Ramon C Sun1,2, Philip G Board1 y Anneke C Blackburn1*
1 Molecular Genetics Group, Department of Translational Biosciences, John Curtin School of Medical Research, Building 131, Australian National University, P.O. Box 334, Canberra ACT 0200, AUSTRALIA
2 Department of Radiation Oncology, Stanford School of Medicine, Stanford CA 94305 USA.
1*Correspondencia: [email protected]
© 2011 Sun et al; licenciatario BioMed Central Ltd. Este es un artículo de Acceso Abierto distribuido bajo los términos de la Licencia de Atribución Creative Commons (http://creativecommons.org/licenses/by/2.0), que permite el uso, distribución y reproducción sin restricciones en cualquier medio, siempre que se cite adecuadamente el trabajo original.
Recibido: 3 de mayo de 2011
Aceptado: 18 de noviembre de 2011
Publicado: 18 de noviembre de 2011
Resumen
Antecedentes
Las células cancerosas tienen un perfil metabólico diferente al de las células normales. El efecto Warburg (aumento de la glucólisis aeróbica) y la glutaminólisis (aumento de la actividad mitocondrial a partir del catabolismo de la glutamina) son características bien conocidas del cáncer y van acompañadas de una mayor producción de lactato, una membrana mitocondrial hiperpolarizada y una mayor producción de especies reactivas del oxígeno.
Métodos
En este estudio nos dirigimos al efecto Warburg con dicloroacetato (DCA) y al aumento de la actividad mitocondrial de la glutaminólisis con trióxido de arsénico (ATO) en células de cáncer de mama, midiendo la proliferación celular, la muerte celular y las características mitocondriales.
Resultados
La combinación de DCA y ATO fue más eficaz para inhibir la proliferación celular e inducir la muerte celular que cualquiera de los dos fármacos por separado. Examinamos el efecto de estos tratamientos sobre el potencial de membrana mitocondrial, la producción de especies reactivas del oxígeno y los niveles de ATP y hemos identificado nuevos mecanismos moleculares dentro de la mitocondria tanto para la ATO como para el DCA: la ATO reduce la función mitocondrial a través de la inhibición de la citocromo C oxidasa (complejo IV de la cadena de transporte de electrones) mientras que el DCA regula al alza la expresión de la subunidad b de la ATP sintasa. La potenciación de la citotoxicidad de la ATO por el DCA está correlacionada con una fuerte supresión de la expresión de c-Myc y HIF-1a, y una disminución de la expresión de la proteína de supervivencia Bcl-2. Conclusión
Este estudio es el primero en demostrar que dirigirse a dos marcadores metabólicos clave del cáncer es una estrategia anticancerígena eficaz con potencial terapéutico.
Palabras clave
Dicloroacetato, cáncer de mama, cadena de transporte de electrones, mitocondrias, trióxido de arsénico
Introducción
El trióxido de arsénico (ATO) se utiliza como agente terapéutico desde hace más de 2000 años. Originario de China [1], actualmente se utiliza contra la leucemia promieloide aguda (LPA) en pacientes que han recaído tras el tratamiento con ácido altrans-retinoico/antraciclina y se está promoviendo para el tratamiento de primera línea de la LPA de novo [2-4]. La ATO es conocida como una molécula hiperreactiva y podría potencialmente unirse a grupos tiol en muchas proteínas [2,5]. Su capacidad para unirse a la proteína mutante PML-RAR-α, rica en tioles, producida a partir de una translocación cromosómica en la LPA, la ha convertido en un fármaco eficaz en la LPA [2,5,6]. Se ha demostrado que la ATO induce la apoptosis en una variedad de líneas celulares de cáncer in vitro e in vivo [7,8], pero ha sido difícil considerar la ATO para su uso clínico en tipos de tumores distintos de la LPA debido a la falta de conocimiento de las dianas moleculares que dan lugar a su citotoxicidad. En los últimos 10 años, los cambios fisiológicos dentro de las células cancerosas en respuesta al tratamiento con ATO han sido bien caracterizados, y muchos ensayos clínicos para nuevas aplicaciones de ATO están en marcha [5]. Se ha propuesto que la ATO es una toxina mitocondrial [9]. La ATO puede despolarizar el potencial de membrana mitocondrial (PMM) [10], aumentar la producción intracelular de especies reactivas de oxígeno (ROS) [8] e inducir la apoptosis [8]. La diana propuesta para la ATO que puede lograr estos cambios fenotípicos es el poro de transición mitocondrial (PTM) [11]. Se ha demostrado que la ATO induce la apertura del MTP, lo que induce la liberación de citocromo c y se propone que disipa el MMP y aumenta la liberación de ROS de la mitocondria [12]. Más recientemente, el sistema de la tioredoxina, en particular la tioredoxina reductasa, también se ha identificado como una diana de la ATO que puede contribuir al aumento del estrés oxidativo y a la alteración de la señalización redox tras el tratamiento con ATO de células cancerosas [9,13].
El efecto Warburg es un fenómeno muy extendido que se ha identificado en más del 90% de todas las formas tumorales. Las células que presentan el efecto Warburg adoptan rutas alternativas de homeostasis energética para mantener su fenotipo proliferativo [14]. El premio Nobel Dr. Otto Warburg afirmó que las células cancerosas recurren a la glucólisis o a la fosforilación de sustratos para generar ATP, y suprimen sus actividades mitocondriales [15]. Con tecnologías más avanzadas, estudios recientes han confirmado el aspecto de la producción de ATP de la hipótesis de Warburg, pero han revelado que la actividad mitocondrial no se suprime en las células cancerosas. Al contrario, las mitocondrias desempeñan un papel vital en el suministro de sustratos para mantener la división celular [16].
Recientemente se ha descrito el efecto anticancerígeno de invertir el efecto Warburg y un antiguo fármaco, el dicloroacetato (DCA), que puede redirigir la síntesis de ATP de la glucólisis a la fosforilación oxidativa, ha demostrado una buena actividad anticancerígena tanto in vitro [17-19] como in vivo [20-23]. El DCA es un inhibidor de la piruvato deshidrogenasa cinasa y provoca un aumento de la actividad de la piruvato deshidrogenasa [19]. Esto conduce a un aumento de la conversión de piruvato en acetil-CoA en lugar de ácido láctico, tal y como describe el efecto Warburg, y estimula la respiración mitocondrial al aumentar el suministro de acetil-CoA. En consecuencia, tras el tratamiento con DCA, las células cancerosas mostraron un aumento de los niveles de ROS, despolarización de la MMP in vitro y aumento de la apoptosis tanto in vitro como in vivo [17,20].
Como el DCA puede redirigir sustratos hacia la respiración mitocondrial y la producción de ATP, podría tener una actividad sinérgica con los fármacos anticancerígenos que deterioran la actividad mitocondrial. Proponemos que al invertir el fenotipo glucolítico con DCA y dirigir más piruvato hacia la fosforilación oxidativa mitocondrial, mientras que simultáneamente se dirige a las mitocondrias con ATO, se producirá una alteración grave de la homeostasis energética dentro de las células cancerosas. En este estudio demostramos que la combinación de DCA y ATO inhibe el crecimiento de líneas celulares de cáncer de mama in vitro. Además, hemos identificado nuevos mecanismos moleculares dentro de las mitocondrias que pueden contribuir a la citotoxicidad de la ATO, proporcionando un apoyo adicional para el uso de la ATO contra tumores sólidos.
Materiales y métodos
Reactivos
JC-1, CFSE y H2DCFDA procedían de Invitrogen (Carlsbad, CA, EE.UU.), los kits de ensayo CellTiter-Glo y Caspase-Glo se compraron a Promega Co (San Luis, CA, EE.UU.). El resto de los productos químicos se adquirieron a Sigma Co (San Luis, MO, EE.UU.).
