Cecilie Abildgaard1 , Christina Dahl1 , Astrid L Basse2 , Tao Ma2 und Per Guldberg1*
1 Forschungszentrum der Dänischen Krebsgesellschaft, Kopenhagen, Dänemark2
Abteilung für Biologie, Universität Kopenhagen, Kopenhagen, Dänemark.
Korrespondenz:
[email protected]: 14 September 2020Akzeptiert
: 4. Dezember 2020Veröffentlicht
: 9. Dezember 2020
Zusammenfassung
Hintergrund: Die Fortschritte bei der Behandlung des Melanoms durch gezielte Hemmung des onkogenen BRAF sind aufgrund der Entwicklung einer erworbenen Resistenz begrenzt. Die Beteiligung von BRAFV600E an der metabolischen Umprogrammierung von Melanomzellen liefert eine Begründung für eine gezielte Beeinflussung des Stoffwechsels als therapeutischen Ansatz.
Methoden: Wir untersuchten die Auswirkungen von Dichloracetat (DCA), einem Inhibitor der Pyruvatdehydrogenase-Kinase, auf das Wachstum und die Stoffwechselaktivität menschlicher Melanomzelllinien. Die kombinierte Wirkung von DCA und dem BRAF-Inhibitor Vemurafenib wurde bei BRAFV600E-mutierten Melanom-Zelllinien untersucht. Vemurafenib-resistente Zelllinien wurden in vitro hergestellt und ihre Empfindlichkeit gegenüber DCA getestet.
Ergebnisse: DCA führte zu einer Verringerung der glykolytischen Aktivität und des intrazellulären ATP-Spiegels und hemmte das Zellwachstum. Die gleichzeitige Behandlung von BRAFV600E-mutierten Melanomzellen mit DCA und Vemurafenib führte zu einer stärkeren Verringerung des intrazellulären ATP-Spiegels und des Zellwachstums als eine der beiden Substanzen allein. Darüber hinaus behielten Melanomzellen, die in vitro eine Resistenz gegen Vemurafenib erworben hatten, ihre Empfindlichkeit gegenüber DCA bei.
Schlussfolgerungen: Diese Ergebnisse deuten darauf hin, dass DCA die Wirkung von Vemurafenib durch eine kooperative Abschwächung der Energieproduktion verstärkt. Darüber hinaus könnte die nachgewiesene Beibehaltung der Empfindlichkeit gegenüber DCA bei Melanomzellen mit erworbener Resistenz gegenüber Vemurafenib Auswirkungen auf die Melanombehandlung haben.
Schlüsselwörter: Dichloracetat, Melanom, BRAF, Bioenergetik, Stoffwechsel, ATPAbkürzungen
: (Acetyl-CoA): Acetyl-Coenzym A, (AMPK): AMP-aktivierte Proteinkinase, (DCA): Dichloracetat, (ECAR): Extrazelluläre Übersäuerungsrate, (HEMn-LP): Humane epidermale Melanozyten, (IC50): Halbe maximale Hemmkonzentration, (LKB1): Leberkinase B1, (MITF): Mikrophthalmie-assoziierter Transkriptionsfaktor, (OCR): Sauerstoffverbrauchsrate, (PDH): Pyruvat-Dehydrogenase, (PDK): Pyruvat-Dehydrogenase-Kinase
© 2014 Abildgaard et al.; Lizenznehmer BioMed Central Ltd.
Hintergrund
Ein Kennzeichen von Krebs ist die Umprogrammierung des zellulären Stoffwechsels auf aerobe Glykolyse. Dieses Stoffwechselmuster ist durch eine erhöhte Glukoseaufnahme und eine hochregulierte glykolytische Aktivität mit Fermentation von Glukose zu Milchsäure anstelle eines vollständigen aeroben Abbaus in den Mitochondrien gekennzeichnet. Die aerobe Glykolyse, die auch als Warburg-Effekt bezeichnet wird, ähnelt dem anaeroben Stoffwechsel normaler Zellen, findet jedoch im Zusammenhang mit einer ausreichenden Sauerstoffversorgung statt [1]. Die Umprogrammierung des Stoffwechsels in Krebszellen ist ein hochkomplexer und heterogener Prozess, der durch eine Vielzahl genetischer und nicht-genetischer Strategien zur Überwindung der Energiebeschränkung angetrieben wird [2]-[4].
Das BRAF V600E-Onkogen, das in mehr als 50 % der Melanome vorkommt [5], wurde direkt in die Reprogrammierung des Zellstoffwechsels einbezogen. Die konstitutive Aktivität der BRAF-Mutante reduziert die Expression oxidativer Enzyme und die Anzahl der Mitochondrien, während die Expression glykolytischer Enzyme und die Milchsäureproduktion zunehmen [6],[7]. Darüber hinaus wurde eine molekulare Verbindung zwischen dem RAS-RAF-MEK-ERK-MAPK-Stoffwechselweg und dem durch den Leberkinase B1 (LKB1)-AMP-aktivierten Proteinkinase (AMPK)-Stoffwechselweg vermittelten energetischen Stress-Kontrollpunkt erkannt, was auf eine Rolle von BRAFV600E bei der Vermittlung der Resistenz gegen energetischen Stress hindeutet [8],[9]. BRAF beeinflusst den oxidativen Stoffwechsel über den Mikrophthalmie-assoziierten Transkriptionsfaktor (MITF), der den mitochondrialen Hauptregulator PGC1α steuert [7]. Frühere Studien haben gezeigt, dass Melanome, die PGC1α exprimieren, einen stärker oxidativen Phänotyp aufweisen als PGC1α-negative Melanome [4],[7]. Darüber hinaus wurde gezeigt, dass BRAFV600E die Onkogen-induzierte Seneszenz durch Stoffwechselregulation vermittelt. Dieser Mechanismus beinhaltet eine Erhöhung der Pyruvat-Dehydrogenase (PDH)-Aktivität durch die Unterdrückung der Pyruvat-Dehydrogenase-Kinase (PDK) [10]. PDH steuert die Kopplung zwischen Glykolyse und mitochondrialer Atmung, indem sie den Zufluss von Pyruvat in die Mitochondrien erleichtert und die vollständige Verwertung von Glukose fördert. Die PDK-PDH-Achse ist bei Krebs häufig dysreguliert, wobei eine PDK-Überexpression die Kopplung zwischen den beiden Energiesystemen reduziert und damit zum Warburg-Effekt beiträgt [11],[12]. Auf der Grundlage dieser Erkenntnisse wurde die gezielte Hemmung der PDK als therapeutische Option für das Melanom vorgeschlagen, mit einem möglichen synergistischen Effekt von chemischen BRAFV600E-Inhibitoren wie Vemurafenib [10],[13].
Dichloracetat (DCA) ist ein Inhibitor der vier PDK-Isoformen und wurde früher zur Behandlung von Laktatazidose eingesetzt [14],[15], wobei die Toxizität bei wirksamen Dosen gering war [16],[17]. Mehrere Studien haben gezeigt, dass DCA den Warburg-Effekt in Krebszellen umkehrt und deren Wachstum und Überleben negativ beeinflusst [13],[18]-[21]. Diese Wirkung wurde auf eine Normalisierung des mitochondrialen Membranpotenzials von dem für Krebszellen charakteristischen hyperpolarisierten Zustand zurückgeführt. Die Veränderungen des Membranpotenzials führen zur Wiederöffnung spannungsgesteuerter Anionenkanäle und bewirken nachweislich eine erneute Sensibilisierung für die Apoptose, da die Fähigkeit zur Freisetzung pro-apoptotischer Mediatoren wiederhergestellt wird [18]. Hier haben wir die Wirkung von DCA auf Melanomzellen untersucht. Insbesondere haben wir die zellulären Reaktionen in Bezug auf Stoffwechsel, Bioenergetik, Wachstum, Proliferation und Zelltod in Melanomzelllinien, primären menschlichen Melanozyten und BRAFV600E-mutierten Melanomzellen mit erworbener Resistenz gegenüber Vemurafenib analysiert.
Methoden
Diechemischen Verbindungen
DCA (Natriumdichloracetat) und 2-Desoxy-D-Glukose (2-DG) wurden von Sigma-Aldrich erworben und in dH2O in einer Arbeitskonzentration von 1 M aufgelöst. Vemurafenib (PLX4032) wurde von Selleck Chemicals erworben und in DMSO in einer Arbeitskonzentration von 0,05 M aufgelöst.
