nunc, США) и давали им расти в течение 24 ч. Лечение метформином и другими ингибиторами проводили в течение желаемых периодов времени и концентрациях. Клетки промывали охлажденным стерильным PBS и фиксировали 3,7% раствором параформальдегида при комнатной температуре в течение 10 мин. Затем их пермеабилизировали 0,025% Triton X-100 в PBS в течение 10 мин и затем блокировали 5% BSA в PBS в течение 1 ч при 37<sup>°</sup>C. Клетки инкубировали с 1:100 разведениями первичных антител в блокирующем растворе в течение 2 ч при комнатной температуре и промывали TBST (TBS, содержащий 0,05% Tween-20) не менее пяти раз перед инкубацией с соответствующими мечеными вторичными антителами (1:200) в блокирующем растворе в течение 1 ч при комнатной температуре. После пяти промывок в TBST образцы покрывали монтажной средой, содержащей DAPI (Santa Cruz Biotechnology, США). Слайды запечатывали, рассматривали под конфокальным лазерным сканирующим микроскопом (LSM510 Carl Zeiss, Германия) и получали изображения. Изображения впоследствии обрабатывались с помощью программного обеспечения для анализа изображений LSM.</p>
<p><strong>Иммуногистохимия и гистопатология<br></strong>Иммуногистохимию и гистопатологию проводили согласно Malvi et al. <sup><a href=»#72″>[72]</a></sup>. Вкратце, тонкие срезы опухоли и других органов изготавливали с помощью микротома, фиксировали на предметных стеклах и парафинизировали. Для иммуногистохимии предметные стекла дважды депарафинизировали в растворе ксилола в течение 10 мин, а затем трижды промывали 100%, 95%, 70% и 50% этанолом. Слайды снова промывали дистиллированной водой, затем промывали PBS в течение 5 мин. Для извлечения антигена предметные стекла кипятили в цитратном буфере натрия (0,01 М, pH 4,5) в микроволновой печи в течение 10 мин и давали остыть при комнатной температуре в течение 20 мин. БСА или нормальную козью сыворотку (2%) использовали для блокирования в увлажненной камере или в холодном боксе в течение 1 ч. Слайды зондировали желаемыми антителами (разведение 1:100) в PBST (PBS, содержащий 0,025% Tween-20), содержащем 0,01% БСА, в течение 2 ч при комнатной температуре или на ночь при 4<sup>°</sup>C. Слайды промывали PBST 4 раза в течение 5 мин, затем зондировали совместимыми HRP-конъюгированными вторичными антителами в течение 1 ч. Их снова промывали и окрашивали диаминобензидином (DAB) в течение 10 мин, затем промывали и окрашивали гематоксилином. Слайды промывали водой, обезвоживали абсолютным спиртом, затем покрывали монтажной средой и анализировали на экспрессию нужных молекул. Для гистопатологии депарафинизированные слайды окрашивали гематоксилином и эозином, а микроскопический анализ на плотность клеток, клеточную морфологию, метастазирование, цитотоксичность и некроз проводили патологоанатомы в слепой манере в госпитале KEM в Пуне, Индия.</p>
<p><strong>Анализ клеточной цитотоксичности и выживаемости</strong><br> Приблизительно 5 × 10<sup>3</sup> (B16F10) и 10 × 10<sup>3</sup> (A375 и SKMel28) клеток высевали в каждую лунку 96-луночного планшета для культуры ткани и давали им прилипнуть в течение 24 ч при 37<sup>°</sup>C. Клетки обрабатывали в соответствии с экспериментальными требованиями, а пролиферацию или жизнеспособность оценивали с помощью МТТ-теста. МТТ (50 мкл, 1 мг/мл в DMEM без фенолового красного) добавляли в каждую лунку и инкубировали в течение 4 ч при 37<sup>°</sup>C. Кристаллы формазана солюбилизировали<sup> </sup>в 100 мкл изопропанола путем инкубации при встряхивании при комнатной температуре в течение 5 мин. Абсорбцию измеряли при 570 нм, используя 630 нм в качестве контрольного фильтра. Необработанные клетки считались контролем (100% выживаемость клеток).</p>
