Cecilie Abildgaard1, Christina Dahl1, Astrid L Basse2, Tao Ma2 e Per Guldberg1*
1 Centro di Ricerca della Società Danese per il Cancro, Copenaghen, Danimarca
2 Dipartimento di Biologia, Università di Copenaghen, Copenaghen, Danimarca.
Corrispondenza: [email protected]
Ricevuto: 14 settembre 2020
Accettato: 4 dicembre 2020
Pubblicato: 9 dicembre 2020
Abstract
Premessa: I progressi nel trattamento del melanoma attraverso l’inibizione mirata dell’oncogeno BRAF sono limitati a causa dello sviluppo di resistenza acquisita. Il coinvolgimento di BRAFV600E nella riprogrammazione metabolica delle cellule di melanoma fornisce un razionale per il co-targeting del metabolismo come approccio terapeutico.
Metodi: Abbiamo esaminato gli effetti del dicloroacetato (DCA), un inibitore della piruvato deidrogenasi chinasi, sulla crescita e sull’attività metabolica di linee cellulari di melanoma umano. L’effetto combinato di DCA e dell’inibitore di BRAF vemurafenib è stato studiato in linee cellulari di melanoma BRAFV600E-mutate. Le linee cellulari resistenti al vemurafenib sono state stabilite in vitro e la loro sensibilità al DCA è stata testata.
Risultati: Il DCA ha indotto una riduzione dell’attività glicolitica e dei livelli intracellulari di ATP e ha inibito la crescita cellulare. Il co-trattamento di cellule di melanoma BRAFV600E-mutanti con DCA e vemurafenib ha indotto una maggiore riduzione dei livelli di ATP intracellulare e della crescita cellulare rispetto a uno dei due composti da solo. Inoltre, le cellule di melanoma con resistenza acquisita in vitro al vemurafenib hanno mantenuto la loro sensibilità al DCA.
Conclusioni: Questi risultati suggeriscono che il DCA potenzia l’effetto del vemurafenib attraverso un’attenuazione cooperativa della produzione di energia. Inoltre, la dimostrazione del mantenimento della sensibilità al DCA in cellule di melanoma con resistenza acquisita al vemurafenib potrebbe avere implicazioni per il trattamento del melanoma.
Parole chiave: Dicloroacetato, Melanoma, BRAF, Bioenergetica, Metabolismo, ATP
Abbreviazioni: (Acetil-CoA): Acetil coenzima A, (AMPK): Proteina chinasi attivata dall’AMP, (DCA): Dicloroacetato, (ECAR): Tasso di acidificazione extracellulare, (HEMn-LP): Melanociti epidermici umani(IC50): semimassima concentrazione inibitoria, (LKB1): Chinasi epatica B1, ( MITF): Fattore di trascrizione associato alla microftalmia, ( OCR): Tasso di consumo di ossigeno, ( PDH): Piruvato deidrogenasi, ( PDK): Piruvato deidrogenasi chinasi
© 2014 Abildgaard et al.; licenziatario BioMed Central Ltd.
Sfondo
Un segno distintivo del cancro è la riprogrammazione del metabolismo cellulare verso la glicolisi aerobica. Questo modello metabolico è caratterizzato da un aumento dell’assorbimento di glucosio e da un’attività glicolitica altamente regolata con fermentazione del glucosio in acido lattico invece di una completa decomposizione aerobica nei mitocondri. La glicolisi aerobica, detta anche effetto Warburg, assomiglia al metabolismo anaerobico delle cellule normali, ma si verifica nel contesto di un adeguato apporto di ossigeno [1]. La riprogrammazione del metabolismo nelle cellule tumorali è un processo altamente complesso ed eterogeneo, guidato da un’ampia varietà di strategie genetiche e non genetiche per superare la restrizione energetica [2]-[4].
L’oncogene BRAF V600E, presente in oltre il 50% dei melanomi [5], è stato direttamente coinvolto nella riprogrammazione del metabolismo cellulare. L’attività costitutiva di BRAF mutante riduce l’espressione degli enzimi ossidativi e il numero di mitocondri, mentre aumenta l’espressione degli enzimi glicolitici e la produzione di acido lattico [6],[7]. Inoltre, è stato riconosciuto un legame molecolare tra la via RAS-RAF-MEK-ERK-MAPK e il check-point dello stress energetico mediato dalla via della chinasi epatica B1 (LKB1)-AMP activated protein kinase (AMPK), suggerendo un ruolo di BRAFV600E nel mediare la resistenza allo stress energetico [8],[9]. BRAF influenza il metabolismo ossidativo attraverso il controllo del fattore di trascrizione associato alla microftalmia (MITF), dipendente dal regolatore mitocondriale PGC1α [7]. Studi precedenti hanno dimostrato che i melanomi che esprimono PGC1α hanno un fenotipo più ossidativo rispetto ai melanomi PGC1α-negativi [4],[7]. Inoltre, è stato dimostrato che BRAFV600E media la senescenza indotta dall’oncogene attraverso una regolazione metabolica. Questo meccanismo comporta un aumento dell’attività della piruvato deidrogenasi (PDH) attraverso la soppressione della piruvato deidrogenasi chinasi (PDK) [10]. La PDH controlla l’accoppiamento tra glicolisi e respirazione mitocondriale facilitando l’afflusso di piruvato nei mitocondri, promuovendo l’utilizzo completo del glucosio. L’asse PDK-PDH è spesso disregolato nel cancro, dove la sovraespressione di PDK riduce l’accoppiamento tra i due sistemi energetici, contribuendo così all’effetto Warburg [11],[12]. Sulla base di questi risultati, l’inibizione mirata di PDK è stata proposta come opzione terapeutica per il melanoma, con un possibile effetto sinergico degli inibitori chimici di BRAFV600E, come vemurafenib [10],[13].
Il dicloroacetato (DCA) è un inibitore delle quattro isoforme della PDK ed è stato precedentemente utilizzato per il trattamento dell’acidosi lattica [14],[15], con bassa tossicità a livelli di dose efficaci [16],[17]. Diversi studi hanno dimostrato che il DCA inverte l’effetto Warburg nelle cellule tumorali e influisce negativamente sulla loro crescita e sopravvivenza [13],[18]-[21]. Questo effetto è stato attribuito a una normalizzazione del potenziale di membrana mitocondriale dallo stato iperpolarizzato che caratterizza le cellule tumorali. Le variazioni del potenziale di membrana comportano la riapertura dei canali anionici voltaggio-gati e hanno dimostrato di introdurre una risensibilizzazione all’apoptosi, dovuta alla riacquisita capacità di rilasciare mediatori pro-apoptotici [18]. Qui abbiamo studiato l’effetto del DCA sulle cellule di melanoma. In particolare, abbiamo analizzato le risposte cellulari in termini di metabolismo, bioenergetica, crescita, proliferazione e morte cellulare in linee cellulari di melanoma, melanociti umani primari e cellule di melanoma BRAFV600E-mutanti con resistenza acquisita a vemurafenib.
I metodi
Icomposti chimici
DCA(dicloroacetato di sodio) e 2-Deossi-D-glucosio (2-DG) sono stati acquistati da Sigma-Aldrich e disciolti in dH2Ofino a concentrazioni di partenza di 1 M. Vemurafenib (PLX4032) è stato acquistato da Selleck Chemicals e disciolto in DMSO fino a una concentrazione di partenza di 0,05 M.
Coltura cellulare
Le linee cellulari di melanoma ED-007, ED-013, ED-024, ED-027, ED-029, ED-034, ED-050, ED-070, ED-071, ED-117, ED-140, ED-179 e ED-196 sono state ottenute dall’European Searchable Tumour line Database (ESTDAB, ED) [22]. La linea cellulare di melanoma SK-MEL-28 è stata acquistata dall’ATCC. I melanociti epidermici umani primari (neonatali) da tessuto leggermente pigmentato (HEMn-LP) sono stati acquistati da Invitrogen. Le linee cellulari di melanoma sono state coltivate a 37°C con il 5%di CO2 in terreno RPMI-1640 integrato con il 10% di siero fetale bovino e l’1% di penicillina/streptomicina. Le cellule HEMn-LP sono state coltivate nelle stesse condizioni in terreno 254CF integrato con l’1% di supplemento per la crescita dei melanociti umani (HMGS-2) e 12-O-tetradecanoil-forbolo-13-acetato(TPA; 10 ng/ml). Tutti i terreni e i supplementi sono stati acquistati da Invitrogen.