Cultivo celular
Las líneas celulares se obtuvieron de las siguientes fuentes en los años indicados. Dra. Anna DeFazio, Westmead Millenium Institute, Sydney, Australia: T-47D (2003), BT-20 (2003), MCF-10A (2005) y MCF-10AT1 (2005); Prof Chris Parish, Australian National University, Canberra, Australia: 13762 MAT (2007), MDA-MB-468 (2003) y MDA-MB-231 (2003). Las líneas celulares tienen apariencias coherentes con las morfologías publicadas, pero no han sido autentificadas recientemente. Las células de carcinoma epitelial de mama humano (T-47D) se cultivaron en medio RPMI 1640 suplementado con un 10% de suero bovino fetal en presencia de un 0,1% de PSN (3% de penicilina, 5% de estreptomicina y 5% de neomicina). Las líneas celulares BT-20, MDA-MB-231, MDA-MB-468 y 13762 MAT se mantuvieron en medio DMEM/F12 suplementado con un 10% de FBS y 2 mM de L-glutamina. Para las células MCF-10A y MCF-10AT1 se utilizó medio DMEM/F-12 con 25% de suero de caballo, 0,01% de EGF, 0,28 UI/ml de insulina, 0,01% de toxina del cólera y 0,5 μg/ml de hidrocortisona. Todas las líneas celulares se mantuvieron a 37 °C en un 5% de CO2.
Viabilidad celular
Para la evaluación de la viabilidad celular, las células se sembraron en placas de 96 pocillos a una densidad de 3000 células por pocillo y 8 pocillos por grupo. Tras la exposición a DCA y ATO durante 24 a 72 horas, las células se incubaron durante 3 horas con rojo neutro (30 μg/ml) en medio fresco, después se lavaron con PBS, a lo que siguió la adición de tampón de lisis (ácido acético/metanol, 80%/20%) y se registró la absorbancia a 540 nm. Los resultados se expresan como media ± D.S., los cálculos se realizaron con el paquete informático Prism, se aplicó ANOVA con postest de Tukey y se consideró que P < 0,05 era estadísticamente significativo. Los experimentos se realizaron al menos tres veces y los datos presentados corresponden a un experimento representativo.
Proliferación celular
Se cosecharon células T-47D y se resuspendieron en 1 ml de medio RPMI. Las células se marcaron añadiendo 1 ml de PBS que contenía 5 μM de éster succinimidílico de carboxifluoresceína (CFSE), seguido de 5 minutos de incubación a temperatura ambiente. A continuación, las células marcadas se lavaron dos veces, se contaron y se sembraron a105 células/pocillo en una placa de 12 pocillos. El día del análisis, se recogieron las células T-47D, se lavaron dos veces con PBS y se examinó la intensidad de CFSE mediante FACS. Los resultados se expresan como media ± D.S. (n = 3), los cálculos se realizaron utilizando el paquete de software Prism, se aplicó ANOVA con postest de Tukey y P < 0,05 se consideró estadísticamente significativo. Los experimentos se realizaron al menos tres veces y los datos presentados son de un experimento representativo.
Muerte celular La apoptosis se cuantificó mediante citometría de flujo tras teñir las células con Annexin-V (AV) marcado con FITC (Invitrogen Co.) y yoduro de propidio (PI). Tras el tratamiento farmacológico, se cosecharon las células T-47D y se centrifugaron a 1200 rpm durante 5 min; los gránulos se lavaron dos veces con PBS y luego se resuspendieron en 100 μl de tampón de unión de Annexin-V (0,14 M NaCl, 2,5 mM CaCl2, 0,01 M HEPES pH 7,4). Se añadieron Annexin-V (1 μL) y 5 μl de PI (50 μg/ml) a las muestras y se incubaron en la oscuridad durante 15 min. Las muestras se mantuvieron en hielo después de la incubación hasta que se realizó el análisis FACS. Los resultados se expresan como media ± D.S. (n = 3), los cálculos se realizaron utilizando el paquete de software Prism, se aplicó ANOVA con postest de Tukey y P < 0,05 se consideró estadísticamente significativo. Los experimentos se realizaron al menos tres veces y los datos presentados corresponden a un experimento representativo.
Generación de ROS
Para evaluar los niveles intracelulares de ROS, las células se colocaron en placas de 12 pocillos con una densidad celular de 1 ×105 células por pocillo y se trataron con fármacos durante 12 horas. se añadió al medio 2′, 7′-dihidroclorofluroresceinacetato (H2DCFDA) a una concentración final de 10 μM y se dejaron teñir las células durante 1 hora en la oscuridad. Tras la tinción conH2DCFDA, se tripsinizaron las células, se lavaron dos veces y se resuspendieron en 100 μl de PBS. La intensidad de H2DCFDAse examinó mediante FACS. Los resultados se expresan como media ± D.S. (n = 3), los cálculos se realizaron con el paquete de software Prism, se aplicó ANOVA con postprueba de Tukey y P < 0,05 se consideró estadísticamente significativo. Los experimentos se realizaron al menos tres veces y los datos presentados corresponden a un experimento representativo.
Concentración de ATP y actividad de lacaspasa
El nivel interno de ATP y la actividad de la caspasa en las células T-47D se evaluaron utilizando los kits de ensayo CellTiter-Glo y Caspase-Glo 3/7 (Promega Corp., Madison, WI) de acuerdo con las instrucciones del fabricante. Las células T-47D se cultivaron en ausencia y presencia de fármacos durante 12 horas en placas opacas blancas de 96 pocillos (4 pocillos por grupo). Se añadieron volúmenes iguales de reactivos CellTiter-Glo, tras lo cual las muestras se incubaron durante 15 minutos en un agitador a temperatura ambiente. La luminiscencia se registró con el luminómetro de microplacas Glomax (Promega Co., Madison, WI) siguiendo el protocolo CellTiter preestablecido. Los resultados se expresan como media ± D.S. (n = 4), los cálculos se realizaron con el paquete de software Prism, se aplicó ANOVA con postprueba de Tukey y se consideró que P < 0,05 era estadísticamente significativo. Los experimentos se realizaron al menos tres veces y los datos presentados corresponden a un experimento representativo.
Potencial de membrana mitocondrial
De forma similar a la medición de ROS, las células se colocaron en placas de 12 pocillos con una densidad celular de 1 ×105 células por pocillo y se trataron con fármacos durante 12 horas. se añadió 5, 5′, 6, 6′-tetracloro-1, 1′, 3, 3′-tetraetilbenzimidazol-carbocianina yoduro (JC-1) en el medio a una concentración final de 0,2 μM y se dejaron teñir las células durante 30 min en la oscuridad. Tras la tinción con JC-1, se tripsinizaron las células, se lavaron dos veces con PBS y se resuspendieron en 100 μl de PBS. La intensidad de JC-1 se examinó mediante FACS. Los resultados se expresan como media ± D.S. (n = 3), los cálculos se realizaron utilizando el paquete de software Prism, se aplicó ANOVA con postest de Tukey y P < 0,05 se consideró estadísticamente significativo. Los experimentos se realizaron al menos tres veces y los datos presentados corresponden a un experimento representativo.
Actividad de la citocromo C ox idasa
El ensayo de la citocromo C oxidasa se evaluó según el método publicado anteriormente [24]. Brevemente, tras el tratamiento farmacológico en placas de 96 pocillos (8 pocillos por grupo), las células T-47D se permeabilizaron con 50 μl de saponina al 0,01%, seguido de la adición de 100 μl de medio de sustrato (4 mM de tetrahidrocloruro de 3,3-diaminobencidina (DAB), 100 μM de citocromo C reducido, 2 μg/ml de catalasa en fosfato de Na 0,1 M, pH 7,0). La absorbancia a 450 nm se midió inmediatamente después de la adición del medio de sustrato y se monitorizó durante 30 min. Los resultados se expresan como media ± D.S. (n = 8), los cálculos se realizaron utilizando el paquete de software Prism, se aplicó ANOVA con postest de Tukey y P < 0,05 se consideró estadísticamente significativo. Los experimentos se realizaron al menos tres veces y los datos presentados corresponden a un experimento representativo.