<p><strong>Определение апоптоза путем окрашивания аннексином V<br></strong>Клетки высевали при плотности приблизительно 3 × 10<sup>5</sup> клеток в 35-миллиметровые планшеты и давали расти в течение 24 ч. Клетки обрабатывали оксаматом и DCA либо отдельно, либо вместе с метформином в течение 48 ч или в соответствии с экспериментальными требованиями. Клетки собирали трипсинизацией и обрабатывали для проточной цитометрии. Апоптоз определяли путем двойного окрашивания аннексином V и PI с помощью набора для анализа апоптоза (BD Bioscience, США) в соответствии с инструкциями производителя.</p>
<p><strong>Анализ клеточного цикла<br></strong>Клетки высевали при плотности приблизительно 3 × 10<sup>5</sup> клеток в 35 мм планшеты и давали расти в течение 24 ч. Клетки обрабатывали метформином, оксаматом и другими ингибиторами гликолиза по отдельности или вместе, как указано, в течение 48 ч или в соответствии с экспериментальными требованиями. Клетки собирали трипсинизацией и обрабатывали для проточной цитометрии. Вкратце, клетки промывали охлажденным PBS и фиксировали в 70% этаноле на льду в течение 30 мин. После обработки РНКазой (200 мкг/мл) в течение 30 мин при 37°C, 50 мкг/мл PI добавляли к гранулам клеток и инкубировали в темноте в течение 30 мин на льду. Флуоресценцию PI регистрировали через фильтр 585 нм в проточном цитометре (FACS Calibur, Becton Dickinson, Калифорния, США). Данные анализировали с помощью программного обеспечения Cell Quest Pro для 10 000 клеток.</p>
<p><strong>Долгосрочный анализ выживаемости клеток<br></strong>Клетки (5 × 10<sup>2</sup>) помещали в 12-луночные планшеты на 24 ч. Клетки обрабатывали без или с 25 мМ или 50 мМ оксаматом и 10 или 20 мМ DCA вместе с метформином и продолжали в течение 48 ч. Затем среду заменяли свежей средой без лекарств. Планшеты инкубировали еще 10-15 дней при 37°C в CO<sub>2</sub> инкубаторе со сменой среды каждые 2-3 дня. Затем клетки фиксировали (3% параформальдегид и 0,02% глутаральдегид в PBS) и окрашивали 0,05% кристаллическим фиолетовым.</p>
<p><strong>Анализ утилизации глюкозы<br></strong>Клетки (3 × 10<sup>5</sup>) культивировали в среде DMEM, содержащей 25 мМ глюкозы. Через 24 ч среду заменяли соответствующей средой, содержащей возрастающую концентрацию метформина (0,1 мМ, 0,5 мМ, 1 мМ и 2 мМ) или ротенона в течение 24 ч, и остаточную глюкозу, присутствующую в отработанной среде, контролировали с помощью набора для определения глюкозы на основе GOD-POD (Spinreact, Испания) в соответствии с инструкциями производителя. Для измерения утилизации глюкозы в присутствии гликолитических ингибиторов клетки обрабатывали 50 мМ оксаматом, 100 мкМ флуретином и 20 мМ DCA, либо отдельно, либо вместе с 2 мМ метформином или 100 мкМ фенформином. Потребленную глюкозу оценивали путем вычитания оставшейся глюкозы в среде из исходной концентрации в контрольной среде (450 мг/дл). Эксперименты проводились как минимум в трех экземплярах, значения нормировались на общее количество клеток.</p>
<p><strong>Анализ оценки лактата<br></strong>Концентрацию лактата в отработанной среде, собранной из клеток, обработанных метформином, оксаматом и другими гликолитическими ингибиторами или без них, определяли с помощью коммерчески доступного набора для оценки лактата (Spinreact, Испания) в соответствии с инструкциями производителя. Вкратце, культуральную среду или сыворотку разводили в 0,9% физиологическом растворе в 10 раз и добавляли 10 мкл образца в каждую лунку 96-луночного ИФА-планшета. 150 мкл реагента из комплекта добавляли в каждую лунку, содержащую образец, оставляя неизрасходованную среду и реагент в качестве пустого места, и регистрировали абсорбцию при 405 нм с помощью спектрофотометра (Thermo-Scientific, США). Эксперименты проводились как минимум в трех экземплярах, а итоговые значения нормировались на общее количество клеток.</p>