Analisi metabolica
La caratterizzazione metabolica è stata eseguita su linee cellulari di melanoma e melanociti umani primari utilizzando un analizzatore di flusso extracellulare Seahorse XF96 (Seahorse Bioscience, Billerica, MA), che esegue misure in tempo reale del tasso di acidificazione extracellulare (ECAR) e del tasso di consumo di ossigeno (OCR). È stato progettato un saggio per studiare la capacità dei sistemi energetici mitocondriale e glicolitico. L’ECAR e l’OCR sono stati misurati in condizioni basali e durante l’aggiunta successiva di cinque modulatori metabolici: L’inibitore dell’ATP sintasi, oligomicina (1 μM); il permeabilizzatore della membrana mitocondriale, carbonilcianuro-4-(trifluorometossi)fenilidrazone (FCCP) (1 μM); gli inibitori della respirazione mitocondriale, rotenone (1 μM) e antimicina A (1 μM); e l’inibitore glicolitico, 2-DG (100 mM). Il kit XF Cell Mito Stress, contenente oligomicina, FCCP, rotenone e antimicina A, è stato acquistato da Seahorse Bioscience.
Misurazioni dell’ATP
I livelli intracellulari di ATP sono stati misurati con il sistema ATPlite, 1 step Luminescence Assay System (Perkin Elmer), un metodo basato sulla reazione dell’ATP con luciferasi e D-luciferina. Le cellule sono state seminate in triplicato con 10.000 cellule per pozzetto e trattate con i composti indicati e con il veicolo di controllo per 2 o 24 ore. La luminescenza è stata misurata con il lettore di micropiastre Spectra Max Gemini EM (Molecular Devices) e normalizzata ai livelli di fondo.
Saggio del violetto di cristallo
Per valutare l’effetto dei composti studiati sulla crescita cellulare è stato applicatounsaggio del violetto di cristallo. Le cellule sono state seminate in duplicato a una densità adeguata e poi trattate con DCA, vemurafenib, i due composti combinati e il veicolo di controllo. Il terreno e i composti di trattamento sono stati sostituiti ogni 48 ore. L’esperimento è stato ripetuto tre volte in modo indipendente. Per terminare l’esperimento, il terreno e le cellule non attaccate sono state rimosse e le cellule rimanenti sono state lavate in PBS e fissate con glutaraldeide per 15 minuti. Le cellule fissate sono state incubate con una soluzione di cristalvioletto (0,1% cristalvioletto, 20% CH3OH) per 1 ora. La quantità di colorante assunta dal monostrato, proporzionale al numero di cellule vitali attaccate al fondo del pozzetto, è stata quantificata estraendo il colore con acido acetico al 10% e misurando l’assorbanza a una lunghezza d’onda di 595 nm. La correlazione lineare tra l’assorbanza e il numero di cellule è stata verificata eseguendo una curva standard. La crescita cellulare relativa è stata determinata normalizzando ai controlli non trattati dopo la sottrazione dello sfondo (senza cellule).
Saggio di proliferazione cellulare
Le cellule di melanoma sono state seminate in triplicato con 500-1.000 cellule per pozzetto e trattate con DCA alle concentrazioni indicate e con il controllo veicolo per 96 ore. La proliferazione è stata quindi misurata rilevando BrdU dopo 12 ore di incorporazione nel DNA cellulare. La procedura è stata condotta secondo il protocollo fornito con il BrdU Cell Proliferation Assay Kit (Cell Signaling Technology®).
Rilevamento dell’apoptosi con Annexin V-FITC
Il rilevamento dell’apoptosi è stato eseguito utilizzando un kit di rilevamento dell’apoptosi con Annexin V-FITC (BD Bioscience), secondo il protocollo fornito. Le cellule sono state raccolte e lavate due volte in PBS freddo. Le cellule sono state poi trasferite in un’altra provetta, centrifugate e risospese nel tampone di legame. Dalla risospensione, 5 ×105 cellule sono state trasferite in provette FACS e colorate con Annexin V-FITC e ioduro di propidio (PI). Dopo 30 minuti di incubazione, la citometria a flusso è stata eseguita su uno strumento Cytomics FC 500 MPL (Beckman Coulter). Le cellule non colorate sono state incluse come controllo.
Induzione della resistenza acquisita in vitroalvemurafenib
La resistenza acquisita al vemurafenib è stata indotta in sette colture derivate da quattro linee cellulari di melanoma BRAFV600E-mutanti e sensibili al vemurafenib (ED-013, ED-071, ED-196 e SK-MEL-28). Le cellule sono state coltivate in concentrazioni crescenti di vemurafenib finché non sono cresciute stabilmente in una concentrazione superiore all’IC50 e sono state quindi mantenute in terreno contenente vemurafenib.
Pirosequenziamento
Il pirosequenziamento degli hotspot di mutazione in BRAF e NRAS è stato eseguito su una piattaforma PyroMark Q24 (Qiagen), utilizzando i reagenti PyroMark Gold Q24 (Qiagen). Le sequenze dei primer sono elencate nel file aggiuntivo 1: Tabella S1.
| Sito target | Nome del primer | Sequenza del primer (5′-3′) |
| BRAF V600E | BRAF-F1 | [Btn]-TTCATGAAGACCTCACAGTAAAA |
| BRAF-R1 | GGCCAAAAATTTAATCAGTGGAA | |
| BRAF-S1 | CCACTCCATCGAGATTT | |
| NRAS Q61K,L,R | NRAS-F1 | [Btn]-ACCCCCAGGATTCTTACAGAAA |
| NRAS-R1 | CGCAAATGACTTGCTATTGA | |
| NRAS-S1 | TCATGGCACTGTACTCTT |
Analisi dell’espressione di PGC1α
L’RNA totale è stato isolato utilizzando il kit RNeasy mini (Qiagen) e il cDNA è stato sintetizzato con il kit SuperScript™ III Reverse Transcriptase (Invitrogen). Come primer per la sintesi del cDNA sono stati utilizzati oligo dT24 e random hexamers. L’espressione genica di PGC1α è stata determinata con la PCR quantitativa in tempo reale su Roche LightCycler 2.0 utilizzando il kit LigthCycler FastStart DNA MasterPLUS SYBR Green I (Roche). Le sequenze dei primer erano: PPARGC1A_2241F: 5′-GCTGTACTTTTGTGGACGCA-3′ e PPARGC1A_2306R: 5′-GGAAGCAGGGTCAAAGTCAT-3′. L’espressione è stata normalizzata rispetto all’espressione del gene housekeeping RPLP0. Le sequenze dei primer erano: RPLP0_433F: 5′-ACTAAAATCTCCAGGGCACC-3′ e RPLP0_547R: 5′-ATGACCAGCCCAAAGGAGAA-3′. Le due linee cellulari di melanoma ED-050 e SK-MEL-28 sono state incluse come controlli positivi e negativi, rispettivamente [4].
Analisi statistica
Le differenze tra serie di dati indipendenti sono state determinate con il test t di Student. Per l’analisi statistica della varianza tra i diversi trattamenti (controllo con veicolo, DCA, vemurafenib e la combinazione di DCA e vemurafenib) è stata utilizzata l’ANOVA a una via per campioni appaiati. Per determinare la significatività statistica è stato utilizzato il test di confronto multiplo HSD (honest significance difference) di Tukey. Il coefficiente di correlazione di Pearson è stato utilizzato per determinare la correlazione tra la sensibilità ai DCA e i parametri metabolici. Un valore di 1 indicava una correlazione positiva, 0 nessuna correlazione e -1 una correlazione negativa.
Gli esperimenti condotti in questo studio hanno coinvolto solo linee cellulari disponibili in commercio e pertanto non hanno richiesto l’approvazione del comitato etico.