Actividad dePDH
La actividad de PDH se midió utilizando el kit de ensayo de microplaca de actividad enzimática de PDH de MitoSciences (#MSP18, MitoSciences, Oregon USA). Para medir el efecto de los fármacos sobre la actividad de la PDH en las células, las células T-47D se trataron con fármacos en medio durante 3 horas, tras lo cual se lavaron y resuspendieron en PBS. Se prepararon extractos celulares y se analizó la actividad de la PDH a una concentración de 15 mg de proteína/ml, de acuerdo con las instrucciones del kit. Para medir la inhibición directa de PDH por ATO, se aisló PDH de células T-47D no tratadas aplicando extractos celulares a la placa de captura de anticuerpos. Tras lavar la placa, se añadió a los pocillos la solución de ensayo que contenía el fármaco y se analizó inmediatamente la actividad PDH. Los resultados se expresan como media ± D.S. (n = 4), los cálculos se realizaron utilizando el paquete de software Prism, se aplicó ANOVA con postest de Tukey y P < 0,05 se consideró estadísticamente significativo. Los experimentos se realizaron al menos tres veces y los datos presentados corresponden a un experimento representativo.
Inmunotransferencia y análisis densitométrico
Las células (1 ×106 ) se colocaron en frascos de cultivo de tejidos T25 y se trataron con ATO o DCA durante 12 horas. Se prepararon lisados celulares añadiendo 500 μl del reactivo de extracción de proteínas de mamíferos MPER® (Thermo Scientific, IL, EE.UU.). La inmunotransferencia se realizó como se ha descrito previamente [25], utilizando los anticuerpos para c-Myc (Roche, IN, EE.UU., clon 9E10), HIF-1α (Abcam, Cambridge, Reino Unido, #ab82832), Bcl-2 (Abcam #ab692), ATP sintasa β-subunidad (Abcam #ab14730) y β-actina (Abcam #ab8227). Los Western blots se detectaron mediante quimioluminiscencia y exposición de película de rayos X. Las imágenes se adquirieron con un escáner plano CanoScan 8600F y se cuantificaron con el programa informático ImageJ (versión 1.4, NIH, EE.UU.) y se normalizaron a β-actina en cada carril. Los resultados se agruparon a partir de 3 experimentos distintos y se expresaron como media ± desviación estándar (n = 3); los cálculos se realizaron con el paquete informático Prism; se aplicó ANOVA con la prueba posterior de Tukey y se consideró que P < 0,05 era estadísticamente significativo.
Resultados
ElDCA y el ATO juntos son más eficaces para reducir la proliferación celular e inducir la muerte celular
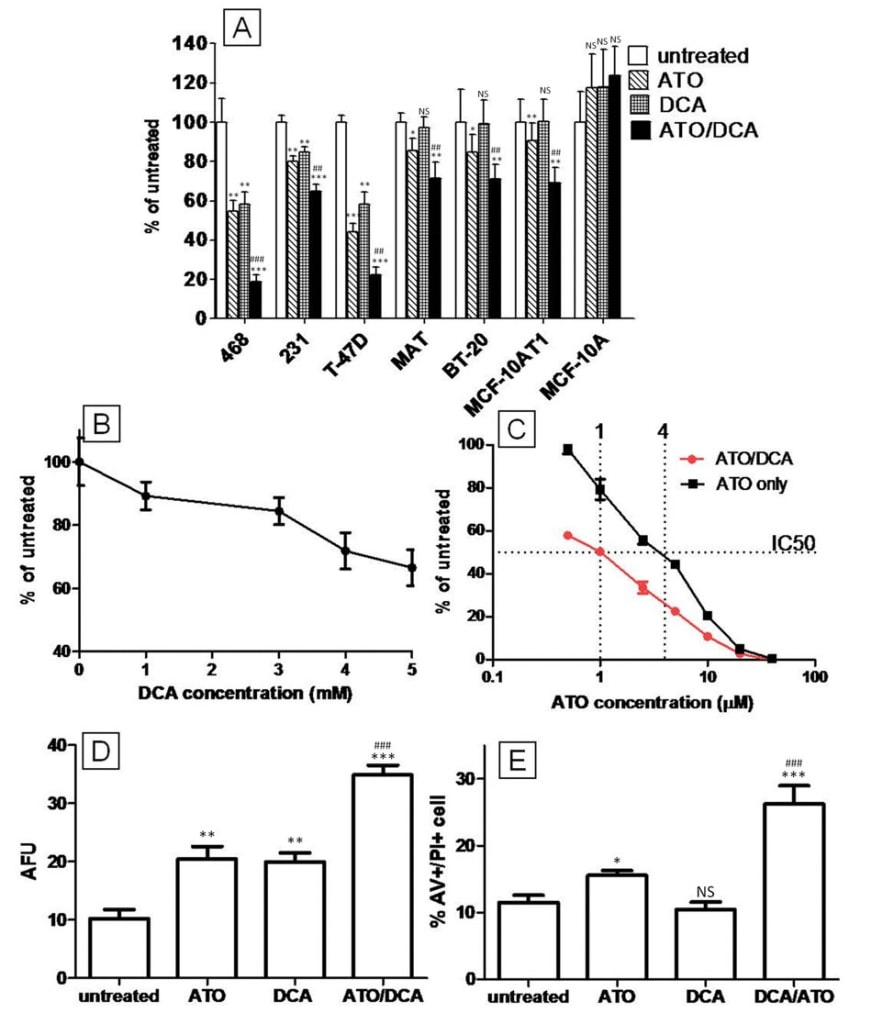
Para investigar el efecto combinado del ATO y el DCA en la inhibición del crecimiento celular, se trataron líneas celulares de cáncer de mama durante varios días con ambos fármacos y se evaluó el número total de células mediante el ensayo de viabilidad de células rojas neutras. El panel de líneas celulares representa los principales subtipos de cáncer de mama humano (luminal (T-47D), basal A (MDAMB-468, BT-20), basal B (MDA-MB-231)), o tienen otra relevancia como modelos experimentales (13762MAT – adenocarcinoma mamario de rata sensible al DCA in vivo [22], MCF10AT1 – derivado maligno de células inmortalizadas MCF-10A). Las células MDA-MB-468, MDA-MB-231 y T-47D mostraron una reducción significativa que oscilaba entre el 10% y el 40% en el número total de células tras 72 horas de tratamiento con 5 mM de DCA (Figura 1A), mientras que las células 13762 MAT, BT-20 y MCF-10AT1 no respondieron durante el periodo de tratamiento. Todas las líneas celulares cancerosas ensayadas fueron sensibles al ATO pero a diferentes concentraciones. Se observó una reducción del número total de células en BT-20, T-47D, MCF-10AT1 y MDA-MB-468 con sólo 5 μM de tratamiento con ATO, sin embargo se necesitaron 15 μM de ATO para conseguir la misma eficacia en las líneas celulares MDA-MB-231 y 13726 MAT. Llamativamente, DCA y ATO en combinación mostraron un mayor efecto que cualquiera de los fármacos por separado en todas las líneas celulares de cáncer probadas. En los casos en que ATO y DCA fueron eficaces como agentes únicos para reducir el número de células, el efecto del tratamiento combinado fue aproximadamente igual a la suma de los efectos de los fármacos individuales (T-47D, MDA-MB-468 y MDA-MB-231). El DCA por sí solo no redujo el número de células, pero fue capaz de potenciar la inhibición del crecimiento de la ATO entre 2 y 3 veces (13762 MAT, BT-20 y MCF10AT1) (Figura 1A). La línea celular no cancerosa MCF-10A no mostró reducción del número de células tras el tratamiento con ATO (15 μM), DCA (5 mM) o combinado.
El efecto de ATO y DCA se examinó más a fondo con células T-47D, una de las líneas celulares más sensibles al DCA solo. La respuesta de las células T-47D al DCA fue dependiente de la dosis y, tras 72 horas, las células tratadas con 5 mM de DCA tenían un 42 ± 6% menos de células que el cultivo de control (Figura 1B). La ATO sola (5 μM) redujo el número total de células en un 56 ± 4% y las células tratadas tanto con DCA como con ATO mostraron una mayor disminución en comparación con el grupo tratado sólo con ATO (Figura 1C). La curva dosis-respuesta mostró que el tratamiento combinado de DCA (5 mM) y ATO puede reducir el IC50 a 0,25 veces el de ATO solo (Figura 1C). Este efecto se produce dentro del rango de concentración alcanzado clínicamente para ATO (hasta 5-7 μM [26]).
