Ramon C Sun1,2, Philip G Board1 et Anneke C Blackburn1*
1 Molecular Genetics Group, Department of Translational Biosciences, John Curtin School of Medical Research, Building 131, Australian National University, P.O. Box 334, Canberra ACT 0200, AUSTRALIE
2 Department of Radiation Oncology, Stanford School of Medicine, Stanford CA 94305 USA.
1*Correspondance: [email protected]
© 2011 Sun et al ; licensee BioMed Central Ltd. Il s’agit d’un article en libre accès distribué selon les termes de la licence Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), qui permet l’utilisation, la distribution et la reproduction sans restriction sur tout support, à condition que l’œuvre originale soit correctement citée.
Reçu : 3 mai 2011
Accepté : 18 novembre 2011
Publié : 18 novembre 2011
Résumé
Contexte
Les cellules cancéreuses ont un profil métabolique différent de celui des cellules normales. L’effet Warburg (augmentation de la glycolyse aérobie) et la glutaminolyse (augmentation de l’activité mitochondriale due au catabolisme de la glutamine) sont des caractéristiques bien connues du cancer et s’accompagnent d’une production accrue de lactate, d’une membrane mitochondriale hyperpolarisée et d’une production accrue d’espèces réactives de l’oxygène.
Méthodes
Dans cette étude, nous ciblons l’effet Warburg avec le dichloroacétate (DCA) et l’augmentation de l’activité mitochondriale de la glutaminolyse avec le trioxyde d’arsenic (ATO) dans des cellules de cancer du sein, en mesurant la prolifération cellulaire, la mort cellulaire et les caractéristiques mitochondriales.
Résultats
L’association du DCA et de l’ATO s’est avérée plus efficace pour inhiber la prolifération cellulaire et induire la mort cellulaire que l’un ou l’autre médicament seul. Nous avons examiné l’effet de ces traitements sur le potentiel de la membrane mitochondriale, la production d’espèces réactives de l’oxygène et les niveaux d’ATP et avons identifié de nouveaux mécanismes moléculaires au sein de la mitochondrie pour l’ATO et le DCA : l’ATO réduit la fonction mitochondriale par l’inhibition de la cytochrome C oxydase (complexe IV de la chaîne de transport des électrons) tandis que le DCA régule à la hausse l’expression de la sous-unité b de l’ATP synthase. La potentialisation de la cytotoxicité de l’ATO par le DCA est corrélée à une forte suppression de l’expression de c-Myc et HIF-1a, et à une diminution de l’expression de la protéine de survie Bcl-2. Conclusion
Cette étude est la première à démontrer que le fait de cibler deux caractéristiques métaboliques clés du cancer constitue une stratégie anticancéreuse efficace et potentiellement thérapeutique.
Mots clés
Dichloroacétate, cancer du sein, chaîne de transport des électrons, mitochondries, trioxyde d’arsenic
Introduction
Le trioxyde d’arsenic (ATO) est utilisé comme agent thérapeutique depuis plus de 2000 ans. Originaire de Chine [1], il est actuellement utilisé contre la leucémie aiguë promyéloïde (LAP) chez les patients qui ont rechuté après un traitement à base d’acide alltrans-rétinoïque et d’anthracycline, et il est promu comme traitement de première intention de la LAP de novo [2-4]. L’ATO est connu comme une molécule hyper-réactive et peut potentiellement se lier aux groupes thiol de nombreuses protéines [2,5]. Sa capacité à se lier à la protéine mutante PML-RAR-α riche en thiol, produite à partir d’une translocation chromosomique dans la LPA, en a fait un médicament efficace dans la LPA [2,5,6]. Il a été démontré que l’ATO induit l’apoptose dans une variété de lignées cellulaires cancéreuses in vitro et in vivo [7,8], mais il a été difficile d’envisager une utilisation clinique de l’ATO dans des types de tumeurs autres que l’APL en raison du manque de connaissances sur les cibles moléculaires qui entraînent sa cytotoxicité. Au cours des 10 dernières années, les changements physiologiques au sein des cellules cancéreuses en réponse au traitement par l’ATO ont été bien caractérisés, et de nombreux essais cliniques pour de nouvelles applications de l’ATO sont en cours [5]. L’ATO a été proposée comme une toxine mitochondriale [9]. L’ATO peut dépolariser le potentiel de la membrane mitochondriale (MMP) [10], augmenter la production d’espèces réactives de l’oxygène (ROS) intracellulaires [8] et induire l’apoptose [8]. La cible proposée pour l’ATO qui peut réaliser ces changements phénotypiques est le pore de transition mitochondrial (PTM) [11]. Il a été démontré que l’ATO induit l’ouverture du PTM, ce qui induit la libération de cytochrome c et est proposé pour dissiper la MMP et augmenter la libération de ROS par les mitochondries [12]. Plus récemment, le système de la thiorédoxine, en particulier la thiorédoxine réductase, a également été identifié comme une cible de l’ATO qui peut contribuer à l’augmentation du stress oxydatif et à la modification de la signalisation redox après le traitement des cellules cancéreuses par l’ATO [9,13].
L’effet Warburg est un phénomène très répandu qui a été identifié dans plus de 90 % de toutes les formes de tumeurs. Les cellules qui présentent l’effet Warburg empruntent des voies alternatives d’homéostasie énergétique pour maintenir leur phénotype prolifératif [14]. Le Dr Otto Warburg, lauréat du prix Nobel, a déclaré que les cellules cancéreuses s’appuient sur la glycolyse ou la phosphorylation du substrat pour générer de l’ATP, et suppriment leurs activités mitochondriales [15]. Grâce à des technologies plus avancées, des études récentes ont confirmé l’aspect production d’ATP de l’hypothèse de Warburg, mais ont révélé que l’activité mitochondriale n’est pas supprimée dans les cellules cancéreuses. Au contraire, les mitochondries jouent un rôle vital en fournissant des substrats pour maintenir la division cellulaire [16].
L’effet anticancéreux de l’inversion de l’effet Warburg a été décrit récemment et un ancien médicament, le dichloroacétate (DCA), qui peut rediriger la synthèse d’ATP de la glycolyse vers la phosphorylation oxydative, a démontré une bonne activité anticancéreuse in vitro [17-19] et in vivo [20-23]. Le DCA est un inhibiteur de la pyruvate déshydrogénase kinase et entraîne une augmentation de l’activité de la pyruvate déshydrogénase [19]. Cela entraîne une conversion accrue du pyruvate en acétyl-CoA plutôt qu’en acide lactique, comme le décrit l’effet Warburg, et stimule la respiration mitochondriale en augmentant l’apport d’acétyl-CoA. Par conséquent, après un traitement au DCA, les cellules cancéreuses ont montré des niveaux accrus de ROS, une dépolarisation de la MMP in vitro et une apoptose accrue à la fois in vitro et in vivo [17,20].
Comme le DCA peut rediriger les substrats vers la respiration mitochondriale et la production d’ATP, il pourrait avoir une activité synergique avec les médicaments anticancéreux qui altèrent l’activité mitochondriale. Nous proposons qu’en inversant le phénotype glycolytique avec le DCA et en dirigeant davantage de pyruvate vers la phosphorylation oxydative mitochondriale, tout en ciblant simultanément les mitochondries avec l’ATO, une grave perturbation de l’homéostasie énergétique se produira dans les cellules cancéreuses. Dans cette étude, nous montrons qu’en combinaison, le DCA et l’ATO agissent ensemble pour inhiber la croissance des lignées cellulaires du cancer du sein in vitro. En outre, nous avons identifié de nouveaux mécanismes moléculaires au sein des mitochondries qui peuvent contribuer à la cytotoxicité de l’ATO, ce qui apporte un soutien supplémentaire à l’utilisation de l’ATO contre les tumeurs solides.
Matériel et méthodes
Réactifs
JC-1, CFSE et H2DCFDA proviennent d’Invitrogen (Carlsbad, CA, USA), les kits de dosage CellTiter-Glo et Caspase-Glo ont été achetés chez Promega Co (San Luis, CA, USA). Le reste des produits chimiques a été acheté auprès de Sigma Co (St. Louis, MO, USA).
