Ramon C Sun1,2, Philip G Board1 en Anneke C Blackburn1*
1 Molecular Genetics Group, Department of Translational Biosciences, John Curtin School of Medical Research, Building 131, Australian National University, P.O. Box 334, Canberra ACT 0200, AUSTRALIA
2 Department of Radiation Oncology, Stanford School of Medicine, Stanford CA 94305 USA.
1*Correspondentie: [email protected]
© 2011 Sun et al; licentiehouder BioMed Central Ltd. Dit is een Open Access artikel verspreid onder de voorwaarden van de Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), die onbeperkt gebruik, distributie en reproductie toestaat in elk medium, op voorwaarde dat het originele werk correct wordt geciteerd.
Ontvangen: 3 mei 2011
Geaccepteerd: 18 november 2011
Gepubliceerd: 18 november 2011
Abstract
Achtergrond
Kankercellen hebben een ander metabool profiel dan normale cellen. Het Warburg effect (verhoogde aerobe glycolyse) en glutaminolyse (verhoogde mitochondriale activiteit van glutamine katabolisme) zijn bekende kenmerken van kanker en gaan gepaard met verhoogde lactaat productie, hypergepolariseerde mitochondriale membraan en verhoogde productie van reactieve zuurstofspecies.
Methoden
In deze studie richten wij ons op het Warburg-effect met dichlooracetaat (DCA) en de verhoogde mitochondriale activiteit van glutaminolyse met arseentrioxide (ATO) in borstkankercellen, waarbij wij de celproliferatie, celdood en mitochondriale kenmerken meten.
Resultaten
De combinatie van DCA en ATO was doeltreffender in het remmen van de celproliferatie en het induceren van celdood dan een van beide geneesmiddelen alleen. Wij onderzochten het effect van deze behandelingen op de mitochondriale membraanpotentiaal, de productie van reactieve zuurstofspecies en de ATP-niveaus en identificeerden nieuwe moleculaire mechanismen binnen de mitochondriën voor zowel ATO als DCA: ATO vermindert de mitochondriale functie door remming van cytochroom C-oxidase (complex IV van de elektronentransportketen), terwijl DCA de expressie van de ATP-synthase b-subeenheid verhoogt. De versterking van ATO-cytotoxiciteit door DCA is gecorreleerd met een sterke onderdrukking van de expressie van c-Myc en HIF-1a, en een verminderde expressie van het overlevingseiwit Bcl-2. Conclusie
Deze studie is de eerste die aantoont dat het aanpakken van twee belangrijke metabolische kenmerken van kanker een effectieve antikankerstrategie is met therapeutisch potentieel.
Trefwoorden
Dichlooracetaat, borstkanker, elektronentransportketen, mitochondriën, arseentrioxide
Inleiding
Arseentrioxide (ATO) wordt al meer dan 2000 jaar gebruikt als therapeutisch middel. Afkomstig uit China [1], wordt het momenteel gebruikt tegen acute promyeloïde leukemie (APL) bij patiënten die hervallen zijn na een behandeling met alltrans-retinoïnezuur/anthracycline, en wordt het gepromoot voor eerstelijnstherapie van de novo APL [2-4]. ATO staat bekend als een hyperreactieve molecule en zou kunnen binden aan thiolgroepen in vele eiwitten [2,5]. Zijn vermogen om te binden aan het thiolrijke, gemuteerde eiwit PML-RAR-α dat ontstaat door een chromosoomtranslocatie bij APL, heeft het tot een effectief geneesmiddel bij APL gemaakt [ 2,5,6]. Er is aangetoond dat ATO in vitro en in vivo apoptose induceert in verschillende kankercellijnen [7,8], maar het is moeilijk gebleken ATO te overwegen voor klinisch gebruik in andere tumortypes dan APL, wegens gebrek aan kennis van de moleculaire doelwitten die de cytotoxiciteit ervan veroorzaken. In de afgelopen 10 jaar zijn de fysiologische veranderingen binnen kankercellen in reactie op de behandeling met ATO goed gekarakteriseerd, en er lopen vele klinische proeven voor nieuwe toepassingen van ATO [5]. ATO is voorgesteld als een mitochondriaal toxine [9]. ATO kan de mitochondriale membraanpotentiaal (MMP) depolariseren [10], de intracellulaire productie van reactieve zuurstofspecies (ROS) verhogen [8], en apoptose induceren [8]. Het voorgestelde doelwit voor ATO dat deze fenotypische veranderingen kan bewerkstelligen is de mitochondriale overgangspoort (MTP) [11]. Van ATO is aangetoond dat het de opening van de MTP induceert, waardoor cytochroom c vrijkomt en wordt voorgesteld de MMP af te voeren en de afgifte van ROS uit de mitochondriën te verhogen [12]. Meer recentelijk is ook het thioredoxinesysteem, met name thioredoxinereductase, geïdentificeerd als een doelwit van ATO dat kan bijdragen tot verhoogde oxidatieve stress en veranderde redoxsignalering na behandeling van kankercellen met ATO [9,13].
