Ramon C Sun1,2, Philip G Board1 e Anneke C Blackburn1*
1 Gruppo di Genetica Molecolare, Dipartimento di Bioscienze Traslazionali, John Curtin School of Medical Research, Building 131, Australian National University, P.O. Box 334, Canberra ACT 0200, AUSTRALIA
2 Dipartimento di Radioterapia Oncologica, Stanford School of Medicine, Stanford CA 94305 USA.
1*Corrispondenza: [email protected]
© 2011 Sun et al; licenziatario BioMed Central Ltd. Questo è un articolo ad accesso libero distribuito secondo i termini della Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), che ne consente l’uso, la distribuzione e la riproduzione illimitata su qualsiasi supporto, a condizione che l’opera originale sia adeguatamente citata.
Ricevuto: 3 maggio 2011
Accettato: 18 novembre 2011
Pubblicato: 18 novembre 2011
Abstract
Background
Le cellule tumorali hanno un profilo metabolico diverso rispetto alle cellule normali. L’effetto Warburg (aumento della glicolisi aerobica) e la glutaminolisi (aumento dell’attività mitocondriale dovuta al catabolismo della glutammina) sono caratteristiche ben note del cancro e sono accompagnate da un aumento della produzione di lattato, da un’iperpolarizzazione della membrana mitocondriale e da una maggiore produzione di specie reattive dell’ossigeno.
Metodi
In questo studio abbiamo mirato all’effetto Warburg con il dicloroacetato (DCA) e all’aumento dell’attività mitocondriale della glutaminolisi con il triossido di arsenico (ATO) in cellule di cancro al seno, misurando la proliferazione cellulare, la morte cellulare e le caratteristiche mitocondriali.
Risultati
La combinazione di DCA e ATO è stata più efficace nell’inibire la proliferazione cellulare e nell’indurre la morte cellulare rispetto a uno dei due farmaci da solo. Abbiamo esaminato l’effetto di questi trattamenti sul potenziale di membrana mitocondriale, sulla produzione di specie reattive dell’ossigeno e sui livelli di ATP e abbiamo identificato nuovi meccanismi molecolari all’interno dei mitocondri sia per l’ATO che per il DCA: l’ATO riduce la funzione mitocondriale attraverso l’inibizione della citocromo C ossidasi (complesso IV della catena di trasporto degli elettroni) mentre il DCA aumenta l’espressione della subunità b dell’ATP sintasi. Il potenziamento della citotossicità dell’ATO da parte del DCA è correlato a una forte soppressione dell’espressione di c-Myc e HIF-1a e a una riduzione dell’espressione della proteina di sopravvivenza Bcl-2. Conclusioni
Questo studio è il primo a dimostrare che puntare su due caratteristiche metaboliche chiave del cancro è una strategia antitumorale efficace con un potenziale terapeutico.
Parole chiave
Dicloroacetato, cancro al seno, catena di trasporto degli elettroni, mitocondri, triossido di arsenico
Introduzione
Il triossido di arsenico (ATO) è stato utilizzato come agente terapeutico per oltre 2000 anni. Originario della Cina [1], viene attualmente utilizzato contro la leucemia promieloide acuta (APL) in pazienti che hanno avuto una ricaduta dopo la terapia con acido allotrans-retinoico/antraciclina e viene promosso per la terapia di prima linea della APL de novo [2-4]. L’ATO è noto come molecola iperreattiva e potenzialmente in grado di legarsi ai gruppi tiolici di molte proteine [2,5]. La sua capacità di legarsi alla proteina mutante PML-RAR-α, ricca di tioli e prodotta da una traslocazione cromosomica nell’APL, lo ha reso un farmaco efficace nell’APL [2,5,6]. È stato dimostrato che l’ATO induce l’apoptosi in diverse linee cellulari tumorali in vitro e in vivo [7,8], ma è stato difficile considerare l’ATO per l’uso clinico in tipi di tumore diversi dall’APL a causa della mancanza di conoscenza dei bersagli molecolari che determinano la sua citotossicità. Negli ultimi 10 anni, i cambiamenti fisiologici all’interno delle cellule tumorali in risposta al trattamento con ATO sono stati ben caratterizzati e sono in corso numerosi studi clinici per nuove applicazioni di ATO [5]. L’ATO è stato proposto come tossina mitocondriale [9]. L’ATO può depolarizzare il potenziale di membrana mitocondriale (MMP) [10], aumentare la produzione intracellulare di specie reattive dell’ossigeno (ROS) [8] e indurre apoptosi [8]. Il bersaglio proposto per l’ATO in grado di ottenere questi cambiamenti fenotipici è il poro di transizione mitocondriale (MTP) [11]. È stato dimostrato che l’ATO induce l’apertura del poro di transizione mitocondriale (MTP), che induce il rilascio di citocromo c e si propone di dissipare l’MMP e aumentare il rilascio di ROS dai mitocondri [12]. Più recentemente, anche il sistema della tioredoxina, in particolare la tioredoxina reduttasi, è stato identificato come un bersaglio dell’ATO che può contribuire all’aumento dello stress ossidativo e all’alterazione della segnalazione redox dopo il trattamento con ATO delle cellule tumorali [9,13].
L’effetto Warburg è un fenomeno molto diffuso che è stato identificato in oltre il 90% di tutte le forme tumorali. Le cellule che presentano l’effetto Warburg adottano vie alternative di omeostasi energetica per mantenere il loro fenotipo proliferativo [14]. Il premio Nobel Otto Warburg ha affermato che le cellule tumorali si affidano alla glicolisi o alla fosforilazione dei substrati per generare ATP e sopprimono le loro attività mitocondriali [15]. Grazie a tecnologie più avanzate, studi recenti hanno confermato l’aspetto della produzione di ATP dell’ipotesi di Warburg, ma hanno rivelato che l’attività mitocondriale non è soppressa nelle cellule tumorali. Al contrario, i mitocondri svolgono un ruolo vitale nel fornire substrati per mantenere la divisione cellulare [16].
L’effetto antitumorale dell’inversione dell’effetto Warburg è stato descritto di recente e un vecchio farmaco, il dicloroacetato (DCA), che può reindirizzare la sintesi di ATP dalla glicolisi alla fosforilazione ossidativa, ha dimostrato una buona attività antitumorale sia in vitro [17-19] che in vivo [20-23]. Il DCA è un inibitore della piruvato deidrogenasi chinasi e determina un aumento dell’attività della piruvato deidrogenasi [19]. Questo porta a una maggiore conversione del piruvato in acetil-CoA piuttosto che in acido lattico, come descritto dall’effetto Warburg, e stimola la respirazione mitocondriale aumentando l’apporto di acetil-CoA. Di conseguenza, dopo il trattamento con DCA, le cellule tumorali hanno mostrato un aumento dei livelli di ROS, una depolarizzazione delle MMP in vitro e un aumento dell’apoptosi sia in vitro che in vivo [17,20].
Poiché il DCA può reindirizzare i substrati verso la respirazione mitocondriale e la produzione di ATP, potrebbe avere un’attività sinergica con i farmaci antitumorali che compromettono l’attività mitocondriale. Proponiamo che, invertendo il fenotipo glicolitico con il DCA e indirizzando una maggiore quantità di piruvato verso la fosforilazione ossidativa mitocondriale, e contemporaneamente colpendo i mitocondri con l’ATO, si verifichi una grave alterazione dell’omeostasi energetica nelle cellule tumorali. In questo studio abbiamo dimostrato che la combinazione di DCA e ATO inibisce la crescita di linee cellulari di cancro al seno in vitro. Inoltre, abbiamo identificato nuovi meccanismi molecolari all’interno dei mitocondri che possono contribuire alla citotossicità dell’ATO, fornendo un ulteriore supporto all’uso dell’ATO contro i tumori solidi.
Materiali e metodi
Reagenti
JC-1, CFSE e H2DCFDA sono stati acquistati da Invitrogen (Carlsbad, CA, USA), i kit per i saggi CellTiter-Glo e Caspase-Glo sono stati acquistati da Promega Co (San Luis, CA, USA). Il resto delle sostanze chimiche è stato acquistato da Sigma Co (St. Louis, MO, USA).
Coltura cellulare
Le linee cellulari sono state ottenute dalle seguenti fonti negli anni indicati. Dott.ssa Anna DeFazio, Westmead Millenium Institute, Sydney, Australia: T-47D (2003), BT-20 (2003), MCF-10A (2005) e MCF-10AT1 (2005); Prof. Chris Parish, Australian National University, Canberra, Australia: 13762 MAT (2007), MDA-MB-468 (2003) e MDA-MB-231 (2003). Le linee cellulari hanno un aspetto coerente con le morfologie pubblicate, ma non sono state autenticate di recente. Le cellule di carcinoma epiteliale mammario umano (T-47D) sono state coltivate in terreno RPMI 1640 integrato con il 10% di siero fetale bovino in presenza dello 0,1% di PSN (3% penicillina, 5% streptomicina e 5% neomicina). Le linee cellulari BT-20, MDA-MB-231, MDA-MB-468 e 13762 MAT sono state mantenute in terreno DMEM/F12 integrato con il 10% di FBS e 2 mM di L-glutamina. Per le cellule MCF-10A e MCF-10AT1 è stato utilizzato un terreno DMEM/F-12 con 25% di siero di cavallo, 0,01% di EGF, 0,28 UI/ml di insulina, 0,01% di tossina di colera e 0,5 μg/ml di idrocortisone. Tutte le linee cellulari sono state mantenute a 37°C in 5% di CO2.
Vitalità cellulare
Per la valutazione della vitalità cellulare, le cellule sono state piastrate in piastre da 96 pozzetti a una densità di 3000 cellule per pozzetto e 8 pozzetti per gruppo. Dopo l’esposizione a DCA e ATO per 24-72 ore, le cellule sono state incubate per 3 ore con rosso neutro (30 μg/ml) in terreno fresco, quindi lavate con PBS, seguite dall’aggiunta di tampone di lisi (acido acetico/metanolo, 80%/20%) e l’assorbanza a 540 nm è stata registrata. I risultati sono espressi come media ± S.D., i calcoli sono stati eseguiti utilizzando il pacchetto software Prism, è stata applicata l’ANOVA con post-test di Tukey e P < 0,05 è stato considerato statisticamente significativo. Gli esperimenti sono stati eseguiti almeno tre volte e i dati presentati sono relativi a un esperimento rappresentativo.