Zellkultur
Die Melanomzelllinien ED-007, ED-013, ED-024, ED-027, ED-029, ED-034, ED-050, ED-070, ED-071, ED-117, ED-140, ED-179 und ED-196 wurden von der European Searchable Tumour line Database (ESTDAB, ED) [22] bezogen. Die Melanomzelllinie SK-MEL-28 wurde von ATCC bezogen. Primäre humane epidermale Melanozyten (neonatal) aus leicht pigmentiertem Gewebe (HEMn-LP) wurden von Invitrogen bezogen. Die Melanomzelllinien wurden bei 37 °C und 5 %CO2 in RPMI-1640-Medium kultiviert, das mit 10 % fötalem Rinderserum und 1 % Penicillin/Streptomycin ergänzt wurde. HEMn-LP-Zellen wurden unter denselben Bedingungen in 254CF-Medium kultiviert, das mit 1 % humanem Melanozyten-Wachstumszusatz (HMGS-2) und 12-O-Tetradecanoyl-Phorbol-13-Acetat (TPA; 10 ng/ml) ergänzt wurde. Alle Medien und Zusätze wurden von Invitrogen bezogen.
Stoffwechselanalyse
Die Stoffwechselcharakterisierung wurde an Melanomzelllinien und primären menschlichen Melanozyten mit einem Seahorse XF96 Extracellular Flux Analyzer (Seahorse Bioscience, Billerica, MA) durchgeführt, der Echtzeitmessungen der extrazellulären Säuerungsrate (ECAR) und der Sauerstoffverbrauchsrate (OCR) vornimmt. Es wurde ein Test entwickelt, um die Kapazität der mitochondrialen und glykolytischen Energiesysteme zu untersuchen. Die ECAR- und OCR-Werte wurden unter basalen Bedingungen und während der aufeinanderfolgenden Zugabe von fünf Stoffwechselmodulatoren gemessen: Der ATP-Synthase-Inhibitor Oligomycin (1 μM); der mitochondriale Membranpermeabilisator Carbonylcyanid-4-(trifluormethoxy)phenylhydrazon (FCCP) (1 μM); die Inhibitoren der mitochondrialen Atmung Rotenon (1 μM) und Antimycin A (1 μM); und der glykolytische Inhibitor 2-DG (100 mM). Das XF Cell Mito Stress Kit, das Oligomycin, FCCP, Rotenon und Antimycin A enthält, wurde von Seahorse Bioscience erworben.
ATP-Messungen
Der intrazelluläre ATP-Spiegel wurde mit dem ATPlite, 1 step Luminescence Assay System (Perkin Elmer) gemessen, einer Methode, die auf der Reaktion von ATP mit Luciferase und D-Luciferin basiert. Die Zellen wurden in dreifacher Ausführung mit 10.000 Zellen pro Vertiefung ausgesät und 2 oder 24 Stunden lang mit den angegebenen Verbindungen und der Vehikelkontrolle behandelt. Die Lumineszenz wurde mit dem Spectra Max Gemini EM Lumineszenz-Mikroplattenlesegerät (Molecular Devices) gemessen und auf die Hintergrundwerte normalisiert.
Kristallviolett-Assay
Um die Wirkung der untersuchten Verbindungen auf das Zellwachstum zu bewerten, wurdeeinKristallviolett-Assay durchgeführt. Die Zellen wurden in Duplikaten in einer geeigneten Dichte ausgesät und dann mit DCA, Vemurafenib, den beiden Verbindungen in Kombination und der Vehikelkontrolle behandelt. Das Medium und die Behandlungssubstanzen wurden alle 48 Stunden ausgetauscht. Der Versuch wurde dreimal unabhängig voneinander wiederholt. Um den Versuch zu beenden, wurden das Medium und die ungebundenen Zellen entfernt, die verbleibenden Zellen in PBS gewaschen und 15 Minuten lang mit Glutaraldehyd fixiert. Die fixierten Zellen wurden 1 Stunde lang mit Kristallviolettlösung (0,1% Kristallviolett, 20% CH3OH) inkubiert. Die Menge des von der Monoschicht aufgenommenen Farbstoffs, die proportional zur Anzahl der lebensfähigen Zellen am Boden der Vertiefung ist, wurde durch Extraktion der Farbe mit 10%iger Essigsäure und Messung der Absorption bei einer Wellenlänge von 595 nm quantifiziert. Die lineare Korrelation zwischen der Absorption und der Anzahl der Zellen wurde durch die Erstellung einer Standardkurve überprüft. Das relative Zellwachstum wurde durch Normalisierung auf die unbehandelten Kontrollen nach Subtraktion des Hintergrunds (ohne Zellen) bestimmt.
Zellproliferationstest
Melanomzellen wurden in dreifacher Ausführung mit 500-1.000 Zellen pro Vertiefung ausgesät und 96 Stunden lang mit DCA in den angegebenen Konzentrationen und der Vehikelkontrolle behandelt. Die Proliferation wurde dann durch den Nachweis von BrdU nach 12 Stunden des Einbaus in die zelluläre DNA gemessen. Das Verfahren wurde gemäß dem Protokoll des BrdU Cell Proliferation Assay Kits (Cell Signaling Technology®) durchgeführt.
Annexin V-FITC-Apoptose-Nachweis
Der Apoptose-Nachweis wurde mit einem Annexin V-FITC-Apoptose-Nachweiskit (BD Bioscience) nach dem mitgelieferten Protokoll durchgeführt. Die Zellen wurden entnommen und zweimal in kaltem PBS gewaschen. Die Zellen wurden dann in ein anderes Röhrchen überführt, geschleudert und in Bindungspuffer resuspendiert. Von der Resuspension wurden 5 ×105 Zellen in FACS-Röhrchen überführt und mit Annexin V-FITC und Propidiumjodid (PI) angefärbt. Nach 30 Minuten Inkubation wurde die Durchflusszytometrie auf einem Cytomics FC 500 MPL Gerät (Beckman Coulter) durchgeführt. Ungefärbte Zellen wurden als Kontrolle herangezogen.
Induktion der in vitro erworbenen Vemurafenib-Resistenz
Die erworbene Resistenz gegen Vemurafenib wurde in sieben Kulturen induziert, die von vier BRAFV600E-mutierten, Vemurafenib-empfindlichen Melanomzelllinien (ED-013, ED-071, ED-196 und SK-MEL-28) stammen. Die Zellen wurden in steigenden Konzentrationen von Vemurafenib kultiviert, bis sie in einer Konzentration oberhalb der IC50 stetig wuchsen, und wurden dann in Vemurafenib-haltigem Medium gehalten.
Pyrosequenzierung
Die Pyrosequenzierung von Mutationshotspots in BRAF und NRAS wurde auf einer PyroMark Q24-Plattform (Qiagen) unter Verwendung von PyroMark Gold Q24-Reagenzien (Qiagen) durchgeführt. Die Primer-Sequenzen sind in Zusatzdatei 1: Tabelle S1 aufgeführt.
| Zielort | Primer-Name | Primer-Sequenz (5′-3′) |
| BRAF V600E | BRAF-F1 | [Btn]-TTCATGAAGACCTCACAGTAAAAA |
| BRAF-R1 | GGCCAAAAATTTAATCAGTGGAA | |
| BRAF-S1 | CCACTCCATCGAGATTT | |
| NRAS Q61K,L,R | NRAS-F1 | [Btn]-ACCCCCAGGATTCTTACAGAAA |
| NRAS-R1 | CGCAAATGACTTGCTATTATTGA | |
| NRAS-S1 | TCATGGCACTGTACTCTT |
PGC1α-Expressionsanalyse
Die gesamte RNA wurde mit dem RNeasy mini kit (Qiagen) isoliert und die cDNA wurde mit dem SuperScript™ III Reverse Transcriptase kit (Invitrogen) synthetisiert. Oligo dT24 und zufällige Hexamere wurden als Primer für die cDNA-Synthese verwendet. Die Genexpression von PGC1α wurde mit quantitativer Echtzeit-PCR auf dem Roche LightCycler 2.0 unter Verwendung des LigthCycler FastStart DNA MasterPLUS SYBR Green I Kits (Roche) bestimmt. Die Primer-Sequenzen waren: PPARGC1A_2241F: 5′-GCTGTACTTTTGTGGACGCA-3′ und PPARGC1A_2306R: 5′-GGAAGCAGGGTCAAAGTCAT-3′. Die Expression wurde auf die Expression des Housekeeping-Gens RPLP0 normalisiert. Die Primer-Sequenzen waren: RPLP0_433F: 5′-ACTAAAATCTCCAGGGGCACC-3′ und RPLP0_547R: 5′-ATGACCAGCCCAAAGGAGAA-3′. Die beiden Melanomzelllinien ED-050 und SK-MEL-28 wurden als Positiv- bzw. Negativkontrollen verwendet [4].