Culture cellulaire
Les lignées cellulaires ont été obtenues auprès des sources suivantes au cours des années indiquées. Dr Anna DeFazio, Westmead Millenium Institute, Sydney, Australie : T-47D (2003), BT-20 (2003), MCF-10A (2005) et MCF-10AT1 (2005) ; Prof. Chris Parish, Australian National University, Canberra, Australie : 13762 MAT (2007), MDA-MB-468 (2003) et MDA-MB-231 (2003). Les lignées cellulaires ont des apparences conformes aux morphologies publiées, mais n’ont pas été authentifiées récemment. Les cellules de carcinome épithélial mammaire humain (T-47D) ont été cultivées dans un milieu RPMI 1640 complété par 10 % de sérum bovin fœtal en présence de 0,1 % de PSN (3 % de pénicilline, 5 % de streptomycine et 5 % de néomycine). Les lignées cellulaires BT-20, MDA-MB-231, MDA-MB-468 et 13762 MAT ont été maintenues dans un milieu DMEM/F12 supplémenté avec 10% de FBS et 2 mM L-glutamine. Le milieu DMEM/F-12 avec 25 % de sérum de cheval, 0,01 % d’EGF, 0,28 UI/ml d’insuline, 0,01 % de toxine cholérique et 0,5 μg/ml d’hydrocortisone a été utilisé pour les cellules MCF-10A et MCF-10AT1. Toutes les lignées cellulaires ont été maintenues à 37°C dans 5% de CO2.
Viabilité cellulaire
Pour l’évaluation de la viabilité cellulaire, les cellules ont été placées dans des plaques à 96 puits à une densité de 3000 cellules par puits et 8 puits par groupe. Après exposition au DCA et à l’ATO pendant 24 à 72 heures, les cellules ont été incubées pendant 3 heures avec du rouge neutre (30 μg/ml) dans des milieux frais, puis lavées avec du PBS, suivi de l’ajout d’un tampon de lyse (acide acétique/méthanol, 80%/20%) et l’absorbance à 540 nm a été enregistrée. Les résultats sont exprimés en moyenne ± S.D., les calculs ont été effectués à l’aide du progiciel Prism, l’ANOVA avec post-test de Tukey a été appliquée et P < 0,05 a été considéré comme statistiquement significatif. Les expériences ont été réalisées au moins trois fois et les données présentées sont celles d’une expérience représentative.
Prolifération cellulaire
Les cellules T-47D ont été récoltées et remises en suspension dans 1 ml de milieu RPMI. Les cellules ont été marquées par l’ajout de 1 ml de PBS contenant 5 μM d’ester succinimidylique de carboxyfluorescéine (CFSE), suivi d’une incubation de 5 min à température ambiante. Les cellules marquées ont ensuite été lavées deux fois, comptées et ensemencées à105 cellules/puits dans une plaque de 12 puits. Le jour de l’analyse, les cellules T-47D ont été récoltées et lavées deux fois avec du PBS, puis l’intensité du CFSE a été examinée par FACS. Les résultats sont exprimés en moyenne ± S.D (n = 3), les calculs ont été effectués à l’aide du progiciel Prism, l’ANOVA avec post-test de Tukey a été appliquée et P < 0,05 a été considéré comme statistiquement significatif. Les expériences ont été réalisées au moins trois fois et les données présentées sont celles d’une expérience représentative.
Mort cellulaire L’apoptose a été quantifiée par cytométrie en flux après coloration des cellules avec de l’Annexin-V (AV) marquée au FITC (Invitrogen Co.) et de l’iodure de propidium (PI). Après le traitement médicamenteux, les cellules T-47D ont été récoltées et centrifugées à 1200 rpm pendant 5 min ; les culots ont été lavés deux fois avec du PBS, puis remis en suspension dans 100 μl de tampon de liaison de l’Annexin-V (0,14 M NaCl, 2,5 mM CaCl2, 0,01 M HEPES pH 7,4). L’Annexin-V (1 μL) et 5 μl de PI (50 μg/ml) ont été ajoutés aux échantillons et incubés dans l’obscurité pendant 15 min. Les échantillons ont été conservés sur la glace après l’incubation jusqu’à ce que l’analyse FACS soit effectuée. Les résultats sont exprimés en moyenne ± S.D (n = 3), les calculs ont été effectués à l’aide du progiciel Prism, l’ANOVA avec post-test de Tukey a été appliquée et P < 0,05 a été considéré comme statistiquement significatif. Les expériences ont été réalisées au moins trois fois et les données présentées sont celles d’une expérience représentative.
Génération de ROS
Pour l’évaluation des niveaux de ROS intracellulaires, les cellules ont été placées dans des plaques à 12 puits avec une densité cellulaire de 1 ×105 cellules par puits et traitées avec des médicaments pendant 12 heures. du 2′, 7′-dihydrochlorofluroresceinacetate (H2DCFDA) a été ajouté au milieu à une concentration finale de 10 μM et les cellules ont été autorisées à être colorées pendant 1 heure dans l’obscurité. Après la coloration auH2DCFDA, les cellules ont été trypsinées, lavées deux fois et remises en suspension dans 100 μl de PBS. L’intensité duH2DCFDAa été examinée en utilisant le FACS. Les résultats sont exprimés en moyenne ± S.D (n = 3), les calculs ont été effectués à l’aide du progiciel Prism, l’ANOVA avec post-test de Tukey a été appliquée et P < 0,05 a été considéré comme statistiquement significatif. Les expériences ont été réalisées au moins trois fois et les données présentées sont celles d’une expérience représentative.
Concentration d’ATP et activité des caspases
Le niveau d’ATP interne et l’activité des caspases dans les cellules T-47D ont été évalués à l’aide des kits de dosage CellTiter-Glo et Caspase-Glo 3/7 (Promega Corp., Madison, WI) conformément aux instructions du fabricant. Les cellules T-47D ont été cultivées en l’absence et en présence de médicaments pendant 12 heures dans des plaques blanches opaques à 96 puits (4 puits par groupe). Des volumes égaux de réactifs CellTiter-Glo ont été ajoutés, puis les échantillons ont été incubés pendant 15 minutes sur un agitateur à température ambiante. La luminescence a été enregistrée à l’aide du luminomètre Glomax pour microplaques (Promega Co., Madison, WI) selon le protocole CellTiter prédéfini. Les résultats sont exprimés en moyenne ± S.D (n = 4), les calculs ont été effectués à l’aide du progiciel Prism, l’ANOVA avec post-test de Tukey a été appliquée et P < 0,05 a été considéré comme statistiquement significatif. Les expériences ont été réalisées au moins trois fois et les données présentées sont celles d’une expérience représentative.
Potentiel de la membrane mitochondriale
Comme pour la mesure des ROS, les cellules ont été placées dans des plaques à 12 puits avec une densité cellulaire de 1 ×105 cellules par puits et traitées avec des médicaments pendant 12 heures. de l’iodure de carbocyanine de 5, 5′, 6, 6′-tétrachloro-1, 1′, 3, 3′-tétraéthylbenzimidazol (JC-1) a été ajouté dans le milieu à une concentration finale de 0,2 μM et les cellules ont été autorisées à être colorées pendant 30 min dans l’obscurité. Après la coloration JC-1, les cellules ont été trypsinées, lavées deux fois avec du PBS et remises en suspension dans 100 μl de PBS. L’intensité du JC-1 a été examinée en utilisant le FACS. Les résultats sont exprimés en moyenne ± S.D (n = 3), les calculs ont été effectués à l’aide du progiciel Prism, l’ANOVA avec post-test de Tukey a été appliquée et P < 0,05 a été considéré comme statistiquement significatif. Les expériences ont été réalisées au moins trois fois et les données présentées sont celles d’une expérience représentative.
Activité de la cytochrome C oxydase
Le dosage de la cytochrome C oxydase a été évalué selon la méthode publiée précédemment [24]. Brièvement, après traitement médicamenteux dans des plaques 96 puits (8 puits par groupe), les cellules T-47D ont été perméabilisées avec 50 μl de saponine à 0,01 %, puis 100 μl de milieu de substrat (4 mM de tétrahydrochlorure de 3,3-diaminobenzidine (DAB), 100 μM de cytochrome C réduit, 2 μg/ml de catalase dans du phosphate de Na 0,1 M, pH 7,0). L’absorbance à 450 nm a été mesurée immédiatement après l’ajout du milieu de substrat et suivie pendant 30 min. Les résultats sont exprimés en moyenne ± S.D (n = 8), les calculs ont été effectués à l’aide du progiciel Prism, l’ANOVA avec post-test de Tukey a été appliquée et P < 0,05 a été considéré comme statistiquement significatif. Les expériences ont été réalisées au moins trois fois et les données présentées sont celles d’une expérience représentative.