Proliferazione cellulare
Le cellule T-47D sono state raccolte e risospese in 1 ml di terreno RPMI. Le cellule sono state marcate con l’aggiunta di 1 ml di PBS contenente 5 μM di carbossifluoresceina succinimidil estere (CFSE), seguita da 5 minuti di incubazione a temperatura ambiente. Le cellule marcate sono state quindi lavate due volte, contate e seminate a105 cellule/pozzetto in una piastra da 12 pozzetti. Il giorno dell’analisi, le cellule T-47D sono state raccolte e lavate due volte con PBS, quindi l’intensità del CFSE è stata esaminata mediante FACS. I risultati sono espressi come media ± S.D. (n = 3), i calcoli sono stati eseguiti con il pacchetto software Prism, è stata applicata l’ANOVA con post-test Tukey e P < 0,05 è stato considerato statisticamente significativo. Gli esperimenti sono stati eseguiti almeno tre volte e i dati presentati si riferiscono a un esperimento rappresentativo.
L’apoptosi è stata quantificata mediante citometria a flusso dopo aver colorato le cellule con Annexina-V (AV) marcata con FITC (Invitrogen Co.) e ioduro di propidio (PI). Dopo il trattamento farmacologico, le cellule T-47D sono state raccolte e centrifugate a 1200 rpm per 5 minuti; il pellet è stato lavato due volte con PBS e poi risospeso in 100 μl di tampone di legame dell’Annexina-V (0,14 M NaCl, 2,5 mM CaCl2, 0,01 M HEPES pH 7,4). L’Annexina-V (1 μl) e 5 μl di PI (50 μg/ml) sono stati aggiunti ai campioni e incubati al buio per 15 minuti. I campioni sono stati tenuti in ghiaccio dopo l’incubazione fino all’analisi FACS. I risultati sono espressi come media ± S.D. (n = 3), i calcoli sono stati eseguiti utilizzando il pacchetto software Prism, è stata applicata l’ANOVA con post-test di Tukey e P < 0,05 è stato considerato statisticamente significativo. Gli esperimenti sono stati eseguiti almeno tre volte e i dati presentati sono relativi a un esperimento rappresentativo.
Generazione di ROS
Per la valutazione dei livelli di ROS intracellulari, le cellule sono state placcate in piastre da 12 pozzetti con densità cellulare di 1 ×105 cellule per pozzetto e trattate con farmaci per 12 ore. il 2′, 7′-diidroclorofluroresceinacetato (H2DCFDA) è stato aggiunto al terreno di coltura a una concentrazione finale di 10 μM e le cellule sono state lasciate colorare per 1 ora al buio. Dopo la colorazionecon H2DCFDA, le cellule sono state tripsinizzate, lavate due volte e risospese in 100 μl di PBS. L’intensità diH2DCFDAè stata esaminata mediante FACS. I risultati sono espressi come media ± S.D. (n = 3), i calcoli sono stati eseguiti con il pacchetto software Prism, è stata applicata l’ANOVA con post-test Tukey e P < 0,05 è stato considerato statisticamente significativo. Gli esperimenti sono stati eseguiti almeno tre volte e i dati presentati sono relativi a un esperimento rappresentativo.
Concentrazione di ATP e attività della caspasi
Il livello interno di ATP e l’attività della caspasi nelle cellule T-47D sono stati valutati utilizzando i kit CellTiter-Glo e Caspase-Glo 3/7 (Promega Corp., Madison, WI) secondo le istruzioni del produttore. Le cellule T-47D sono state coltivate in assenza e in presenza di farmaci per 12 ore in piastre opache bianche da 96 pozzetti (4 pozzetti per gruppo). Sono stati aggiunti volumi uguali di reagenti CellTiter-Glo, dopodiché i campioni sono stati incubati per 15 minuti su un agitatore a temperatura ambiente. La luminescenza è stata registrata con il luminometro per micropiastre Glomax (Promega Co., Madison, WI) secondo il protocollo CellTiter preimpostato. I risultati sono espressi come media ± S.D. (n = 4), i calcoli sono stati eseguiti con il pacchetto software Prism, è stata applicata l’ANOVA con post-test Tukey e P < 0,05 è stato considerato statisticamente significativo. Gli esperimenti sono stati eseguiti almeno tre volte e i dati presentati sono relativi a un esperimento rappresentativo.
Potenziale di membrana mitocondriale
Analogamente alla misurazione dei ROS, le cellule sono state placcate in piastre da 12 pozzetti con densità cellulare di 1 ×105 cellule per pozzetto e trattate con farmaci per 12 ore. il 5, 5′, 6, 6′-tetracloro-1, 1′, 3, 3′-tetraetilbenzimidazolo-carbocianina ioduro (JC-1) è stato aggiunto al terreno di coltura a una concentrazione finale di 0,2 μM e le cellule sono state lasciate colorare per 30 minuti al buio. Dopo la colorazione con JC-1, le cellule sono state tripsinizzate, lavate due volte con PBS e risospese in 100 μl di PBS. L’intensità di JC-1 è stata esaminata mediante FACS. I risultati sono espressi come media ± S.D. (n = 3), i calcoli sono stati eseguiti utilizzando il pacchetto software Prism, è stata applicata l’ANOVA con post-test di Tukey e P < 0,05 è stato considerato statisticamente significativo. Gli esperimenti sono stati eseguiti almeno tre volte e i dati presentati sono relativi a un esperimento rappresentativo.
Attività della citocromo C ossidasi
Il saggio della citocromo C ossidasi è stato valutato in base al metodo precedentemente pubblicato [24]. In breve, dopo il trattamento farmacologico in piastre da 96 pozzetti (8 pozzetti per gruppo), le cellule T-47D sono state permeabilizzate con 50 μl di saponina allo 0,01%, seguite dall’aggiunta di 100 μl di terreno di substrato (4 mM di 3,3-diaminobenzidina tetracloruro (DAB), 100 μM di citocromo C ridotto, 2 μg/ml di catalasi in 0,1 M Na fosfato, pH 7,0). L’assorbanza a 450 nm è stata misurata subito dopo l’aggiunta del substrato e monitorata per 30 minuti. I risultati sono espressi come media ± S.D. (n = 8), i calcoli sono stati eseguiti utilizzando il pacchetto software Prism, è stata applicata l’ANOVA con post-test di Tukey e P < 0,05 è stato considerato statisticamente significativo. Gli esperimenti sono stati eseguiti almeno tre volte e i dati presentati sono relativi a un esperimento rappresentativo.
Attività della PDH
L’attività della PDH è stata misurata utilizzando il kit per micropiastre MitoSciences PDH (#MSP18, MitoSciences, Oregon USA). Per misurare l’effetto dei farmaci sull’attività della PDH nelle cellule, le cellule T-47D sono state trattate con farmaci nei mezzi di coltura per 3 ore, dopodiché sono state lavate e risospese in PBS. Gli estratti cellulari sono stati preparati e dosati per l’attività della PDH a una concentrazione di 15 mg di proteine/ml, secondo le istruzioni del kit. Per misurare l’inibizione diretta della PDH da parte dell’ATO, la PDH è stata isolata dalle cellule T-47D non trattate applicando gli estratti cellulari alla piastra di cattura degli anticorpi. Dopo aver lavato la piastra, la soluzione di dosaggio contenente il farmaco è stata aggiunta ai pozzetti e quindi è stata immediatamente analizzata l’attività della PDH. I risultati sono espressi come media ± S.D. (n = 4), i calcoli sono stati eseguiti utilizzando il pacchetto software Prism, è stata applicata l’ANOVA con post-test Tukey e P < 0,05 è stato considerato statisticamente significativo. Gli esperimenti sono stati eseguiti almeno tre volte e i dati presentati si riferiscono a un esperimento rappresentativo.
Immunoblotting e analisi densitometrica
Le cellule (1 ×106 ) sono state poste in piastre di coltura tissutale T25 e sono state trattate con ATO o DCA per 12 ore. I lisati cellulari sono stati preparati con l’aggiunta di 500 μl del reagente per l’estrazione delle proteine di mammifero MPER® (Thermo Scientific, IL, USA). L’immunoblotting è stato eseguito come descritto in precedenza [25], utilizzando gli anticorpi per c-Myc (Roche, IN, USA, clone 9E10), HIF-1α (Abcam, Cambridge, UK, #ab82832), Bcl-2 (Abcam #ab692), ATP sintasi β-subunità (Abcam #ab14730) e β-actina (Abcam #ab8227). I Western blot sono stati rilevati utilizzando la chemiluminescenza e l’esposizione di pellicole a raggi X. Le immagini sono state acquisite con uno scanner piano CanoScan 8600F e quantificate con il software ImageJ (versione 1.4, NIH, USA) e standardizzate per la β-actina in ogni corsia. I risultati sono stati raggruppati da 3 esperimenti separati ed espressi come media ± S.D. (n = 3), i calcoli sono stati eseguiti utilizzando il pacchetto software Prism, è stata applicata l’ANOVA con post-test Tukey e P < 0,05 è stato considerato statisticamente significativo.
Risultati
DCA e ATO insieme sono più efficaci nel ridurre la proliferazione cellulare e nell’indurre la morte cellulare
Per studiare l’effetto combinato di ATO e DCA nell’inibire la crescita cellulare, le linee cellulari di carcinoma mammario sono state trattate per diversi giorni con entrambi i farmaci e il numero totale di cellule è stato valutato utilizzando il saggio di vitalità cellulare in rosso neutro. Il gruppo di linee cellulari rappresenta i principali sottotipi di carcinoma mammario umano (luminale (T-47D), basale A (MDAMB-468, BT-20), basale B (MDA-MB-231)), o ha altra rilevanza come modello sperimentale (13762MAT – adenocarcinoma mammario di ratto sensibile al DCA in vivo [22], MCF10AT1 – derivato maligno di cellule immortalizzate MCF-10A). Le cellule MDA-MB-468, MDA-MB-231 e T-47D hanno mostrato una riduzione significativa, compresa tra il 10% e il 40%, del numero totale di cellule dopo 72 ore di trattamento con DCA 5 mM (Figura 1A), mentre le cellule 13762 MAT, BT-20 e MCF-10AT1 non hanno risposto durante il periodo di trattamento. Tutte le linee cellulari tumorali analizzate sono risultate sensibili all’ATO, ma a concentrazioni diverse. Una riduzione del numero totale di cellule è stata osservata in BT-20, T-47D, MCF-10AT1 e MDA-MB-468 con un trattamento con ATO di soli 5 μM, mentre sono stati necessari 15 μM di ATO per ottenere la stessa efficacia nelle linee cellulari MDA-MB-231 e 13726 MAT. È sorprendente notare che DCA e ATO in combinazione hanno mostrato un effetto maggiore rispetto a uno dei due farmaci da solo in tutte le linee cellulari tumorali testate. Nei casi in cui ATO e DCA sono stati efficaci come agenti singoli nel ridurre il numero di cellule, l’effetto del trattamento combinato è stato approssimativamente pari alla somma degli effetti dei singoli farmaci (T-47D, MDA-MB-468 e MDA-MB-231). Se il DCA da solo non ha mostrato alcuna riduzione del numero di cellule, è stato comunque in grado di potenziare l’inibizione della crescita di ATO di 2-3 volte (13762 MAT, BT-20 e MCF10AT1) (Figura 1A). La linea cellulare non cancerosa MCF-10A non ha mostrato alcuna riduzione del numero di cellule dopo ATO (15 μM), DCA (5 mM) o trattamento combinato.