Statistische Analyse
Unterschiede zwischen unabhängigen Datensätzen wurden mit dem Student’s t-Test ermittelt. Für die statistische Analyse der Varianz zwischen verschiedenen Behandlungen (Vehikelkontrolle, DCA, Vemurafenib und die Kombination aus DCA und Vemurafenib) wurde eine einseitige ANOVA mit angepassten Stichproben verwendet. Zur Bestimmung der statistischen Signifikanz wurde der Tukey-Vergleichstest (HSD) verwendet. Der Pearson-Korrelationskoeffizient wurde verwendet, um die Korrelation zwischen der DCA-Empfindlichkeit und den Stoffwechselparametern zu bestimmen. Ein Wert von 1 bedeutete eine positive Korrelation, 0 keine Korrelation und -1 eine negative Korrelation.
Bei den in dieser Studie durchgeführten Experimenten wurden ausschließlich handelsübliche Zelllinien verwendet, so dass keine Genehmigung der Ethikkommission erforderlich war.
Ergebnisse
Metabolische Charakterisierung von Melanomzelllinien und primären Melanozyten
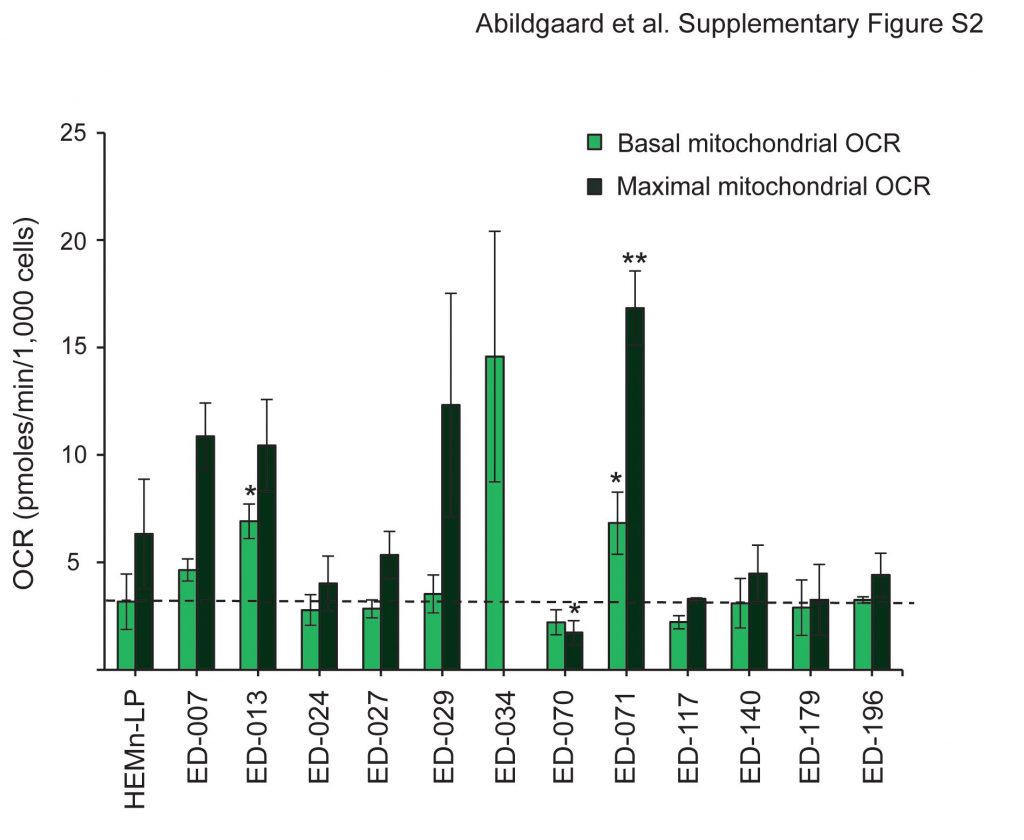
Die metabolische Profilierung von 12 Melanomzelllinien und primären menschlichen Melanozyten (HEMn-LP) wurde mit dem Seahorse FX96-Analysegerät durchgeführt. Dieses Gerät führt Echtzeitmessungen der extrazellulären Säuerungsrate (ECAR) und der Sauerstoffverbrauchsrate (OCR) durch, die indirekte Messgrößen für die glykolytische Aktivität bzw. die mitochondriale Atmung sind [23]. Die Messungen wurden unter Basalbedingungen und während der aufeinanderfolgenden Zugabe von fünf Stoffwechselmodulatoren durchgeführt (Abbildung 1A). Im Vergleich zu normalen Melanozyten wiesen 11 der 12 Zelllinien höhere glykolytische Raten auf, wie aus den höheren basalen glykolytischen ECARs hervorgeht, was zeigt, dass der Warburg-Effekt ein allgemeines Merkmal von Melanomzellen ist. Außerdem wiesen neun der Zelllinien im Vergleich zu Melanozyten höhere maximale glykolytische Kapazitäten auf (Abbildung 1B). Mit wenigen Ausnahmen gab es keine signifikanten Unterschiede in der basalen und maximalen mitochondrialen Atmung zwischen Melanozyten und Melanomzellen (Zusatzdatei 2: Abbildung S2). Gemäß den OCR-zu-ECAR-Verhältnissen (OCR/ECAR; Abbildung 1C) war der relative Beitrag der mitochondrialen Atmung zur ATP-Produktion bei 10 der Melanomzelllinien im Vergleich zu Melanozyten geringer.
.
.
Die mitochondriale Effizienz (ATP-Kopplung) und Leistung (respiratorisches Kontrollverhältnis) wurden anhand von Messungen zweier Parameter für die mitochondriale Funktion geschätzt: Protonenleck und maximale mitochondriale OCR [24]. Das Protonenleck wurde nach Zugabe des ATP-Synthase-Inhibitors Oligomycin und die maximale mitochondriale OCR nach Zugabe des mitochondrialen Entkopplers FCCP bestimmt. Diese Analyse zeigte eine signifikant niedrigere ATP-Kopplung in Melanomzellen im Vergleich zu Melanozyten (p < 0,05; ED-007: p = 0,07), was auf ein höheres Protonenleck und eine weniger effiziente ATP-Produktion im Verhältnis zum Sauerstoffverbrauch schließen lässt (Abbildung 1D). Darüber hinaus wiesen sechs der Melanom-Zelllinien auch signifikant niedrigere Atmungskontrollverhältnisse als Melanozyten auf (p?<?0,01, Abbildung 1E), was auf eine schlechte mitochondriale Leistung hindeutet.