Activité de la PDH
L’activité de la PDH a été mesurée à l’aide du kit de dosage en microplaque de l’activité enzymatique de la PDH de MitoSciences (#MSP18, MitoSciences, Oregon USA). Pour mesurer l’effet des médicaments sur l’activité de la PDH dans les cellules, les cellules T-47D ont été traitées avec des médicaments dans le milieu pendant 3 heures, après quoi elles ont été lavées et remises en suspension dans du PBS. Des extraits cellulaires ont été préparés et dosés pour l’activité de la PDH à une concentration de 15 mg de protéines/ml, conformément aux instructions du kit. Pour mesurer l’inhibition directe de la PDH par l’ATO, la PDH a été isolée à partir de cellules T-47D non traitées en appliquant les extraits cellulaires sur la plaque de capture d’anticorps. Après avoir lavé la plaque, une solution de dosage contenant le médicament a été ajoutée dans les puits, puis l’activité de la PDH a été immédiatement mesurée. Les résultats sont exprimés en moyenne ± S.D (n = 4), les calculs ont été effectués à l’aide du progiciel Prism, l’ANOVA avec post-test de Tukey a été appliquée et P < 0,05 a été considéré comme statistiquement significatif. Les expériences ont été réalisées au moins trois fois et les données présentées sont celles d’une expérience représentative.
Immunoblotting et analyse densitométrique
Des cellules (1 ×106 ) ont été placées dans des flacons de culture tissulaire T25 et ont été traitées avec de l’ATO ou du DCA pendant 12 heures. Les lysats cellulaires ont été préparés par l’ajout de 500 μl du réactif d’extraction des protéines de mammifères MPER® (Thermo Scientific, IL, USA). L’immunoblotting a été réalisé comme décrit précédemment [25], en utilisant les anticorps pour c-Myc (Roche, IN, USA, clone 9E10), HIF-1α (Abcam, Cambridge, UK, #ab82832), Bcl-2 (Abcam #ab692), ATP synthase β-sous-unité (Abcam #ab14730) et β-actine (Abcam #ab8227). Les Western blots ont été détectés par chimiluminescence et exposition d’un film radiographique. Les images ont été acquises à l’aide d’un scanner à plat CanoScan 8600F, et quantifiées à l’aide du logiciel ImageJ (version 1.4, NIH, USA) et normalisées par rapport à la β-actine dans chaque voie. Les résultats ont été regroupés à partir de 3 expériences distinctes et exprimés en moyenne ± S.D (n = 3), les calculs ont été effectués à l’aide du progiciel Prism, l’ANOVA avec post-test de Tukey a été appliquée et P < 0,05 a été considéré comme statistiquement significatif.
Résultats
L‘association du DCA et de l’ATO est plus efficace pour réduire la prolifération cellulaire et induire la mort cellulaire
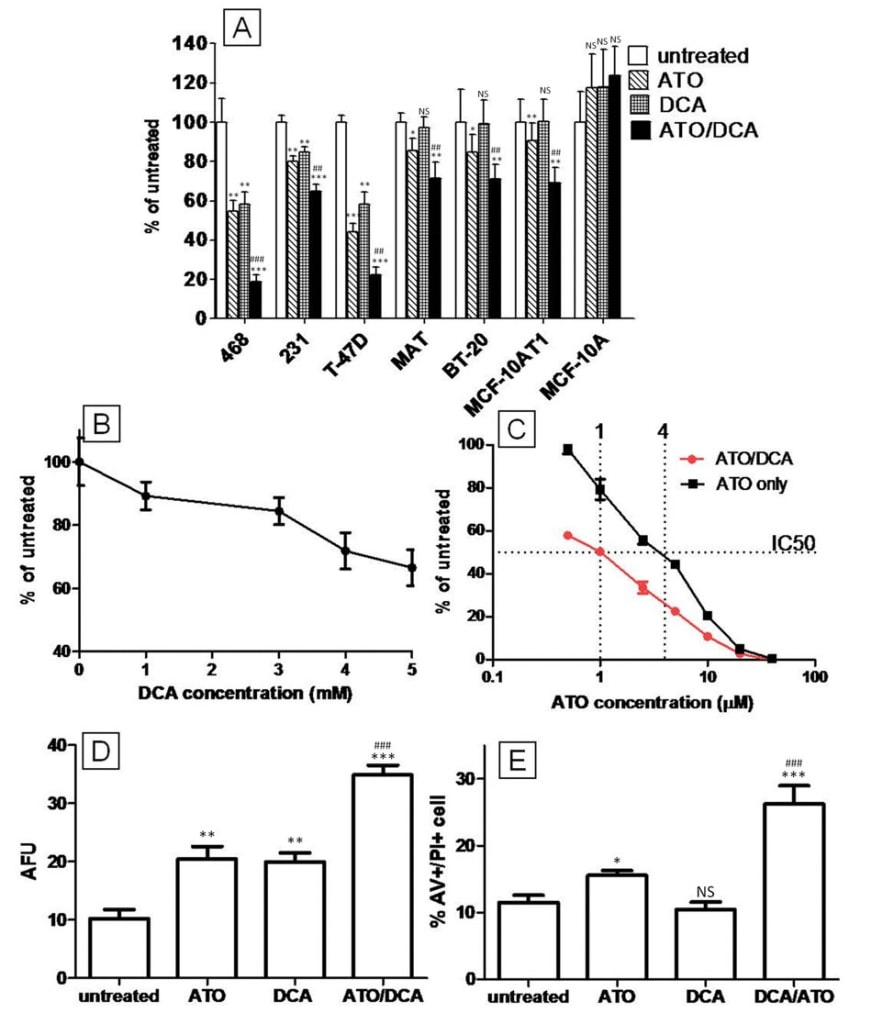
Pour étudier l’effet combiné de l’ATO et du DCA sur l’inhibition de la croissance cellulaire, des lignées cellulaires de cancer du sein ont été traitées pendant plusieurs jours avec les deux médicaments et le nombre total de cellules a été évalué à l’aide du test de viabilité cellulaire au rouge neutre. Le panel de lignées cellulaires représente les principaux sous-types de cancer du sein humain (luminal (T-47D), basal A (MDAMB-468, BT-20), basal B (MDA-MB-231)), ou présente un autre intérêt en tant que modèle expérimental (13762MAT – adénocarcinome mammaire de rat sensible au DCA in vivo [22], MCF10AT1 – dérivé malin des cellules immortalisées MCF-10A). Les cellules MDA-MB-468, MDA-MB-231 et T-47D ont toutes montré une réduction significative allant de 10 à 40 % du nombre total de cellules après 72 heures de traitement avec 5 mM de DCA (figure 1A), tandis que les cellules 13762 MAT, BT-20 et MCF-10AT1 n’ont pas réagi pendant la période de traitement. Toutes les lignées cellulaires cancéreuses testées étaient sensibles à l’ATO, mais à des concentrations différentes. Une réduction du nombre total de cellules a été observée dans les lignées cellulaires BT-20, T-47D, MCF-10AT1 et MDA-MB-468 avec seulement 5 μM d’ATO, mais 15 μM d’ATO ont été nécessaires pour obtenir la même efficacité dans les lignées cellulaires MDA-MB-231 et 13726 MAT. Il est frappant de constater que le DCA et l’ATO combinés ont montré un effet plus important que l’un ou l’autre médicament seul dans toutes les lignées cellulaires cancéreuses testées. Lorsque l’ATO et le DCA étaient efficaces en tant qu’agents individuels pour réduire le nombre de cellules, l’effet du traitement combiné était approximativement égal à la somme des effets des médicaments individuels (T-47D, MDA-MB-468 et MDA-MB-231). Alors que le DCA seul n’a pas montré de réduction du nombre de cellules, il a tout de même été capable d’augmenter l’inhibition de la croissance de l’ATO de 2 à 3 fois (13762 MAT, BT-20 et MCF10AT1) (Figure 1A). La lignée cellulaire non cancéreuse, MCF-10A, n’a montré aucune réduction du nombre de cellules après l’ATO (15 μM), le DCA (5 mM) ou un traitement combiné.
L’effet de l’ATO et du DCA a été examiné plus avant avec les cellules T-47D, l’une des lignées cellulaires les plus sensibles au DCA seul. La réponse des cellules T-47D au DCA était dose-dépendante et après 72 heures, les cellules traitées au DCA 5 mM contenaient 42 ± 6 % de cellules en moins que la culture témoin (figure 1B). L’ATO seul (5 μM) a réduit le nombre total de cellules de 56 ± 4 % et les cellules traitées à la fois par le DCA et l’ATO ont montré une diminution supplémentaire par rapport au groupe ATO seul (figure 1C). La courbe dose-réponse a montré que le traitement combiné de DCA (5 mM) et d’ATO peut réduire la CI50 à 0,25 fois celle de l’ATO seul (Figure 1C). Cet effet se produit dans la plage de concentration atteinte cliniquement pour l’ATO (jusqu’à 5-7 μM [26]).