L’effetto di ATO e DCA è stato ulteriormente esaminato con le cellule T-47D, una delle linee cellulari più sensibili al solo DCA. La risposta delle cellule T-47D al DCA è stata dose-dipendente e dopo 72 ore le cellule trattate con DCA 5 mM avevano il 42 ± 6% di cellule in meno rispetto alla coltura di controllo (Figura 1B). L’ATO da solo (5 μM) ha ridotto il numero totale di cellule del 56 ± 4% e le cellule trattate sia con DCA che con ATO hanno mostrato un’ulteriore diminuzione rispetto al gruppo con solo ATO (Figura 1C). La curva dose-risposta ha mostrato che il trattamento combinato di DCA (5 mM) e ATO può ridurre l’IC50 a 0,25 volte quello di ATO da solo (Figura 1C). Questo effetto si verifica all’interno dell’intervallo di concentrazione raggiunto clinicamente per ATO (fino a 5-7 μM [26]).
Il saggio di proliferazione CFSE ha dimostrato che le cellule trattate con ATO (5 μM) o DCA (5 mM) hanno emesso una fluorescenza CFSE significativamente più alta (rispettivamente 2,1 volte e 2,2 volte) dopo 72 ore di trattamento, indicando l’inibizione della crescita. Le cellule trattate con ATO e DCA hanno mostrato un aumento di 3,4 volte dell’intensità della CFSE rispetto alle cellule non trattate, indicando che i farmaci hanno lavorato insieme per inibire la proliferazione cellulare (Figura 1D).
L’effetto di ATO e DCA sulla morte delle cellule T-47D è stato valutato utilizzando la doppia colorazione AV e PI e le cellule sono state analizzate con il cell sorting a fluorescenza. Il solo DCA (5 mM) non ha indotto la morte cellulare nelle cellule T-47D (Figura 1E), analogamente all’effetto del DCA sulle cellule MAT 13762 riportato in precedenza [22]. Anche l’ATO (5 μM) non ha indotto la morte cellulare dopo 12 ore di trattamento, tuttavia le cellule trattate sia con ATO 5 μM che con DCA 5 mM hanno mostrato un piccolo (15%) aumento della popolazione AV+/PI+ (13,2 ± 0,6% di cellule apoptotiche rispetto all’11,5 ± 1,0% per ATO/DCA vs. non trattate rispettivamente, p = 0,07), suggerendo che il DCA potrebbe potenziare gli effetti apoptotici dell’ATO. A concentrazioni più elevate e con 48 ore di trattamento, ATO (20 μM) ha aumentato la quantità di morte cellulare del 35 ± 8% (P = 0,029) rispetto alla coltura non trattata (Figura 1E). La combinazione di 5 mM di DCA con il trattamento con 20 μM di ATO ha determinato un aumento di 4 volte della popolazione AV+/PI+ rispetto al solo ATO, indicando che il DCA può potenziare la morte cellulare indotta da ATO nelle cellule di carcinoma mammario T-47D (Figura 1E).
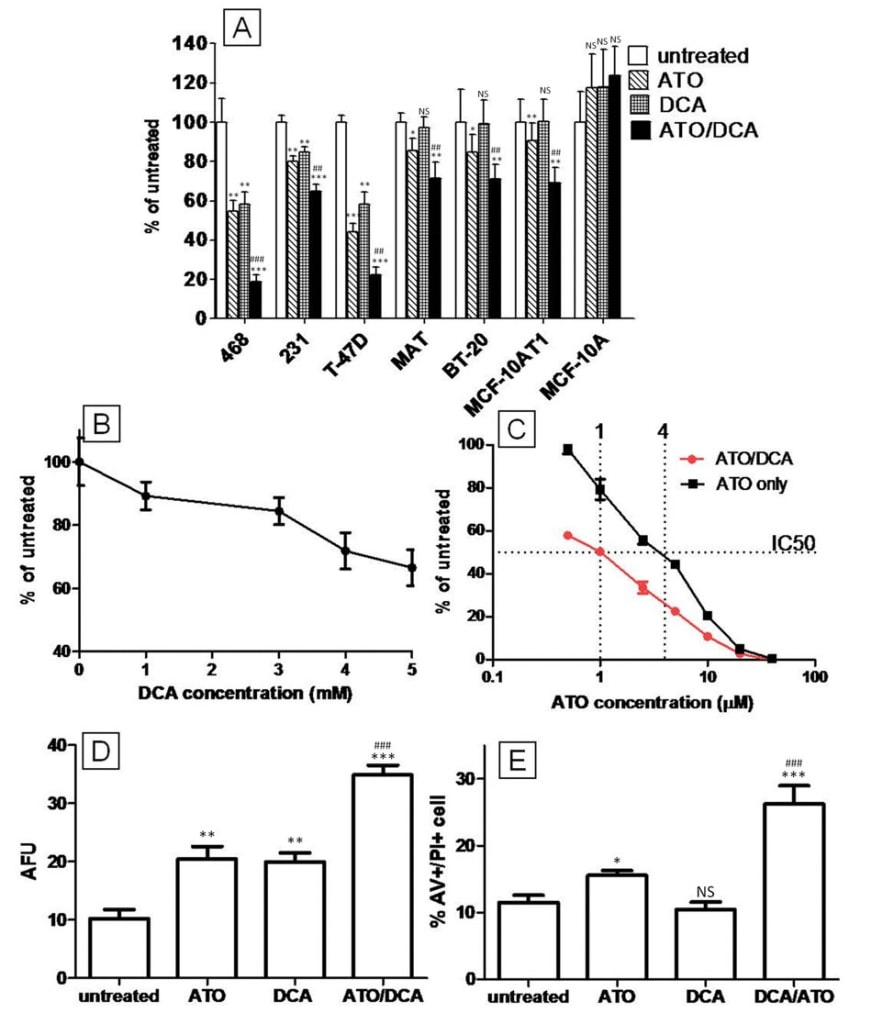
L’ATO e il DCA agiscono insieme sulla depolarizzazione dell’MMP, ma hanno effetti opposti sull’induzione della produzione di ATP e ROS
È stato dimostrato che sia l’ATO che il DCA alterano la funzione mitocondriale, depolarizzando l’MMP e aumentando la produzione di ROS [20]. Pertanto, questi parametri, insieme al livello di ATP, sono stati studiati nel tentativo di determinare se contribuiscono al potenziamento degli effetti antitumorali del trattamento combinato ATO/DCA. Misurato mediante colorazione JC-1, il numero di cellule T-47D con una MMP iperpolarizzata (quadrante superiore destro) è stato significativamente ridotto dal trattamento con DCA (5 mM) o ATO (5 μM e 20 μM) (t = 12 ore) (rispettivamente 28 ± 6%, 38 ± 4% e 69 ± 7% di riduzione rispetto alle cellule di controllo non trattate), coerentemente con i dati precedentemente pubblicati [20,27]. La combinazione di ATO (5 μM o 20 μM) con 5 mM di DCA ha portato a una diminuzione ancora maggiore (rispettivamente 53 ± 9% e 77 ± 12%) rispetto alle cellule non trattate (Figura 2A), dimostrando che DCA e ATO possono lavorare insieme per depolarizzare la MMP. I livelli di ATP sono diminuiti sia a basse che ad alte dosi di ATO (rispettivamente 14 ± 3% e 32 ± 5%) dopo 12 ore di trattamento, mentre le cellule trattate con DCA hanno mostrato un aumento del 6 ± 1% dei livelli di ATP (Figura 2B e 2C). Alla dose elevata di ATO, il DCA non è stato in grado di aumentare la produzione di ATP (Figura 2B), mentre alla dose bassa di ATO, le cellule trattate con DCA e ATO hanno mostrato un piccolo aumento della produzione di ATP rispetto al trattamento con il solo ATO (Figura 2C). Questi dati indicano che DCA e ATO agiscono sulla produzione di ATP attraverso bersagli distinti all’interno delle cellule.