Het Warburg-effect is een wijdverbreid verschijnsel dat is vastgesteld bij meer dan 90% van alle tumoren. Cellen die het Warburg-effect vertonen nemen alternatieve routes van energiehomeostase aan om hun proliferatief fenotype in stand te houden [14]. Nobelprijswinnaar Dr. Otto Warburg stelde dat kankercellen vertrouwen op glycolyse of substraatfosforylering om ATP te genereren, en hun mitochondriale activiteiten onderdrukken [15]. Met meer geavanceerde technologieën hebben recente studies het ATP-productieaspect van de Warburg-hypothese bevestigd, maar aangetoond dat de mitochondriale activiteit in kankercellen niet wordt onderdrukt. Integendeel, mitochondriën spelen een vitale rol bij het leveren van substraten om de celdeling in stand te houden [16].
Het anti-kanker effect van het omkeren van het Warburg effect is onlangs beschreven en een oud geneesmiddel dichlooracetaat (DCA), dat de ATP-synthese kan omleiden van glycolyse naar oxidatieve fosforylering, heeft een goede anti-kanker activiteit aangetoond, zowel in vitro [17-19] als in vivo [20-23]. DCA is een pyruvaat dehydrogenase kinase remmer, en resulteert in een verhoogde pyruvaat dehydrogenase activiteit [19]. Dit leidt tot een verhoogde omzetting van pyruvaat in acetyl-CoA in plaats van melkzuur, zoals beschreven door het Warburg-effect, en stimuleert de mitochondriale ademhaling door de toevoer van acetyl-CoA te vergroten. Bijgevolg vertoonden kankercellen na behandeling met DCA verhoogde niveaus van ROS, depolarisatie van het MMP in vitro en verhoogde apoptose, zowel in vitro als in vivo [17,20].
Aangezien DCA substraten kan omleiden naar mitochondriale ademhaling en ATP-productie, zou het een synergetische werking kunnen hebben met anti-kankermedicijnen die de mitochondriale activiteit aantasten. Wij stellen voor dat door het glycolytische fenotype om te keren met DCA en meer pyruvaat om te leiden naar mitochondriale oxidatieve fosforylering, terwijl tegelijkertijd de mitochondriën worden aangepakt met ATO, een ernstige verstoring van de energiehomeostase in kankercellen zal optreden. In deze studie tonen wij aan dat de combinatie van DCA en ATO de groei van borstkankercellijnen in vitro remt. Verder hebben wij nieuwe moleculaire mechanismen binnen de mitochondriën geïdentificeerd die kunnen bijdragen tot de cytotoxiciteit van ATO, hetgeen extra steun biedt voor het gebruik van ATO tegen vaste tumoren.
Materialen en methoden
Reagentia
JC-1, CFSE en H2DCFDA waren afkomstig van Invitrogen (Carlsbad, CA, USA), CellTiter-Glo en Caspase-Glo assay kits werden gekocht van Promega Co (San Luis, CA, USA). De rest van de chemicaliën werd gekocht bij Sigma Co (St. Louis, MO, USA).
Celkweek
Cellijnen werden verkregen van de volgende bronnen in de aangegeven jaren. Dr. Anna DeFazio, Westmead Millenium Institute, Sydney, Australië: T-47D (2003), BT-20 (2003), MCF-10A (2005) en MCF-10AT1 (2005); Prof. Chris Parish, Australian National University, Canberra, Australië: 13762 MAT (2007), MDA-MB-468 (2003) en MDA-MB-231 (2003). De cellijnen hebben verschijningsvormen die overeenkomen met gepubliceerde morfologieën, maar zijn recentelijk niet geauthenticeerd. Menselijke borstepitheelcarcinoomcellen (T-47D) werden gekweekt in RPMI 1640 medium aangevuld met 10% foetaal runderserum in aanwezigheid van 0,1% PSN (3% penicilline, 5% streptomycine en 5% neomycine). BT-20, MDA-MB-231, MDA-MB-468 en 13762 MAT-cellijnen werden gehandhaafd in DMEM/F12-medium aangevuld met 10% FBS en 2 mM L-glutamine. DMEM/F-12 medium met 25% paardenserum, 0,01% EGF, 0,28 IE/ml insuline, 0,01% cholera toxine en 0,5 μg/ml hydrocortison werd gebruikt voor MCF-10A en MCF-10AT1 cellen. Alle cellijnen werden onderhouden bij 37°C in 5% CO2.
Levensvatbaarheid van de cellen
Voor de beoordeling van de levensvatbaarheid van de cellen werden de cellen uitgezet in 96 wells platen bij een dichtheid van 3000 cellen per well en 8 wells per groep. Na blootstelling aan DCA en ATO gedurende 24 tot 72 uur werden de cellen gedurende 3 uur geïncubeerd met neutraalrood (30 μg/ml) in verse media, vervolgens gewassen met PBS, gevolgd door toevoeging van lysisbuffer (azijnzuur/methanol, 80%/20%) en de absorptie bij 540 nm werd geregistreerd. De resultaten worden uitgedrukt als gemiddelde ± S.D., berekeningen werden uitgevoerd met het Prism softwarepakket, ANOVA met Tukey post test werd toegepast en P < 0,05 werd beschouwd als statistisch significant. De experimenten werden ten minste driemaal uitgevoerd en de gepresenteerde gegevens zijn van één representatief experiment.