Risultati
Caratterizzazione metabolica di linee cellulari di melanoma e melanociti primari
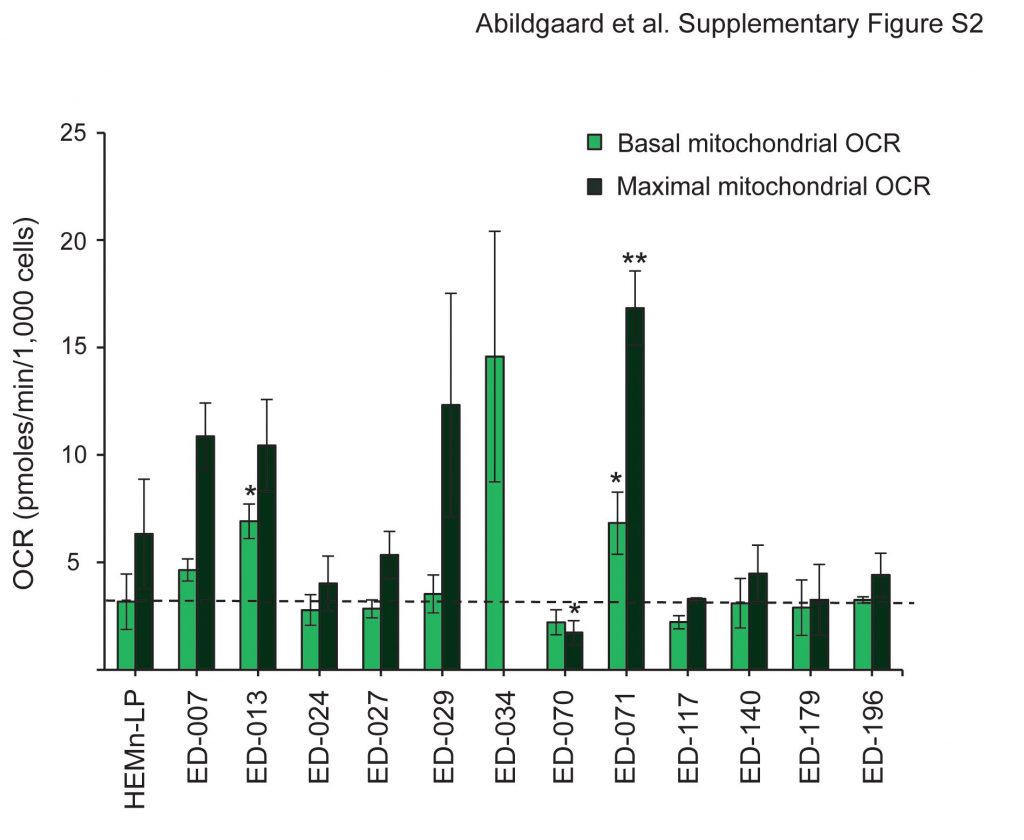
La profilazione metabolica di 12 linee cellulari di melanoma e di melanociti umani primari (HEMn-LP) è stata eseguita utilizzando l’analizzatore Seahorse FX96. Questo strumento esegue misure in tempo reale del tasso di acidificazione extracellulare (ECAR) e del tasso di consumo di ossigeno (OCR), che sono misure indirette dell’attività glicolitica e della respirazione mitocondriale, rispettivamente [23]. Le misurazioni sono state eseguite in condizioni basali e durante l’aggiunta successiva di cinque modulatori metabolici (Figura 1A). Rispetto ai melanociti normali, 11 linee cellulari su 12 presentavano tassi glicolitici più elevati, come indicato da ECAR glicolitici basali più alti, a dimostrazione che l’effetto Warburg è una caratteristica generale delle cellule di melanoma. Inoltre, nove delle linee cellulari hanno mostrato capacità glicolitiche massime più elevate rispetto ai melanociti (Figura 1B). Con poche eccezioni, non ci sono state differenze significative nella respirazione mitocondriale basale e massima tra melanociti e cellule di melanoma (File aggiuntivo 2: Figura S2). In base ai rapporti OCR-to-ECAR (OCR/ECAR; Figura 1C), il contributo relativo della respirazione mitocondriale alla produzione di ATP è risultato inferiore in 10 linee cellulari di melanoma rispetto ai melanociti.
.
.
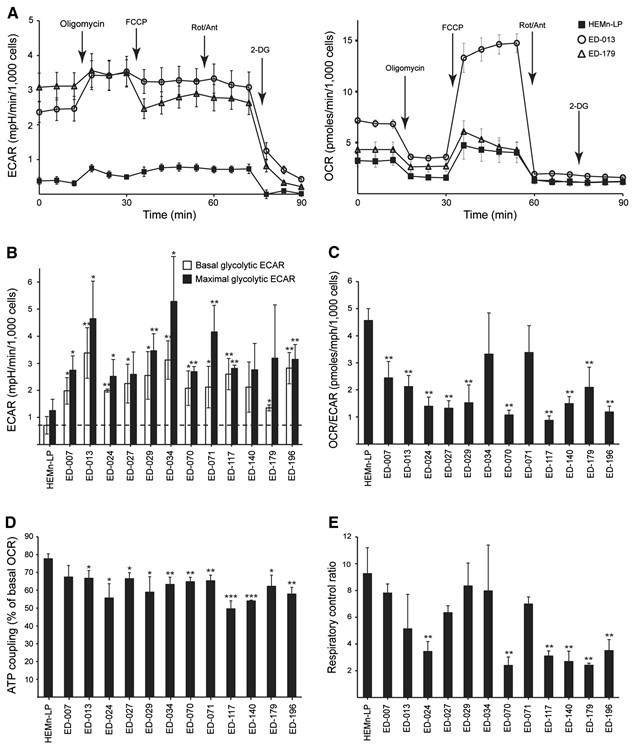
L’efficienza mitocondriale (accoppiamento ATP) e le prestazioni (rapporto di controllo respiratorio) sono state stimate in base alle misurazioni di due parametri della funzione mitocondriale: la perdita di protoni e l’OCR mitocondriale massimo [24]. Il leak protonico è stato determinato dopo l’aggiunta dell’inibitore dell’ATP sintasi oligomicina, mentre l’OCR mitocondriale massimo è stato determinato dopo l’aggiunta del disaccoppiatore mitocondriale FCCP. Questa analisi ha mostrato un accoppiamento ATP significativamente inferiore nelle cellule di melanoma rispetto ai melanociti (p < 0,05; ED-007: p = 0,07), suggerendo una maggiore perdita di protoni e una produzione meno efficiente di ATP rispetto al livello di consumo di ossigeno (Figura 1D). Inoltre, sei delle linee cellulari di melanoma presentavano anche rapporti di controllo respiratorio significativamente più bassi rispetto ai melanociti (p?<?0.01, Figura 1E), indicativi di una scarsa performance mitocondriale.
IlDCA sposta il metabolismo verso la respirazione mitocondriale e riduce i livelli di ATP
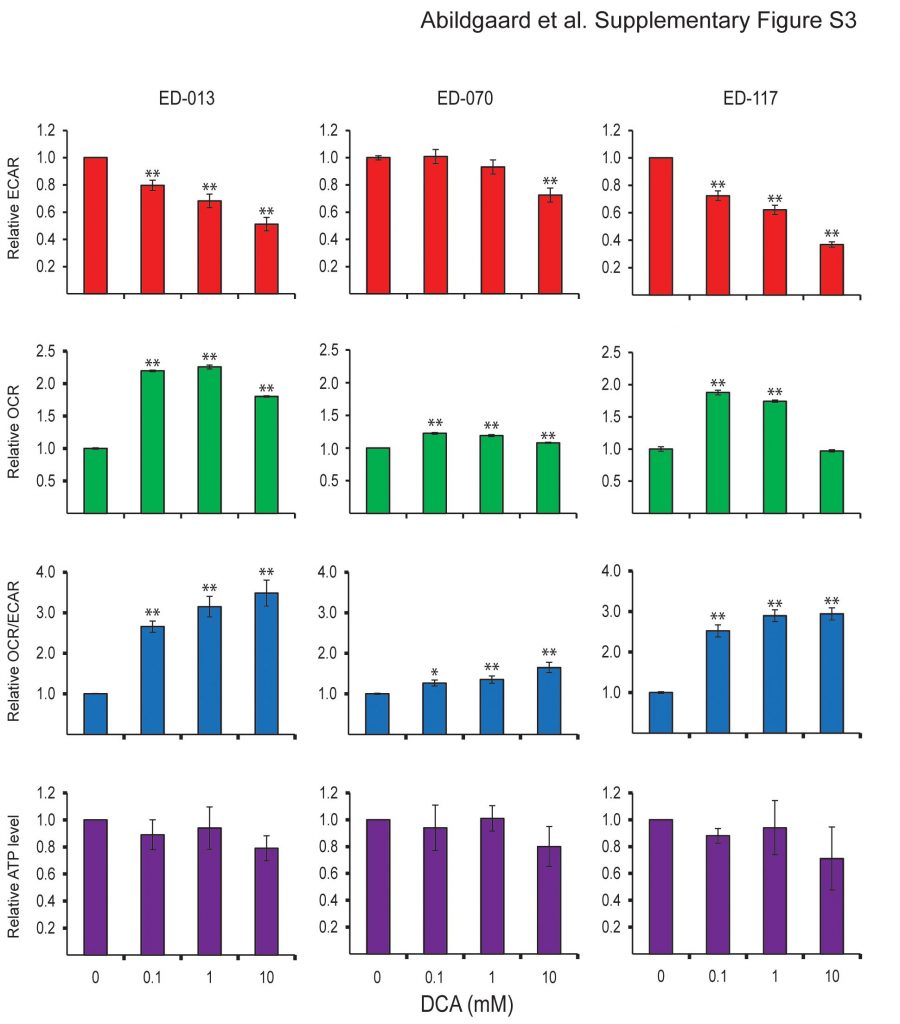
Per determinare l’effetto del DCA sul metabolismo delle cellule di melanoma, abbiamo analizzato il pannello di 12 linee cellulari utilizzando l’analizzatore Seahorse XF96. Dopo il trattamento con 10 mM di DCA per 2 ore, tutte le linee cellulari hanno risposto con una riduzione dell’ECAR e un aumento dell’OCR (Figura 2A), indicando uno spostamento verso la respirazione mitocondriale. La risposta dell’ECAR è stata simile tra le linee cellulari, mentre c’è stata una grande variazione nella risposta dell’OCR (Figura 2A). Le variazioni relative di ECAR, OCR e OCR/ECAR in risposta al DCA sono risultate dipendenti dalla concentrazione (File aggiuntivo 3: Figura S3).