<p><strong>трансфекция siRNA<br></strong>Специфическая siRNA против LDHA была приобретена у Santa Cruz Biotechnology (США). Трансфекцию проводили с помощью Lipofectamine 2000 (Life Technologies, США) в соответствии с инструкциями производителя. Вкратце, клетки высевали при приблизительно 60%-ной конфлюентности. На следующий день среду заменяли на OptiMEM (Life Technologies, США) и выдерживали в течение 3 ч. Необходимые siRNA растворяли в буферах, поставляемых вместе с ними. Lipofectamine2000 и siRNA разводили отдельно в OptiMEM и инкубировали в течение 5 мин. После этого разбавленные реагенты смешивали и инкубировали в течение 30 мин при комнатной температуре. Полученный осадок оставляли на клетках на 6 ч, после чего добавляли свежий DMEM, дополненный 10% FBS, и инкубировали еще 24-36 ч. Эффективность трансфекции оценивали путем одновременной трансфекции плазмиды pEGFPN1. Иммуноблоттинг проводили для проверки ингибирования экспрессии соответствующих генов.</p>
<p><strong>Приготовление богатой митохондриями фракции из клеток и тканей<br></strong>Богатую митохондриями фракцию готовили из культивируемых клеток и опухолей или нормальных тканей, как сообщалось ранее<sup><a href=»#73″> [73]</a></sup>. Вкратце, клетки (1 × 10<sup>6</sup>) высевали в 10-см планшеты и трипсинизировали после обработки нужными ингибиторами в течение 24 ч. Суспензию клеток подвергали трем циклам замораживания-оттаивания в гипотоническом буфере (20 мМ фосфат калия). Эту суспензию центрифугировали при 50 000 об/мин в течение 1 ч для получения супернатанта, богатого митохондриями. Аналогично, для выделения митохондрий из тканей замороженные ткани нарезали на мелкие кусочки и гомогенизировали в буфере для гомогенизации (0,5 М Трис буфер pH 7,5, 100 мМ EGTA и 250 мМ сахарозы), затем проводили циклическое замораживание-оттаивание и центрифугировали при 50 000 об/мин в течение 1 ч. Супернатант собирали как митохондриальную фракцию и использовали для определения митохондриальной функции и активности фермента OXPHOS.</p>
<p><strong>Энзимные анализы<br></strong>Клетки гомогенизировали в гипотоническом (20 мМ) калий-фосфатном буфере (pH 7,5), содержащем коктейль ингибиторов протеаз (Roche, Германия), вихревым методом и лизировали путем трех циклов замораживания и оттаивания. Для приготовления экстракта из тканей образцы опухолей измельчали на мелкие кусочки и гомогенизировали в буфере для гомогенизации (0,1 М Трис, 0,1 М KCl, 350 мМ ЭДТА и 1 М сахарозы, pH 7,5) с помощью тканевого гомогенизатора. Гомогенаты осветляли центрифугированием при 10 000 об/мин в течение 10 мин при 4<sup>°</sup>C и хранили на льду до проведения анализов.</p>
<p><strong><em>Оценка активности ЛДГ:</em> </strong> Активность лактатдегидрогеназы в лизатах клеток или экстракте опухоли и сыворотке определяли с помощью набора для определения активности ЛДГ (Spinreact, Испания) в соответствии с инструкциями производителя.</p>
<p><em><strong>Анализ активности комплекса I:</strong></em> Анализ фермента комплекса I митохондриальной OXPHOS проводили, как описано в другом месте<sup><a href=»#73″> [73]</a></sup>. Вкратце, богатую митохондриями фракцию гомогената клеток или ткани (20-50 мкг белка из гомогената ткани или 10-20 мкг богатой митохондриями фракции) добавляли к 700 мкл дистиллированной воды, взятой в кювету объемом 1 мл, затем добавляли 100 мкл калий-фосфатного буфера (0,5 М, pH 7,5), 60 мкл BSA без жирных кислот (50 мг/мл), 30 мкл KCN (10 мМ) и 10 мкл NADH (10 мМ). Конечный объем доводили до 994 мкл с помощью дистиллированной воды, перемешивали, переворачивая кюветы, и получали показания базовой линии при 340 нм в течение 2 мин. Реакцию начинали добавлением 6 мкл децилубихинона (10 мМ, Sigma, США), перемешивали, переворачивая кюветы. Снижение поглощения при 340 нм отслеживали в течение 10 мин. Аналогичная процедура была проведена для расчета активности комплекса I в присутствии 2 мМ метформина и 100 мкМ фенформина. Ротенон (10 мкМ) использовали в качестве положительного контроля. Конечные значения были нормализованы с общим содержанием клеточного белка.</p>