Celproliferatie
T-47D-cellen werden geoogst en geresuspendeerd in 1 ml RPMI-medium. De cellen werden gelabeld door toevoeging van 1 ml PBS met 5 μM carboxyfluoresceïne succinimidylester (CFSE), gevolgd door 5 minuten incubatie bij kamertemperatuur. Gelabelde cellen werden vervolgens tweemaal gewassen, geteld en uitgezaaid bij105 cellen/well in een plaat met 12 putjes. Op de dag van de analyse werden de T-47D-cellen geoogst en tweemaal gewassen met PBS, waarna de CFSE-intensiteit werd onderzocht met FACS. Resultaten worden uitgedrukt als gemiddelde ± S.D. (n = 3), berekeningen werden uitgevoerd met het Prism softwarepakket, ANOVA met Tukey posttest werd toegepast en P < 0,05 werd beschouwd als statistisch significant. De experimenten werden ten minste driemaal uitgevoerd en de gepresenteerde gegevens zijn van één representatief experiment.
celdood Apoptose werd gekwantificeerd door flowcytometrie na kleuring van de cellen met FITC-gelabeld Annexine-V (AV) (Invitrogen Co.) en propidiumjodide (PI). Na behandeling met geneesmiddelen werden T-47D-cellen geoogst en gecentrifugeerd bij 1200 rpm gedurende 5 minuten; de pellets werden tweemaal gewassen met PBS en vervolgens geresuspendeerd in 100 μl Annexine-V-bindingsbuffer (0,14 M NaCl, 2,5 mM CaCl2, 0,01 M HEPES pH 7,4). Annexine-V (1 μl) en 5 μl PI (50 μg/ml) werden aan de monsters toegevoegd en gedurende 15 minuten in het donker geïncubeerd. De monsters werden na incubatie op ijs bewaard totdat de FACS-analyse werd uitgevoerd. De resultaten worden uitgedrukt als gemiddelde ± S.D. (n = 3), berekeningen werden uitgevoerd met het softwarepakket Prism, ANOVA met Tukey post-test werd toegepast en P < 0,05 werd beschouwd als statistisch significant. De experimenten werden ten minste driemaal uitgevoerd en de gepresenteerde gegevens zijn van één representatief experiment.
ROS-generatie
Voor de beoordeling van intracellulaire ROS-niveaus werden cellen uitgezet in platen met 12 putjes met een celdichtheid van 1 ×105 cellen per putje en gedurende 12 uur behandeld met geneesmiddelen. 2′, 7′-dihydrochlorofluroresceinacetaat (H2DCFDA) werd aan het medium toegevoegd in een eindconcentratie van 10 uM en de cellen mochten 1 uur in het donker worden gekleurd. NaH2DCFDA-kleuringwerden de cellen getrypsiniseerd, tweemaal gewassen en geresuspendeerd in 100 μl PBS.De H2DCFDA-intensiteitwerd onderzocht met behulp van FACS. Resultaten worden uitgedrukt als gemiddelde ± S.D. (n = 3), berekeningen werden uitgevoerd met het Prism softwarepakket, ANOVA met Tukey posttest werd toegepast en P < 0,05 werd beschouwd als statistisch significant. De experimenten werden ten minste driemaal uitgevoerd en de gepresenteerde gegevens zijn van één representatief experiment.
ATP-concentratie en Caspase-activiteit
Het interne ATP-niveau en de caspase-activiteit in T-47D-cellen werden beoordeeld met behulp van CellTiter-Glo en Caspase-Glo 3/7 assay kits (Promega Corp., Madison, WI) volgens de instructies van de fabrikant. T-47D-cellen werden gedurende 12 uur in afwezigheid en aanwezigheid van geneesmiddelen gekweekt in witte ondoorzichtige 96 wells platen (4 wells per groep). Gelijke volumes CellTiter-Glo-reagentia werden toegevoegd, waarna de monsters gedurende 15 minuten werden geïncubeerd op een schudapparaat bij kamertemperatuur. De luminescentie werd geregistreerd met de Glomax microplaat-luminometer (Promega Co., Madison, WI) volgens het vooraf ingestelde CellTiter-protocol. De resultaten worden uitgedrukt als gemiddelde ± S.D. (n = 4), berekeningen werden uitgevoerd met het softwarepakket Prism, ANOVA met Tukey post-test werd toegepast en P < 0,05 werd beschouwd als statistisch significant. De experimenten werden ten minste driemaal uitgevoerd en de gepresenteerde gegevens zijn van één representatief experiment.
Mitochondriaal membraanpotentieel
Net als bij de meting van ROS werden de cellen uitgezet in platen met 12 putjes met een celdichtheid van 1 ×105 cellen per putje en gedurende 12 uur behandeld met geneesmiddelen. 5, 5′, 6, 6′-tetrachloor-1, 1′, 3, 3′-tetraethylbenzimidazol-carbocyaninejodide (JC-1) werd aan het medium toegevoegd in een eindconcentratie van 0,2 uM en de cellen mochten 30 minuten in het donker worden gekleurd. Na JC-1 kleuring werden de cellen getrypsiniseerd, tweemaal gewassen met PBS en geresuspendeerd in 100 μl PBS. De intensiteit van JC-1 werd onderzocht met behulp van FACS. Resultaten worden uitgedrukt als gemiddelde ± S.D. (n = 3), berekeningen werden uitgevoerd met het Prism softwarepakket, ANOVA met Tukey posttest werd toegepast en P < 0,05 werd beschouwd als statistisch significant. De experimenten werden ten minste driemaal uitgevoerd en de gepresenteerde gegevens zijn van één representatief experiment.