DCA verlagert den Stoffwechsel in Richtung mitochondrialer Atmung und reduziert den ATP-Gehalt
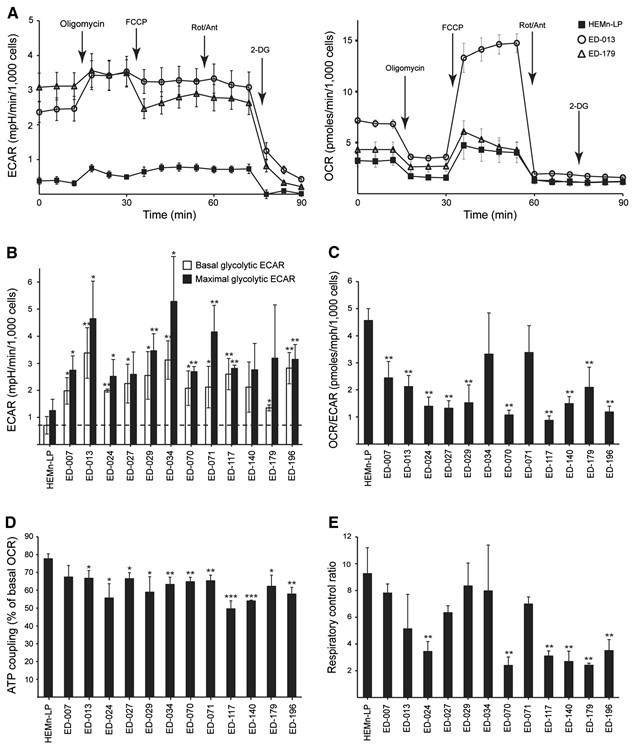
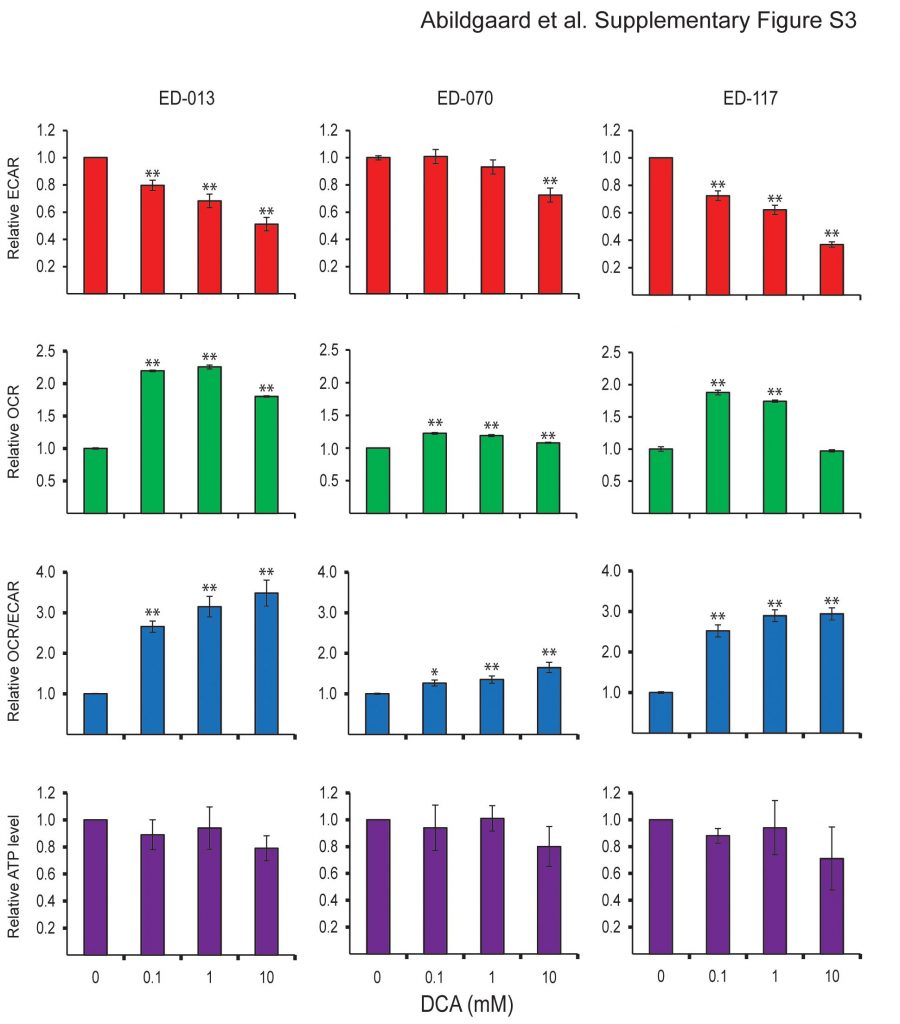
Um die Wirkung von DCA auf den Stoffwechsel von Melanomzellen zu bestimmen, analysierten wir das Panel von 12 Zelllinien mit dem Seahorse XF96-Analysegerät. Nach einer 2-stündigen Behandlung mit 10 mM DCA reagierten alle Zelllinien mit einer Verringerung der ECAR und einem Anstieg der OCR (Abbildung 2A), was auf eine Verlagerung der mitochondrialen Atmung hinweist. Die ECAR-Antwort war bei allen Zelllinien ähnlich, während es bei der OCR-Antwort große Unterschiede gab (Abbildung 2A). Die relativen Veränderungen von ECAR, OCR und OCR/ECAR als Reaktion auf DCA waren konzentrationsabhängig (Zusätzliche Datei 3: Abbildung S3).
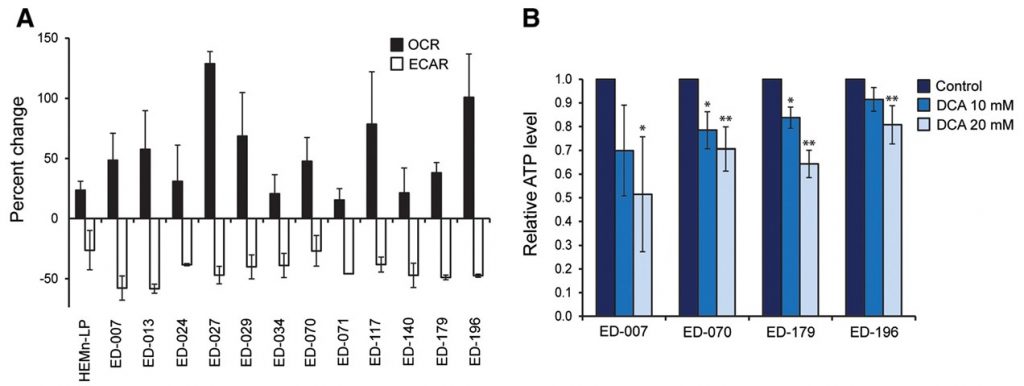
Um festzustellen, ob die Verschiebung des Stoffwechsels die energetische Homöostase beeinträchtigt, haben wir den ATP-Gehalt in Melanomzellen nach einer Behandlung mit 0,1, 1 oder 10 mM DCA für 2 Stunden gemessen. Alle drei getesteten Zelllinien waren in der Lage, den ATP-Spiegel nach der Behandlung mit niedrigen DCA-Konzentrationen (0,1-1 mM) aufrechtzuerhalten, während bei Kulturen, die mit 10 mM DCA behandelt wurden, eine Tendenz zur Verringerung des ATP-Spiegels beobachtet wurde (Additional file 3: Abbildung S3). Wurden die Zellen 24 Stunden lang mit DCA (10 oder 20 mM) behandelt, wurde ein signifikanter konzentrationsabhängiger ATP-Abfall beobachtet (Abbildung 2B), was auf eine allmähliche Erschöpfung des Stoffwechselsystems hinweist.
DCA reduziert das Wachstum von Melanomzellen unabhängig vom Status des genetischen Treibers und der PGC1α-Expression
Um eine mögliche klinische Anwendung von DCA zur Behandlung von Melanomen zu bewerten, haben wir seine Auswirkungen auf verschiedene Parameter des Zellwachstums untersucht. Alle Zelllinien und primären Melanozyten wurden 96 Stunden lang mit einer Reihe von DCA-Konzentrationen (0,5-100 mM) behandelt (siehe Abbildung 3A für repräsentative Ergebnisse). Alle Zelllinien zeigten eine konzentrationsabhängige Verringerung des Wachstums mit IC50-Werten im Bereich von 9-38 mM (Tabelle 1), verglichen mit einem IC50-Wert von 70 mM für primäre Melanozyten (p < 0,001). Es gab keine Korrelation zwischen der Reaktion auf DCA und dem BRAF/NRAS-Mutationsstatus oder den Expressionswerten von PGC1α (Tabelle 1).
| Zelllinie | DCA IC50(mM)4 | BRAF/ NRAS-Status1 | PGC1α-Expression2,4 |
|---|---|---|---|
| ED-070 | 8.9 ± 0.6*** | NRASQ61L | 1.0 ± 0.2 |
| ED-007 | 12.2 ± 2.2*** | WT3 | 0.6 ± 0.0 |
| ED-071 | 12.3 ± 3.6*** | BRAFV600E | 0.0 ± 0.0 |
| ED-034 | 12.7 ± 1.7*** | BRAFL597S | 1.0 ± 0.2 |
| ED-013 | 14.4 ± 2.0*** | BRAFV600E | 1.9 ± 0.3 |
| ED-027 | 17.7 ± 2.1*** | BRAFV600E | 0.5 ± 0.1 |
| SK-MEL-28 | 20.0 ± 4.5*** | BRAFV600E | 0.0 ± 0.0 |
| ED-179 | 20.6 ± 1.8*** | NRASQ61R | 0.3 ± 0.0 |
| ED-024 | 21.9 ± 1.6*** | NRASQ61L | 0.0 ± 0.0 |
| ED-140 | 23.9 ± 2.0*** | WT | 0.4 ± 0.1 |
| ED-050 | 24.1 ± 3.1*** | WT | 2.7 ± 0.5 |
| ED-029 | 29.7 ± 5.1*** | BRAFV600K | 1.3 ± 0.2 |
| ED-196 | 35.8 ± 3.2*** | BRAFV600E | 1.5 ± 0.5 |
| ED-117 | 37.6 ± 2.2*** | BRAFV600E | 1.2 ± 0.1 |
| HEMn-LP | 69.1 ± 6.4 | WT | 1 |
| ED-013-R1 | 12.6 ± 3.0*** | BRAFV600E | |
| ED-013-R2 | 13.6 ± 2.4*** | BRAFV600E | |
| ED-071-R1 | 12.2 ± 0.9*** | BRAFV600E | |
| ED-071-R2 | 13.8 ± 3.7*** | BRAFV600E | |
| SK-MEL-28-R1 | 23.1 ± 4.6*** | BRAFV600E | |
| SK-MEL-28-R2 | 26.2 ± 8.0** | BRAFV600E |
< 0,01; ***p < 0,001 im Vergleich zu HEMn-LP.