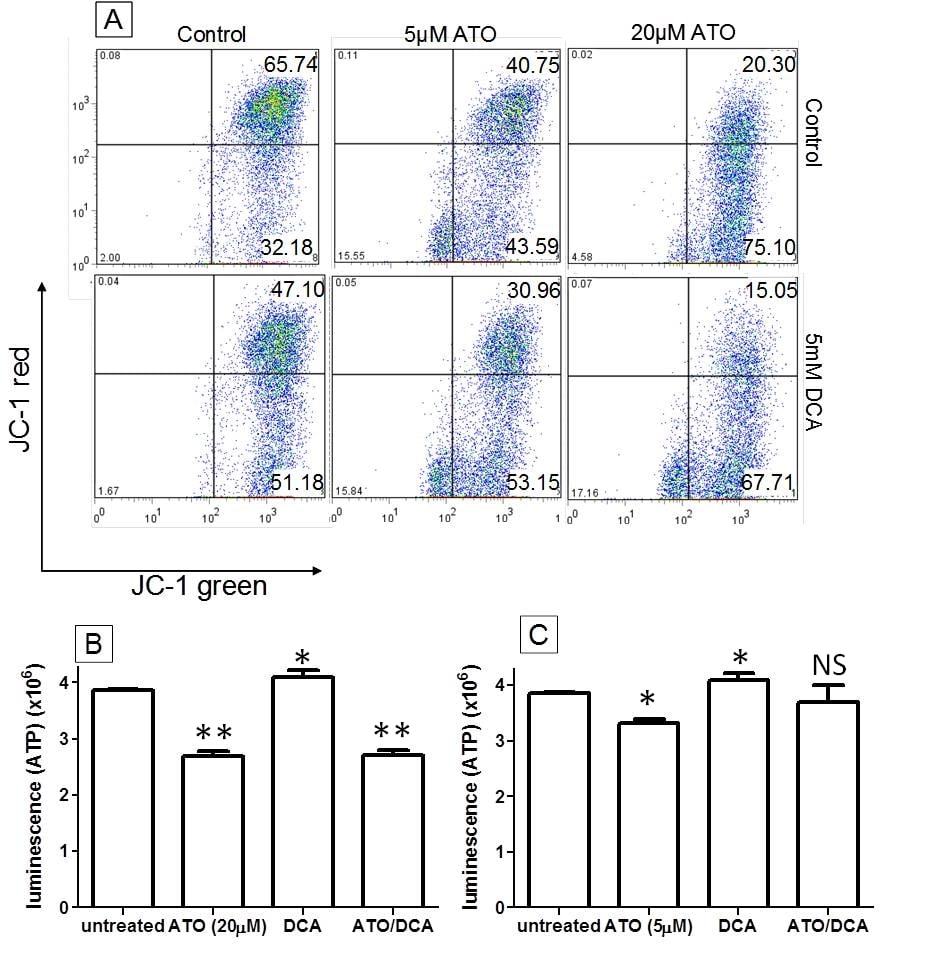
L’aumento dei livelli intracellulari di ROS è stato proposto come causa della citotossicità dell’ATO [2,9]. L’effetto combinato di ATO e DCA sui ROS è stato studiato utilizzando il colorante fluorescente H2DCFDA. Le cellule trattate con 20 μM di ATO o 5 mM di DCA hanno mostrato livelli elevati di ROS intracellulari (rispettivamente 2,8 e 1,9 volte) (Figura 3A), simili ai risultati pubblicati in precedenza [27], mentre le cellule trattate con ATO e DCA hanno mostrato un ulteriore aumento (5,2 volte) della produzione di ROS (Figura 3A). Al contrario, l’ATO a bassa concentrazione (5 μM) ha indotto una diminuzione di 0,5 volte della produzione di ROS (Figura 3B). Questo dato è in contrasto con quanto riportato in precedenza sulla produzione di ROS e il trattamento con ATO, pertanto sono stati analizzati la dose-risposta e l’andamento temporale della produzione di ROS dopo il trattamento con ATO. Ciò ha rivelato che concentrazioni inferiori di ATO hanno ridotto la produzione intracellulare di ROS, mentre un’elevata quantità di ATO ha indotto la produzione di ROS (Figura 3C). Analogamente, il trattamento con 10 μM di ATO ha determinato una notevole diminuzione della produzione di ROS nelle prime 4 ore, prima che i livelli di ROS aumentassero dopo 8 ore fino a raggiungere il livello riportato da altri (Figura 3D). Il trattamento combinato delle cellule con 5 mM di DCA e 5 μM di ATO ha determinato una produzione intermedia di ROS (Figura 3B) simile a quella delle cellule non trattate.
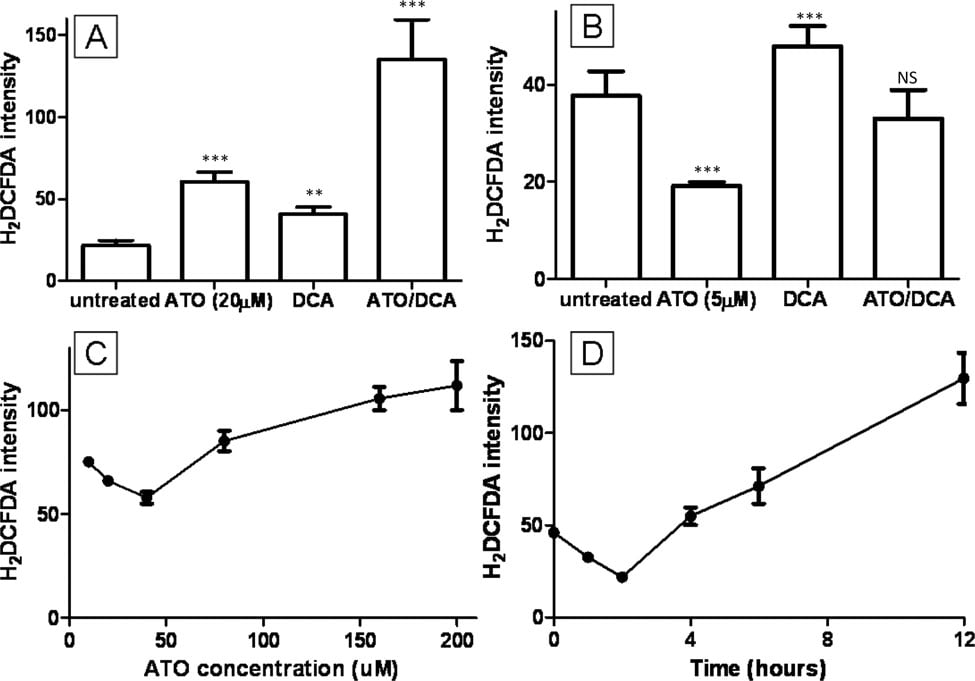
Gli effetti intermedi del trattamento combinato DCA/ATO sui livelli di ATP e ROS possono essere spiegati da effetti concorrenti sull’attività della PDH. Il DCA ha come bersaglio la PDK e quindi dovrebbe aumentare l’attività della PDH, mentre è stato riportato che l’ATO inibisce la PDH sia direttamente tramite la reazione con i tioli vicinali della PDH sia indirettamente tramite l’aumento della produzione di perossido di idrogeno [28]. Per esaminare gli effetti di ATO e/o DCA sull’attività della PDH, cellule intatte o PDH isolate da cellule T-47D sono state trattate con i farmaci e l’attività della PDH è stata determinata. Come previsto, il trattamento di 3 ore delle cellule intatte con DCA ha determinato un aumento dell’attività della PDH, mentre l’ATO (fino a 20 μM) non ha alterato l’attività della PDH delle cellule intatte (Figura 4A). Al contrario, il DCA non ha modificato l’attività della PDH isolata, ma alte concentrazioni di ATO sono state in grado di inibire l’attività della PDH (Figura 4b). Pertanto, nelle cellule T-47D, l’ATO non ha diminuito l’attività della PDH alle concentrazioni utilizzate in questo studio.
L’ATO è un inibitore del complesso IV della catena di trasporto degli elettroni
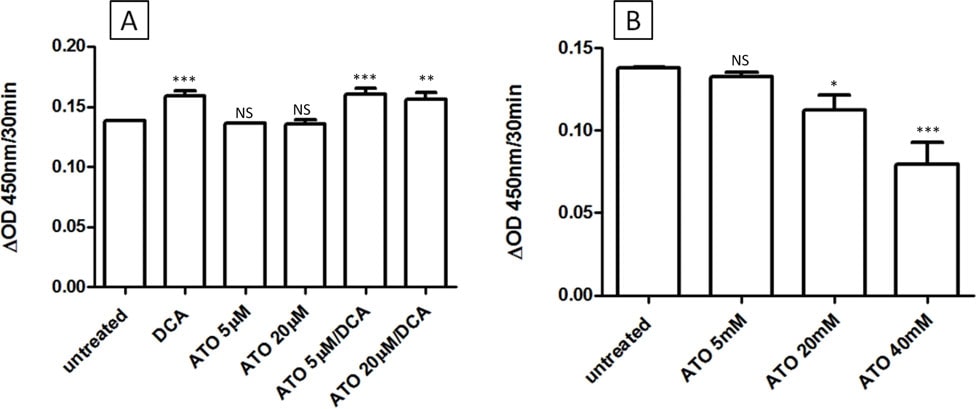
La catena di trasporto degli elettroni (ETC) è situata nella membrana interna dei mitocondri e svolge un ruolo fondamentale nella produzione di energia. L’ETC è responsabile della generazione del gradiente protonico per lo spazio interno della membrana dei mitocondri per mantenere la MMP per la produzione di ATP [29]. L’ETC è anche documentato come il principale sito di produzione di ROS all’interno delle cellule come risultato della perdita di elettroni dal complesso I e III [30]. Data la sua capacità di ridurre i ROS, depolarizzare la MMP e diminuire la produzione di ATP contemporaneamente, abbiamo ipotizzato che l’ATO operi come inibitore dell’ETC. Un gruppo di inibitori dell’ETC è stato confrontato con ATO (5 μM) per verificare se inducono gli stessi cambiamenti fenotipici di ATO nelle cellule T-47D. Il rotenone (0,1 μM, complesso I), il dietiltrifluoroacetone (TTFA) (10 μM, complesso II), l’antimicina (0,1 μM, complesso III) e il NaCN (10 mM, complesso IV) sono stati utilizzati per trattare le cellule T-47D e i livelli di ROS, MMP e ATP sono stati confrontati con quelli delle cellule trattate con ATO. Tutti gli inibitori hanno dimostrato di essere in grado di depolarizzare la MMP (Figura 5A) e di ridurre i livelli di ATP (Figura 5B) in modo simile all’ATO. Tuttavia, a differenza dell’ATO, il rotenone, l’antimicina e il TTFA non sono riusciti a ridurre la produzione di ROS nelle cellule, aumentando invece i ROS da 3 a 6 volte dopo 12 ore di trattamento (Figura 5C). Le cellule trattate con NaCN (10 mM), invece, hanno mostrato una diminuzione del 52% della produzione di ROS (Figura 5C), simile al trattamento con ATO. Sulla base di questi dati, abbiamo concluso che nelle cellule di carcinoma mammario T47D l’inibizione del complesso IV dell’ETC, ma non dei complessi I-III, determina una riduzione dei ROS; è quindi probabile che i cambiamenti fenotipici indotti dall’ATO siano causati dall’inibizione del complesso IV (citocromo C ossidasi) dell’ETC. A conferma di ciò, l’attività della citocromo C ossidasi nelle cellule T-47D è stata misurata spettrofotometricamente. Il saggio della citocromo C ossidasi ha dimostrato chiaramente che, mentre il DCA non ha avuto alcun effetto sull’attività del complesso IV, l’ATO (5 μM, 5 min) può inibire l’attività del complesso IV, confermando la nostra ipotesi (Figura 5D). Risultati simili sono stati ottenuti dopo trattamenti farmacologici di 10 minuti, 3 ore e 12 ore. La combinazione di ATO e DCA ha mostrato un’inibizione enzimatica simile a quella del solo ATO.