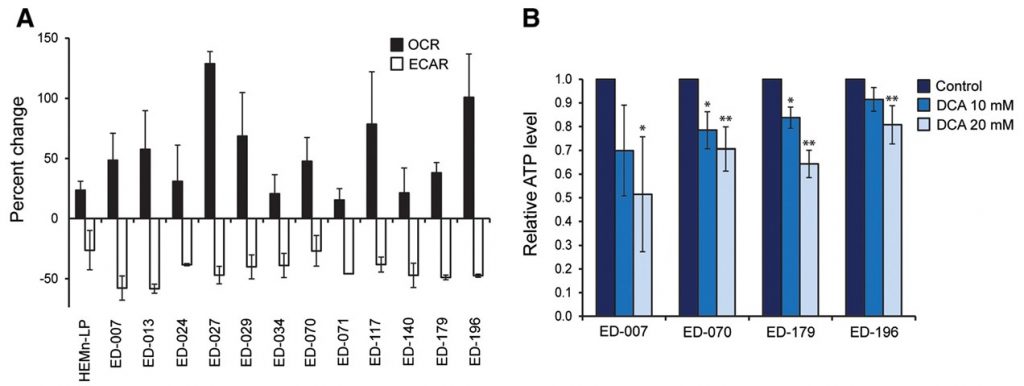
Per determinare se lo spostamento del metabolismo interferisse con l’omeostasi energetica, abbiamo misurato i livelli di ATP nelle cellule di melanoma dopo il trattamento con DCA 0,1, 1 o 10 mM per 2 ore. Tutte e tre le linee cellulari testate sono state in grado di sostenere i livelli di ATP dopo il trattamento con basse concentrazioni di DCA (0,1-1 mM), mentre una tendenza alla riduzione dell’ATP è stata osservata nelle colture trattate con 10 mM di DCA (File aggiuntivo 3: Figura S3). Quando le cellule sono state trattate con DCA (10 o 20 mM) per 24 ore, è stata osservata una significativa diminuzione dell’ATP in funzione della concentrazione (Figura 2B), indicando un graduale esaurimento del sistema metabolico.
IlDCA riduce la crescita delle cellule di melanoma indipendentemente dallo stato genetico-driver e dall’espressione di PGC1α
Per valutare una possibile applicazione clinica del DCA per il trattamento del melanoma, abbiamo studiato i suoi effetti su diversi parametri di crescita cellulare. L’intero gruppo di linee cellulari e i melanociti primari sono stati trattati con un range di concentrazioni di DCA (0,5-100 mM) per 96 ore (vedi Figura 3A per i risultati rappresentativi). Tutte le linee cellulari hanno mostrato una riduzione della crescita dipendente dalla concentrazione, con valori IC50 compresi nell’intervallo 9-38 mM (Tabella 1), rispetto a un valore IC50 di 70 mM per i melanociti primari (p < 0,001). Non vi è stata alcuna correlazione tra la risposta al DCA e lo stato di mutazione BRAF/NRAS o i livelli di espressione di PGC1α (Tabella 1).
| Linea cellulare | DCA IC50(mM)4 | StatoBRAF/ NRASS1 | Espressione di PGC1α2,4 |
|---|---|---|---|
| ED-070 | 8.9 ± 0.6*** | NRASQ61L | 1.0 ± 0.2 |
| ED-007 | 12.2 ± 2.2*** | WT3 | 0.6 ± 0.0 |
| ED-071 | 12.3 ± 3.6*** | BRAFV600E | 0.0 ± 0.0 |
| ED-034 | 12.7 ± 1.7*** | BRAFL597S | 1.0 ± 0.2 |
| ED-013 | 14.4 ± 2.0*** | BRAFV600E | 1.9 ± 0.3 |
| ED-027 | 17.7 ± 2.1*** | BRAFV600E | 0.5 ± 0.1 |
| SK-MEL-28 | 20.0 ± 4.5*** | BRAFV600E | 0.0 ± 0.0 |
| ED-179 | 20.6 ± 1.8*** | NRASQ61R | 0.3 ± 0.0 |
| ED-024 | 21.9 ± 1.6*** | NRASQ61L | 0.0 ± 0.0 |
| ED-140 | 23.9 ± 2.0*** | WT | 0.4 ± 0.1 |
| ED-050 | 24.1 ± 3.1*** | WT | 2.7 ± 0.5 |
| ED-029 | 29.7 ± 5.1*** | BRAFV600K | 1.3 ± 0.2 |
| ED-196 | 35.8 ± 3.2*** | BRAFV600E | 1.5 ± 0.5 |
| ED-117 | 37.6 ± 2.2*** | BRAFV600E | 1.2 ± 0.1 |
| HEMn-LP | 69.1 ± 6.4 | WT | 1 |
| ED-013-R1 | 12.6 ± 3.0*** | BRAFV600E | |
| ED-013-R2 | 13.6 ± 2.4*** | BRAFV600E | |
| ED-071-R1 | 12.2 ± 0.9*** | BRAFV600E | |
| ED-071-R2 | 13.8 ± 3.7*** | BRAFV600E | |
| SK-MEL-28-R1 | 23.1 ± 4.6*** | BRAFV600E | |
| SK-MEL-28-R2 | 26.2 ± 8.0** | BRAFV600E |
**p < 0,01; ***p < 0,001 rispetto a HEMn-LP.
1BRAF/NRASsono in accordo con i risultati pubblicati[25].
2Relativiall’espressione di PGC1α nei melanociti normali.
3wildtype.
4Ivalori IC50 diDCAe l’espressione di PGC1α rappresentano le medie di tre misurazioni indipendenti ± deviazione standard.