1BRAF/NRAS-Statusentspricht den veröffentlichten Ergebnissen [25].
2Bezogenauf die PGC1α-Expression in normalen Melanozyten.
3Wildtyp.
4DCA-IC50-Werteund PGC1α-Expression stellen die Mittelwerte von drei unabhängigen Messungen ± Standardabweichung dar.
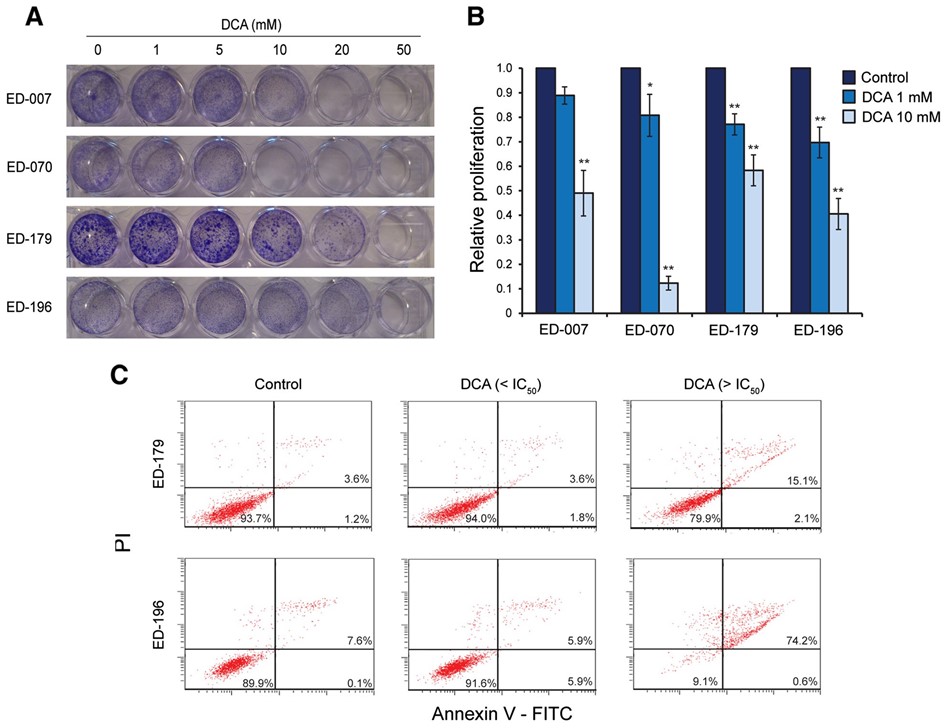
Um die Auswirkungen von DCA auf das Zellwachstum weiter zu charakterisieren, haben wir den Einbau von BrdU in Zellen gemessen, die 96 Stunden lang mit 1 oder 10 mM DCA behandelt wurden. Wie in Abbildung 3B dargestellt, reagierten alle vier getesteten Zelllinien mit einer verringerten Proliferation im Bereich von 11-30 % bei 1 mM und von 42-88 % bei 10 mM. Wir haben auch die apoptotische Reaktion auf DCA durch eine durchflusszytometrische Analyse der Annexin-V-Spiegel gemessen. Bei DCA-Konzentrationen unterhalb der IC50 stieg die Zahl der Annexin V-positiven Zellen nach 96 Stunden und bis zu 3 Wochen nicht an. Im Gegensatz dazu erhöhte sich bei der Behandlung mit Konzentrationen oberhalb der IC50 die Zahl der Zellen, die sowohl für Annexin V als auch für PI positiv waren, was auf eine Induktion des Zelltods bereits nach 96 Stunden hindeutet (Abbildung 3C).
DCA potenziert die Wirkung von Vemurafenib auf BRAFV600E-mutierte Melanomzellen
Um zu untersuchen, ob DCA zur Verbesserung der Wirksamkeit chemischer BRAF-Inhibitoren bei der Behandlung von Melanomen eingesetzt werden kann, haben wir verschiedene Kombinationen von DCA und Vemurafenib auf das Zellwachstum getestet. Die 96-stündige Behandlung von vier BRAFV600E-mutierten Zelllinien mit Vemurafenib (0,05-5 μM) ergab IC50-Werte von 0,5 bis 4,5 μM, was mit Daten aus früheren Studien übereinstimmt [26]. Als wir primäre Melanozyten der gleichen Behandlung aussetzten, erreichten wir den IC50-Wert für diese Zellen nicht, selbst mit der höchsten getesteten Konzentration (5 μM), was die Spezifität der Verbindung bestätigt.
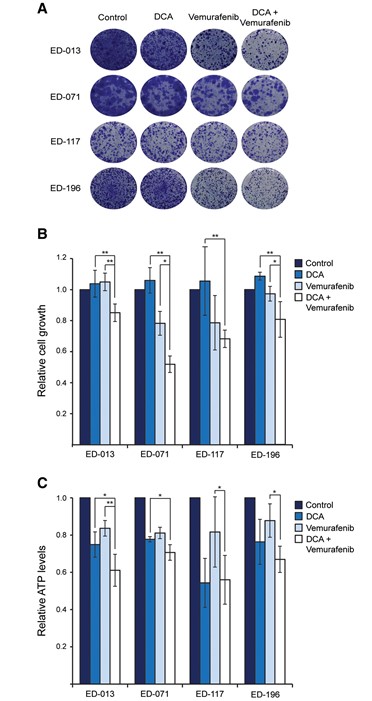
Wurden die Zellen mit 1 mM DCA in Kombination mit niedrigen Konzentrationen von Vemurafenib (<IC50) behandelt, war die Verringerung des Zellwachstums stärker ausgeprägt als bei DCA oder Vemurafenib allein (p < 0,05; Abbildung 4A, B). DCA verstärkte die Wirkung von Vemurafenib in ED-117-Zellen nicht, was auf die inhärente Resistenz dieser Zellen gegenüber DCA (IC50 38 mM; Tabelle 1) zurückzuführen sein könnte. Bei IC50-Konzentrationen verursachten sowohl DCA als auch Vemurafenib eine Verringerung des intrazellulären ATP-Spiegels, wenn sie als Einzelwirkstoffe eingesetzt wurden, und eine weitere Verringerung, wenn sie in Kombination eingesetzt wurden, obwohl dies nicht für alle Zelllinien statistisch signifikant war (Abbildung 4C).
Vemurafenib-resistente Zelllinien haben eine verbesserte oxidative Kapazität und bleiben empfindlich gegenüber DCA
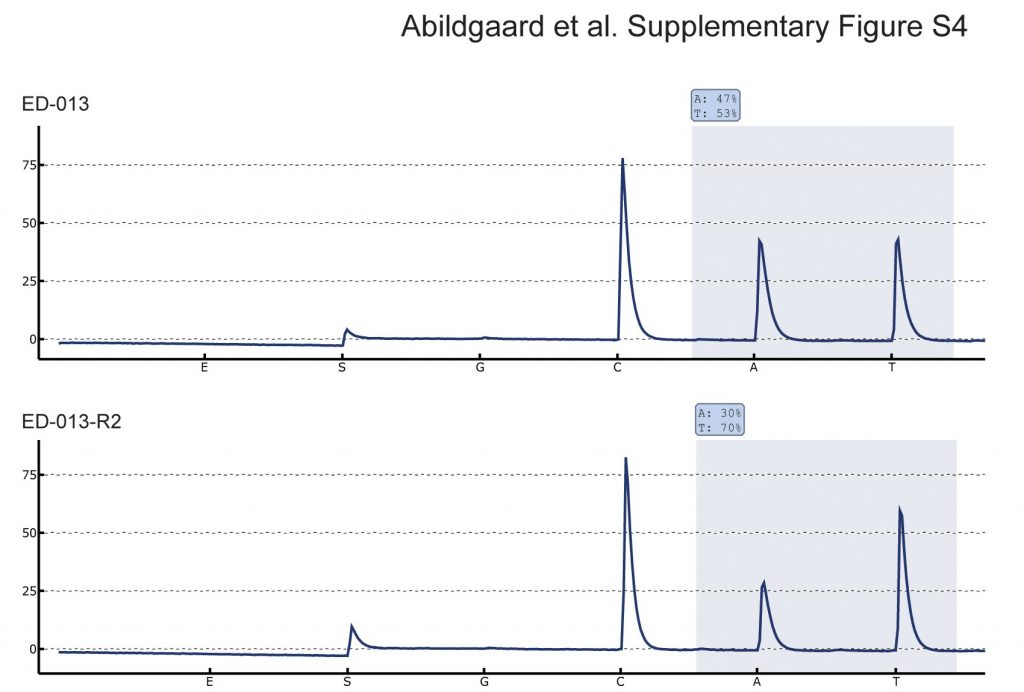
Aus vier BRAFV600E-mutierten Zelllinien wurden sieben Kulturen Vemurafenib-resistenter Zellen erzeugt, indem die Zellen steigenden Konzentrationen von Vemurafenib ausgesetzt wurden. Die Zellen galten als resistent, wenn sie bei einer Vemurafenib-Konzentration oberhalb der IC50 kontinuierlich vermehrt werden konnten. Aus allen sieben Kulturen wurde DNA isoliert und auf BRAF-Kopienzahlzuwachs und sekundäre NRAS-Mutationen untersucht, zwei gut beschriebene Mechanismen der erworbenen Resistenz gegen Vemurafenib [27],[28]. Die Pyrosequenzierung ergab einen Anstieg des Verhältnisses von BRAF V600E zu BRAF WT in einer der resistenten Zelllinien (ED-013-R2) im Vergleich zur elterlichen Zelllinie (Additional file 4: Abbildung S4). Bei den übrigen resistenten Zelllinien wurden keine BRAF- oder NRAS-Veränderungen festgestellt.