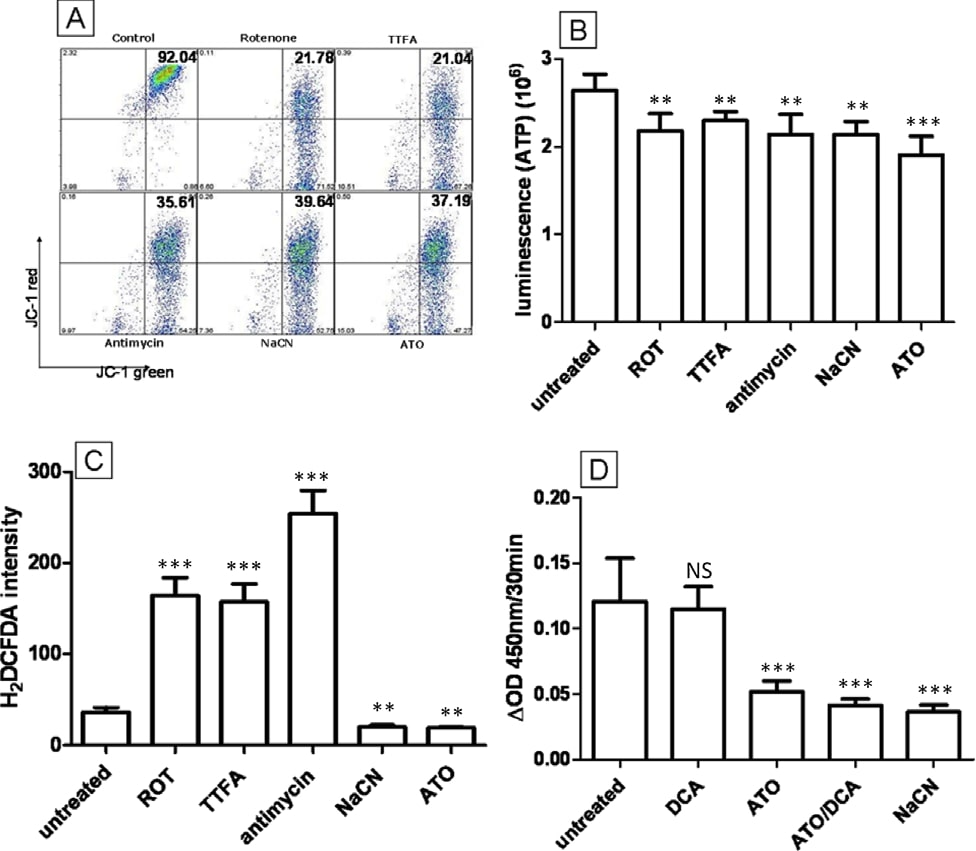
Gliinibitori del poro di transizione mitocondriale bloccano parzialmente l’attivazione delle caspasi indotta da ATO, ma non hanno alcun effetto su ROS, ATP e MMP
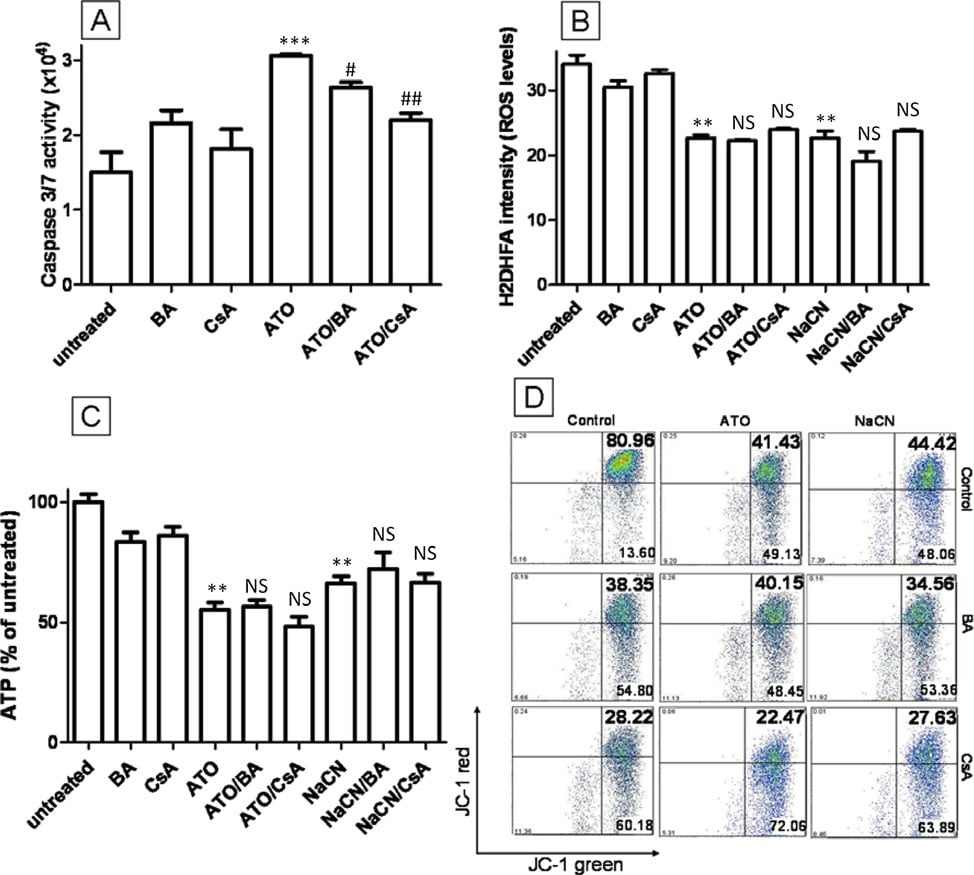
Un meccanismo che è stato precedentemente descritto come la chiave della citotossicità di ATO è l’apertura del poro di transizione mitocondriale (MTP). È stato dimostrato che l’ATO induce l’apertura del MTP in mitocondri isolati e può aumentare il rilascio di citocromo C [11] e indurre apoptosi nucleare in sistemi cellulari liberi [31]. È stato proposto che l’apertura del MTP da parte dell’ATO induca la permeabilizzazione mitocondriale, dissipi la MMP e aumenti i ROS intracellulari. Per verificare se l’MTP sia importante per la tossicità dell’ATO nelle cellule T-47D, l’acido bongkrekico (BA) e la ciclosporina A (CsA), entrambi potenti bloccanti dei pori dell’MTP [32], sono stati combinati con l’ATO per vedere se possono inibire i cambiamenti intracellulari indotti dall’ATO. L’aggiunta di 5 μM di CsA o 50 μM di BA alle cellule trattate con ATO (5 μM) non ha interferito con la capacità di ATO di ridurre la produzione di ROS dopo 12 ore di trattamento, di diminuire la produzione di ATP o di depolarizzare la MMP (P > 0,05) (Figura 6B, C e 6D). Dati simili si osservano per il trattamento con NaCN e CsA/BA (P > 0,05) (Figura 6B, C e 6D). Per confermare l’attività di CsA e BA nelle nostre condizioni sperimentali, è stata valutata l’attività della caspasi 3/7, un marcatore di apoptosi a valle dell’apertura del MTP, attivato dal rilascio di citocromo C, dopo il trattamento con ATO ad alta concentrazione (Figura 6A). Il trattamento con ATO (20 μM) ha raddoppiato l’attività della caspasi 3/7 rispetto alle cellule non trattate (2,1 volte, Figura 6A) e questo effetto è stato ridotto da BA e CsA (rispettivamente 1,7 volte e 1,4 volte), indicando che CsA o BA possono bloccare l’apertura della MTP nelle cellule T-47D. Questi dati dimostrano chiaramente che, sebbene l’ATO (20 μM) possa indurre l’apertura dell’MTP, questo meccanismo non può spiegare le alterazioni di ROS, MMP e ATP che si verificano durante il trattamento con 5 μM di ATO. Concludiamo che queste alterazioni sono probabilmente dovute all’inibizione del complesso ETC IV.
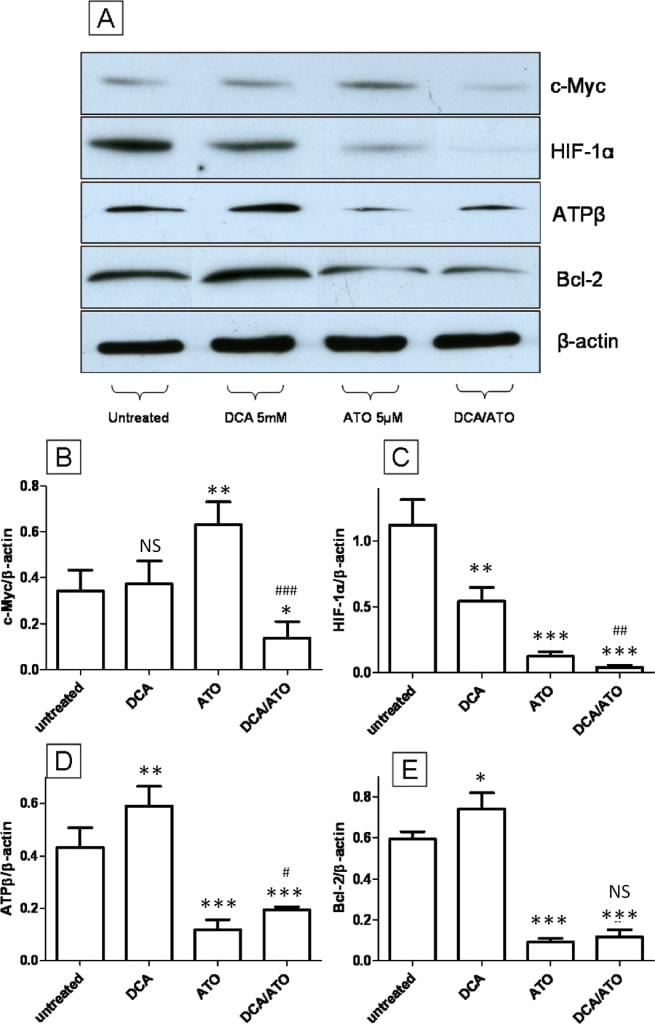
DCA e ATO hanno effetti opposti sull’espressione della βsubunitàdell’ATP sintasi e di Bcl-2, mentre lavorano in modo cooperativo alla down-regolazione dei livelli proteici di c-Myc e HIF-1α
Abbiamo esaminato l’effetto del trattamento con 5 mM di DCA e 5 μM di ATO (12 ore) sull’espressione di c-Myc e HIF1α, due importanti fattori di trascrizione noti per regolare l’effetto Warburg [33] e l’attività mitocondriale [34], e sull’espressione delle proteine mitocondriali ATP sintasi β subunità (ATPβ) e Bcl-2. L’immunoblotting per c-Myc non ha mostrato cambiamenti dopo il trattamento con DCA, mentre l’ATO ha indotto l’up-regolazione di c-Myc (Figura 7A e 7B), tuttavia l’ATO in combinazione con il DCA ha ridotto il livello di c-Myc rispetto al controllo non trattato. I livelli di HIF-1α, invece, hanno mostrato una riduzione dopo il trattamento con DCA e ATO. La combinazione di ATO e DCA ha mostrato un’ulteriore riduzione dei livelli di HIF-1α rispetto ai gruppi trattati con un solo agente e ai gruppi di controllo (Figura 7A e 7C). Bcl-2 è un membro della famiglia di proteine BH3 che si lega direttamente a Bad e Bax e previene l’apoptosi [35]. L’immunoblotting eseguito per l’ATPβ ha rilevato che il trattamento con DCA ha aumentato la regolazione dell’ATPβ, mentre l’ATO ha mostrato un effetto opposto, abbassando i livelli di ATPβ (Figura 7A e 7D); il trattamento combinato con ATO e DCA ha mostrato una risposta intermedia rispetto ai gruppi trattati con ATO o DCA. L’immunoblotting per la proteina pro-sopravvivenza Bcl-2 ha mostrato un’up-regulation dopo DCA, una down-regulation per il trattamento con ATO e una risposta intermedia dalla combinazione dei due farmaci (Figura 7A e 7E). Questi risultati sono molto simili a quelli dei livelli di ATP e della produzione di ROS, dove DCA e ATO hanno avuto effetti opposti (Figura 2C e 3B). I risultati di Bcl-2 e ATPβ sono direttamente correlati alla capacità di DCA e ATO di influenzare la produzione di ROS e i livelli di ATP.