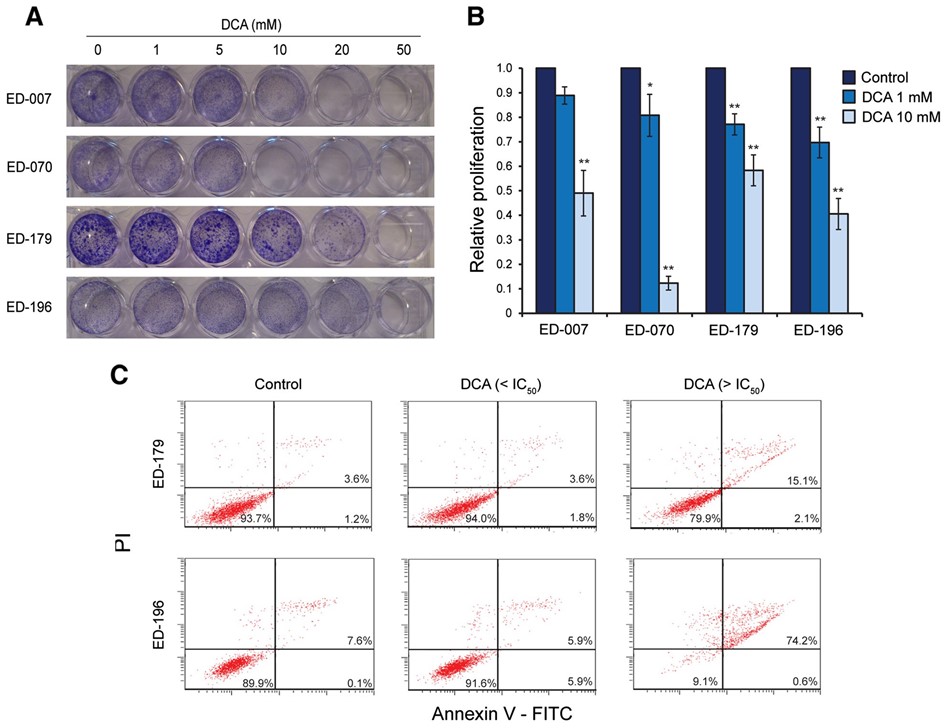
Per caratterizzare ulteriormente gli effetti del DCA sulla crescita cellulare, abbiamo misurato l’incorporazione di BrdU in cellule trattate con 1 o 10 mM di DCA per 96 ore. Come mostrato nella Figura 3B, tutte e quattro le linee cellulari testate hanno risposto con una riduzione della proliferazione, nell’ordine dell’11-30% a 1 mM e del 42-88% a 10 mM. Abbiamo anche misurato la risposta apoptotica al DCA mediante l’analisi citometrica a flusso dei livelli di annexina V. A concentrazioni di DCA inferiori all’IC50, il numero di cellule annexin V-positive non è aumentato dopo 96 ore e fino a 3 settimane. Al contrario, il trattamento con concentrazioni superiori all’IC50 ha aumentato il numero di cellule positive sia all’annexina V che al PI, indicando l’induzione della morte cellulare già dopo 96 ore (Figura 3C).
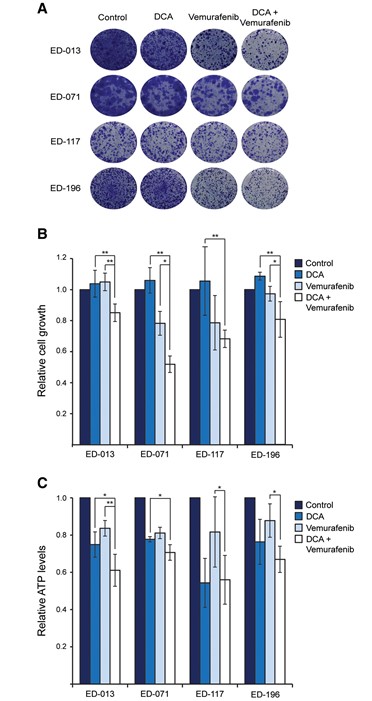
IlDCA potenzia l’effetto del vemurafenib sulle cellule di melanoma BRAFV600E-mutanti
Per verificare se il DCA possa essere utilizzato per migliorare l’efficacia degli inibitori chimici di BRAF nel trattamento del melanoma, abbiamo testato varie combinazioni di DCA e vemurafenib sulla crescita cellulare. Il trattamento di quattro linee cellulari BRAFV600E-mutanti con vemurafenib (0,05-5 μM) per 96 ore ha rivelato valori di IC50 da 0,5 a 4,5 μM, coerentemente con i dati di studi precedenti [26]. Esponendo i melanociti primari allo stesso trattamento, non abbiamo raggiunto il valore IC50 per queste cellule, nemmeno con la concentrazione più alta testata (5 μM), confermando la specificità del composto.
Quando le cellule sono state trattate con 1 mM di DCA in combinazione con basse concentrazioni di vemurafenib (<IC50), la riduzione della crescita cellulare è stata più pronunciata rispetto a quella ottenuta con DCA o vemurafenib da soli (p <0,05; Figura 4A, B). Il DCA non ha potenziato l’effetto di vemurafenib nelle cellule ED-117, il che può essere attribuito alla resistenza intrinseca di queste cellule al DCA (IC50 38 mM; Tabella 1). Alle concentrazioni IC50, sia il DCA che il vemurafenib hanno causato una riduzione dei livelli intracellulari di ATP quando sono stati usati come agenti singoli e un’ulteriore riduzione quando sono stati usati in combinazione, anche se non statisticamente significativa per tutte le linee cellulari (Figura 4C).
Lelinee cellulari resistenti al vemurafenib hanno una migliore capacità ossidativa e mantengono la sensibilità al DCA
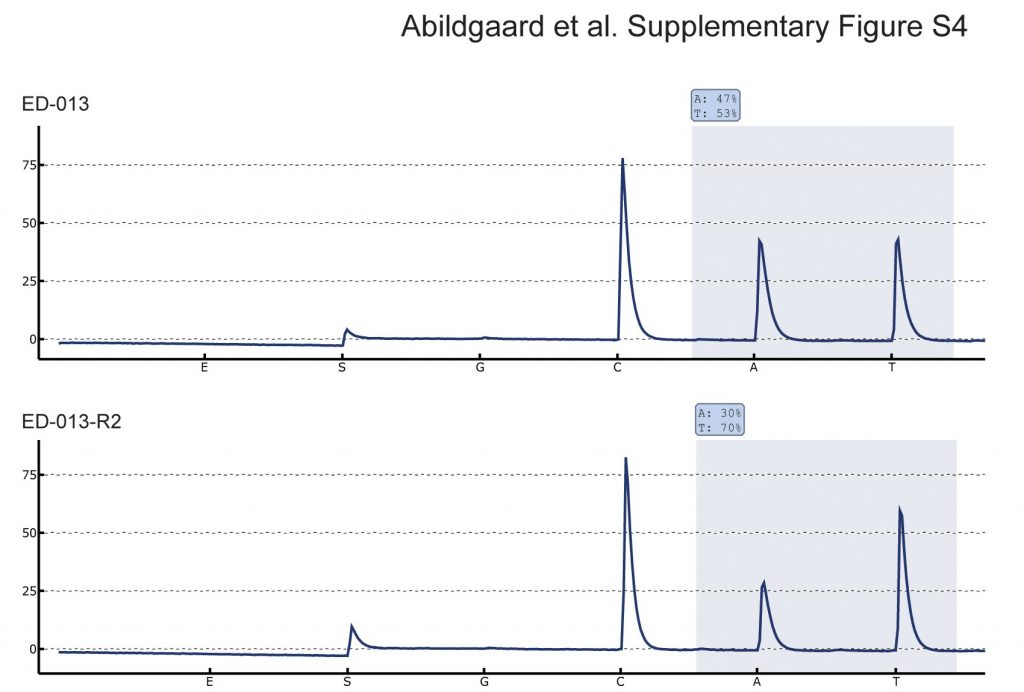
Sette colture di cellule resistenti al vemurafenib sono state generate da quattro linee cellulari BRAFV600E-mutanti esponendo le cellule a concentrazioni crescenti di vemurafenib. Le cellule sono state considerate resistenti quando potevano essere continuamente propagate a una concentrazione di vemurafenib superiore all’IC50. Il DNA è stato isolato da tutte e sette le colture e analizzato per l’aumento del numero di copie di BRAF e per le mutazioni secondarie di NRAS, che sono due meccanismi ben descritti di resistenza acquisita a vemurafenib [27],[28]. Il pirosequenziamento ha rivelato un aumento del rapporto tra BRAF V600E e BRAF WT in una delle linee cellulari resistenti (ED-013-R2) rispetto alla linea cellulare parentale (File aggiuntivo 4: Figura S4). Nelle restanti linee cellulari resistenti non sono state riscontrate alterazioni di BRAF o NRAS.