Die metabolische Charakterisierung von zwei der resistenten Zelllinien (ED-013-R1 und ED-196-R) mit dem Seahorse XF96-Analysegerät zeigte, dass beide resistenten Zelllinien ein verändertes metabolisches Profil mit einer signifikant erhöhten maximalen Atmungskapazität aufwiesen (Abbildung 5A), aber keine Veränderungen in der basalen respiratorischen OCR, ATP-Kopplung oder nicht-mitochondrialen OCR. Interessanterweise wiesen die vemurafenib-resistenten Zelllinien, als sie auf ihre Empfindlichkeit gegenüber DCA getestet wurden, alle ähnliche IC50-Werte auf wie die elterlichen Zelllinien (Tabelle 1).
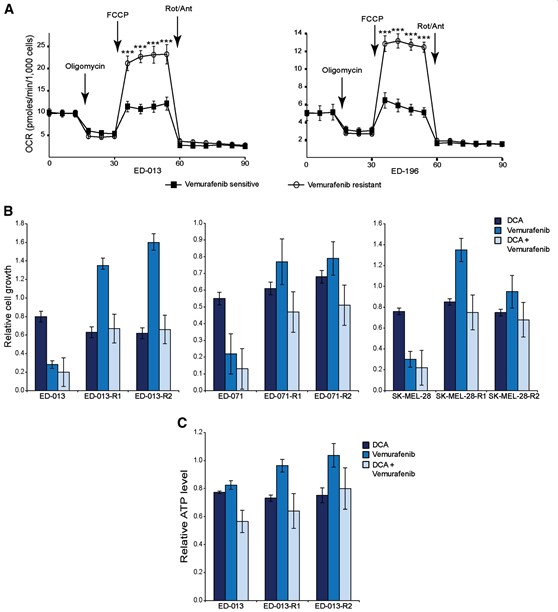
Abbildung 5Bzeigt die Empfindlichkeit der Vemurafenib-resistenten Zelllinien gegenüber DCA und Vemurafenib als Einzelwirkstoffe oder in Kombination mit den elterlichen Zelllinien. Das Wachstum der resistenten Zelllinien war in Anwesenheit von Vemurafenib nach 96 Stunden leicht verringert, unbeeinflusst oder sogar erhöht, während die Empfindlichkeit gegenüber DCA sowohl in Anwesenheit als auch in Abwesenheit von Vemurafenib ähnlich wie bei den Elternzellen war (Abbildung 5B). Ähnliche Ergebnisse wurden bei der Messung der Veränderungen des ATP-Spiegels nach einer 24-stündigen Behandlung mit denselben Medikamenten erzielt (Abbildung 5C). Insgesamt zeigen diese Daten, dass die Vemurafenib-resistenten Melanomzellen trotz der Veränderung ihres Stoffwechselprofils weiterhin empfindlich auf DCA reagieren.
Diskussion
Die gezielte Stoffwechseltherapie bei Krebserkrankungen konzentrierte sich bisher in erster Linie auf die Energieversorgung durch Hemmung der Glykolyse. Die Erkenntnis, dass Mitochondrien aktiv zum Fortschreiten des Melanoms beitragen können, hat jedoch die Aufmerksamkeit auf den oxidativen Stoffwechsel als potenzielles therapeutisches Ziel gelenkt [10],[13],[29]. DCA fördert die PDK-abhängige Aktivierung der PDH und kehrt die Laktatproduktion zugunsten des Pyruvateinstroms in die Mitochondrien um [15],[18]. Durch diesen Mechanismus verbessert DCA die Kopplung zwischen Glykolyse und mitochondrialer Atmung, was bei Zellen mit einer mangelhaften Kopplung, wie z. B. Krebszellen, größere Auswirkungen hat [18]. Alle in unserer Studie untersuchten Melanomzelllinien reagierten auf DCA mit einer verringerten Laktatproduktion und einer erhöhten OCR. Diese Verlagerung auf die mitochondriale Atmung sollte die Substratnutzung optimieren und zu einer effizienteren Energieausbeute führen, führte aber stattdessen zu einem deutlichen Rückgang der ATP-Werte trotz einer unbeeinflussten oder sogar erhöhten mitochondrialen ATP-Kopplung. Die beobachtete Verringerung der ECAR als Reaktion auf DCA deutet darauf hin, dass die Hemmung der Glykolyse ein wesentlicher Faktor für den Energiemangel sein könnte. Ein glykolysehemmender Mechanismus von DCA wurde bisher nicht beschrieben. Es wurde jedoch nachgewiesen, dass die Pyruvatkinase, die letzte ATP-produzierende Stelle im glykolytischen Stoffwechselweg, durch Acetyl-Coenzym A (Acetyl-CoA) negativ reguliert wird [30]. Da die PDH-Aktivierung direkt die Bildung von Acetyl-CoA erhöht [31], könnte dies die DCA-vermittelte Hemmung der Glykolyse erklären. Die strukturelle Ähnlichkeit zwischen DCA und Pyruvat [32] könnte auch auf eine direkte Hemmung der Glykolyse durch DCA hindeuten, möglicherweise durch einen allosterischen Rückkopplungsmechanismus.
Die metabolische Reaktion auf DCA ging mit einer verringerten Proliferation der Melanomzellen einher, unabhängig vom genetischen Treiberstatus und den metabolischen Profilen dieser Zellen. Mehrere frühere Studien haben eine apoptotische Wirkung von DCA auf Krebszellen nachgewiesen [13],[18],[19],[32]-[34]. In Übereinstimmung mit unseren Ergebnissen wurde die apoptotische Reaktion jedoch erst bei Konzentrationen ausgelöst, die zu hoch waren, um klinisch relevant zu sein [32]. Um die klinische Relevanz von DCA für die Melanombehandlung weiter zu erforschen, untersuchten wir die Wirksamkeit dieses Wirkstoffs in Kombination mit dem BRAF-Inhibitor Vemurafenib. Diese Experimente zeigten eine potenzierende Wirkung von DCA auf die Wachstumshemmung von BRAFV600E-mutierten Melanomzellen. Bei niedrigen DCA-Konzentrationen, die allein keine Wirkung auf das Zellwachstum hatten, hatte die Kombination mit niedrigen Vemurafenib-Konzentrationen eine deutlich stärkere wachstumshemmende Wirkung als Vemurafenib allein. Diese potenzierende Wirkung von DCA spiegelte sich auch in der Verringerung des ATP-Spiegels wider. Biochemische Analysen haben gezeigt, dass BRAFV600E in der Lage ist, den LKB1-AMPK-Energiesensorweg zu entkoppeln, was die Resistenz gegenüber Energiemangel fördert und eine apoptotische Reaktion verhindert [8],[9]. Die Behandlung mit BRAF-Inhibitoren stellt diesen Weg wieder her [35] und kann daher die Reaktion auf Verbindungen, die die ATP-Bildung verringern, verstärken. Sowohl DCA als auch Vemurafenib unterdrücken die glykolytische Aktivität in Melanomzellen und machen sie damit abhängiger von der mitochondrialen Atmung [6]. Da die Glykolyse einen großen Teil der gesamten Energieproduktion in diesen Zellen ausmacht, wird durch die Hemmung dieses Prozesses das oxidative System zur ATP-Produktion stark beansprucht. Die geringere Leistung der Mitochondrien in Melanomzellen könnte die Unfähigkeit dieser Zellen erklären, das ATP-Niveau in Gegenwart von DCA und Vemurafenib aufrechtzuerhalten. Der kooperative Effekt dieser Verbindungen bei der Senkung des ATP-Spiegels deutet darauf hin, dass die energetische Schwelle, die den Wachstumsstillstand oder Zelltod in Melanomzellen fördert, mit niedrigeren Konzentrationen von Vemurafenib in Gegenwart von DCA erreicht werden kann.