El ensayo de proliferación CFSE demostró que las células tratadas con ATO (5 μM) o DCA (5 mM) emitían una fluorescencia CFSE significativamente mayor (2,1 veces y 2,2 veces respectivamente) tras 72 horas de tratamiento, lo que indica una inhibición del crecimiento. Las células tratadas con ATO y DCA mostraron un aumento de 3,4 veces en la intensidad de CFSE en comparación con las células no tratadas, lo que indica que los fármacos actuaron conjuntamente en la inhibición de la proliferación celular (Figura 1D).
El efecto de ATO y DCA sobre la muerte celular de T-47D se evaluó mediante tinción doble de AV y PI y las células se analizaron mediante clasificación celular fluorescente. El DCA solo (5 mM) no indujo la muerte celular en las células T-47D (Figura 1E), similar al efecto del DCA en las células 13762 MAT descrito anteriormente [22]. La ATO (5 μM) tampoco indujo la muerte celular tras 12 horas de tratamiento; sin embargo, las células tratadas tanto con 5 μM de ATO como con 5 mM de DCA mostraron un pequeño aumento (15%) de la población AV+/PI+ (13,2 ± 0,6% de células apoptóticas frente a 11,5 ± 1,0% para ATO/DCA frente a no tratadas respectivamente, p = 0,07), lo que sugiere que el DCA puede estar potenciando los efectos apoptóticos de la ATO. A concentraciones más altas y con 48 horas de tratamiento, ATO (20 μM) aumentó la cantidad de muerte celular en un 35 ± 8% (p = 0,029) en comparación con el cultivo no tratado (Figura 1E). La combinación de 5 mM de DCA con 20 μM de tratamiento con ATO produjo un aumento 4 veces mayor de la población AV+/PI+ en comparación con la ATO sola, lo que indica que el DCA puede potenciar la muerte celular inducida por la ATO en las células de cáncer de mama T-47D (Figura 1E).
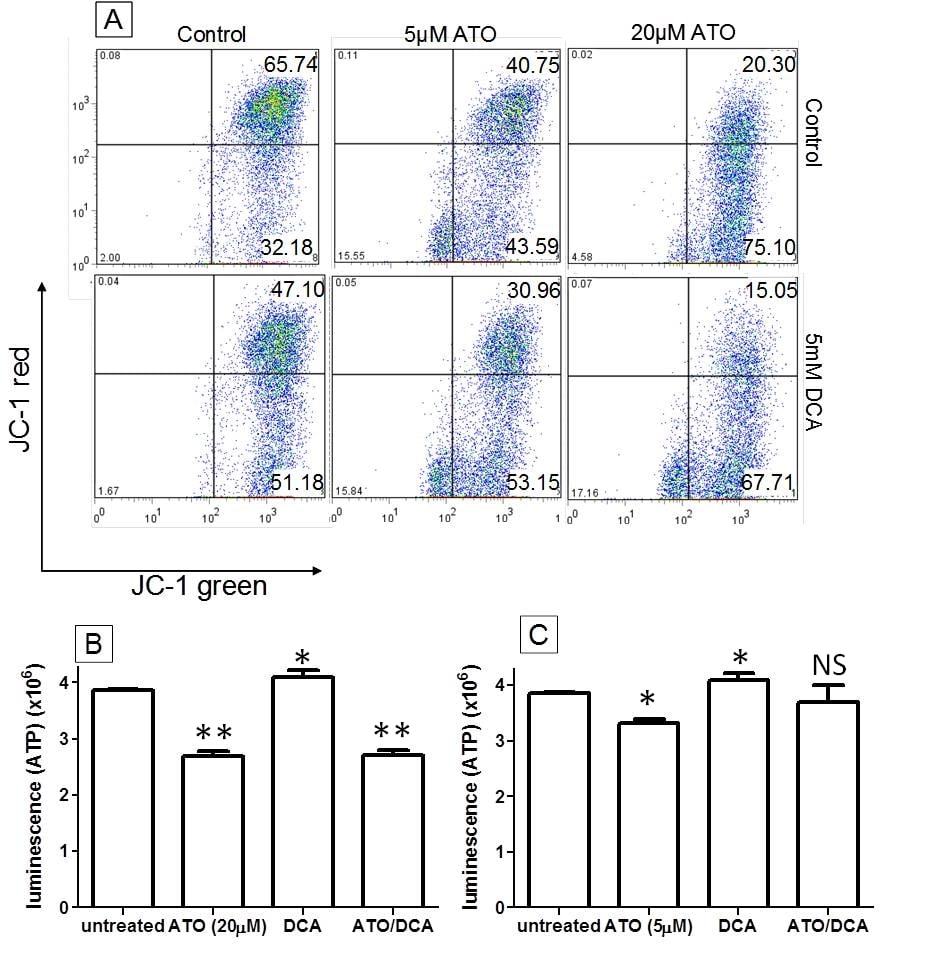
La ATOy el DCA actúan conjuntamente en la despolarización del MMPpero tienen efectos contrarios en la inducción de la producción de ATP y ROS
Se ha demostrado que tanto la ATO como el DCA alteran la función mitocondrial, despolarizando el MMP y aumentando la producción de ROS [20]. Por lo tanto, se estudiaron estos parámetros junto con el nivel de ATP en un esfuerzo por determinar si contribuyen a potenciar los efectos anticancerígenos del tratamiento combinado ATO/DCA. Medido mediante tinción con JC-1, el número de células T-47D con un MMP hiperpolarizado (cuadrante superior derecho) disminuyó significativamente con el tratamiento con DCA (5 mM) o ATO (5 μM y 20 μM) (t = 12 horas) (28 ± 6%, 38 ± 4% y 69 ± 7% de disminución, respectivamente, en comparación con las células control no tratadas), en consonancia con los datos publicados anteriormente [20,27]. La combinación de ATO (5 μM o 20 μM) con 5 mM de DCA condujo a una disminución aún mayor (53 ± 9% y 77 ± 12% respectivamente) en comparación con las células no tratadas (Figura 2A), lo que demuestra que DCA y ATO pueden trabajar juntos en la despolarización de la MMP. Los niveles de ATP disminuyeron tanto con dosis bajas como altas de ATO (14 ± 3% y 32 ± 5% respectivamente) tras 12 horas de tratamiento, mientras que las células tratadas con DCA mostraron un aumento del 6 ± 1% en los niveles de ATP (Figura 2B y 2C). A la dosis alta de ATO, el DCA fue incapaz de aumentar la producción de ATP (Figura 2B), mientras que a la dosis baja de ATO, las células tratadas con DCA y ATO combinados mostraron un pequeño aumento en la producción de ATP respecto al tratamiento con ATO solo (Figura 2C). Estos datos indican que el DCA y la ATO afectan a la producción de ATP a través de objetivos distintos dentro de las células.