Discussione
Nell’ultimo decennio, l’ATO è stato utilizzato efficacemente per trattare sia i pazienti con APL di nuova diagnosi che quelli con recidiva, con pazienti che hanno mostrato una remissione completa dopo il trattamento con ATO a basso dosaggio [4]. La capacità antitumorale di ATO non è limitata all’APL [36] , e molti altri tumori in modelli animali hanno dimostrato di essere sensibili al trattamento con ATO [37,38]. Tuttavia, la mancanza di informazioni sui siti d’azione della citotossicità dell’ATO in tipi di tumore diversi dall’APL ha limitato la sua diffusione per il trattamento di altri tipi di cancro fino agli ultimi anni [5].
In questo studio abbiamo dimostrato che il complesso IV dell’ETC è un bersaglio per ATO. Da un gruppo di inibitori dell’ETC, la diminuzione dei ROS, la riduzione della produzione di ATP e la depolarizzazione delle MMP causate dal trattamento con ATO a basse dosi delle cellule T-47D sono state riprodotte solo dal cianuro, un inibitore del complesso IV ben caratterizzato (Figura 5). La misurazione diretta dell’attività della citocromo C ossidasi in cellule intere trattate con ATO ha confermato la capacità di ATO di inibire direttamente questa attività enzimatica. Il complesso della citocromo C ossidasi dell’ETC è noto per la presenza di residui di cisteina strettamente distanziati tra loro [39], che probabilmente reagiscono con l’ATO.
La maggior parte delle cellule tumorali presenta livelli di ROS regolati al rialzo rispetto allecellulenormali [40] e l’ETC è considerata la principale fonte di ROS intracellulari. La riduzione dei ROS fino al 60% con NaCN o ATO ha dimostrato che il complesso IV è responsabile della maggior parte della produzione di ROS nelle cellule T-47D. Lo stress ossidativo dovuto all’elevata produzione di ROS è stato descritto come un’arma a doppio taglio. È stato dimostrato che un moderato aumento dei ROS è associato a un aumento del potenziale proliferativo [41], aumento degli enzimi antiossidanti come la glutatione transferasi [42], superossido dismutasi e catalasi [43] e l’aumento di proteine pro-sopravvivenza come Bcl-2 [42] e survivin [44]. Tuttavia, quantità intollerabili di ROS finiscono per portare alla morte cellulare [45]. La riduzione dei ROS dopo 12 ore di trattamento con 5 μM di ATO è direttamente correlata anche a una diminuzione dei livelli di Bcl-2 (Figura 7E), suggerendo che le cellule T-47D si trovano in uno stato di resistenza apoptotica a causa degli elevati livelli di ROS. La continua inibizione del complesso IV dell’ETC porterà alla depolarizzazione dell’MMP e all’inibizione della produzione di ATP. La morte cellulare che consegue a questa disregolazione mitocondriale darà origine agli elevati ROS osservati dopo 4 ore o a concentrazioni più elevate di ATO (Figura 3C e 3D) come riportato da altri [46] dopo il trattamento con ATO. Pertanto, l’elevato ROS è una conseguenza, non la causa, della morte cellulare indotta da ATO.
Il bersaglio mitocondriale proposto in precedenza per ATO è il MTP. Zheng et al. hanno dimostrato che l’ATO induce l’apertura del MTP in mitocondri isolati di fegato di ratto [11], portando alla proposta che l’MTP sia responsabile del rilascio di ROS e della depolarizzazione dell’MMP osservata durante il trattamento con arsenico e della conseguente apoptosi [47]. Il nostro studio non esclude la MTP come bersaglio dell’ATO. I nostri dati indicano che l’inibizione della MTP tramite CsA e BA può bloccare parzialmente l’attivazione della caspasi 3/7 indotta da ATO ad alta concentrazione; tuttavia, i bloccanti della MTP non sono riusciti a inibire la depolarizzazione della MMP e la riduzione dei livelli di ROS e ATP causati da ATO a bassa concentrazione. Pertanto, sebbene l’ATO possa aprire la MTP, sono necessari bersagli alternativi per spiegare l’azione dell’ATO a basse concentrazioni. È stato dimostrato che l’inibizione del sistema della tioredoxina si verifica nelle cellule MCF-7 dopo il trattamento con ATO a 2-5 μM e può mediare alcuni effetti cellulari di ATO a basse concentrazioni [13], tuttavia questo meccanismo porterebbe a un aumento dello stress ossidativo e non può spiegare la diminuzione dei ROS osservata nello studio attuale.
Il DCA, in quanto inibitore della piruvato deidrogenasi chinasi, può invertire l’effetto glicolitico e ha dimostrato significative proprietà antitumorali sia in vitro che in vivo [17-23]. Alcuni studi mostrano un aumento dell’apoptosi con il DCA [17,18,20]. Tuttavia, i nostri studi sul cancro al seno e quelli di Stockwin et al. indicano che il DCA agisce come agente citostatico piuttosto che citotossico in una serie di linee cellulari, tranne che a concentrazioni molto elevate [22,48]. Michelakis ha recentemente riportato livelli sierici di DCA pari a 0,5 mM in pazienti affetti da glioblastoma che assumevano 6,25 mg/kg per via orale due volte al giorno [23], quindi le concentrazioni clinicamente rilevanti sono probabilmente comprese nell’intervallo 0,5-5 mM. È stato dimostrato che il DCA induce un piccolo ma significativo aumento dell’attività della caspasi 3/7 anche quando non è stata osservata apoptosi ed è stato proposto che il DCA possa sensibilizzare le cellule tumorali nei confronti degli agenti citotossici [22]. Lo studio attuale ha confermato questa ipotesi dimostrando che il DCA ha sensibilizzato le cellule T-47D al trattamento con ATO, come si evince dalla doppia colorazione AV+/PI+ (Figura 1E e testo). L’ATO è stato scelto come agente da testare per le sue proprietà antimitocondriali, in quanto abbiamo proposto che, invertendo il fenotipo glicolitico con il DCA e indirizzando una maggiore quantità di piruvato verso la fosforilazione ossidativa mitocondriale, e colpendo contemporaneamente i mitocondri con l’ATO, si sarebbe visto un effetto cooperativo con questa combinazione di farmaci. Il principio di questa strategia a doppio bersaglio è supportato dai risultati di due recenti pubblicazioni che hanno rilevato che le cellule con difetti mitocondriali (ad esempio rho (0) o dopo trattamento con inibitori mitocondriali non farmacologici come il rotenone) mostrano una maggiore sensibilità agli effetti di inibizione della crescita del DCA [48,49]. Sebbene l’efficacia di questa strategia a doppio bersaglio debba ancora essere dimostrata in vivo, gli effetti della combinazione potenziata ATO/DCA sono stati osservati sia sull’inibizione della crescita che sull’apoptosi a concentrazioni di farmaco in range clinicamente rilevanti. Alcuni effetti all’estremità inferiore dell’intervallo di concentrazioni non sono stati di grande entità (ad esempio, aumento del 15% delle cellule apoptotiche), tuttavia l’impatto in vivo per settimane piuttosto che per ore di trattamento merita ulteriori indagini.
È stato ben documentato che il DCA può alterare il comportamento mitocondriale, vale a dire depolarizzare l’MMP [18,20]. Tuttavia, questo fenomeno non è stato ben spiegato. Abbiamo osservato che il DCA può aumentare l’ATP mentre depolarizza la MMP nelle cellule T-47D (Figura 2). Sulla base di questi dati, abbiamo proposto che il DCA alteri la funzione mitocondriale attraverso il complesso V dell’ETC, l’ATP sintasi. L’ATP sintasi è l’ultima fase dell’ETC e utilizza il MMP generato dall’ETC per produrre ATP. L’ATPb è downregolato nei tumori del polmone, del cervello, del seno e del tratto gastrico [50], il che potrebbe spiegare l’iperpolarizzazione della membrana mitocondriale che si riscontra in molte cellule tumorali [51]. Abbiamo ipotizzato che il DCA possa aumentare l’attività dell’ATP sintasi sia attraverso una regolazione allosterica dovuta alla sua omologia con il piruvato, sia attraverso l’aumento dei livelli di proteina ATP sintasi in seguito all’inversione dell’effetto Warburg e alla diminuzione dei livelli di HIF-1α. L’immunoblotting ha dimostrato chiaramente che l’ATPβ è up-regolato dopo il trattamento con DCA (Figura 7D), suggerendo che il DCA può aumentare l’attività dell’ATP sintasi, che a sua volta contribuisce all’aumento della produzione di ATP e all’esaurimento della MMP.