In früheren Studien wurde untersucht, ob Stoffwechselmodulatoren die therapeutische Wirkung von BRAF-Inhibitoren bei der Behandlung von Melanomen verbessern können. Die Kombination von PLX4720 (einem Vemurafenib-Analogon) mit einem der beiden antidiabetischen Biguanide, Metformin und Phenformin, zeigte eine synergistische Hemmung der Lebensfähigkeit von Melanomzellen [35],[36]. Beide Wirkstoffe beeinträchtigen die ATP-Synthese durch Hemmung der Aktivität des mitochondrialen Komplexes I, was zu einer Verringerung des Verhältnisses von ATP zu ADP und zur Aktivierung des LKB1-AMPK-Signalwegs zur Unterdrückung des Wachstums führt [35],[36]. Im Gegensatz zu DCA stimulieren Metformin und Phenformin beide die Glykolyse und die Milchsäureproduktion [37],[38], was die wachstumsfördernde Wirkung von Metformin auf einige Melanomzelllinien erklären könnte, wenn es als Einzelwirkstoff eingesetzt wird. Darüber hinaus lagen die Konzentrationen, bei denen Metformin wirksam war, über einem therapeutisch relevanten Niveau [35]. Phenformin war deutlich wirksamer als Metformin [36], wurde jedoch mit einem hohen Risiko einer Laktatazidose in Verbindung gebracht [39] und wurde in vielen Ländern vom Markt für die Behandlung von Typ-2-Diabetes genommen. DCA hingegen verstärkte nachweislich die Wirkung von Vemurafenib bei Konzentrationen von bis zu 1 mM und hatte bei der Verabreichung an Patienten nur wenige unerwünschte Wirkungen [17],[19],[40]. Diese Ergebnisse deuten auf ein therapeutisches Potenzial von DCA als Co-Wirkstoff für die Behandlung von BRAFV600E-mutierten Melanomen mit Vemurafenib hin. Dies wurde noch verstärkt durch den Nachweis, dass die Empfindlichkeit gegenüber DCA in Melanomzelllinien mit erworbener Resistenz gegen Vemurafenib erhalten blieb. Obwohl die resistenten Zellen ein verändertes Stoffwechselprofil mit deutlich erhöhter maximaler mitochondrialer Atmung aufwiesen, wie auch von Corazao-Rozas et al. gezeigt [41], waren sie genauso empfindlich gegenüber DCA wie die Vemurafenib-empfindlichen Stammzellen. Daher könnte DCA möglicherweise eine Strategie darstellen, um das Auftreten von Vemurafenib-toleranten Subpopulationen während der Erstbehandlung zu verhindern und dadurch die Entwicklung einer Resistenz hinauszuzögern oder zu verhindern.
Schlussfolgerungen
Wir haben hier ein genaueres Verständnis der Auswirkungen von DCA auf den Stoffwechsel und das Wachstum von Melanomzellen vermittelt. Die Fähigkeit von DCA, den ATP-Spiegel und das Melanomwachstum zu senken, scheint die Wirkung von Vemurafenib zu verstärken, einem Medikament, das bereits in der Klinik zur Behandlung von BRAFV600E-mutierten metastatischen Melanomen eingesetzt wird. Wichtig ist, dass Melanomzellen mit erworbener Resistenz gegen Vemurafenib ihre Empfindlichkeit gegenüber DCA beibehielten. Diese Ergebnisse sollten zur weiteren Erforschung dieser Wirkstoffkombination und der In-vivo-Anwendung von DCA ermutigen.
Zusätzliche Dateien
Zusätzliche Datei 1: Tabelle S1.(33K, doc)
Pyrosequenzierungs-Primer für die Amplifikation und Sequenzierung von BRAF- und NRAS-Mutationshotspots. Vorwärts-, Rückwärts- und Sequenzierungsprimer werden mit F, R bzw. S bezeichnet.
Zusätzliche Datei 2: Abbildung S2.(8.5M, tiff)
Basale und maximale mitochondriale OCR-Werte für Melanomzelllinien und humane epidermale Melanozyten (HEMn-LP). Die nach Zugabe von Rotenon/Antimycin A gemessene OCR (nicht-mitochondriale OCR) wurde von allen Werten subtrahiert. Die gestrichelte Linie zeigt die basale OCR von HEMn-LP. Die angegebenen Werte sind Mittelwerte von drei unabhängigen Messungen ± Standardabweichung. Zur Bestimmung der Unterschiede zwischen HEMn-LP und den Melanom-Zelllinien wurde der Students t-Test verwendet (*p < 0,05; **p < 0,01; ***p < 0,001). Die maximale mitochondriale OCR für ED-034 wurde nicht angegeben, da sie in vier unabhängigen Experimenten sehr stark variierte und zwischen 6,19 und 58,36 pmol/min/1.000 Zellen lag.
Zusätzliche Datei 3: Abbildung S3.(12M, tiff)
Stoffwechselreaktion von Melanomzellen auf DCA Relative Reaktion der ECAR-, OCR-, OCR/ECAR- und ATP-Spiegel nach 2-stündiger Behandlung mit DCA (0,1, 1 und 10 mM). Die Fehlerbalken in den ersten drei Feldern stellen die Standardabweichungen von drei wiederholten Messungen an sechs parallelen Proben dar. Die Fehlerbalken im unteren Feld stellen die Standardabweichung von drei unabhängigen Experimenten dar. Für die statistische Analyse wurde eine einseitige ANOVA mit übereinstimmenden Stichproben verwendet, und die statistische Signifikanz wurde mit Tukey’s HSD-Test bestimmt (*p < 0,05; **p < 0,01).
Zusätzliche Datei 4: Abbildung S4.(5.6M, tiff)
Allelstatus von BRAF. Pyrosequenzierung der BRAF c.1799 T > A Mutationsstelle in ED-013 und dem Vemurafenib-resistenten Derivat ED-013-R2. Die erhöhte BRAF V600E-zu-BRAF WT verhältnis in ED-013-R2 deutet auf einen Anstieg der Kopienzahl hin, was die Resistenz gegen Vemurafenib erklären könnte.
Konkurrierende Interessen
Die Autoren erklären, dass sie keine konkurrierenden Interessen haben.
Beiträge der Autoren
PG und CA planten und organisierten die Studie. CA führte den Großteil der Experimente und die Verarbeitung der Daten durch. CD plante und führte den Zellproliferationsversuch durch und half bei der Interpretation der Ergebnisse. AB und TM halfen bei der Vorbereitung und Optimierung des Designs für die Stoffwechselanalyse mit dem Seahorse XF-Instrument. CA, AB und TM diskutierten und interpretierten die Ergebnisse der Stoffwechselanalyse. CA und PG schrieben das Manuskript mit Beiträgen und Korrekturen von CD, AB und TM. Das endgültige Manuskript wurde von allen Autoren gelesen und genehmigt.
Danksagung
Wir danken Professor Karsten Kristiansen (Fachbereich Biologie, Universität Kopenhagen) und Associate Professor Jacob B. Hansen (Fachbereich Biomedizinische Wissenschaften, Universität Kopenhagen) für die Bereitstellung der Geräte und des technischen Fachwissens in Bezug auf die Seahorse XF-Analyse. Diese Studie wurde durch Zuschüsse der Dänischen Krebsgesellschaft und der Dänischen Krebsforschungsstiftung unterstützt.
REFERENZEN
1 Warburg O, Wind F, Negelein E: Der Metabolismus von Tumoren im Körper. J Gen Physiol. 1927, 8: 519-530. 10.1085/jgp.8.6.519.2 Gatenby RA, Gillies RJ: Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004, 4: 891-899. 10.1038/nrc1478.
3 Vander Heiden MG, Cantley LC, Thompson CB: Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009, 324: 1029-1033. 10.1126/science.1160809.
4 Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P: PGC1alpha expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013, 23: 287-301. 10.1016/j.ccr.2012.11.020.
5 vDavies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J: Mutationen des BRAF-Gens bei menschlichem Krebs. Nature. 2002, 417: 949-954. 10.1038/nature00766.
6 Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C: Dysfunktionale oxidative Phosphorylierung macht maligne Melanomzellen süchtig nach Glykolyse, angetrieben durch das (V600E)BRAF-Onkogen. Oncotarget. 2013, 4: 584-599.
7 Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, Arany Z, Widlund HR: Onkogenes BRAF reguliert den oxidativen Stoffwechsel über PGC1alpha und MITF. Cancer Cell. 2013, 23: 302-315. 10.1016/j.ccr.2013.02.003.