Le test de prolifération CFSE a démontré que les cellules traitées par l’ATO (5 μM) ou le DCA (5 mM) émettaient une fluorescence CFSE significativement plus élevée (respectivement 2,1 fois et 2,2 fois) après 72 heures de traitement, indiquant une inhibition de la croissance. Les cellules traitées à la fois par l’ATO et le DCA ont présenté une augmentation de 3,4 fois de l’intensité du CFSE par rapport aux cellules non traitées, ce qui indique que les médicaments ont agi conjointement pour inhiber la prolifération cellulaire (figure 1D).
L’effet de l’ATO et du DCA sur la mort des cellules T-47D a été évalué par une double coloration AV et PI et les cellules ont été analysées par triage cellulaire fluorescent. Le DCA seul (5 mM) n’a pas induit de mort cellulaire dans les cellules T-47D (figure 1E), ce qui est similaire à l’effet du DCA sur les cellules 13762 MAT rapporté précédemment [22]. L’ATO (5 μM) n’a pas non plus induit la mort cellulaire après 12 heures de traitement, mais les cellules traitées à la fois par 5 μM d’ATO et 5 mM de DCA ont présenté une faible augmentation (15 %) de la population AV+/PI+ (13,2 ± 0,6 % de cellules apoptotiques contre 11,5 ± 1,0 % pour ATO/DCA par rapport aux cellules non traitées respectivement, p = 0,07), ce qui suggère que le DCA pourrait renforcer les effets apoptotiques de l’ATO. À des concentrations plus élevées et avec 48 heures de traitement, l’ATO (20 μM) a augmenté la quantité de mort cellulaire de 35 ± 8 % (P = 0,029) par rapport à la culture non traitée (figure 1E). La combinaison de 5 mM de DCA avec un traitement par ATO 20 μM a entraîné une augmentation 4 fois plus importante de la population AV+/PI+ par rapport à l’ATO seul, ce qui indique que le DCA peut potentialiser la mort cellulaire induite par l’ATO dans les cellules cancéreuses du sein T-47D (Figure 1E).
L’ATO et le DCA agissent ensemble sur la dépolarisation de la MMP mais ont des effets contraires sur l’induction de la production d’ATP et de ROS
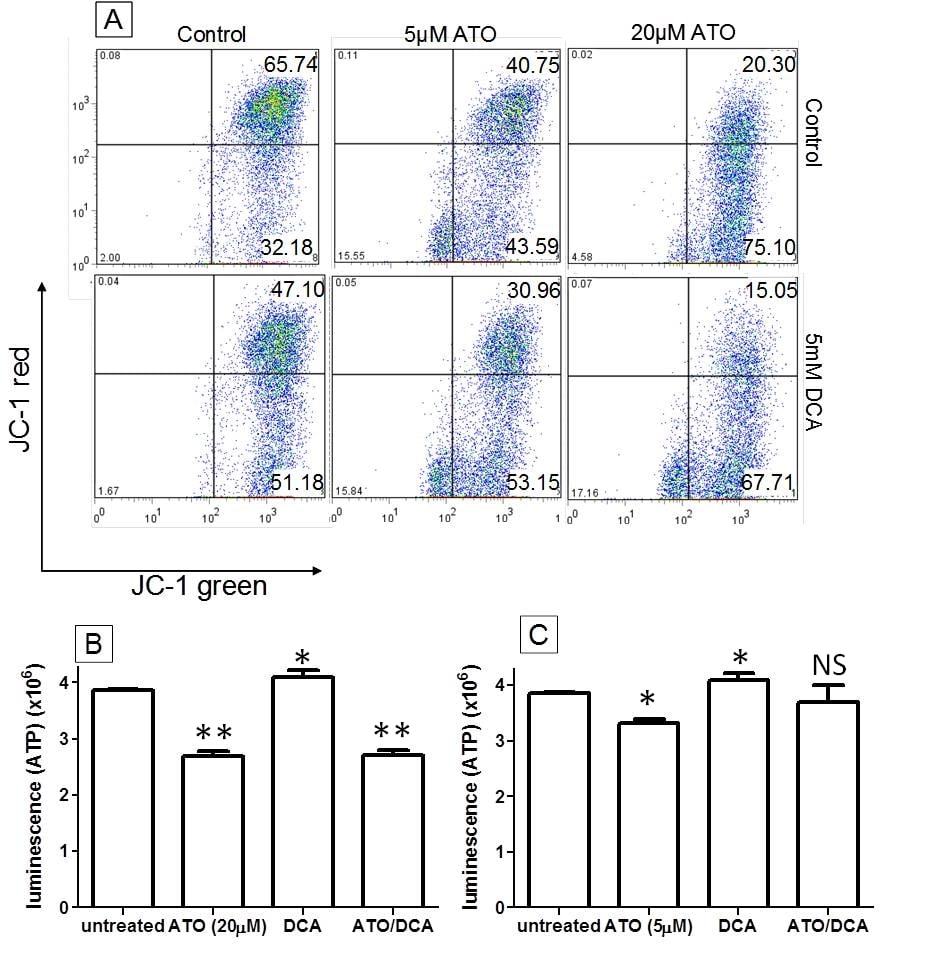
Il a été démontré que l’ATO et le DCA modifient tous deux la fonction mitochondriale, en dépolarisant la MMP et en augmentant la production de ROS [20]. C’est pourquoi ces paramètres, ainsi que le niveau d’ATP, ont été étudiés afin de déterminer s’ils contribuent aux effets anticancéreux accrus du traitement combiné ATO/DCA. Mesuré par la coloration JC-1, le nombre de cellules T-47D présentant une MMP hyperpolarisée (quadrant supérieur droit) a été significativement diminué par le traitement au DCA (5 mM) ou à l’ATO (5 μM et 20 μM) (t = 12 heures) (28 ± 6 %, 38 ± 4 % et 69 ± 7 % de diminution respectivement par rapport aux cellules témoins non traitées), conformément aux données publiées précédemment [20,27]. La combinaison de l’ATO (5 μM ou 20 μM) avec 5 mM de DCA a entraîné une diminution encore plus importante (53 ± 9 % et 77 ± 12 % respectivement) par rapport aux cellules non traitées (figure 2A), ce qui démontre que le DCA et l’ATO peuvent agir ensemble pour dépolariser la MMP. Les niveaux d’ATP ont été réduits par l’ATO à faible et à forte dose (14 ± 3 % et 32 ± 5 % respectivement) après 12 heures de traitement, tandis que les cellules traitées au DCA ont montré une augmentation de 6 ± 1 % des niveaux d’ATP (figures 2B et 2C). À la dose élevée d’ATO, le DCA n’a pas réussi à augmenter la production d’ATP (figure 2B), alors qu’à la faible dose d’ATO, les cellules traitées avec le DCA et l’ATO combinés ont montré une légère augmentation de la production d’ATP par rapport au traitement à l’ATO seul (figure 2C). Ces données indiquent que le DCA et l’ATO affectent la production d’ATP via des cibles distinctes à l’intérieur des cellules.