Cytochroom C oxidase activiteit
Cytochroom C oxidase assay werd beoordeeld op basis van de eerder gepubliceerde methode [24]. Kortom, na behandeling met geneesmiddelen in 96 wells platen (8 wells per groep) werden T-47D-cellen gepermeabiliseerd met 50 μl 0,01% saponine, gevolgd door toevoeging van 100 μl substraatmedium (4 mM 3,3-diaminobenzidine tetrahydrochloride (DAB), 100 μM gereduceerd cytochroom C, 2 μg/ml catalase in 0,1 M Na fosfaat, pH 7,0). De absorptie bij 450 nm werd onmiddellijk na toevoeging van het substraat gemeten en gedurende 30 minuten gecontroleerd. De resultaten worden uitgedrukt als gemiddelde ± S.D. (n = 8), berekeningen werden uitgevoerd met het softwarepakket Prism, ANOVA met Tukey posttest werd toegepast en P < 0,05 werd beschouwd als statistisch significant. De experimenten werden ten minste driemaal uitgevoerd en de gepresenteerde gegevens zijn van één representatief experiment.
PDH-activiteit
De PDH-activiteit werd gemeten met de MitoSciences PDH-enzymactiviteit microplaat assay kit (#MSP18, MitoSciences, Oregon USA). Voor het meten van het effect van geneesmiddelen op de PDH-activiteit in de cellen werden T-47D-cellen gedurende 3 uur behandeld met geneesmiddelen in de media, waarna zij werden gewassen en opnieuw gesuspendeerd in PBS. Celextracten werden bereid en getest op PDH-activiteit in een concentratie van 15 mg eiwit/ml, volgens de instructies van de kit. Voor het meten van directe remming van PDH door ATO werd PDH geïsoleerd uit onbehandelde T-47D-cellen door celextracten aan te brengen op de antilichaamvangplaat. Na het wassen van de plaat werd de testoplossing met geneesmiddel aan de putjes toegevoegd en vervolgens onmiddellijk getest op activiteit PDH. De resultaten worden uitgedrukt als gemiddelde ± S.D. (n = 4), berekeningen werden uitgevoerd met het softwarepakket Prism, ANOVA met Tukey-posttest werd toegepast en P < 0,05 werd beschouwd als statistisch significant. De experimenten werden ten minste driemaal uitgevoerd en de gepresenteerde gegevens zijn van één representatief experiment.
Immunoblotting en densitometrische analyse
Cellen (1 ×106 ) werden uitgezet in T25 weefselkweekkolven en gedurende 12 uur behandeld met ATO of DCA. Cellysaten werden bereid door toevoeging van 500 μl MPER® mammalian protein extraction reagens (Thermo Scientific, IL, USA). Immunoblotting werd uitgevoerd zoals eerder beschreven [25], met de antilichamen voor c-Myc (Roche, IN, USA, kloon 9E10), HIF-1α (Abcam, Cambridge, UK, #ab82832), Bcl-2 (Abcam #ab692), ATP synthase β-subeenheid (Abcam #ab14730) en β-actine (Abcam #ab8227). Western blots werden gedetecteerd met chemiluminescentie en belichting van röntgenfilm. Beelden werden verkregen met een CanoScan 8600F flatbed scanner, en gekwantificeerd met de ImageJ software (versie 1.4, NIH, USA) en gestandaardiseerd naar β-actine in elke lane. De resultaten werden samengevoegd uit 3 afzonderlijke experimenten en uitgedrukt als gemiddelde ± S.D. (n = 3), berekeningen werden uitgevoerd met het softwarepakket Prism, ANOVA met Tukey post-test werd toegepast en P < 0,05 werd beschouwd als statistisch significant.