La caratterizzazione metabolica di due linee cellulari resistenti (ED-013-R1 e ED-196-R) mediante l’analizzatore Seahorse XF96 ha mostrato che entrambe le linee cellulari resistenti presentavano un profilo metabolico trasformato con un aumento significativo della capacità respiratoria massima (Figura 5A), ma nessun cambiamento nell’OCR respiratorio basale, nell’accoppiamento ATP o nell’OCR non mitocondriale. È interessante notare che, quando sono state testate per la sensibilità al DCA, le linee cellulari resistenti al vemurafenib avevano tutte valori di IC50 simili a quelli delle linee cellulari parentali (Tabella 1).
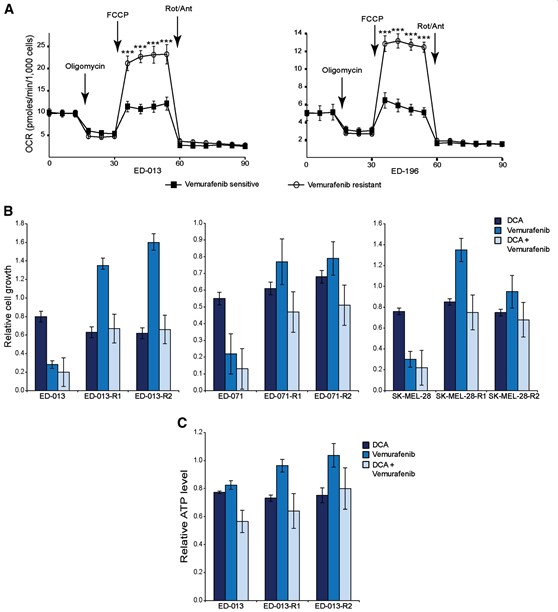
La Figura 5Billustra la sensibilità delle linee cellulari resistenti al vemurafenib al DCA e al vemurafenib, come agenti singoli o in combinazione, rispetto alle linee cellulari parentali. La crescita delle linee cellulari resistenti è risultata leggermente ridotta, inalterata o addirittura aumentata in presenza di vemurafenib dopo 96 ore, mentre la sensibilità al DCA era simile a quella delle cellule parentali, sia in presenza che in assenza di vemurafenib (Figura 5B). Risultati simili sono stati ottenuti misurando le variazioni dei livelli di ATP dopo il trattamento con gli stessi farmaci per 24 ore (Figura 5C). Nel complesso, questi dati dimostrano che le cellule di melanoma resistenti al vemurafenib hanno mantenuto la sensibilità al DCA, nonostante il cambiamento del loro profilo metabolico.
Discussione
La terapia metabolica mirata per il cancro si è concentrata principalmente sul bersaglio dell’approvvigionamento energetico attraverso l’inibizione della glicolisi. Tuttavia, il riconoscimento che i mitocondri possono contribuire attivamente alla progressione del melanoma ha aumentato l’attenzione sul metabolismo ossidativo come potenziale bersaglio terapeutico [10],[13],[29]. Il DCA promuove l’attivazione PDK-dipendente della PDH, invertendo la produzione di lattato a favore dell’afflusso di piruvato nei mitocondri [15],[18]. Attraverso questo meccanismo, il DCA migliora l’accoppiamento tra glicolisi e respirazione mitocondriale, con un impatto maggiore sulle cellule con un accoppiamento carente, come le cellule tumorali [18]. Tutte le linee cellulari di melanoma esaminate nel nostro studio hanno risposto al DCA con una riduzione della produzione di lattato e un aumento dell’OCR. Questo spostamento verso la respirazione mitocondriale avrebbe dovuto ottimizzare l’utilizzo dei substrati e portare a un rendimento energetico più efficiente, ma ha invece portato a un calo significativo dei livelli di ATP nonostante un accoppiamento mitocondriale ATP inalterato o addirittura aumentato. La riduzione osservata dell’ECAR in risposta al DCA suggerisce che l’inibizione della glicolisi potrebbe essere uno dei principali fattori di deprivazione energetica. Un meccanismo di inibizione della glicolisi da parte del DCA non è stato descritto in precedenza. Tuttavia, è stato dimostrato che la piruvato chinasi, l’ultimo sito che produce ATP nella via glicolitica, è regolata negativamente dall’acetil coenzima A (acetil-CoA) [30]. Poiché l’attivazione della PDH aumenta direttamente la formazione di acetil-CoA [31], ciò potrebbe spiegare l’inibizione della glicolisi mediata dal DCA. La somiglianza strutturale tra DCA e piruvato [32] potrebbe anche implicare un’inibizione diretta della glicolisi da parte del DCA, forse attraverso un meccanismo di feedback allosterico.
La risposta metabolica al DCA è stata accompagnata da una riduzione della proliferazione delle cellule di melanoma, indipendentemente dallo stato di driver genetico e dai profili metabolici di queste cellule. Diversi studi precedenti hanno dimostrato un effetto apoptotico del DCA sulle cellule tumorali [13],[18],[19],[32]-[34]. Tuttavia, in accordo con i nostri risultati, la risposta apoptotica è stata attivata solo a concentrazioni troppo elevate per essere clinicamente rilevanti [32]. Per esplorare ulteriormente la rilevanza clinica del DCA nel trattamento del melanoma, abbiamo esaminato l’efficacia di questo agente in combinazione con l’inibitore BRAF vemurafenib. Questi esperimenti hanno dimostrato un effetto potenziante del DCA sull’inibizione della crescita delle cellule di melanoma BRAFV600E-mutanti. A basse concentrazioni di DCA che da sole non avevano alcun effetto sulla crescita cellulare, la combinazione con basse concentrazioni di vemurafenib aveva un effetto di riduzione della crescita significativamente più forte rispetto al vemurafenib da solo. Questo effetto potenziante del DCA si è riflesso anche nella riduzione dei livelli di ATP. L’analisi biochimica ha dimostrato la capacità di BRAFV600E di disaccoppiare la via di rilevamento energetico LKB1-AMPK, promuovendo la resistenza alla deprivazione energetica e prevenendo una risposta apoptotica [8],[9]. Il trattamento con inibitori di BRAF ripristina questa via [35] e può, quindi, potenziare la risposta ai composti che riducono la generazione di ATP. Sia il DCA che il vemurafenib sopprimono l’attività glicolitica nelle cellule di melanoma, rendendole così più dipendenti dalla respirazione mitocondriale [6]. Poiché la glicolisi rappresenta un’ampia frazione della produzione totale di energia in queste cellule, l’inibizione di questo processo comporterà un’elevata richiesta di produzione di ATP da parte del sistema ossidativo. Le minori prestazioni dei mitocondri nelle cellule di melanoma potrebbero spiegare l’incapacità di queste cellule di sostenere i livelli di ATP in presenza di DCA e vemurafenib. L’effetto cooperativo di questi composti nell’abbassare i livelli di ATP suggerisce che la soglia energetica che promuove l’arresto della crescita o la morte cellulare nelle cellule di melanoma può essere raggiunta con concentrazioni inferiori di vemurafenib in presenza di DCA.
Studi precedenti hanno esaminato la capacità dei modulatori metabolici di migliorare l’effetto terapeutico degli inibitori di BRAF per il trattamento del melanoma. La combinazione di PLX4720 (un analogo di vemurafenib) con una delle due biguanidi antidiabetiche, metformina e fenformina, ha mostrato un’inibizione sinergica della vitalità delle cellule di melanoma [35],[36]. Entrambi gli agenti compromettono la sintesi di ATP attraverso l’inibizione dell’attività del complesso I mitocondriale, con conseguente riduzione del rapporto ATP/ADP e attivazione della via LKB1-AMPK per sopprimere la crescita [35],[36]. A differenza del DCA, la metformina e la fenformina stimolano entrambe la glicolisi e la produzione di acido lattico [37],[38], il che potrebbe spiegare gli effetti di stimolazione della crescita della metformina su alcune linee cellulari di melanoma quando viene utilizzata come agente singolo. Inoltre, le concentrazioni alle quali la metformina era efficace erano superiori a un livello terapeuticamente rilevante [35]. La fenformina era significativamente più potente della metformina [36], ma è stata associata a un elevato rischio di acidosi lattica [39] ed è stata ritirata dal mercato per il trattamento del diabete di tipo 2 in molti Paesi. Il DCA, invece, ha dimostrato di potenziare l’effetto del vemurafenib a concentrazioni fino a 1 mM e in precedenza ha dimostrato di avere pochi effetti avversi quando somministrato ai pazienti [17],[19],[40]. Questi risultati alludono a un potenziale terapeutico del DCA come co-farmaco per il trattamento con vemurafenib del melanoma BRAFV600E-mutante. Ciò è stato rafforzato dalla dimostrazione che la sensibilità al DCA è stata mantenuta in linee cellulari di melanoma con resistenza acquisita al vemurafenib. Sebbene le cellule resistenti mostrassero un profilo metabolico alterato con un aumento significativo della respirazione mitocondriale massima, come dimostrato anche da Corazao-Rozas et al. [41], erano sensibili al DCA come le cellule parentali sensibili al vemurafenib. Pertanto, il DCA potrebbe fornire una strategia per prevenire la comparsa di sottopopolazioni tolleranti al vemurafenib durante il trattamento iniziale e quindi posticipare o prevenire lo sviluppo della resistenza.