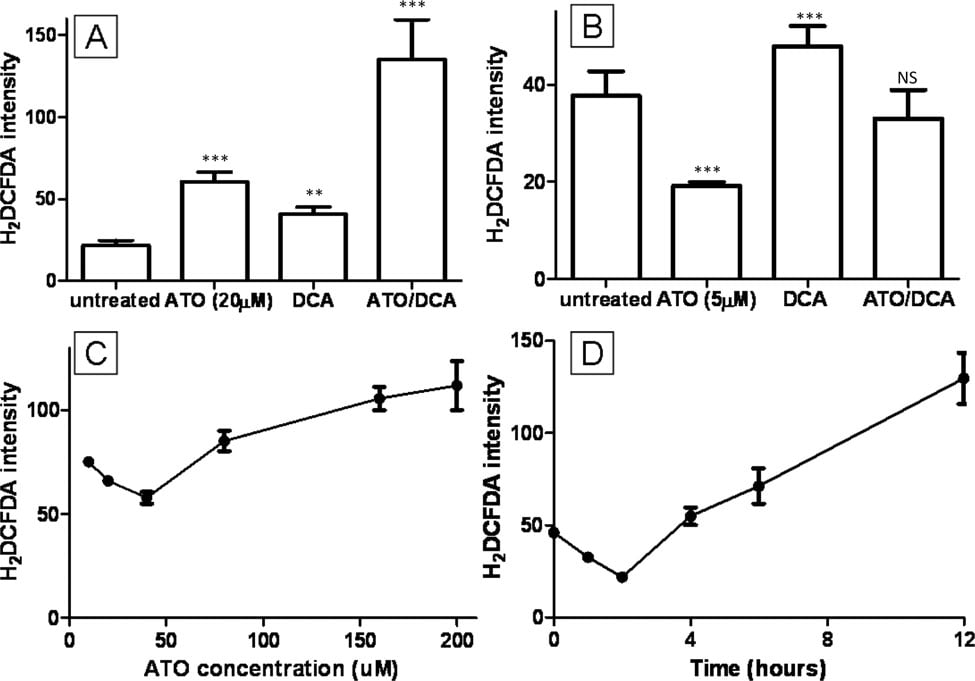
L’augmentation des niveaux de ROS intracellulaires a été proposée comme étant la raison de la cytotoxicité de l’ATO [2,9]. L’effet combiné de l’ATO et du DCA sur les ROS a été étudié à l’aide du colorant fluorescent H2DCFDA. Les cellules traitées avec 20 μM d’ATO ou 5 mM de DCA ont montré des niveaux élevés de ROS intracellulaires (respectivement 2,8 et 1,9 fois) (figure 3A), ce qui est similaire aux résultats précédemment publiés [27], tandis que les cellules traitées à la fois avec l’ATO et le DCA ont montré une augmentation supplémentaire (5,2 fois) de la production de ROS.(figure 3A). En revanche, l’ATO à faible concentration (5 μM) a induit une diminution de 0,5 fois de la production de ROS (figure 3B). Cela était contraire aux rapports précédents sur la production de ROS et le traitement par ATO, donc la dose-réponse et le cours du temps de la production de ROS après le traitement par ATO ont été analysés. Cela a révélé que des concentrations plus faibles d’ATO diminuaient la production intracellulaire de ROS, tandis qu’une concentration élevée d’ATO induisait la production de ROS (figure 3C). De même, le traitement par 10 μM d’ATO a entraîné une diminution notable de la production de ROS au cours des 4 premières heures avant que les niveaux de ROS n’augmentent après 8 heures pour atteindre le niveau rapporté par d’autres (figure 3D). Le traitement combiné des cellules avec 5 mM de DCA et 5 μM d’ATO a entraîné une production intermédiaire de ROS (figure 3B) similaire à celle des cellules non traitées.
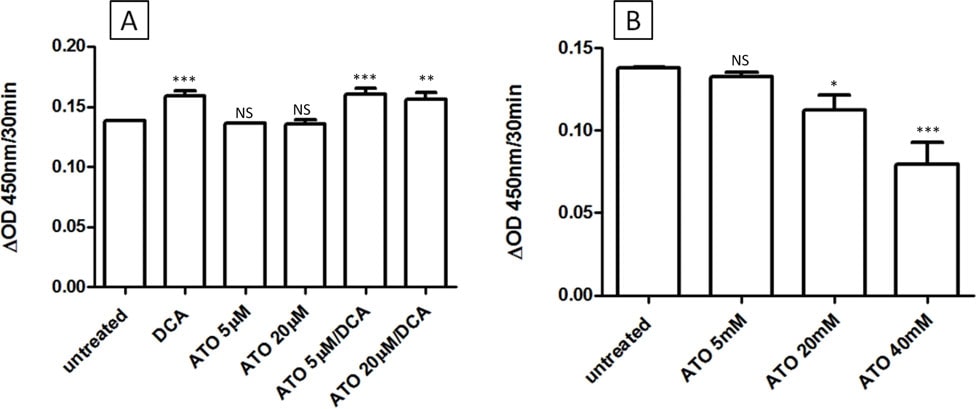
Les effets intermédiaires du traitement combiné DCA/ATO sur les niveaux d’ATP et de ROS peuvent s’expliquer par des effets concurrents sur l’activité de la PDH. Le DCA cible la PDK et devrait donc augmenter l’activité de la PDH, tandis que l’ATO inhibe la PDH soit directement par réaction avec les thiols vicinaux de la PDH, soit indirectement par augmentation de la production de peroxyde d’hydrogène [28]. Pour examiner les effets de l’ATO et/ou du DCA sur l’activité de la PDH, des cellules intactes ou des PDH isolées de cellules T-47D ont été traitées avec le médicament et l’activité de la PDH a été déterminée. Comme prévu, le traitement de 3 h de cellules intactes par le DCA a entraîné une augmentation de l’activité de la PDH, tandis que l’ATO (jusqu’à 20 μM) n’a pas modifié l’activité de la PDH des cellules intactes (figure 4A). En revanche, le DCA n’a pas modifié l’activité de la PDH isolée, mais des concentrations élevées d’ATO ont été capables d’inhiber l’activité de la PDH (figure 4b). Ainsi, dans les cellules T-47D, l’ATO n’a pas diminué l’activité de la PDH aux concentrations utilisées dans cette étude.
L‘ATO est un inhibiteur du complexe IV de la chaîne de transport des électrons
La chaîne de transport des électrons (ETC) est située dans la membrane interne de la mitochondrie et joue un rôle essentiel dans la production d’énergie. L’ETC est responsable de la génération du gradient de protons pour l’espace de la membrane interne des mitochondries afin de maintenir la MMP pour la production d’ATP [29]. Le CTE est également considéré comme le principal site de production de ROS dans les cellules en raison de la fuite d’électrons des complexes I et III [30]. En raison de sa capacité à réduire les ROS, à dépolariser la MMP et à diminuer la production d’ATP simultanément, nous avons émis l’hypothèse que l’ATO fonctionne comme un inhibiteur du CTE. Un panel d’inhibiteurs de l’ETC a été comparé à l’ATO (5 μM) pour identifier s’ils induisent les mêmes changements phénotypiques que l’ATO dans les cellules T-47D. La roténone (0,1 μM, complexe I), la thiolytrifluoroacétone (TTFA) (10 μM, complexe II), l’antimycine (0,1 μM, complexe III) et le NaCN (10 mM, complexe IV) ont été utilisés pour traiter les cellules T-47D et les niveaux de ROS, MMP et ATP ont été comparés à ceux des cellules traitées par l’ATO. Tous les inhibiteurs ont démontré leur capacité à dépolariser la MMP (figure 5A) et à réduire les niveaux d’ATP (figure 5B) de façon similaire à l’ATO. Cependant, contrairement à l’ATO, la roténone, l’antimycine et le TTFA n’ont pas réussi à réduire la production de ROS dans les cellules, augmentant au contraire les ROS de 3 à 6 fois après 12 heures de traitement (Figure 5C). Les cellules traitées avec NaCN (10 mM) ont cependant montré une diminution de 52% de la production de ROS (Figure 5C), similaire au traitement par ATO. Sur la base de ces données, nous avons conclu que dans les cellules cancéreuses du sein T47D, l’inhibition du complexe IV du CTE mais pas des complexes I-III entraîne une réduction des ROS, et il est donc probable que les changements phénotypiques induits par l’ATO soient causés par l’inhibition du complexe IV (cytochrome C oxydase) du CTE. Pour confirmer cela, l’activité de la cytochrome C oxydase dans les cellules T-47D a été mesurée par spectrophotométrie. Le dosage de la cytochrome C oxydase a clairement démontré que si le DCA n’avait aucun effet sur l’activité du complexe IV, l’ATO (5 μM, 5 min) peut inhiber cette activité, ce qui confirme notre hypothèse (figure 5D). Des résultats similaires ont été obtenus après 10 min, 3 h et 12 h de traitement médicamenteux. La combinaison de l’ATO et du DCA a montré une inhibition enzymatique similaire à celle de l’ATO seul.