Resultaten
DCA en ATO zijn samen doeltreffender in het verminderen van de celproliferatie en het induceren van celdood
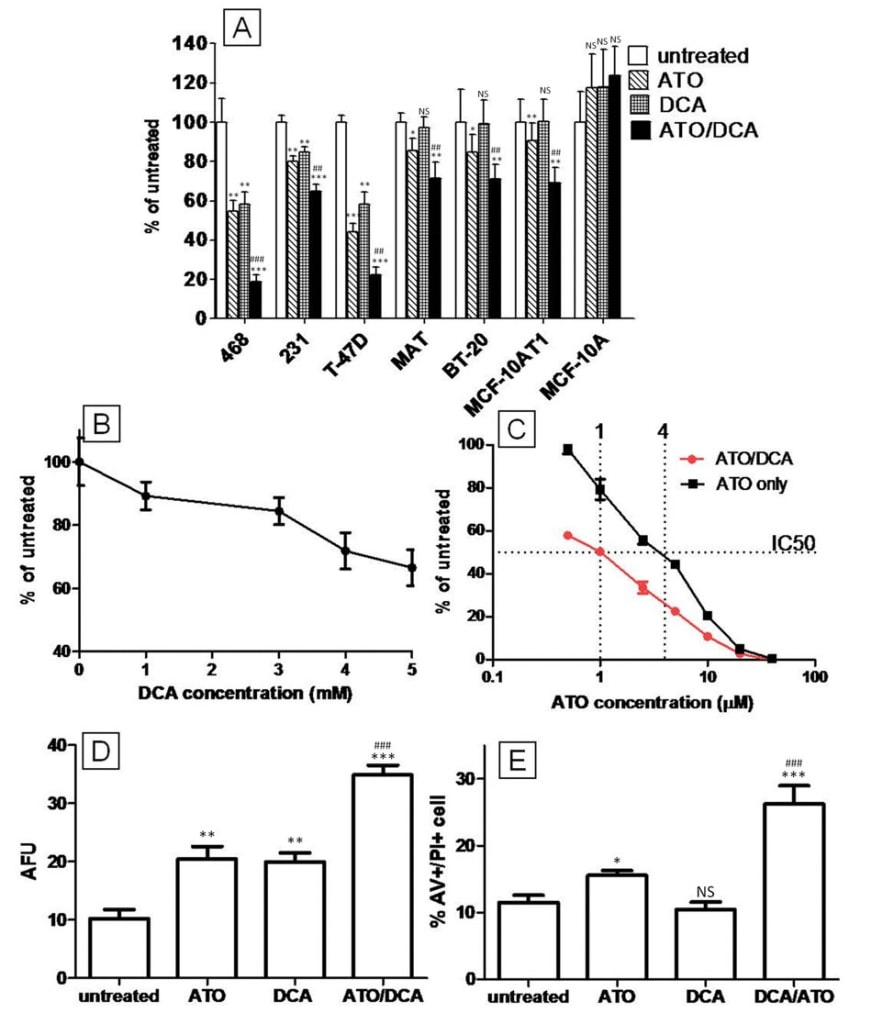
Om het gecombineerde effect van ATO en DCA op de remming van de celgroei te onderzoeken, werden borstkankercellijnen gedurende verscheidene dagen met beide geneesmiddelen behandeld en werd het totale aantal cellen beoordeeld met de neutrale rode-celvitaliteitstest. Het panel van cellijnen vertegenwoordigt de belangrijkste subtypes van menselijke borstkanker (luminaal (T-47D), basaal A (MDAMB-468, BT-20), basaal B (MDA-MB-231)), of zijn anderszins relevant als experimentele modellen (13762MAT – mammadenocarcinoom van ratten dat gevoelig is voor DCA in vivo [22], MCF10AT1 – kwaadaardig derivaat van geïmmortaliseerde cellen MCF-10A). MDA-MB-468, MDA-MB-231 en T-47D cellen vertoonden allemaal een significante vermindering, variërend van 10% tot 40% van het totale aantal cellen na 72 uur behandeling met 5 mM DCA (figuur 1A), terwijl 13762 MAT, BT-20 en MCF-10AT1 cellen niet reageerden tijdens de behandelingsperiode. Alle geteste kankercellijnen waren gevoelig voor ATO, maar bij verschillende concentraties. In BT-20, T-47D, MCF-10AT1 en MDA-MB-468 werd een vermindering van het totale aantal cellen gezien met slechts 5 μM ATO-behandeling, maar 15 μM ATO was nodig om dezelfde efficiëntie te bereiken in MDA-MB-231 en 13726 MAT-cellijnen. Opvallend is dat DCA en ATO in combinatie een groter effect vertoonden dan beide geneesmiddelen alleen in alle geteste kankercellijnen. Waar ATO en DCA als afzonderlijke middelen effectief waren in het verminderen van het aantal cellen, was het effect van de gecombineerde behandeling ongeveer gelijk aan de som van de effecten van de afzonderlijke geneesmiddelen (T-47D, MDA-MB-468 en MDA-MB-231). Waar DCA alleen geen vermindering van het aantal cellen te zien gaf, kon het de groeiremming van ATO nog met 2-3 versterken (13762 MAT, BT-20 en MCF10AT1) (figuur 1A). De niet-kankerachtige cellijn MCF-10A vertoonde geen vermindering van het aantal cellen na ATO (15 μM), DCA (5 mM) of gecombineerde behandeling.
Het effect van ATO en DCA werd verder onderzocht met T-47D cellen, een van de cellijnen die gevoeliger is voor DCA alleen. De reactie van T-47D-cellen op DCA was dosisafhankelijk en na 72 uur hadden de met 5 mM DCA behandelde cellen 42 ± 6% minder cellen dan de controlecultuur (figuur 1B). ATO alleen (5 μM) verminderde het totale aantal cellen met 56 ± 4% en cellen behandeld met zowel DCA als ATO vertoonden een verdere afname in vergelijking met de ATO-groep alleen (figuur 1C). Uit de dosis-responscurve bleek dat de combinatiebehandeling van DCA (5 mM) en ATO de IC50 kan verlagen tot het 0,25-voudige van die van ATO alleen (figuur 1C). Dit effect treedt op binnen het concentratiebereik dat klinisch voor ATO wordt bereikt (tot 5-7 μM [26]).
De CFSE-proliferatietest toonde aan dat ofwel ATO (5 μM) ofwel DCA (5 mM) behandelde cellen significant meer CFSE-fluorescentie uitstraalden (respectievelijk 2,1-voud en 2,2-voud) na 72 uur behandeling, wat wijst op groeiremming. Cellen behandeld met zowel ATO als DCA vertoonden een 3,4-voudige toename van de CFSE-intensiteit in vergelijking met onbehandelde cellen, wat erop wijst dat de geneesmiddelen samenwerkten bij het remmen van de celproliferatie (figuur 1D).