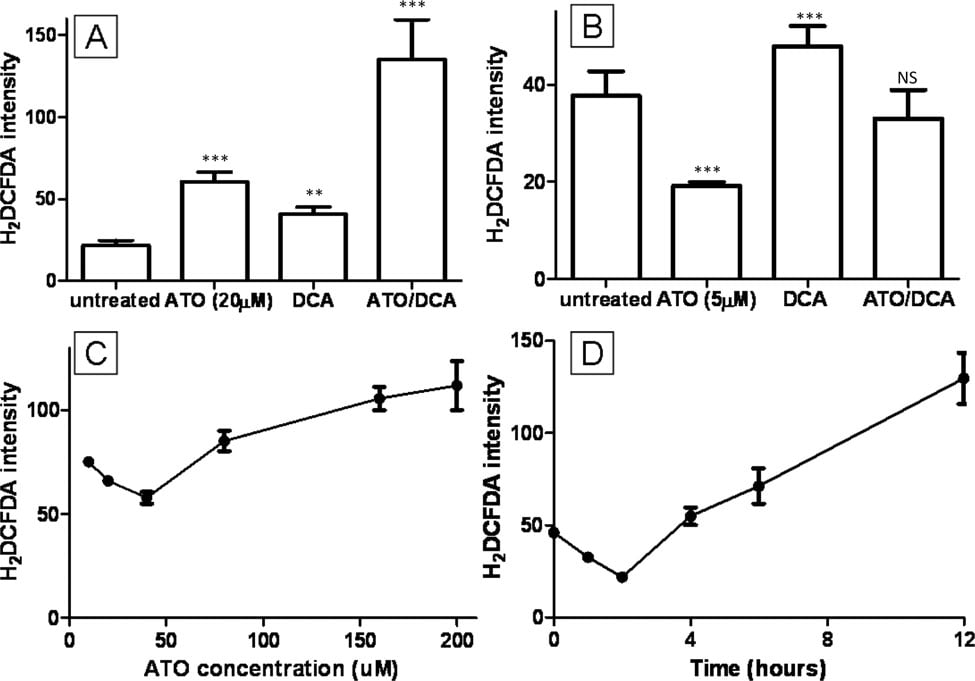
Se ha propuesto que el aumento de los niveles intracelulares de ROS es la causa de la citotoxicidad de la ATO [2,9]. El efecto combinado de ATO y DCA sobre ROS se investigó utilizando la tinción fluorescente H2DCFDA. Las células tratadas con 20 μM de ATO o 5 mM de DCA mostraron niveles intracelulares elevados de ROS (2,8 y 1,9 veces respectivamente) (Figura 3A), similares a los resultados publicados previamente [27], mientras que las células tratadas con ATO y DCA mostraron un aumento adicional (5,2 veces) en la producción de ROS (Figura 3A). Por el contrario, la baja concentración de ATO (5 μM) indujo una disminución de 0,5 veces en la producción de ROS (Figura 3B). Esto era contrario a informes anteriores sobre la producción de ROS y el tratamiento con ATO, por lo que se analizó la relación dosis-respuesta y el curso temporal de la producción de ROS tras el tratamiento con ATO. Esto reveló que las concentraciones más bajas de ATO disminuyeron la producción intracelular de ROS, mientras que las altas de ATO indujeron la producción de ROS (Figura 3C). De forma similar, el tratamiento con 10 μM de ATO provocó una disminución notable de la producción de ROS en las primeras 4 horas, antes de que los niveles de ROS aumentaran después de 8 horas hasta alcanzar el nivel descrito por otros autores (Figura 3D). El tratamiento combinado de células con 5 mM de DCA y 5 μM de ATO dio lugar a una producción intermedia de ROS (Figura 3B) similar a la de las células no tratadas.
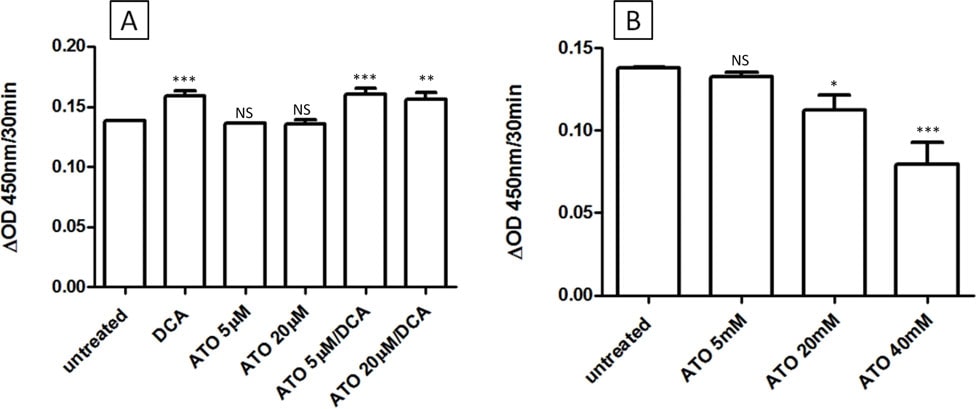
Los efectos intermedios del tratamiento combinado DCA/ATO sobre los niveles de ATP y ROS pueden explicarse por efectos contrapuestos sobre la actividad PDH. El DCA se dirige a la PDK, y por lo tanto debería aumentar la actividad de la PDH, mientras que se ha descrito que la ATO inhibe la PDH directamente por reacción con los tioles vicinales de la PDH o indirectamente a través del aumento de la producción de peróxido de hidrógeno [28]. Para examinar los efectos de ATO y/o DCA en la actividad de PDH, células intactas o PDH aisladas de células T-47D, fueron tratadas con la droga y la actividad de PDH determinada. Como se predijo, el tratamiento de 3 h de células intactas con DCA resultó en un aumento de la actividad PDH, mientras que ATO (hasta 20 μM) no alteró la actividad PDH de las células intactas (Figura 4A). Por el contrario, el DCA no modificó la actividad de la PDH aislada, pero altas concentraciones de ATO fueron capaces de inhibir la actividad de la PDH (Figura 4b). Así pues, en las células T-47D, la ATO no disminuyó la actividad de la PDH a las concentraciones utilizadas en este estudio.
ATO es un inhibidor del complejo IV de la cadena de transporte de electrones
La cadena de transporte de electrones (ETC) se encuentra en la membrana interna de la mitocondria y desempeña un papel vital en la producción de energía. La ETC es responsable de generar el gradiente de protones para que el espacio de la membrana interna de la mitocondria mantenga el MMP para la producción de ATP [29]. El ETC también está documentado como el principal lugar de producción de ROS dentro de las células como resultado de la fuga de electrones de los complejos I y III [30]. Debido a su capacidad para reducir las ERO, despolarizar la MMP y disminuir la producción de ATP simultáneamente, hemos planteado la hipótesis de que la ATO funciona como un inhibidor de la ETC. Se comparó un panel de inhibidores de la ETC con la ATO (5 μM) para identificar si inducen los mismos cambios fenotípicos que la ATO en las células T-47D. Se utilizaron rotenona (0,1 μM, complejo I), thenolytrifluoroacetone (TTFA) (10 μM, complejo II), antimycin (0,1 μM, complejo III) y NaCN (10 mM, complejo IV) para tratar las células T-47D y se compararon los niveles de ROS, MMP y ATP con los de las células tratadas con ATO. Todos los inhibidores demostraron ser capaces de despolarizar las MMP (Figura 5A) y reducir los niveles de ATP (Figura 5B) de forma similar a la ATO. Sin embargo, a diferencia de la ATO, la rotenona, la antimicina y el TTFA no consiguieron reducir la producción de ROS en las células, sino que las elevaron entre 3 y 6 veces tras 12 horas de tratamiento (Figura 5C). Sin embargo, las células tratadas con NaCN (10 mM) mostraron una disminución del 52% en la producción de ROS (Figura 5C), similar al tratamiento con ATO. Basándonos en estos datos concluimos que en las células de cáncer de mama T47D, la inhibición del complejo IV de la ETC pero no de los complejos I-III resulta en una reducción de ROS, y por lo tanto es probable que los cambios fenotípicos inducidos por la ATO sean causados por la inhibición del complejo IV (citocromo C oxidasa) de la ETC. Para confirmarlo, se midió espectrofotométricamente la actividad de la citocromo C oxidasa en células T-47D. El ensayo de citocromo C oxidasa demostró claramente que, mientras que el DCA no tenía ningún efecto sobre la actividad del complejo IV, la ATO (5 μM, 5 min) puede inhibir la actividad del complejo IV, confirmando nuestra hipótesis (Figura 5D). Se obtuvieron resultados similares tras los tratamientos farmacológicos de 10 min, 3 h y 12 h. La combinación de ATO y DCA mostró una inhibición enzimática similar a la de ATO solo.