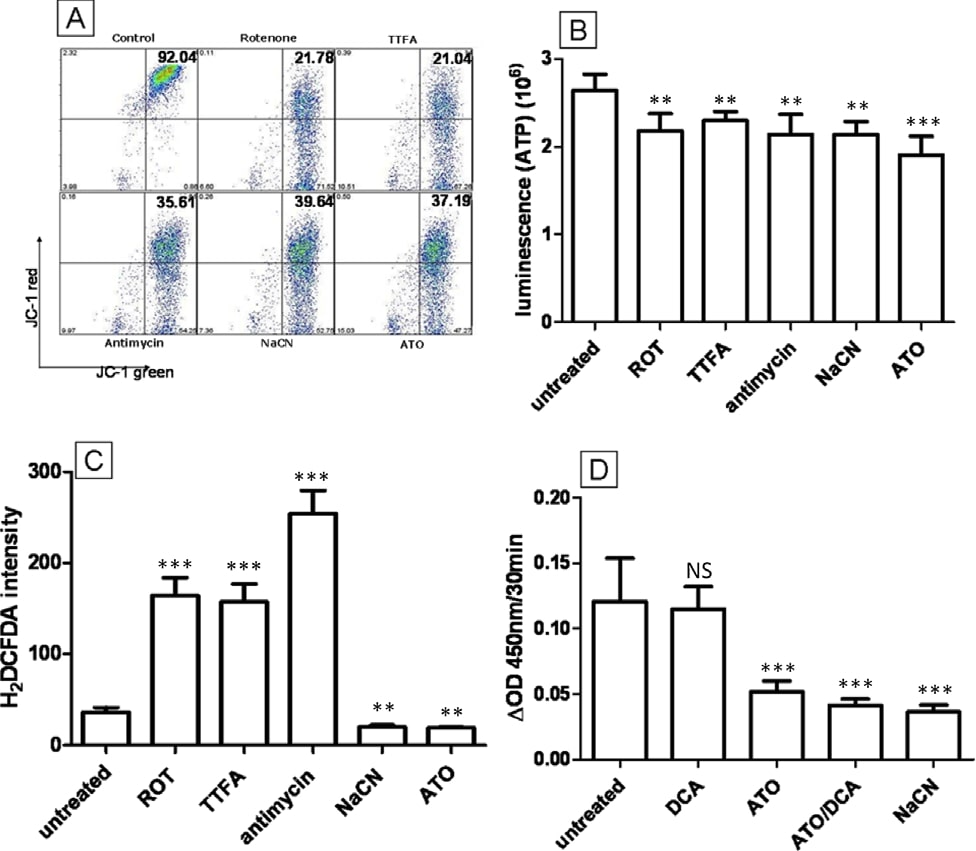
Lesinhibiteurs du pore de transition mitochondrial ont partiellement bloqué l’activation des caspases induite par l’ATO mais n’ont eu aucun effet sur les ROS, l’ATP et la MMP
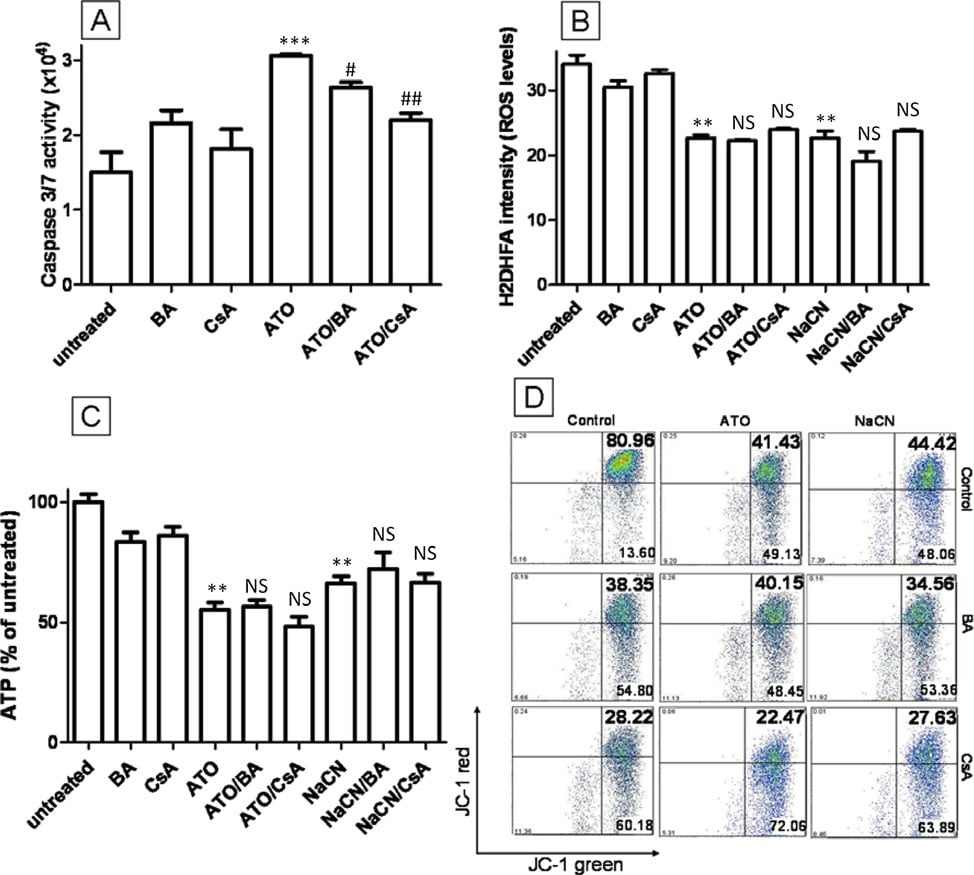
Un mécanisme qui a été précédemment décrit comme la clé de la cytotoxicité de l’ATO est l’ouverture du pore de transition mitochondrial (MTP). Il a été démontré que l’ATO induit l’ouverture du PTM dans des mitochondries isolées et peut augmenter la libération de cytochrome C [11] et induire l’apoptose nucléaire dans des systèmes sans cellules [31]. Il a été proposé que l’ouverture de la MTP par l’ATO induise une perméabilisation mitochondriale, dissipe la MMP et augmente les ROS intracellulaires. Pour vérifier si le MTP est important pour la toxicité de l’ATO dans les cellules T-47D, l’acide bongkrekique (BA) et la cyclosporine A (CsA), deux puissants bloqueurs de pores MTP [32], ont été associés à l’ATO pour voir s’ils peuvent inhiber les changements intracellulaires induits par l’ATO. L’ajout de 5 μM de CsA ou de 50 μM de BA aux cellules traitées par l’ATO (5 μM) n’a pas interféré avec la capacité de l’ATO à réduire la production de ROS après 12 heures de traitement, à diminuer la production d’ATP ou à dépolariser le MTP (P > 0,05) (figure 6B, C et 6D). Des données similaires sont observées pour le traitement au NaCN et à la CsA/BA (P > 0,05) (Figure 6B, C et 6D). Pour confirmer l’activité de la CsA et du BA dans nos conditions expérimentales, l’activité de la caspase 3/7, un marqueur d’apoptose en aval de l’ouverture du MTP, activé par la libération de cytochrome C, a été évaluée après un traitement par ATO à haute concentration (Figure 6A). Le traitement par ATO (20 μM) a doublé l’activité de la caspase 3/7 par rapport aux cellules non traitées (2,1 fois, figure 6A) et cet effet a été diminué par le BA et la CsA (1,7 fois et 1,4 fois, respectivement), ce qui indique que la CsA ou le BA peuvent bloquer l’ouverture du MTP dans les cellules T-47D. Ces données démontrent clairement que, bien que l’ATO (20 μM) puisse induire l’ouverture du MTP, ce mécanisme ne peut pas expliquer les modifications des ROS, des MMP et de l’ATP survenant lors du traitement par l’ATO 5 μM. Nous concluons que ces altérations sont probablement dues à l’inhibition du complexe IV du CTE.