Conclusioni
Abbiamo fornito una comprensione più elaborata degli effetti del DCA sul metabolismo e sulla crescita delle cellule di melanoma. La capacità del DCA di ridurre i livelli di ATP e la crescita del melanoma sembra potenziare l’effetto di vemurafenib, un farmaco già utilizzato in clinica per il trattamento dei melanomi metastatici BRAFV600E-mutanti. È importante notare che le cellule di melanoma con resistenza acquisita al vemurafenib hanno mantenuto la loro sensibilità al DCA. Questi risultati dovrebbero incoraggiare ulteriori indagini su questa combinazione di farmaci e sull’applicazione in vivo del DCA.
File aggiuntivi
File aggiuntivo 1: Tabella S1.(33K, doc)
Primer di pirosequenziamento per l’amplificazione e il sequenziamento degli hotspot di mutazione BRAF e NRAS. I primer forward, reverse e di sequenziamento sono indicati rispettivamente con F, R e S.
File aggiuntivo 2:Figura S2.(8.5M, tiff)
Valori di OCR mitocondriale basale e massima per linee cellulari di melanoma e melanociti epidermici umani (HEMn-LP). L’OCR misurata dopo l’aggiunta di rotenone/antimicina A (OCR non mitocondriale) è stata sottratta da tutti i valori. La linea tratteggiata indica l’OCR basale di HEMn-LP. I valori indicati sono le medie di tre misurazioni indipendenti ± deviazione standard. Il test t di Student è stato utilizzato per determinare le differenze tra HEMn-LP e le linee cellulari di melanoma (*p < 0,05; **p < 0,01; ***p < 0,001). L’OCR mitocondriale massimo per ED-034 non è stato indicato a causa di una variazione molto ampia tra quattro esperimenti indipendenti, che variava da 6,19 a 58,36 pmoli/min/1.000 cellule.
Additional file 3: Figura S3.(12M, tiff)
Risposta metabolica delle cellule di melanoma al DCA Risposta relativa dei livelli di ECAR, OCR, OCR/ECAR e ATP dopo il trattamento con DCA (0,1, 1 e 10 mM) per 2 ore. Le barre di errore nei primi tre pannelli rappresentano le deviazioni standard di tre misurazioni ripetute di sei campioni paralleli. Le barre di errore nel pannello inferiore rappresentano la deviazione standard di tre esperimenti indipendenti. Per l’analisi statistica è stata utilizzata l’ANOVA a una via e il test HSD di Tukey per determinare la significatività statistica (*p < 0,05; **p < 0,01).
File aggiuntivo 4: Figura S4.(5.6M, tiff)
Stato allelico di BRAF. Pirosequenziamento del sito di mutazione BRAF c.1799 T > A in ED-013 e nel derivato resistente a vemurafenib ED-013-R2. L’aumento del rapporto BRAF V600E-BRAF WT in ED-013-R2 indica un aumento del numero di copie, che potrebbe spiegare la resistenza a vemurafenib.
Interessi in gioco
Gli autori dichiarano di non avere interessi in competizione.
Contributi degli autori
PG e CA hanno pianificato e organizzato lo studio. CA ha eseguito la maggior parte degli esperimenti e l’elaborazione dei dati. CD ha pianificato ed eseguito il saggio di proliferazione cellulare e ha contribuito all’interpretazione dei risultati. AB e TM hanno contribuito alla preparazione e all’ottimizzazione del progetto per l’analisi metabolica sullo strumento Seahorse XF. CA, AB e TM hanno discusso e interpretato i risultati dell’analisi metabolica. CA e PG hanno scritto il manoscritto con i contributi e le modifiche di CD, AB e TM. Il manoscritto finale è stato letto e approvato da tutti gli autori.
Ringraziamenti
Ringraziamo il professor Karsten Kristiansen (Dipartimento di Biologia dell’Università di Copenaghen) e il professore associato Jacob B. Hansen (Dipartimento di Scienze Biomediche dell’Università di Copenaghen) per aver fornito le attrezzature e le competenze tecniche per l’analisi Seahorse XF. Questo studio è stato sostenuto da sovvenzioni della Danish Cancer Society e della Danish Cancer Research Foundation.
RIFERIMENTI
1 Warburg O, Wind F, Negelein E: Il metabolismo dei tumori nell’organismo. J Gen Physiol. 1927, 8: 519-530. 10.1085/jgp.8.6.519.2 Gatenby RA, Gillies RJ: Perché i tumori hanno una glicolisi aerobica elevata? Nat Rev Cancer. 2004, 4: 891-899. 10.1038/nrc1478.
3 Vander Heiden MG, Cantley LC, Thompson CB: Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009, 324: 1029-1033. 10.1126/science.1160809.
4 Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, Clish CB, Granter SR, Widlund HR, Spiegelman BM, Puigserver P: L’espressione di PGC1alpha definisce un sottogruppo di tumori di melanoma umano con una maggiore capacità mitocondriale e resistenza allo stress ossidativo. Cancer Cell. 2013, 23: 287-301. 10.1016/j.ccr.2012.11.020.
5 vDavies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J: Mutazioni del gene BRAF nel cancro umano. Nature. 2002, 417: 949-954. 10.1038/nature00766.
6 Hall A, Meyle KD, Lange MK, Klima M, Sanderhoff M, Dahl C, Abildgaard C, Thorup K, Moghimi SM, Jensen PB, Bartek J, Guldberg P, Christensen C: La fosforilazione ossidativa disfunzionale rende le cellule di melanoma maligno dipendenti dalla glicolisi guidata dall’oncogene (V600E)BRAF. Oncotarget. 2013, 4: 584-599.
7 Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, Wargo JA, Song JS, Fisher DE, Arany Z, Widlund HR: BRAF oncogenico regola il metabolismo ossidativo attraverso PGC1alpha e MITF. Cancer Cell. 2013, 23: 302-315. 10.1016/j.ccr.2013.02.003.
8 Esteve-Puig R, Canals F, Colome N, Merlino G, Recio JA: Uncoupling of the LKB1-AMPKalpha energy sensor pathway by growth factors and oncogenic BRAF.PLoS One 2009, 4:e4771…,
9 Zheng B, Jeong JH, Asara JM, Yuan YY, Granter SR, Chin L, Cantley LC: B-RAF oncogenico regola negativamente il soppressore tumorale LKB1 per promuovere la proliferazione delle cellule di melanoma. Mol Cell. 2009, 33: 237-247. 10.1016/j.molcel.2008.12.026.
10 Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS: A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013, 498: 109-112. 10.1038/nature12154.
11 Baumunk D, Reichelt U, Hildebrandt J, Krause H, Ebbing J, Cash H, Miller K, Schostak M, Weikert S: Parametri di espressione dei geni della via metabolica piruvato deidrogenasi chinasi-1 (PDK-1) e DJ-1/PARK7 nel carcinoma a cellule renali (RCC). World J Urol. 2013, 31: 1191-1196. 10.1007/s00345-012-0874-5.
12 Hur H, Xuan Y, Kim YB, Lee G, Shim W, Yun J, Ham IH, Han SU: Espressione della piruvato deidrogenasi chinasi-1 nel cancro gastrico come potenziale bersaglio terapeutico. Int J Oncol. 2013, 42: 44-54.
13 Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, de Lassalle EM, Maboudou P, Formstecher P, Polakowska R, Mortier L, Marchetti P: L’inattivazione dell’asse di segnalazione HIF-1alfa/PDK3 spinge il melanoma verso il metabolismo ossidativo mitocondriale e potenzia l’attività terapeutica dei pro-ossidanti. Cancer Res. 2012, 72: 5035-5047. 10.1158/0008-5472.CAN-12-0979.