8 Esteve-Puig R, Canals F, Colome N, Merlino G, Recio JA: Uncoupling of the LKB1-AMPKalpha energy sensor pathway by growth factors and oncogenic BRAF.PLoS One 2009, 4:e4771,
9 Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC: Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Mol Cell. 2009, 33: 237-247. 10.1016/j.molcel.2008.12.026.
10 Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS: A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013, 498: 109-112. 10.1038/nature12154.
11 Baumunk D, Reichelt U, Hildebrandt J, Krause H, Ebbing J, Cash H, Miller K, Schostak M, Weikert S: Expressionsparameter der Stoffwechselweg-Gene Pyruvat-Dehydrogenase-Kinase-1 (PDK-1) und DJ-1/PARK7 im Nierenzellkarzinom (RCC). World J Urol. 2013, 31: 1191-1196. 10.1007/s00345-012-0874-5.
12 Hur H, Xuan Y, Kim YB, Lee G, Shim W, Yun J, Ham IH, Han SU: Expression von Pyruvat-Dehydrogenase-Kinase-1 in Magenkrebs als potenzielles therapeutisches Ziel. Int J Oncol. 2013, 42: 44-54.
13 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, de Lassalle EM, Maboudou P, Formstecher P, Polakowska R, Mortier L, Marchetti P: Die Inaktivierung der HIF-1alpha/PDK3-Signalachse treibt das Melanom zum mitochondrialen oxidativen Stoffwechsel und potenziert die therapeutische Wirkung von Pro-Oxidantien. Cancer Res. 2012, 72: 5035-5047. 10.1158/0008-5472.CAN-12-0979.
14 Stacpoole PW, Lorenz AC, Thomas RG, Harman EM: Dichloroacetate in the treatment of lactic acidosis. Ann Intern Med. 1988, 108: 58-63. 10.7326/0003-4819-108-1-58.
15 Stacpoole PW: The pharmacology of dichloroacetate. Metabolism. 1989, 38: 1124-1144. 10.1016/0026-0495(89)90051-6.
16 Michelakis ED, Webster L, Mackey JR: Dichloracetat (DCA) als potenzielle metabolische Zieltherapie für Krebs. Br J Cancer. 2008, 99: 989-994. 10.1038/sj.bjc.6604554.
17 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, O’Brien RG, Perkins LA, Quisling RG, Shroads AL, Shuster JJ, Silverstein JH, Theriaque DW, Valenstein E: Kontrollierter klinischer Versuch mit Dichloracetat zur Behandlung der kongenitalen Laktatazidose bei Kindern. Pediatrics. 2006, 117: 1519-1531. 10.1542/peds.2005-1226.
18 Bonnet S, Archer SL, Lalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED: Eine Mitochondrien-K+-Kanal-Achse ist bei Krebs unterdrückt und ihre Normalisierung fördert die Apoptose und hemmt das Krebswachstum. Cancer Cell. 2007, 11: 37-51. 10.1016/j.ccr.2006.10.020.
19 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC: Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010, 2: 31-34. 10.1126/scitranslmed.3000677.
20 Wong JY, Huggins GS, Debidda M, Munshi NC, De VI: Dichloracetat induziert Apoptose in Endometriumkarzinomzellen. Gynecol Oncol. 2008, 109: 394-402. 10.1016/j.ygyno.2008.01.038.
21 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ: Dichloracetat (DCA) sensibilisiert sowohl Wildtyp- als auch überexprimierende Bcl-2 Prostatakrebszellen in vitro für Strahlung. Prostate. 2008, 68: 1223-1231. 10.1002/pros.20788.
22 Robinson J, Roberts CH, Dodi IA, Madrigal JA, Pawelec G, Wedel L, Marsh SG: The European searchable tumour line database. Cancer Immunol Immunother. 2009, 58: 1501-1506. 10.1007/s00262-008-0656-5.
23 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA: Multiparameter metabolic analysis reveals a close link between attenuated mitochondrial bioenergetic function and enhanced glycolysis dependency in human tumor cells. Am J Physiol Cell Physiol. 2007, 292: C125-C136. 10.1152/ajpcell.00247.2006.
24 Brand MD, Nicholls DG: Assessing mitochondrial dysfunction in cells. Biochem J. 2011, 435: 297-312. 10.1042/BJ20110162.
25 Dahl C, Christensen C, Jonsson G, Lorentzen A, Skjodt ML, Borg A, Pawelec G, Guldberg P: Mutual Exclusivity Analysis of Genetic and Epigenetic Drivers in Melanoma Identifies a Link Between p14ARF and RARbeta Signaling. Mol Cancer Res. 2013, 11: 1166-1178. 10.1158/1541-7786.MCR-13-0006.
26 Sondergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S, Sazegar H, MacConaill LE, Barretina JG, Kehoe SM, Attar N, von EE, Zuckerman JE, Chmielowski B, Comin-Anduix B, Koya RC, Mischel PS, Lo RS, Ribas A: Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032.J Transl Med 2010, 8:39,
27 Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS: Melanome erwerben Resistenz gegen B-RAF(V600E)-Inhibition durch RTK- oder N-RAS-Hochregulierung. Nature. 2010, 468: 973-977. 10.1038/nature09626.
28 Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, Sosman JA, Kefford RF, Long GV, Nelson SF, Ribas A, Lo RS: Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance.Nat Commun 2012, 3:724,
29 de Barbi MM, Vincent G, Fayewicz SL, Bateman NW, Hood BL, Sun M, Suhan J, Duensing S, Yin Y, Sander C, Kirkwood JM, Becker D, Conrads TP, Van HB, Moschos SJ: Mitochondrial respiration-an important therapeutic target in melanoma.PLoS One 2012, 7:e40690,
30 Gilbert RJ, Klein RA:Pyruvate kinase: a carnitine-regulated site of ATP production in Trypanosoma brucei brucei. Comp Biochem Physiol B. 1984, 78: 595-599.
31 Kobayashi K, Neely JR: Mechanismus der Aktivierung der Pyruvatdehydrogenase durch erhöhte Herzarbeit. J Mol Cell Cardiol. 1983, 15: 369-382. 10.1016/0022-2828(83)90321-8.
32 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL:Sodium dichloroacetate selectively targets cells with defects in the mitochondrial ETC. Int J Cancer. 2010, 127: 2510-2519. 10.1002/ijc.25499.
33 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG:Dichloracetat induziert Apoptose und Zellzyklus-Stillstand in kolorektalen Krebszellen. Br J Cancer. 2010, 102: 1746-1752. 10.1038/sj.bjc.6605701.
34 Tong J, Xie G, He J, Li J, Pan F, Liang H: Synergistic antitumor effect of dichloroacetate in combination with 5-fluorouracil in colorectal cancer.J Biomed Biotechnol 2011, 2011:740564..,
35 Niehr F, Von EE, Attar N, Guo D, Matsunaga D, Sazegar H, Ng C, Glaspy JA, Recio JA, Lo RS, Mischel PS, Comin-Anduix B, Ribas A: Combination therapy with vemurafenib (PLX4032/RG7204) and metformin in melanoma cell lines with distinct driver mutations.J Transl Med 2011, 9:76,
36 Yuan P, Ito K, Perez-Lorenzo R, Del GC, Lee JH, Shen CH, Bosenberg MW, McMahon M, Cantley LC, Zheng B:Phenformin steigert den therapeutischen Nutzen der BRAFV600E-Hemmung beim Melanom. Proc Natl Acad Sci U S A. 2013, 110: 18226-18231. 10.1073/pnas.1317577110.
37 Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB:Systemic treatment with the antidiabetic drug metformin selectively impaired p53-deficient tumor cell growth. Cancer Res. 2007, 67: 6745-6752. 10.1158/0008-5472.CAN-06-4447.
38 Choi YW, Lim IK:Sensitization of metformin-cytotoxicity by dichloroacetate via reprogramming glucose metabolism in cancer cells. Cancer Lett. 2014, 346: 300-8. 10.1016/j.canlet.2014.01.015.
39 Bergman U, Boman G, Wiholm BE:Epidemiology of adverse drug reactions to phenformin and metformin. Br Med J. 1978, 2: 464-466. 10.1136/bmj.2.6135.464.
40DunbarEM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW:Phase-1-Studie von Dichloracetat (DCA) bei Erwachsenen mit wiederkehrenden bösartigen Hirntumoren. Invest New Drugs. 2013, 32: 452-64. 10.1007/s10637-013-0047-4.
41 Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C, Garcon G, Malet-Martino M, Balayssac S, Rocchi S, Savina A, Formstecher P, Mortier L, Kluza J, Marchetti P:Mitochondrialer oxidativer Stress ist die Achillesferse von Melanomzellen, die gegen Braf-Mutanten-Inhibitor resistent sind. Oncotarget. 2013, 4: 1986-1998.