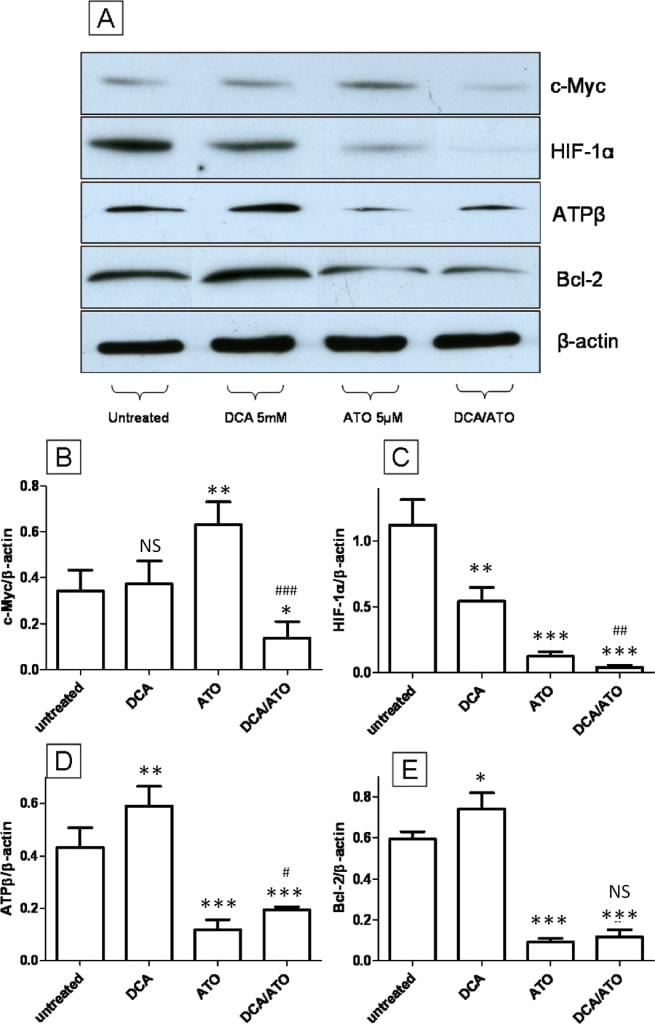
LeDCA et l’ATO ont des effets opposés sur l’expression de la sous-unité βde l’ATP synthase et de Bcl-2 tout en travaillant de manière coopérative à la régulation à la baisse des niveaux de protéines c-Myc et HIF-1α
Nous avons examiné l’effet d’un traitement par 5 mM de DCA et 5 μM d’ATO (12 heures) sur l’expression de c-Myc et HIF1α, deux facteurs de transcription majeurs connus pour réguler l’effet Warburg [33] et l’activité mitochondriale [34], et sur l’expression des protéines mitochondriales ATP synthase β sous-unité (ATPβ) et Bcl-2. L’immunoblotting pour c-Myc n’a montré aucun changement après le traitement avec le DCA alors que l’ATO a induit la régulation à la hausse de c-Myc (Figure 7A et 7B), cependant l’ATO en combinaison avec le DCA a réduit le niveau de c-Myc par rapport au contrôle non traité. Les niveaux de HIF-1α ont par contre montré une régulation à la baisse après le traitement par DCA et ATO. La combinaison de l’ATO et du DCA a montré une régulation encore plus faible de HIF-1α par rapport aux groupes traités par un seul agent et aux groupes témoins (Figure 7A et 7C). Bcl-2 est un membre de la famille des protéines BH3 qui se lie directement à Bad et Bax et prévient l’apoptose [35]. L’immunoblotting réalisé pour l’ATPβ a permis de constater que le traitement par DCA régulait à la hausse l’ATPβ, tandis que l’ATO présentait un effet opposé, en abaissant les niveaux d’ATPβ (Figure 7A et 7D), et le traitement combiné par ATO et DCA présentait une réponse intermédiaire par rapport aux groupes traités par ATO ou DCA. L’immunoblotting pour la protéine pro-survie Bcl-2 a montré une régulation à la hausse après le DCA, une régulation à la baisse pour le traitement par l’ATO, et une réponse intermédiaire de la combinaison des deux médicaments (Figure 7A et 7E). Ces résultats sont également très similaires aux niveaux d’ATP et à la production de ROS où le DCA et l’ATO ont eu des effets opposés (Figure 2C et 3B). Les résultats concernant Bcl-2 et ATPβ sont directement corrélés à la capacité du DCA et de l’ATO à influencer la production de ROS et les niveaux d’ATP.
Discussion
Au cours de la dernière décennie, l’ATO a été utilisé efficacement pour traiter les patients atteints de LPA nouvellement diagnostiqués ou en rechute, les patients présentant une rémission complète après un traitement à faible dose d’ATO [4]. La capacité anticancéreuse de l’ATO n’est pas limitée à l’APL [36] et de nombreuses autres tumeurs dans des modèles animaux se sont révélées sensibles au traitement par l’ATO [37,38]. Cependant, le manque d’information sur les sites d’action de la cytotoxicité de l’ATO dans des types de tumeurs autres que le LPA a limité son utilisation pour le traitement d’autres cancers jusqu’à ces dernières années [5] [5]. <p>Cette recherche a été soutenue par une subvention de la National Breast Cancer Foundation Australia (AB), et par le NHMRC 366787 R.D. Wright Career Development Award (AB). PB est soutenu par le NHRMC et l’Australian National University. RS a été soutenu par une bourse de troisième cycle de l’Université nationale australienne</p> <p>1 Groupe de génétique moléculaire, Département des biosciences translationnelles, École de recherche médicale John Curtin, bâtiment 131, Université nationale australienne, B.P. 334, Canberra ACT 0200, AUSTRALIE. 2 Département de radio-oncologie, École de médecine de Stanford, Stanford CA 94305 USA.</p> <p>Les auteurs déclarent ne pas avoir d’intérêts concurrents.<br></p>
Dans cette étude, nous avons démontré que le complexe IV de l’ETC est une cible pour l’ATO. Parmi un panel d’inhibiteurs de l’ETC, la diminution des ROS, la diminution de la production d’ATP et la dépolarisation des MMP provoquées par le traitement des cellules T-47D par l’ATO à faible dose n’ont été reproduites que par le cyanure, un inhibiteur du complexe IV bien caractérisé (Figure 5). La mesure directe de l’activité de la cytochrome C oxydase dans les cellules entières traitées par l’ATO a confirmé la capacité de l’ATO à inhiber directement cette activité enzymatique. Le complexe cytochrome C oxydase de l’ETC est connu pour avoir des résidus cystéine très rapprochés [39] [39]et ceux-ci sont susceptibles de réagir avec l’ATO.
La plupart des cellules cancéreuses ont des niveaux de ROS régulés à la hausse par rapport auxcellulesnormales [40] [40] et le CTE est considéré comme la principale source de ROS intracellulaires. La réduction des ROS jusqu’à 60% par le NaCN ou l’ATO, a démontré que le complexe IV est responsable de la majorité de la production de ROS dans les cellules T-47D. Le stress oxydatif lié à une production élevée de ROS a été décrit comme une arme à double tranchant. Il a été démontré qu’une augmentation modérée des ROS est associée à un potentiel prolifératif accru [41] [41], à une augmentation des enzymes antioxydantes comme la glutathion transférase [42]la superoxyde dismutase et la catalase [43] et une augmentation des protéines prosurvivantes telles que Bcl-2 [42] et la survivine [44]. Cependant, des quantités intolérables de ROS finissent par entraîner la mort cellulaire [45]. La réduction des ROS après 12 heures de traitement par 5 μM d’ATO est également directement corrélée à une diminution des niveaux de Bcl-2 (figure 7E), ce qui suggère que les cellules T-47D sont dans un état de résistance apoptotique en raison des niveaux élevés de ROS. L’inhibition continue du complexe IV du CTE entraînera la dépolarisation de la MMP et l’inhibition de la production d’ATP. La mort cellulaire qui découle de ce dérèglement mitochondrial donnera lieu à l’augmentation des ROS observée après 4 heures ou à une concentration plus élevée d’ATO (figure 3C et 3D), comme l’ont rapporté d’autres chercheurs [46] après un traitement à l’ATO. Ainsi, l’augmentation des ROS est une conséquence, et non la cause, de la mort cellulaire induite par l’ATO.
La cible mitochondriale précédemment proposée pour l’ATO est la PTM. Zheng et al ont démontré que l’ATO induit l’ouverture de la PTM dans les mitochondries isolées du foie de rat [11]ce qui a conduit à la proposition que la MTP est responsable de la libération de ROS et de la dépolarisation de la MMP observées pendant le traitement à l’arsenic, et de l’apoptose qui s’ensuit [47]. Notre étude n’exclut pas la MTP comme cible de l’ATO. Nos données indiquent que l’inhibition de la MTP par la CsA et le BA peut partiellement bloquer l’activation de la caspase 3/7 induite par l’ATO à forte concentration, mais les bloqueurs de la MTP n’ont pas réussi à inhiber la dépolarisation de la MMP et la réduction des niveaux de ROS et d’ATP causées par l’ATO à faible concentration. Ainsi, bien que l’ATO puisse ouvrir le MTP, d’autres cibles sont nécessaires pour expliquer l’action de l’ATO à de faibles concentrations. Il a été démontré que l’inhibition du système de la thiorédoxine se produit dans les cellules MCF-7 après un traitement avec de l’ATO de 2 à 5 μM et qu’elle peut servir de médiateur à certains effets cellulaires de l’ATO à faible concentration [13]cependant, ce mécanisme conduirait à une augmentation du stress oxydatif et ne peut pas expliquer la diminution des ROS observée dans l’étude actuelle.