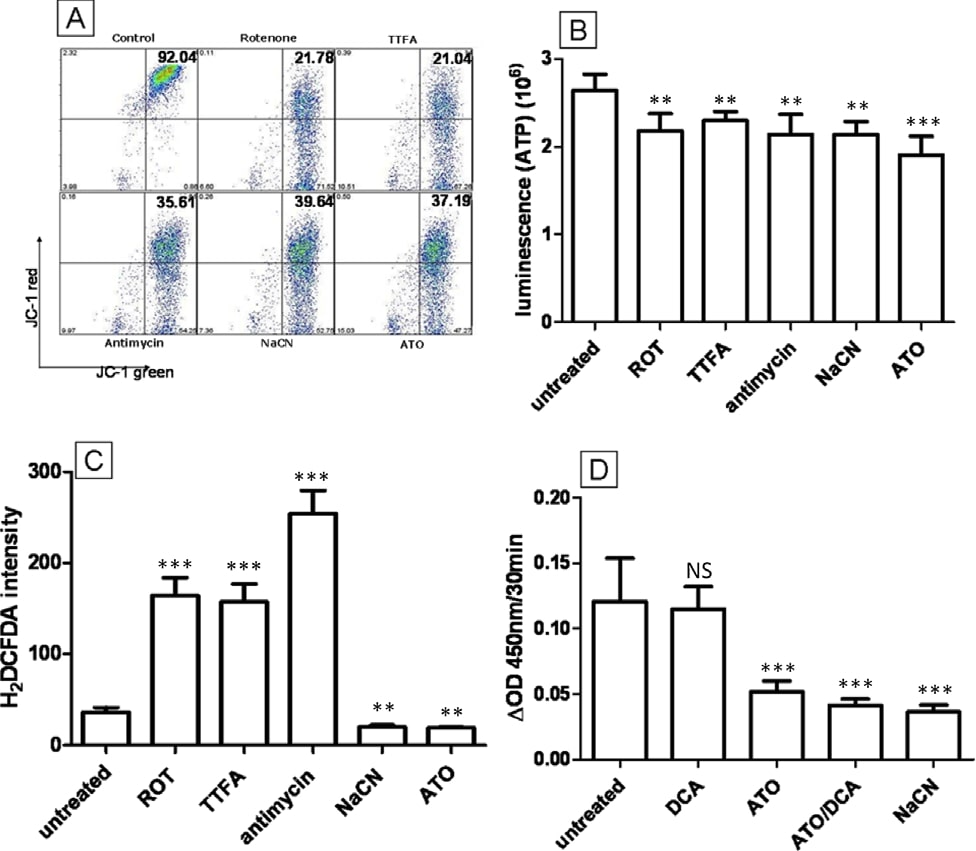
Los inhibidores del poro de transición mitocondrialbloquearon parcialmente la activación de caspasas inducida por ATO, pero no tuvieron efecto sobre ROS, ATP y MMP
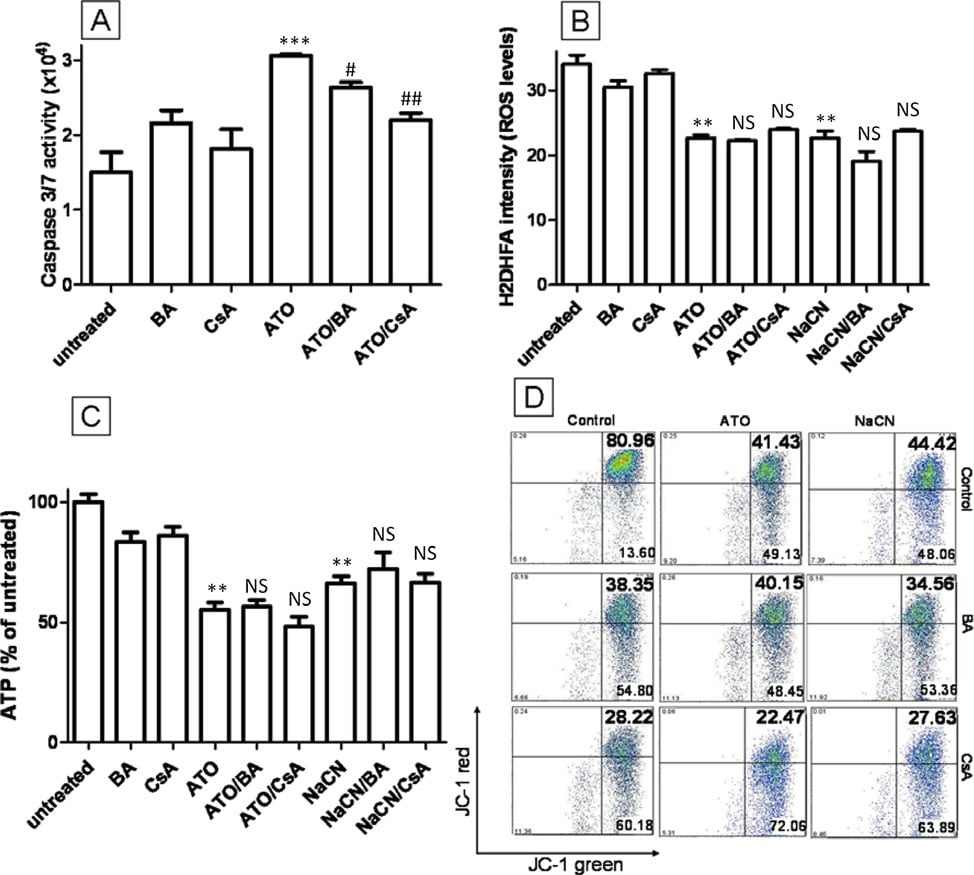
Un mecanismo que se ha descrito previamente como la clave de la citotoxicidad de ATO es la apertura del poro de transición mitocondrial (MTP). Se ha demostrado que la ATO induce la apertura del MTP en mitocondrias aisladas y puede aumentar la liberación de citocromo C [11] e inducir la apoptosis nuclear en sistemas libres de células [31]. Se ha propuesto que la apertura de la MTP por la ATO induce la permeabilización mitocondrial, disipa la MMP y aumenta las ROS intracelulares. Para comprobar si la MTP es importante para la toxicidad de la ATO en las células T-47D, se combinaron el ácido bongkrekico (BA) y la ciclosporina A (CsA), ambos potentes bloqueadores del poro de la MTP [32], con la ATO para ver si pueden inhibir los cambios intracelulares inducidos por la ATO. La adición de 5 μM de CsA o 50 μM de BA a las células tratadas con ATO (5 μM) no interfirió en la capacidad de la ATO para reducir la producción de ROS tras 12 horas de tratamiento, disminuir la producción de ATP o despolarizar la MMP (P > 0,05) (Figura 6B, C y 6D). Se observan datos similares para el tratamiento con NaCN y CsA/BA (P > 0,05) (Figura 6B, C y 6D). Para confirmar la actividad de CsA y BA en nuestras condiciones experimentales, se evaluó la actividad de la caspasa 3/7, un marcador de apoptosis corriente abajo de la apertura de la MTP, activada por la liberación de citocromo C, tras el tratamiento con ATO a altas concentraciones (Figura 6A). El tratamiento con ATO (20 μM) duplicó la actividad de la caspasa 3/7 en comparación con las células no tratadas (2,1 veces, Figura 6A) y este efecto disminuyó con BA y CsA (1,7 veces y 1,4 veces, respectivamente), lo que indica que CsA o BA pueden bloquear la apertura de la MTP en las células T-47D. Estos datos demuestran claramente que aunque la ATO (20 μM) puede inducir la apertura de la MTP, este mecanismo no puede explicar los cambios de ROS, MMP y ATP que se producen durante el tratamiento con 5 μM de ATO. Concluimos que estas alteraciones se deben probablemente a la inhibición del complejo ETC IV.