Het effect van ATO en DCA op T-47D celdood werd beoordeeld met AV en PI dubbele kleuring en de cellen werden geanalyseerd met behulp van fluorescerende celsortering. DCA alleen (5 mM) induceerde geen celdood in T-47D-cellen (figuur 1E), vergelijkbaar met het effect van DCA op 13762 MAT-cellen dat eerder is gerapporteerd [22]. ATO (5 μM) induceerde ook geen celdood na 12 uur behandeling, maar cellen behandeld met zowel 5 μM ATO als 5 mM DCA vertoonden een kleine (15%) toename van de AV+/PI+ populatie (13,2 ± 0,6% apoptotische cellen vergeleken met 11,5 ± 1,0% voor ATO/DCA versus onbehandeld respectievelijk, p = 0,07), wat suggereert dat DCA de apoptotische effecten van ATO kan versterken. Bij hogere concentraties en met 48 uur behandeling verhoogde ATO (20 μM) de hoeveelheid celdood met 35 ± 8% (P = 0,029) in vergelijking met de onbehandelde kweek (figuur 1E). De combinatie van 5 mM DCA met 20 uM ATO-behandeling resulteerde in een 4-voudige toename van de AV+/PI+-populatie in vergelijking met ATO alleen, hetgeen erop wijst dat DCA de celdood geïnduceerd door ATO in T-47D borstkankercellen kan versterken (figuur 1E).
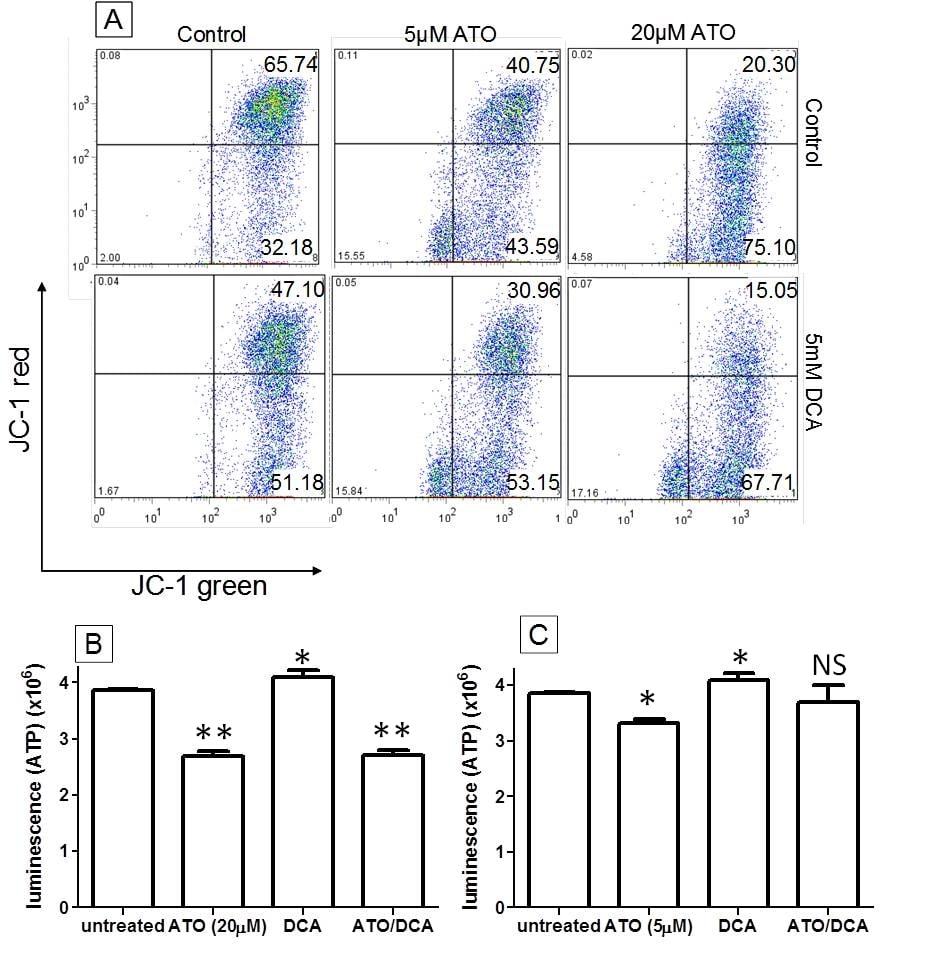
ATO en DCA werken samen bij het depolariseren van de MMP, maar hebben tegengestelde effecten bij het induceren van ATP- en ROS-productie
Van zowel ATO als DCA is aangetoond dat zij de mitochondriale functie veranderen, waarbij de MMP wordt gedepolariseerd en de ROS-productie toeneemt [20]. Daarom werden deze parameters samen met het ATP-niveau bestudeerd in een poging te bepalen of zij bijdragen tot de versterkte antikankereffecten van een gecombineerde behandeling met ATO/DCA. Gemeten met JC-1 kleuring, werd het aantal T-47D cellen met een gehyperpolariseerd MMP (kwadrant rechtsboven) significant verminderd door behandeling met DCA (5 mM) of ATO (5 μM en 20 μM) (t = 12 uur) (respectievelijk 28 ± 6%, 38 ± 4% en 69 ± 7% vermindering in vergelijking met onbehandelde controlecellen), in overeenstemming met eerder gepubliceerde gegevens [20,27]. De combinatie van ATO (5 μM of 20 μM) met 5 mM DCA leidde tot een nog grotere afname (respectievelijk 53 ± 9% en 77 ± 12%) in vergelijking met de onbehandelde cellen (figuur 2A), waaruit blijkt dat DCA en ATO samen kunnen werken bij het depolariseren van de MMP. ATP-niveaus werden verminderd door zowel lage als hoge dosis ATO (respectievelijk 14 ± 3% en 32 ± 5%) na 12 uur behandeling, terwijl met DCA behandelde cellen een toename van 6 ± 1% van de ATP-niveaus vertoonden (figuur 2B en 2C). Bij de hoge dosis ATO was DCA niet in staat de ATP-productie te verhogen (figuur 2B), terwijl bij de lage dosis ATO de met DCA en ATO gecombineerde behandelde cellen een kleine toename van de ATP-productie vertoonden ten opzichte van de ATO-behandeling alleen (figuur 2C). Deze gegevens wijzen erop dat DCA en ATO de ATP-productie beïnvloeden via afzonderlijke doelen in de cellen.