Le DCA, en tant qu’inhibiteur de la pyruvate déshydrogénase kinase, peut inverser l’effet glycolytique et a démontré d’importantes propriétés anticancéreuses tant in vitro qu’in vivo [17-23]. Certaines études montrent une augmentation de l’apoptose avec le DCA [17,18,20]. Cependant, nos études sur le cancer du sein et celles de Stockwin et al. indiquent que le DCA agit comme un agent cytostatique plutôt que cytotoxique sur une gamme de lignées cellulaires, sauf à des concentrations très élevées [22,48] [22,48]. Michelakis a récemment signalé que les concentrations sériques minimales de DCA étaient de 0,5 mM chez des patients atteints de glioblastome prenant 6,25 mg/kg par voie orale deux fois par jour [23]les concentrations cliniquement pertinentes se situent donc probablement entre 0,5 et 5 mM. Il a été démontré que le DCA induit une augmentation faible mais significative de l’activité de la caspase 3/7, même si l’apoptose n’a pas été observée, et il a été proposé que le DCA puisse sensibiliser les cellules cancéreuses aux agents cytotoxiques [22]. L’étude actuelle a confirmé cette hypothèse en montrant que le DCA sensibilisait les cellules T-47D au traitement par l’ATO, comme le montre la double coloration AV+/PI+ (figure 1E et texte). L’ATO a été choisi comme agent à tester en raison de ses propriétés anti-mitochondriales, car nous avons proposé qu’en inversant le phénotype glycolytique avec le DCA et en dirigeant davantage de pyruvate vers la phosphorylation oxydative mitochondriale, tout en ciblant simultanément les mitochondries avec l’ATO, un effet coopératif serait observé avec cette combinaison de médicaments. Le principe de cette stratégie de double ciblage est étayé par les résultats de deux publications récentes qui ont montré que les cellules présentant des défauts mitochondriaux (par exemple rho (0) ou après traitement avec des inhibiteurs mitochondriaux non médicamenteux tels que la roténone) sont plus sensibles aux effets d’inhibition de la croissance du DCA [48,49]. Bien que l’efficacité de cette stratégie de double ciblage doive encore être démontrée in vivo, les effets de la combinaison ATO/DCA renforcée ont été observés à la fois sur l’inhibition de la croissance et sur l’apoptose à des concentrations de médicaments dans des plages cliniquement pertinentes. Certains effets à l’extrémité inférieure de la gamme de concentrations n’étaient pas importants (par exemple, une augmentation de 15 % des cellules apoptotiques), mais l’impact in vivo sur des semaines plutôt que des heures de traitement justifie une étude plus approfondie.
Il est bien documenté que le DCA peut modifier le comportement des mitochondries, c’est-à-dire dépolariser la MMP [18,20]. Cependant, ce phénomène n’a pas été bien expliqué. Nous avons observé que le DCA peut augmenter l’ATP tout en dépolarisant la MMP dans les cellules T-47D (figure 2). Sur la base de ces données, nous avons proposé que le DCA modifie la fonction mitochondriale par le biais du complexe V du CTE, l’ATP synthase. L’ATP synthase est la dernière étape du CTE et elle utilise la MMP générée par le CTE pour produire de l’ATP. L’ATPb est régulée à la baisse dans les cancers du poumon, du cerveau, du sein et de l’estomac [50]ce qui pourrait expliquer la membrane mitochondriale hyperpolarisée que l’on retrouve dans de nombreuses cellules cancéreuses [51]. Nous avons émis l’hypothèse que le DCA peut augmenter l’activité de l’ATP synthase soit par une régulation allostérique en raison de son homologie avec le pyruvate, soit par une augmentation des niveaux de protéine ATP synthase après l’inversion de l’effet Warburg et la diminution des niveaux de HIF-1α. L’immunoblotting a clairement démontré que l’ATPβ est régulé à la hausse après le traitement par le DCA (figure 7D), ce qui suggère que le DCA peut augmenter l’activité de l’ATP synthase, qui contribue à son tour à une production accrue d’ATP et à l’épuisement de la MMP.
HIF-1α et c-Myc sont deux facteurs de transcription oncogènes majeurs connus pour réguler le métabolisme dans les cellules cancéreuses. HIF-1α peut réguler à la hausse les enzymes de la voie glycolytique et l’acide lactique déshydrogénase, soutenant ainsi l’effet Warburg dans les cellules cancéreuses[33] [33]. Cela augmente non seulement la production d’ATP, mais aussi l’approvisionnement en précurseurs tels que le glucose-6-phophate et le fructose-6-phosphate pour la production d’acides nucléiques par la voie du pentose phosphate [16]par ailleurs, c-Myc joue non seulement un rôle central dans la promotion de la transition du cycle cellulaire de la phase G1 à la phase S en régulant les cyclines, leurs kinases et leurs inhibiteurs, mais elle régule également les composants protéiques du CTE tels que COXI-IV et contribue à augmenter l’activité mitochondriale [52] [52]. Ainsi, la combinaison de la surexpression de c-Myc et de HIF-1α est importante pour induire l’effet Warburg tout en augmentant l’activité mitochondriale, soutenant la plaque tournante de la glutamine-TCA qui est essentielle pour l’anabolisme des acides aminés et des acides gras nécessaires à la division cellulaire [16]. Cette signature métabolique contribue à la carcinogenèse et au phénotype malin de nombreuses tumeurs [33,34]. L’ATO et le DCA peuvent tous deux diminuer de manière significative les niveaux de HIF-1α, ce qui suggère une réduction de l’effet Warburg (figure 7C). Alors que le DCA seul n’avait aucun effet sur les niveaux de c-Myc, de façon surprenante, l’ATO a régulé à la hausse les niveaux de c-Myc de façon significative (Figure 7B). Il peut s’agir d’une boucle de rétroaction positive, où les cellules essaient d’augmenter l’activité du CTE après l’inhibition du complexe IV. Il est frappant de constater que le DCA a inversé l’effet de l’ATO sur le c-Myc, et que la combinaison ATO/DCA a fortement réprimé l’expression du c-Myc, ce qui correspond à la capacité de ces agents à travailler ensemble pour réduire la prolifération cellulaire et induire la mort cellulaire (Figure 1).
L’efficacité de l’ATO contre la LEMP a été attribuée à plusieurs caractéristiques particulières de cette tumeur maligne : différenciation par l’inactivation de PML-RARα, faible capacité antioxydante des cellules de la LEMP à se protéger contre les niveaux élevés de ROS et accumulation du médicament en raison d’une osmorégulation altérée [5] [5]. De nombreux essais sont en cours pour l’utilisation de l’ATO dans une série de tumeurs malignes solides, bien que peu de résultats aient été publiés jusqu’à présent. Les résultats disponibles suggèrent que l’ATO n’est pas très efficace en tant qu’agent unique chez les patients atteints de cancer avancé du pancréas, du foie ou de mélanome <a href= »#5″> [5]</a></sup>. Les investigateurs préconisent d’évaluer l’ATO en association avec d’autres médicaments anticancéreux<sup><a href= »#53″> [53,54]</a></sup>, certains essais de phase I étant en cours d’achèvement <sup><a href= »#55″> [55]</a></sup>. Au fur et à mesure que notre compréhension des mécanismes de l’ATO continue de croître, en particulier ses effets à travers le métabolisme du cancer, nous pourrions être mieux en mesure d’exploiter son potentiel anticancéreux in vivo dans de nouvelles combinaisons de médicaments</p>
<h2>Conclusions</h2>
<p>Deux rapports récents ont trouvé que le DCA était plus efficace contre les cellules présentant des défauts mitochondriaux <sup><a href= »#48″>[48,49].</a></sup> Ce rapport est le premier à démontrer que le ciblage de deux aspects du métabolisme – l’inversion de l’effet Warburg avec le DCA tout en inhibant la phosphorylation oxydative avec l’ATO – est clairement une stratégie anticancéreuse efficace in vitro contre les lignées cellulaires du cancer du sein. L’identification de la cytochrome C oxydase comme cible mitochondriale de l’ATO fournit de nouvelles informations mécanistiques pour l’application de l’ATO au traitement de types de tumeurs autres que l’APL. La capacité du DCA à augmenter l’expression de la sous-unité b de l’ATP synthase, en inversant un autre phénotype métabolique cancéreux très répandu, suggère également que le DCA pourrait être pertinent pour un large éventail de types de tumeurs. La capacité du DCA à renforcer les effets cytotoxiques des agents chimiothérapeutiques autres que l’ATO in vitro et in vivo mérite également d’être étudiée plus avant. L’absence de sensibilité de la lignée cellulaire non cancéreuse MCF-10A aux concentrations cliniquement pertinentes d’ATO/DCA testées est également encourageante, et suggère que cette stratégie de traitement devrait être davantage testée contre les tumeurs solides in vivo.</p>
<h2>Liste des abréviations</h2>
<p>LAP : leucémie aiguë promyéloïde ; ATO : trioxyde d’arsenic ; ATPβ : ATP synthase β subunit ; AV : annexin V ; BA : acide bongkrekique ; CFSE : carboxyfluorescein succinimidyl ester ; CsA : cyclosporine A ; DCA : dichloroacetate ; ETC : electron transport chain ; H2DCFDA : 2′, 7′-dihydrochlorofluroréscéine-acétate ; JC-1 : iodure de 5,5′,6,6′-tétrachloro-1,1′, 3,3′-tétraéthylbenzimidazol-carbocyanine ; MMP : potentiel de la membrane mitochondriale ; MTP : pore de transition mitochondrial ; NaCN : cyanure de sodium ; PI : iodure de propidium ; ROS : Espèces réactives de l’oxygène ; TTFA : thenolytrifluoroacetone.</p>
<h2>Remerciements</h2>