HIF-1α e c-Myc sono due importanti fattori di trascrizione oncogeni noti per regolare il metabolismo nelle cellule tumorali. HIF-1α può regolare gli enzimi della via glicolitica e della deidrogenasi dell’acido lattico, sostenendo l’effetto Warburg nelle cellule tumorali [33]. Questo non solo aumenta la produzione di ATP, ma incrementa anche l’apporto di precursori come il glucosio-6-fosfato e il fruttosio-6-fosfato per la produzione di acidi nucleici attraverso la via del pentoso fosfato [16]c-Myc, invece, non solo svolge un ruolo centrale nel promuovere la transizione del ciclo cellulare dalla fase G1 alla fase S, regolando le cicline e le loro chinasi e inibitori, ma regola anche i componenti proteici dell’ETC, come COXI-IV, e contribuisce ad aumentare l’attività mitocondriale [52]. Pertanto, la combinazione della sovraespressione di c-Myc e HIF-1α è importante per indurre l’effetto Warburg e aumentare l’attività mitocondriale, sostenendo l’hub glutammina-TCA, essenziale per l’anabolismo degli aminoacidi e degli acidi grassi necessari per la divisione cellulare [16] [16]. Questa firma metabolica contribuisce alla carcinogenesi e al fenotipo maligno di molti tumori [33,34]. Sia l’ATO che il DCA possono diminuire i livelli di HIF-1α in modo significativo, suggerendo una riduzione dell’effetto Warburg (Figura 7C). Mentre il DCA da solo non ha avuto alcun effetto sui livelli di c-Myc, sorprendentemente l’ATO ha aumentato significativamente i livelli di c-Myc (Figura 7B). Potrebbe trattarsi di un ciclo di feed back positivo, in cui le cellule cercano di aumentare l’attività dell’ETC dopo l’inibizione del complesso IV. È sorprendente che il DCA abbia invertito l’effetto di ATO su c-Myc e che la combinazione di ATO/DCA abbia fortemente represso l’espressione di c-Myc, correlando la capacità di questi agenti di lavorare insieme per ridurre la proliferazione e indurre la morte cellulare (Figura 1).
L’efficacia di ATO contro la PML è stata attribuita a diverse caratteristiche peculiari di questa neoplasia: differenziazione attraverso l’inattivazione di PML-RARα, bassa capacità antiossidante delle cellule PML di proteggersi dagli elevati livelli di ROS e accumulo del farmaco a causa di una compromessa osmoregolazione [5]. Sono in corso numerosi studi sull’uso di ATO in una serie di neoplasie solide, anche se finora sono stati pubblicati pochi risultati. I risultati disponibili suggeriscono che ATO non è altamente efficace come agente singolo in pazienti con cancro avanzato del pancreas, del fegato o melanoma <a href=”#5″> [5]</a></sup>. Gli studiosi sono favorevoli alla valutazione di ATO in combinazione con altri farmaci antitumorali<sup><a href=”#53″> [53,54]</a></sup> con alcuni studi di fase I in fase di completamento <sup><a href=”#55″>[55]</a></sup>. Man mano che la nostra comprensione dei meccanismi dell’ATO continua a crescere, soprattutto per quanto riguarda i suoi effetti sul metabolismo del cancro, potremo essere in grado di sfruttare meglio il suo potenziale antitumorale in vivo in nuove combinazioni di farmaci</p>
<h2>Conclusioni</h2>
<p>Due recenti rapporti hanno riscontrato che il DCA è più efficace contro le cellule con difetti mitocondriali <sup><a href=”#48″>[48,49].</a></sup> Questo rapporto è il primo a dimostrare che il bersaglio di due aspetti del metabolismo – l’inversione dell’effetto Warburg con il DCA e l’inibizione della fosforilazione ossidativa con l’ATO – è chiaramente una strategia antitumorale efficace in vitro contro le linee cellulari di cancro al seno. L’identificazione della citocromo C ossidasi come bersaglio mitocondriale dell’ATO fornisce nuove informazioni meccanicistiche per l’applicazione dell’ATO al trattamento di tipi di tumore diversi dall’APL. La capacità del DCA di aumentare l’espressione della subunità b dell’ATP sintasi, invertendo un altro diffuso fenotipo metabolico del cancro, suggerisce inoltre che il DCA può essere rilevante per un’ampia gamma di tipi di tumore. Anche la capacità del DCA di potenziare gli effetti citotossici di agenti chemioterapici diversi dall’ATO in vitro e in vivo merita ulteriori indagini. Anche la mancanza di sensibilità della linea cellulare non cancerosa MCF-10A alle concentrazioni clinicamente rilevanti di ATO/DCA testate è incoraggiante e suggerisce che questa strategia di trattamento dovrebbe essere ulteriormente testata contro i tumori solidi in vivo.</p>
<h2>Elenco delle abbreviazioni</h2>
<p>APL: leucemia promieloide acuta; ATO: triossido di arsenico; ATPβ: Subunità β dell’ATP sintasi; AV: annexina V; BA: acido bongkrekico; CFSE: carbossifluoresceina succinimidil estere; CsA: ciclosporina A; DCA: dicloroacetato; ETC: catena di trasporto degli elettroni; H2DCFDA: 2′, 7′-diidroclorofluroresceinacetato; JC-1: 5,5′,6,6′-tetracloro-1,1′, 3,3′-tetraetilbenzimidazolo-carbocianina ioduro; MMP: potenziale di membrana mitocondriale; MTP: poro di transizione mitocondriale; NaCN: cianuro di sodio; PI: propidio ioduro; ROS: Specie reattive dell’ossigeno; TTFA: thenolytrifluoroacetone.</p>
<h2>Riconoscimenti</h2>
<p>Questa ricerca è stata sostenuta da una sovvenzione della National Breast Cancer Foundation Australia (AB) e dal premio NHMRC 366787 R.D. Wright Career Development Award (AB). PB è sostenuto dall’NHRMC e dall’Australian National University. RS è stata sostenuta da una borsa di studio post-laurea dell’Australian National University</p>
<h2>Dettagli dell’autore</h2>
<p>1 Gruppo di Genetica Molecolare, Dipartimento di Bioscienze Traslazionali, John Curtin School of Medical Research, Edificio 131, Australian National University, P.O. Box 334, Canberra ACT 0200, AUSTRALIA. 2 Dipartimento di radioterapia oncologica, Stanford School of Medicine, Stanford CA 94305 USA.</p>
<h2>Contributi degli autori</h2>
<p>RS ha progettato e condotto gli esperimenti e ha redatto il manoscritto. PB ha partecipato all’analisi dei dati e alla preparazione del manoscritto. AB ha partecipato alla progettazione e all’analisi dei dati e ha preparato il manoscritto per la pubblicazione. Tutti gli autori hanno letto e approvato il manoscritto finale.</p>
<h2>Interessi contrastanti</h2>
<p>Gli autori dichiarano di non avere interessi in competizione.<br></p>
<p>Ricevuto: 3 maggio 2011 Accettato: 18 novembre 2011<br>Pubblicato: 18 novembre 2011</p>
<h2>REFERENZE</h2>
<br><span id=”1″ class=”referencess blue-text”>1</span> Antman KH: Introduzione: la storia del triossido di arsenico nella terapia del cancro. Oncologist 2001, 6(Suppl 2):1-2.
<br><span id=”2″ class=”referencess blue-text”>2</span> Emadi A, Gore SD: Arsenic trioxide – An old drug rediscovered. Blood Rev 2010, 24:191-199.
<br><span id=”3″ class=”referencess blue-text”>3</span> Powell BL, Moser B, Stock W, Gallagher RE, Willman CL, Stone RM, Rowe JM, Coutre S, Feusner JH, Gregory J, et al: Il triossido di arsenico migliora la sopravvivenza libera da eventi e la sopravvivenza globale negli adulti con leucemia promielocitica acuta: North American Leukemia Intergroup Study C9710. Blood 2010, 116:3751-3757.
<br><span id=”4″ class=”referencess blue-text”>4</span> Wang ZY, Chen Z: Acute promyelocytic leukemia: from highly fatal to highly cureble. Blood 2008, 111:2505-2515.
<br><span id=”5″ class=”referencess blue-text”>5</span> Dilda PJ, Hogg PJ: Arsenical-based cancer drugs. Cancer Treat Rev 2007, 33:542-564.
<br><span id=”6″ class=”referencess blue-text”>6</span> Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, Jin XL, Tang W, Li XS, Xong SM, et al: Studi in vitro sui meccanismi cellulari e molecolari del triossido di arsenico (As2O3) nel trattamento della leucemia promielocitica acuta: L’As2O3 induce l’apoptosi delle cellule NB4 con la downregulation dell’espressione di Bcl2 e la modulazione delle proteine PML-RAR alfa/PML. 1996, 88:1052-1061.
<br><span id=”7″ class=”referencess blue-text”>7</span> Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I: Induzione della morte cellulare autofagica in cellule di glioma maligno da parte del triossido di arsenico. Cancer Res 2003, 63:2103-2108.
<br><span id=”8″ class=”referencess blue-text”>8</span> Nakagawa Y, Akao Y, Morikawa H, Hirata I, Katsu K, Naoe T, Ohishi N, Yagi K: L’apoptosi indotta dal triossido di arsenico attraverso lo stress ossidativo in cellule di linee cellulari di cancro del colon. Life Sciences 2002, 70:2253-2269.
<br><span id=”9″ class=”referencess blue-text”>9</span> Ralph SJ: Arsenic-based antineoplastic drugs and their mechanisms of action. Met Based Drugs 2008, 2008:260146.
<br><span id=”10″ class=”referencess blue-text”>10</span> Cai X, Shen YL, Zhu Q, Jia PM, Yu Y, Zhou L, Huang Y, Zhang JW, Xiong SM, Chen SJ, et al: Arsenic trioxide-induced apoptosis and differentiation are associated respectively with mitochondrial transmembrane potential collapse and retinoic acid signaling paths in acute promyelocytic leukemia. Leukemia 2000, 14:262-270.
<br><span id=”11″ class=”referencess blue-text”>11</span> Zheng Y, Shi Y, Tian C, Jiang C, Jin H, Chen J, Almasan A, Tang H, Chen Q: Ruolo essenziale del canale anionico voltaggio-dipendente (VDAC) nell’apertura del poro di transizione di permeabilità mitocondriale e nel rilascio di citocromo c indotto dal triossido di arsenico. Oncogene 2004, 23:1239-1247.
<br><span id=”12″ class=”referencess blue-text”>12</span> Tian X, Ma X, Qiao D, Ma A, Yan F, Huang X: mCICR è richiesto per l’apertura del poro di transizione di permeabilità indotta dall’As2O3 e il rilascio di citocromo c dai mitocondri. Mol Cell Biochem 2005, 277:33-42.
<br><span id=”13″ class=”referencess blue-text”>13</span> Lu J, Chew EH, Holmgren A: Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci USA 2007, 104:12288-12293.
<br><span id=”14″ class=”referencess blue-text”>14</span> Kim JW, Dang CV: La dolcezza molecolare del cancro e l’effetto Warburg. Cancer Res 2006, 66:8927-8930.