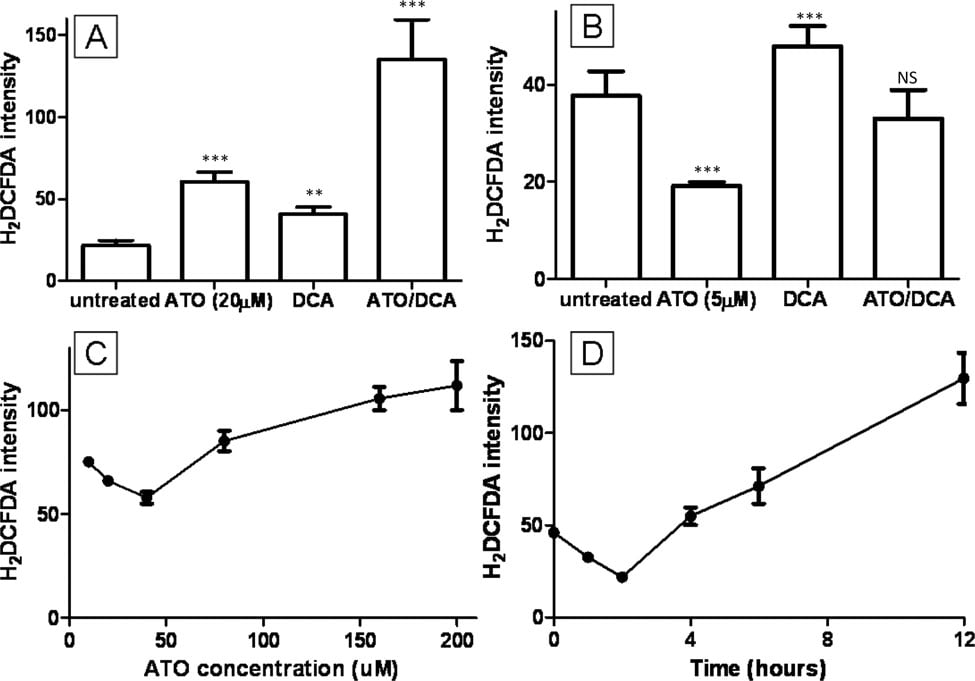
Verhoogde intracellulaire ROS-niveaus zijn voorgesteld als de reden achter ATO-cytotoxiciteit [2,9]. Het gecombineerde effect van ATO en DCA op ROS werd onderzocht met behulp van de fluorescerende kleurstof H2DCFDA. Cellen behandeld met 20 μM ATO of 5 mM DCA vertoonden verhoogde intracellulaire ROS-niveaus (respectievelijk 2,8 en 1,9-voudig) (figuur 3A), vergelijkbaar met eerder gepubliceerde resultaten [27], terwijl cellen behandeld met zowel ATO als DCA een verdere toename (5,2-voudig) van de ROS-productie vertoonden.(figuur 3A). Daarentegen induceerde een lage concentratie ATO (5 μM) een 0,5-voudige daling van de ROS-productie (figuur 3B). Dit was in strijd met eerdere rapporten over ROS-productie en ATO-behandeling, zodat de dosis-respons en het tijdsverloop van de ROS-productie na ATO-behandeling werden geanalyseerd. Hieruit bleek dat lagere concentraties ATO de intracellulaire ROS-productie verminderden, terwijl hoge ATO de ROS-productie induceerde (figuur 3C). Ook 10 uM ATO-behandeling resulteerde in een opmerkelijke daling van de ROS-productie in de eerste 4 uur voordat de ROS-niveaus na 8 uur stegen tot het door anderen gerapporteerde niveau (figuur 3D). Gecombineerde behandeling van cellen met 5 mM DCA en 5 uM ATO resulteerde in een intermediaire ROS-productie (figuur 3B), vergelijkbaar met onbehandelde cellen.