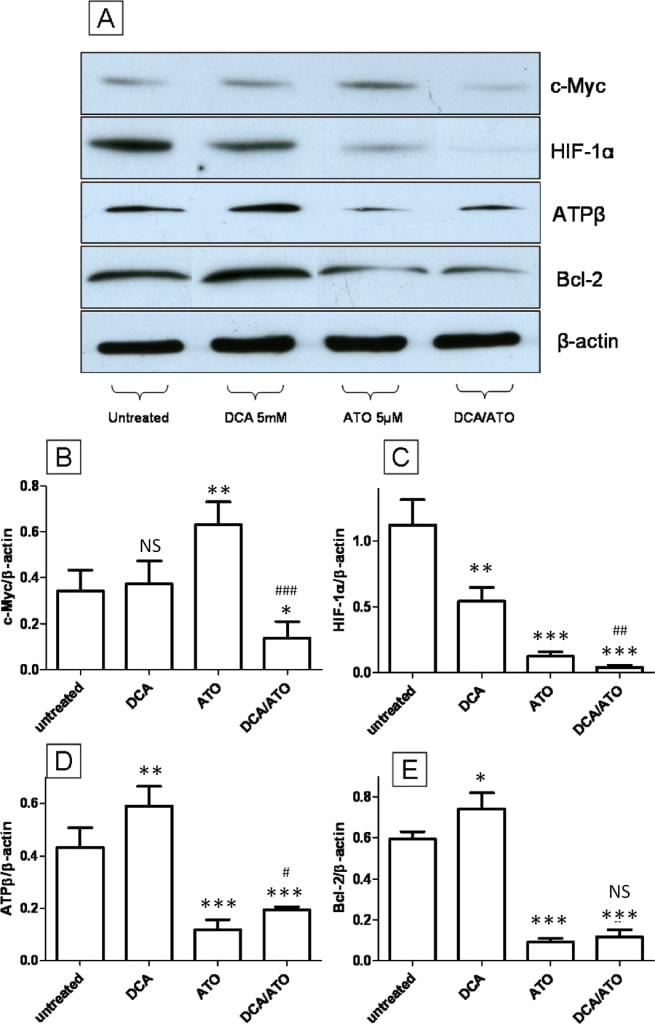
El DCAy la ATOtienen efectos opuestos sobre la expresión de la βsubunidadde la ATP sintasa y de Bcl-2, mientras que trabajan cooperativamente en la regulación a la baja de los niveles de proteínas c -Mycy HIF-1α
Examinamos el efecto del tratamiento con DCA 5 mM y ATO 5 μM (12 horas) sobre la expresión c-Myc y HIF1α, dos importantes factores de transcripción que se sabe que regulan el efecto Warburg [33] y la actividad mitocondrial [34], y sobre la expresión de las proteínas mitocondriales ATP sintasa β subunidad (ATPβ) y Bcl-2. La inmunotransferencia para c-Myc no mostró ningún cambio tras el tratamiento con DCA, mientras que ATO indujo la regulación al alza de c-Myc (Figura 7A y 7B), sin embargo ATO en combinación con DCA redujo el nivel de c-Myc en comparación con el control no tratado. Por otro lado, los niveles de HIF-1α mostraron una regulación a la baja tras el tratamiento con DCA y ATO. La combinación de ATO y DCA mostró una mayor regulación a la baja de HIF-1α en comparación con los grupos tratados con un solo agente y de control (Figura 7A y 7C). Bcl-2 es un miembro de la familia de proteínas BH3 que se une directamente a Bad y Bax y previene la apoptosis [35]. La inmunotransferencia realizada para ATPβ reveló que el tratamiento con DCA regulaba al alza la ATPβ, mientras que el ATO mostraba un efecto opuesto, reduciendo los niveles de ATPβ (Figura 7A y 7D), y el tratamiento combinado de ATO y DCA mostraba una respuesta intermedia en comparación con los grupos tratados con ATO o DCA. La inmunotransferencia para la proteína pro-supervivencia Bcl-2 mostró una regulación al alza tras el DCA, una regulación a la baja para el tratamiento con ATO, y una respuesta intermedia de la combinación de los dos fármacos (Figura 7A y 7E). Estos resultados también son muy similares a los niveles de ATP y la producción de ROS, donde el DCA y el ATO tuvieron efectos opuestos (Figura 2C y 3B). Los resultados de Bcl-2 y ATPβ se correlacionan directamente con la capacidad de DCA y ATO para influir en la producción de ROS y los niveles de ATP.
Discusión
Durante la última década, la ATO se ha utilizado eficazmente para tratar tanto a pacientes recién diagnosticados como a pacientes con LPA recidivante [4]. La capacidad anticancerígena de la ATO no se limita a la LPA [36] y muchos otros tumores en modelos animales han demostrado ser sensibles al tratamiento con ATO [37,38]. Sin embargo, la falta de información sobre los lugares de acción de la citotoxicidad de la ATO en otros tipos de tumores distintos de la LPA ha limitado su uso para el tratamiento de otros tipos de cáncer hasta hace pocos años [5] [5]. <p>Dos estudios recientes han demostrado que el DCA es más eficaz contra las células con defectos mitocondriales <sup><a href=»#48″>[48,49].</a></sup> Este estudio es el primero en demostrar que el tratamiento de dos aspectos del metabolismo – la inversión del efecto Warburg con DCA y la inhibición de la fosforilación oxidativa con ATO – es claramente una estrategia anticancerígena eficaz in vitro contra líneas celulares de cáncer de mama. La identificación de la citocromo C oxidasa como diana mitocondrial de la ATO proporciona información mecanicista novedosa para la aplicación de la ATO al tratamiento de tipos tumorales distintos de la LPA. La capacidad del DCA para aumentar la expresión de la subunidad b de la ATP sintasa, revirtiendo otro fenotipo metabólico generalizado del cáncer, también sugiere que el DCA puede ser relevante para una amplia gama de tipos de tumores. La capacidad del DCA para potenciar los efectos citotóxicos de agentes quimioterapéuticos distintos del ATO in vitro e in vivo también merece una mayor investigación. La falta de sensibilidad de la línea celular no cancerosa MCF-10A a las concentraciones clínicamente relevantes de ATO/DCA ensayadas también es alentadora, y sugiere que esta estrategia de tratamiento debería ensayarse más a fondo contra tumores sólidos in vivo.</p> <p class=»text-align: justify;»> <p>1 Molecular Genetics Group, Department of Translational Biosciences, John Curtin School of Medical Research, Building 131, Australian National University, P.O. Box 334, Canberra ACT 0200, AUSTRALIA. 2 Departamento de Oncología Radioterápica, Facultad de Medicina de Stanford, Stanford CA 94305 EE.UU.</p> <p><strong>Departamento de Oncología Radioterápica</strong>
En este estudio, hemos demostrado que el complejo IV de la ETC es una diana para la ATO. De un panel de inhibidores de la ETC, la disminución de ROS, la disminución de la producción de ATP y la despolarización de la MMP causadas por el tratamiento con dosis bajas de ATO de las células T-47D se reprodujeron únicamente con cianuro, un inhibidor del complejo IV bien caracterizado (Figura 5). La medición directa de la actividad de la citocromo C oxidasa en células enteras tratadas con ATO confirmó la capacidad de la ATO para inhibir directamente esta actividad enzimática. Se sabe que el complejo citocromo C oxidasa de la ETC tiene residuos de cisteína muy próximos entre sí [39]y es probable que reaccionen con la ATO.
La mayoría de las células cancerosas tienen niveles de ROS regulados al alza en comparación con lascélulasnormales [40] y el ETC se considera la principal fuente de ROS intracelular. La reducción de ROS de hasta un 60% por NaCN o ATO, demostró que el complejo IV es responsable de la mayor parte de la producción de ROS en las células T-47D. El estrés oxidativo de la producción elevada de ROS se ha descrito como un arma de doble filo. Se ha demostrado que un aumento moderado de ROS se asocia a un mayor potencial proliferativo [41]el aumento de enzimas antioxidantes como la glutatión transferasa [42], la superóxido dismutasa y la catalasa [43] y el aumento de proteínas prosupervivencia como Bcl-2 [42] y survivina [44]. Sin embargo, cantidades intolerables de ROS conducen finalmente a la muerte celular [45]. La reducción de ROS tras 12 horas de tratamiento con 5 μM de ATO también se correlacionó directamente con una disminución de los niveles de Bcl-2 (Figura 7E), lo que sugiere que las células T-47D se encuentran en un estado de resistencia apoptótica debido a los elevados niveles de ROS. La inhibición continuada del complejo IV del ETC conducirá a la despolarización del MMP y a la inhibición de la producción de ATP. La muerte celular que sigue a esta desregulación mitocondrial dará lugar a las elevadas ERO observadas al cabo de 4 horas o a una mayor concentración de ATO (Figura 3C y 3D), tal como han señalado otros autores [46] tras el tratamiento con ATO. Así pues, la elevación de ROS es una consecuencia, no la causa, de la muerte celular inducida por ATO.