<br><span id=”15″ class=”referencess blue-text”>15</span> Warburg O, Wind F, Negelein E: Ueber den Stoffwechsel von Tumoren im körper. Klinische Wochenschrift 1926, 5:829-832.
<br><span id=”16″ class=”referencess blue-text”>16</span> DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB: Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci 2007, 104:19345-19350.
<br><span id=”17″ class=”referencess blue-text”>17</span> Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I: Il dicloroacetato induce apoptosi nelle cellule del cancro endometriale. Gynecol Oncol 2008, 109:394-402.
<br><span id=”18″ class=”referencess blue-text”>18</span> Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ: Il dicloroacetato (DCA) sensibilizza in vitro alle radiazioni sia le cellule di cancro alla prostata wild-type che quelle che esprimono Bcl-2. Prostata 2008, 68:1223-1231.
<br><span id=”19″ class=”referencess blue-text”>19</span> Papandreou I, Goliasova T, Denko NC: Farmaci antitumorali che colpiscono il metabolismo: il dicloroacetato è il nuovo paradigma? Int J Cancer 2011, 128:1001-1008.
<br><span id=”20″ class=”referencess blue-text”>20</span> Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al: A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11:37-51.
<br><span id=”21″ class=”referencess blue-text”>21</span> Chen Y, Cairns R, Papandreou I, Koong A, Denko NC: Il consumo di ossigeno può regolare la crescita dei tumori, una nuova prospettiva sull’effetto Warburg. PLoS One 2009, 4:e7033.
<br><span id=”22″ class=”referencess blue-text”>22</span> Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC: Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer growth in vitro and in vivo. Breast Cancer Res Treat 2010, 120:253-260.
<br><span id=”23″ class=”referencess blue-text”>23</span> Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al: Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010, 2:31ra34.
<br><span id=”24″ class=”referencess blue-text”>24</span> Chrzanowska-Lightowlers ZMA, Turnbull DM, Lightowlers RN: Un test su piastra microtiter per la citocromo c ossidasi in cellule intere permeabilizzate. Analytical Biochemistry 1993, 214:45-49.
<br><span id=”25″ class=”referencess blue-text”>25</span> Theodoratos A, Tu WJ, Cappello J, Blackburn AC, Matthaei K, Board PG: Phenylalanine-induced leucopenia in genetic and dichloroacetic acid generated deficiency of glutathione transferase Zeta. Biochem Pharmacol 2009, 77:1358-1363.
<br><span id=”26″ class=”referencess blue-text”>26</span> Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al: Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Efficacia clinica e farmacocinetica nei pazienti recidivati. Blood 1997, 89:3354-3360.
<br><span id=”27″ class=”referencess blue-text”>27</span> Maeda H, Hori S, Nishitoh H, Ichijo H, Ogawa O, Kakehi Y, Kakizuka A: Tumor Growth Inhibition by Arsenic Trioxide (As2O3) in the Orthotopic Metastasis Model of Androgen-independent Prostate Cancer. Cancer Res 2001, , 61: 5432-5440.
<br><span id=”28″ class=”referencess blue-text”>28</span> Samikkannu T, Chen CH, Yih LH, Wang AS, Lin SY, Chen TC, Jan KY: Reactive oxygen species are involved in arsenic trioxide inhibition of pyruvate dehydrogenase activity. Chem Res Toxicol 2003, 16:409-414.
<br><span id=”29″ class=”referencess blue-text”>29</span> Campbell MK, Farrell SO: Biochimica. 4 edizione. Thomson learning, Inc.; 2003.
<br><span id=”30″ class=”referencess blue-text”>30</span> Liu Y, Fiskum G, Schubert D: Generazione di specie reattive dell’ossigeno da parte della catena di trasporto degli elettroni mitocondriale. J Neurochem 2002, 80:780-787.
<br><span id=”31″ class=”referencess blue-text”>31</span> Larochette N, Decaudin D, Jacotot E, Brenner C, Marzo I, Susin SA, Zamzami N, Xie Z, Reed J, Kroemer G: L’arsenito induce l’apoptosi attraverso un effetto diretto sul poro di transizione di permeabilità mitocondriale. Exp Cell Res 1999, 249:413-421.
<br><span id=”32″ class=”referencess blue-text”>32</span> Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G: Bax e l’adenina nucleotide translocator cooperano nel controllo mitocondriale dell’apoptosi. Science 1998, 281:2027-2031.
<br><span id=”33″ class=”referencess blue-text”>33</span> Semenza GL: Regolazione del metabolismo delle cellule tumorali da parte dell’hypoxia-inducible factor 1. Semin Cancer Biol 2009, 19:12-16.
<br><span id=”34″ class=”referencess blue-text”>34</span> Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F: The c-Myc target gene network. Seminars in Cancer Biology 2006, 16:253-264. Sun et al. Molecular Cancer 2011, 10:142 http://www.molecular-cancer.com/content/10/1/142 Pagina 14 di 15
<br><span id=”35″ class=”referencess blue-text”>35</span> Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X: Prevenzione dell’apoptosi da parte di Bcl-2: blocco del rilascio di citocromo c dai mitocondri. Science 1997, 275:1129-1132.
<br><span id=”36″ class=”referencess blue-text”>36</span> Carney DA: Arsenic trioxide mechanisms of action-looking beyond acute promyelocytic leukemia. Leuk Lymphoma 2008, 49:1846-1851.
<br><span id=”37″ class=”referencess blue-text”>37</span> Kito M, Matsumoto K, Wada N, Sera K, Futatsugawa S, Naoe T, Nozawa Y, Akao Y: Antitumor effect of arsenic trioxide in murine xenograft model. Cancer Sci 2003, 94:1010-1014.
<br><span id=”38″ class=”referencess blue-text”>38</span> Xu HY, Yang YL, Liu SM, Bi L, Chen SX: Effetto del triossido di arsenico sull’epatocarcinoma umano in topi nudi. World J Gastroenterol 2004, 10:3677-3679.
<br><span id=”39″ class=”referencess blue-text”>39</span> Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, ShinzawaItoh K, Nakashima R, Yaono R, Yoshikawa S: L’intera struttura della citocromo c ossidasi a 13 subunità a 2,8 A. Science 1996, 272:1136-1144.
<br><span id=”40″ class=”referencess blue-text”>40</span> Pelicano H, Carney D, Huang P: ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 2004, 7:97-110.
<br><span id=”41″ class=”referencess blue-text”>41</span> Kamata H, Hirata H: Redox regulation of cellular signalling. Cellular Signalling 1999, 11:1-14.
<br><span id=”42″ class=”referencess blue-text”>42</span> Hayes JD, McLellan LI: Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res 1999, 31:273-300.
<br><span id=”43″ class=”referencess blue-text”>43</span> Nordberg J, Arner ESJ: Specie reattive dell’ossigeno, antiossidanti e sistema della tioredoxina nei mammiferi. Free Radical Biology and Medicine 2001, 31:1287-1312.
<br><span id=”44″ class=”referencess blue-text”>44</span> Yao LL, Wang YG, Cai WJ, Yao T, Zhu YC: Survivin media l’effetto antiapoptotico della stimolazione del recettore δ-opioide nei cardiomiociti. Journal of Cell Science 2007, 120:895-907.
<br><span id=”45″ class=”referencess blue-text”>45</span> Martindale JL, Holbrook NJ: Risposta cellulare allo stress ossidativo: Segnalazioni per il suicidio e la sopravvivenza. J Cell Physiol 2002, 192:1-15.
<br><span id=”46″ class=”referencess blue-text”>46</span> Perkins C, Kim CN, Fang G, Bhalla KN: L’arsenico induce l’apoptosi di cellule di leucemia mieloide umana multiresistente che esprimono Bcr-Abl o sovraesprimono MDR, MRP, Bcl-2 o Bcl-x(L). Blood 2000, 95:1014-1022.
<br><span id=”47″ class=”referencess blue-text”>47</span> Ma XD, Qiao DF, Tian XM, Yan F, Ma AD: Meccanismo di apertura del poro di transizione di permeabilità mitocondriale indotto dal triossido di arsenico. Ai Zheng 2006, 25:17-21.
<br><span id=”48″ class=”referencess blue-text”>48</span> Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL: Il dicloroacetato di sodio (DCA) colpisce selettivamente le cellule con difetti nell’ETC mitocondriale. Int J Cancer 2010.
<br><span id=”49″ class=”referencess blue-text”>49</span> Sanchez-Arago M, Chamorro M, Cuezva JM: La selezione di cellule tumorali con mitocondri repressi innesca la progressione del cancro al colon. Carcinogenesi 2010, 31:567-576.
<br><span id=”50″ class=”referencess blue-text”>50</span> Isidoro A, Martinez M, Fernandez PL, Ortega AD, Santamaria G, Chamorro M, Reed JC, Cuezva JM: Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochemical Journal 2004, 378:17-20.
<br><span id=”51″ class=”referencess blue-text”>51</span> Susin SA, Zamzami N, Kroemer G: Mitochondria as regulators of apoptosis: Nessun dubbio. Biochimica et Biophysica Acta – Bioenergetica 1998, 1366:151-165.
<br><span id=”52″ class=”referencess blue-text”>52</span> Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV: c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc Natl Acad Sci USA 1997, 94:6658-6663.
<br><span id=”53″ class=”referencess blue-text”>53</span> Kim KB, Bedikian AY, Camacho LH, Papadopoulos NE, McCullough C: A phase II trial of arsenic trioxide in patients with metastatic melanoma. Cancer 2005, 104:1687-1692.
<br><span id=”54″ class=”referencess blue-text”>54</span> Subbarayan PR, Lima M, Ardalan B: Terapia con triossido di arsenico/acido ascorbico in pazienti con carcinoma colorettale metastatico refrattario: un’esperienza clinica. Acta Oncol 2007, 46:557-561.
<br><span id=”55″ class=”referencess blue-text”>55</span> Ardalan B, Subbarayan PR, Ramos Y, Gonzalez M, Fernandez A, Mezentsev D, Reis I, Duncan R, Podolsky L, Lee K, et al: A phase I study of 5-fluorouracil/leucovorin and arsenic trioxide for patients with refractory/relapsed colorectal carcinoma. Clin Cancer Res 2010, 16:3019-3027.
<p></p>
<p>Contenuto correlato:</p>
<figure class=”wp-block-embed is-type-wp-embed is-provider-dca-guide wp-block-embed-dca-guide”><div class=”wp-block-embed__wrapper”>
</div></figura>
<figure class=”wp-block-embed is-type-wp-embed is-provider-dca-guide wp-block-embed-dca-guide”><div class=”wp-block-embed__wrapper”>
</div></figura>