<p><strong>Измерение общего клеточного АТФ<br></strong> Уровень АТФ измеряли с помощью коммерчески доступного набора для биолюминесценции АТФ (Roche, Германия) в соответствии с инструкциями производителя. Вкратце, клетки выращивали в присутствии указанных препаратов. Клетки собирали и лизировали в 100 мМ Трис-ЭДТА буфере, содержащем 0,01% NP-40, кипятили в течение 1 мин с последующим циклом замораживания-оттаивания. Люминесценцию регистрировали как для пустого образца, так и для образцов. Для измерения АТФ, образующегося исключительно в результате OXPHOS и гликолиза, клетки обрабатывали олигомицином (10 мкМ) и оксаматом или DCA соответственно с метформином или без него. Эксперименты проводились в трех экземплярах и повторялись не менее одного раза, а конечные значения нормировались на общее содержание белка.</p>
<p><strong>Статистика</strong><br>Статистический анализ проводили с помощью программы Sigma Plot 12.0 (Systat Software Inc., CA, USA). Большинство экспериментов повторяли как минимум один раз в трех экземплярах, если не указано иное. Данные были представлены как среднее ± SD, за исключением указанных экспериментов. Там, где это было необходимо, в экспериментах использовался парный или непарный двухвостный тест Стьюдента <em>t</em>, предполагающий неравную дисперсию, если не указано иное, для расчета p-значения. Значения *<em>p</em> < 0,05, **<em>p</em> < 0,01, ***<em>p</em> < 0,001 означают значительные различия между группами (<em>n</em> > 3 как минимум).</p>
<h2>Примечания</h2>
<p>Авторы благодарят д-ра С.К. Манде, директора NCCS, Пуна, Индия, и д-ра Г.К. Мишру, бывшего директора NCCS, Пуна, Индия, за то, что они очень поддерживали и давали все стимулы для выполнения этой работы. Мы благодарим д-ра Бенуа Виолет (INSERM, Франция) за предоставленные МЭФы, д-ра Махеша Дж Кулкарни (Национальная химическая лаборатория, Индия) за клетки L6 и д-ра Ракеша К Тяги (Университет Джавахарлала Неру, Индия) за предоставленные клетки AML12. Мы также благодарим доктора Vijayakumar MV за помощь в экспериментах на животных и за критическое прочтение рукописи. BC и NM благодарят Совет научных и промышленных исследований (CSIR) Индии; PM и SVS благодарят Университетскую грантовую комиссию (UGC), Индия, за предоставление стипендии. Мы выражаем благодарность за поддержку другим сотрудникам лаборатории и коллегам из NCCS, а также сотрудникам экспериментальной лаборатории для животных, FACS, конфокальной и масс-спектрометрической лаборатории.</p>
<h2>ФИНАНСОВАЯ ПОДДЕРЖКА</h2>
<p>Эта работа была поддержана внутривузовским грантом NCCS, финансируемым Департаментом биотехнологии правительства Индии.</p>
<h2>КОНФЛИКТЫ ИНТЕРЕСОВ</h2>
<p>Авторы заявляют, что у них нет потенциальных конфликтов интересов</p>
<h3>ПРИМЕЧАНИЕ</h3>
<p>Данная работа выполнена в рамках частичного выполнения диссертации на соискание ученой степени доктора философии (B.C.), которая будет представлена в Университет Савитрибай Фуле Пуна, Пуна, Индия.</p>
<p></p>
<h2>REFERENCES</h2>
<br><span id=»1″ class=»referencess blue-text»>1</span> Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007; 445:851-857.
<br><span id=»52 «class=»referencess blue-text»>52</span>. Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I. Дихлорацетат индуцирует апоптоз в клетках рака эндометрия. Gynecol Oncol. 2008; 109:402.
<br><span id=»55″ class=»referencess blue-text»>55</span> Doherty JR, Cleveland JL. Таргетинг метаболизма лактата для терапии рака. J Clin Invest. 2013; 123:3685-3692.
<p></p>