El objetivo mitocondrial propuesto previamente para la ATO es la MTP. Zheng et al demostraron que la ATO induce la apertura de la MTP en mitocondrias aisladas de hígado de rata [11], lo que llevó a proponer que la MTP es responsable de la liberación de ROS y de la despolarización de la MMP observada durante el tratamiento con arsénico, y de la posterior apoptosis [47]. Nuestro estudio no excluye a la MTP como diana de la ATO. Nuestros datos indican que la inhibición de la MTP mediante CsA y BA puede bloquear parcialmente la activación de la caspasa 3/7 inducida por una alta concentración de ATO, sin embargo los bloqueantes de la MTP no consiguieron inhibir la despolarización de la MMP y la reducción de los niveles de ROS y ATP causados por una baja concentración de ATO. Así, aunque la ATO puede abrir la MTP, se requieren dianas alternativas para explicar la acción de la ATO a bajas concentraciones. Se ha demostrado que la inhibición del sistema de tioredoxina se produce en células MCF-7 tras el tratamiento con 2-5 μM de ATO y puede mediar algunos efectos celulares de la ATO a bajas concentraciones [13]sin embargo, este mecanismo conduciría a un aumento del estrés oxidativo y no puede explicar la disminución de ROS observada en el presente estudio.
El DCA, como inhibidor de la piruvato deshidrogenasa cinasa, puede revertir el efecto glucolítico y ha demostrado importantes propiedades anticancerígenas tanto in vitro como in vivo [17-23]. Algunos estudios muestran un aumento de la apoptosis con DCA [17,18,20]. Sin embargo, nuestros estudios en cáncer de mama y los estudios de Stockwin et al indican que el DCA actúa como un agente citostático en lugar de citotóxico en una serie de líneas celulares, excepto a concentraciones muy altas [22,48] [22,48]. Michelakis informó recientemente de que los niveles séricos de DCA eran de 0,5 mM en pacientes con glioblastoma que tomaban 6,25 mg/kg por vía oral dos veces al día [23] [23]por lo que es probable que las concentraciones clínicamente relevantes se sitúen en el intervalo de 0,5-5 mM. Se ha demostrado que el DCA induce un aumento pequeño pero significativo de la actividad de la caspasa 3/7, incluso cuando no se observa apoptosis, y se ha propuesto que el DCA puede sensibilizar las células cancerosas a los agentes citotóxicos [22]. El presente estudio confirmó esta hipótesis al demostrar que el DCA sensibilizó a las células T-47D frente al tratamiento con ATO, tal y como se observó en la doble tinción AV+/PI+ (Figura 1E y texto). La ATO fue seleccionada como agente de ensayo debido a sus propiedades antimitocondriales, ya que propusimos que al invertir el fenotipo glucolítico con DCA y dirigir más piruvato hacia la fosforilación oxidativa mitocondrial, mientras que simultáneamente se dirigía a las mitocondrias con ATO, se observaría un efecto cooperativo con esta combinación de fármacos. El principio de esta estrategia dual está respaldado por los resultados de dos publicaciones recientes que han descubierto que las células con defectos mitocondriales (por ejemplo, rho (0) o después del tratamiento con inhibidores mitocondriales no farmacológicos como la rotenona) muestran una mayor sensibilidad a los efectos inhibidores del crecimiento del DCA [48,49]. Aunque la eficacia de esta estrategia dual aún no se ha demostrado in vivo, los efectos de la combinación ATO/DCA mejorada se observaron tanto en la inhibición del crecimiento como en la apoptosis a concentraciones del fármaco en rangos clínicamente relevantes. Algunos efectos en el extremo inferior del intervalo de concentración no fueron importantes (por ejemplo, un aumento del 15% en las células apoptóticas), sin embargo, el impacto in vivo durante semanas en lugar de horas de tratamiento justifica una mayor investigación.
Está bien documentado que el DCA puede alterar el comportamiento mitocondrial, es decir, despolarizar la MMP [18,20]. Sin embargo, este fenómeno no ha sido bien explicado. Observamos que el DCA puede aumentar el ATP a la vez que despolariza el MMP en células T-47D (Figura 2). Basándonos en estos datos propusimos que el DCA está alterando la función mitocondrial a través del complejo V del ETC, la ATP sintasa. La ATP sintasa es el último paso del ETC y utiliza el MMP generado por el ETC para producir ATP. La ATPb se regula a la baja en los cánceres de pulmón, cerebro, mama y gástrico [50], lo que podría explicar la membrana mitocondrial hiperpolarizada que puede encontrarse en muchas células cancerosas [51]. Hemos planteado la hipótesis de que el DCA puede aumentar la actividad de la ATP sintasa ya sea a través de la regulación alostérica debido a su homología con el piruvato o a través del aumento de los niveles de proteína ATP sintasa tras la inversión del efecto Warburg y la disminución de los niveles de HIF-1α. La inmunotransferencia demostró claramente que la ATPβ está regulada al alza tras el tratamiento con DCA (Figura 7D), lo que sugiere que el DCA puede aumentar la actividad de la ATP sintasa, que a su vez contribuye al aumento de la producción de ATP y al agotamiento de la MMP.
HIF-1α y c-Myc son dos importantes factores de transcripción oncogénicos que regulan el metabolismo en las células cancerosas. HIF-1α puede regular al alza las enzimas de la vía glucolítica y la deshidrogenasa del ácido láctico, manteniendo el efecto Warburg en las células cancerosas [33]. Esto no sólo aumenta la producción de ATP, sino también el suministro de precursores como la glucosa-6-fosfato y la fructosa-6-fosfato para la producción de ácidos nucleicos a través de la vía de las pentosas fosfato [16]por otra parte, c-Myc no sólo desempeña un papel central en la promoción de la transición del ciclo celular de la fase G1 a la fase S mediante la regulación de las ciclinas y sus quinasas e inhibidores, sino que también regula al alza los componentes proteicos del ETC, como la COXI-IV, y ayuda a aumentar la actividad mitocondrial [52] [52]. Por lo tanto, la combinación de la sobreexpresión de c-Myc y HIF-1α es importante para inducir el efecto Warburg y aumentar la actividad mitocondrial, apoyando el eje glutamina-TCA que es esencial para el anabolismo de aminoácidos y ácidos grasos necesarios para la división celular [16] [16]. Esta firma metabólica contribuye a la carcinogénesis y al fenotipo maligno de muchos tumores [33,34]. Tanto el ATO como el DCA pueden disminuir significativamente los niveles de HIF-1α, lo que sugiere una reducción del efecto Warburg (Figura 7C). Mientras que el DCA por sí solo no tuvo ningún efecto sobre los niveles de c-Myc, sorprendentemente, la ATO aumentó significativamente los niveles de c-Myc (Figura 7B). Esto puede ser un bucle de retroalimentación positiva, donde las células están tratando de aumentar la actividad ETC después de la inhibición del complejo IV. Sorprendentemente, el DCA revirtió el efecto de la ATO sobre c-Myc, y la combinación de ATO/DCA reprimió fuertemente la expresión de c-Myc, en correlación con la capacidad de estos agentes para trabajar juntos en la reducción de la proliferación celular y la inducción de la muerte celular (Figura 1).
La eficacia de ATO contra PML se ha atribuido a varias características especiales de esta malignidad – diferenciación a través de la inactivación de PML-RARα, baja capacidad antioxidante de las células PML para protegerse contra los elevados niveles de ROS, y la acumulación del fármaco debido a la alteración de la osmorregulación [5]. Hay muchos ensayos en marcha para el uso de ATO en una serie de neoplasias malignas sólidas, aunque hasta ahora hay pocos resultados publicados. Los resultados disponibles sugieren que la ATO no es muy eficaz como agente único en pacientes con cáncer avanzado de páncreas, hígado o melanoma <a href=»#5″>[5]</a></sup>. Los investigadores abogan por evaluar la ATO en combinación con otros fármacos anticancerosos<sup><a href=»#53″>[53,54]</a></sup>, habiéndose completado algunos ensayos de fase I <sup><a href=»#55″>[55]</a></sup>. A medida que nuestra comprensión de los mecanismos de ATO sigue creciendo, especialmente sus efectos a través del metabolismo del cáncer, podemos ser más capaces de aprovechar su potencial contra el cáncer in vivo en nuevas combinaciones de fármacos</p> <p>
<h2>Conclusiones</h2>