14 Stacpoole PW, Lorenz AC, Thomas RG, Harman EM: Il dicloroacetato nel trattamento dell’acidosi lattica. Ann Intern Med. 1988, 108: 58-63. 10.7326/0003-4819-108-1-58.
15 Stacpoole PW: La farmacologia del dicloroacetato. Metabolismo. 1989, 38: 1124-1144. 10.1016/0026-0495(89)90051-6.
16 Michelakis ED, Webster L, Mackey JR: Il dicloroacetato (DCA) come potenziale terapia a bersaglio metabolico per il cancro. Br J Cancer. 2008, 99: 989-994. 10.1038/sj.bjc.6604554.
17 Stacpoole PW, Kerr DS, Barnes C, Bunch ST, Carney PR, Fennell EM, Felitsyn NM, Gilmore RL, Greer M, Henderson GN, Hutson AD, Neiberger RE, O’Brien RG, Perkins LA, Quisling RG, Shroads AL, Shuster JJ, Silverstein JH, Theriaque DW, Valenstein E: Studio clinico controllato del dicloroacetato per il trattamento dell’acidosi lattica congenita nei bambini. Pediatria. 2006, 117: 1519-1531. 10.1542/peds.2005-1226.
18 Bonnet S, Archer SL, lalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED: L’asse mitocondri-K+ channel è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita del tumore. Cancer Cell. 2007, 11: 37-51. 10.1016/j.ccr.2006.10.020.
19 Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC: Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010, 2: 31-34. 10.1126/scitranslmed.3000677.
20 Wong JY, Huggins GS, Debidda M, Munshi NC, De VI: Il dicloroacetato induce l’apoptosi nelle cellule del cancro endometriale. Gynecol Oncol. 2008, 109: 394-402. 10.1016/j.ygyno.2008.01.038.
21 Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ: il dicloroacetato (DCA) sensibilizza alle radiazioni in vitro sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2. Prostata. 2008, 68: 1223-1231. 10.1002/pros.20788.
22 Robinson J, Roberts CH, Dodi IA, Madrigal JA, Pawelec G, Wedel L, Marsh SG: The European searchable tumour line database. Cancer Immunol Immunother. 2009, 58: 1501-1506. 10.1007/s00262-008-0656-5.
23 Wu M, Neilson A, Swift AL, Moran R, Tamagnine J, Parslow D, Armistead S, Lemire K, Orrell J, Teich J, Chomicz S, Ferrick DA: L’analisi metabolica multiparametrica rivela uno stretto legame tra l’attenuazione della funzione bioenergetica mitocondriale e la maggiore dipendenza dalla glicolisi nelle cellule tumorali umane. Am J Physiol Cell Physiol. 2007, 292: C125-C136. 10.1152/ajpcell.00247.2006.
24 Brand MD, Nicholls DG: Valutare la disfunzione mitocondriale nelle cellule. Biochem J. 2011, 435: 297-312. 10.1042/BJ20110162.
25 Dahl C, Christensen C, Jonsson G, Lorentzen A, Skjodt ML, Borg A, Pawelec G, Guldberg P: Mutual Exclusivity Analysis of Genetic and Epigenetic Drivers in Melanoma Identifies a Link Between p14ARF and RARbeta Signaling. Mol Cancer Res. 2013, 11: 1166-1178. 10.1158/1541-7786.MCR-13-0006.
26 Sondergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S, Sazegar H, MacConaill LE, Barretina JG, Kehoe SM, Attar N, von EE, Zuckerman JE, Chmielowski B, Comin-Anduix B, Koya RC, Mischel PS, Lo RS, Ribas A: Sensibilità differenziale delle linee cellulari di melanoma con mutazione BRAFV600E all’inibitore Raf specifico PLX4032.J Transl Med 2010, 8:39.,
27 Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS: I melanomi acquisiscono resistenza all’inibizione di B-RAF(V600E) attraverso l’upregulation di RTK o N-RAS. Nature. 2010, 468: 973-977. 10.1038/nature09626.
28 Shi H, Moriceau G, Kong X, Lee MK, Lee H, Koya RC, Ng C, Chodon T, Scolyer RA, Dahlman KB, Sosman JA, Kefford RF, Long GV, Nelson SF, Ribas A, Lo RS: Melanoma whole-exome sequencing identifies (V600E)B-RAF amplification-mediated acquired B-RAF inhibitor resistance.Nat Commun 2012, 3:724.,
29 de Barbi MM, Vincent G, Fayewicz SL, Bateman NW, Hood BL, Sun M, Suhan J, Duensing S, Yin Y, Sander C, Kirkwood JM, Becker D, Conrads TP, Van HB, Moschos SJ: La respirazione mitocondriale: un importante bersaglio terapeutico nel melanoma,
30 Gilbert RJ, Klein RA: Piruvato chinasi: un sito di produzione di ATP regolato dalla carnitina in Trypanosoma brucei brucei. Comp Biochem Physiol B. 1984, 78: 595-599.
31 Kobayashi K, Neely JR: Meccanismo di attivazione della piruvato deidrogenasi da parte dell’aumento del lavoro cardiaco. J Mol Cell Cardiol. 1983, 15: 369-382. 10.1016/0022-2828(83)90321-8.
32 Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL: il dicloroacetato di sodio colpisce selettivamente le cellule con difetti nell’ETC mitocondriale. Int J Cancer. 2010, 127: 2510-2519. 10.1002/ijc.25499.
33 Madhok BM, Yeluri S, Perry SL, Hughes TA, Jayne DG: il dicloroacetato induce apoptosi e arresto del ciclo cellulare in cellule di cancro del colon-retto. Br J Cancer. 2010, 102: 1746-1752. 10.1038/sj.bjc.6605701.
34 Tong J, Xie G, He J, Li J, Pan F, Liang H: Effetto antitumorale sinergico del dicloroacetato in combinazione con il 5-fluorouracile nel cancro del colon-retto,
35 Niehr F, Von EE, Attar N, Guo D, Matsunaga D, Sazegar H, Ng C, Glaspy JA, Recio JA, Lo RS, Mischel PS, Comin-Anduix B, Ribas A: Terapia combinata con vemurafenib (PLX4032/RG7204) e metformina in linee cellulari di melanoma con mutazioni driver distinte,
36 Yuan P, Ito K, Perez-Lorenzo R, Del GC, Lee JH, Shen CH, Bosenberg MW, McMahon M, Cantley LC, Zheng B: la fenformina aumenta il beneficio terapeutico dell’inibizione di BRAFV600E nel melanoma. Proc Natl Acad Sci U S A. 2013, 110: 18226-18231. 10.1073/pnas.1317577110.
37 Buzzai M, Jones RG, Amaravadi RK, Lum JJ, DeBerardinis RJ, Zhao F, Viollet B, Thompson CB: Il trattamento sistemico con il farmaco antidiabetico metformina ostacola selettivamente la crescita delle cellule tumorali con deficit di p53. Cancer Res. 2007, 67: 6745-6752. 10.1158/0008-5472.CAN-06-4447.
38 Choi YW, Lim IK: Sensibilizzazione della citotossicità della metformina da parte del dicloroacetato attraverso la riprogrammazione del metabolismo del glucosio nelle cellule tumorali. Cancer Lett. 2014, 346: 300-8. 10.1016/j.canlet.2014.01.015.
39 Bergman U, Boman G, Wiholm BE: Epidemiologia delle reazioni avverse a fenformina e metformina. Br Med J. 1978, 2: 464-466. 10.1136/bmj.2.6135.464.
40DunbarEM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW:Studio di fase 1 del dicloroacetato (DCA) in adulti con tumori cerebrali maligni ricorrenti. Invest New Drugs. 2013, 32: 452-64. 10.1007/s10637-013-0047-4.
41 Corazao-Rozas P, Guerreschi P, Jendoubi M, Andre F, Jonneaux A, Scalbert C, Garcon G, Malet-Martino M, Balayssac S, Rocchi S, Savina A, Formstecher P, Mortier L, Kluza J, Marchetti P: lo stress ossidativo mitocondriale è il tallone d’Achille delle cellule di melanoma resistenti all’inibitore Braf-mutante. Oncotarget. 2013, 4: 1986-1998.