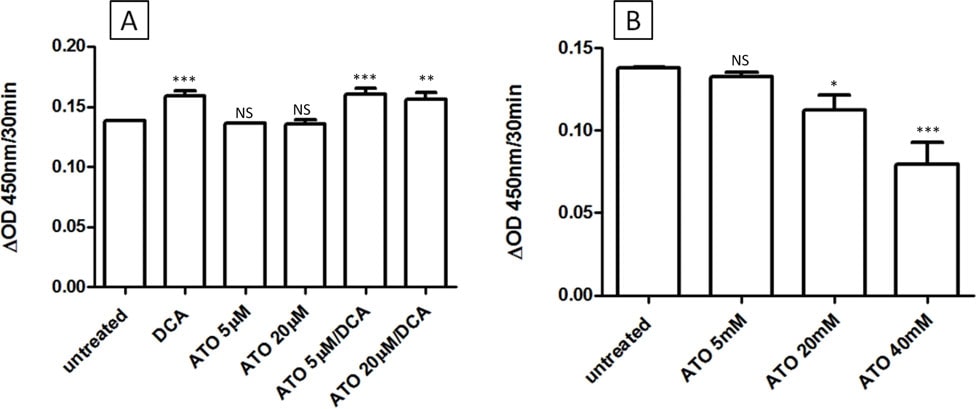
De intermediaire effecten van de combinatiebehandeling met DCA/ATO op de ATP- en ROS-niveaus kunnen worden verklaard door concurrerende effecten op de PDH-activiteit. DCA richt zich op PDK, en zou daarom de PDH-activiteit moeten verhogen, terwijl van ATO is gemeld dat het PDH remt, hetzij direct door reactie met vicinale thiolen op PDH, hetzij indirect via verhoogde waterstofperoxideproductie [28]. Om de effecten van ATO en/of DCA op de PDH-activiteit te onderzoeken, werden intacte cellen of PDH geïsoleerd uit T-47D cellen behandeld met het geneesmiddel en werd de PDH-activiteit bepaald. Zoals voorspeld resulteerde 3 uur behandeling van intacte cellen met DCA in verhoogde PDH-activiteit, terwijl ATO (tot 20 μM) de PDH-activiteit van intacte cellen niet veranderde (figuur 4A). DCA veranderde daarentegen niets aan de activiteit van geïsoleerd PDH, maar hoge concentraties ATO konden de PDH-activiteit remmen (figuur 4b). In T-47D-cellen verminderde ATO dus niet de PDH-activiteit bij de in deze studie gebruikte concentraties.
ATO is een remmer van complex IV van de elektronentransportketen
De elektronentransportketen (ETC) bevindt zich in het binnenste membraan van de mitochondriën en speelt een vitale rol bij de energieproductie. De ETC is verantwoordelijk voor het genereren van de protonengradiënt voor de binnenste membraanruimte van de mitochondriën om het MMP in stand te houden voor de productie van ATP [29]. Het ETC is ook gedocumenteerd als de belangrijkste plaats van ROS-productie in cellen als gevolg van elektronenlekkage uit complex I en III [30]. Vanwege zijn vermogen om tegelijkertijd ROS te verlagen, MMP te depolariseren en de ATP-productie te verminderen, veronderstellen wij dat ATO werkt als een ETC-remmer. Een panel van ETC-remmers werd vergeleken met ATO (5 μM) om na te gaan of zij dezelfde fenotypische veranderingen teweegbrengen als ATO in T-47D-cellen. Rotenone (0,1 μM, complex I), thenolytrifluoroaceton (TTFA) (10 μM, complex II), antimycine (0,1 μM, complex III) en NaCN (10 mM, complex IV) werden gebruikt om T-47D-cellen te behandelen en de ROS-, MMP- en ATP-niveaus werden vergeleken met die van de met ATO behandelde cellen. Alle remmers bleken in staat het MMP te depolariseren (figuur 5A) en het ATP-niveau te verlagen (figuur 5B), net als bij ATO. In tegenstelling tot ATO slaagden rotenon, antimycine en TTFA er echter niet in de ROS-productie in de cellen te verminderen, maar verhoogden ROS na 12 uur behandeling met een factor 3 tot 6 (figuur 5C). Cellen behandeld met NaCN (10 mM) vertoonden echter een 52% daling van de ROS-productie (figuur 5C), vergelijkbaar met ATO-behandeling. Op grond van deze gegevens concluderen wij dat in T47D-borstkankercellen de remming van complex IV van de ETC, maar niet van de complexen I-III, resulteert in een vermindering van ROS, en daarom is het waarschijnlijk dat de fenotypische veranderingen als gevolg van ATO worden veroorzaakt door de remming van complex IV (cytochroom C-oxidase) van de ETC. Om dit te bevestigen werd de cytochroom C-oxidase-activiteit in T-47D-cellen spectrofotometrisch gemeten. De cytochroom C-oxidase-test toonde duidelijk aan dat DCA geen effect had op de activiteit van complex IV, maar dat ATO (5 μM, 5 min) de activiteit van complex IV kan remmen, hetgeen onze hypothese bevestigt (figuur 5D). Soortgelijke resultaten werden verkregen na 10 min, 3 uur en 12 uur behandeling met geneesmiddelen. De combinatie van ATO en DCA vertoonde een soortgelijke enzymremming als ATO alleen.
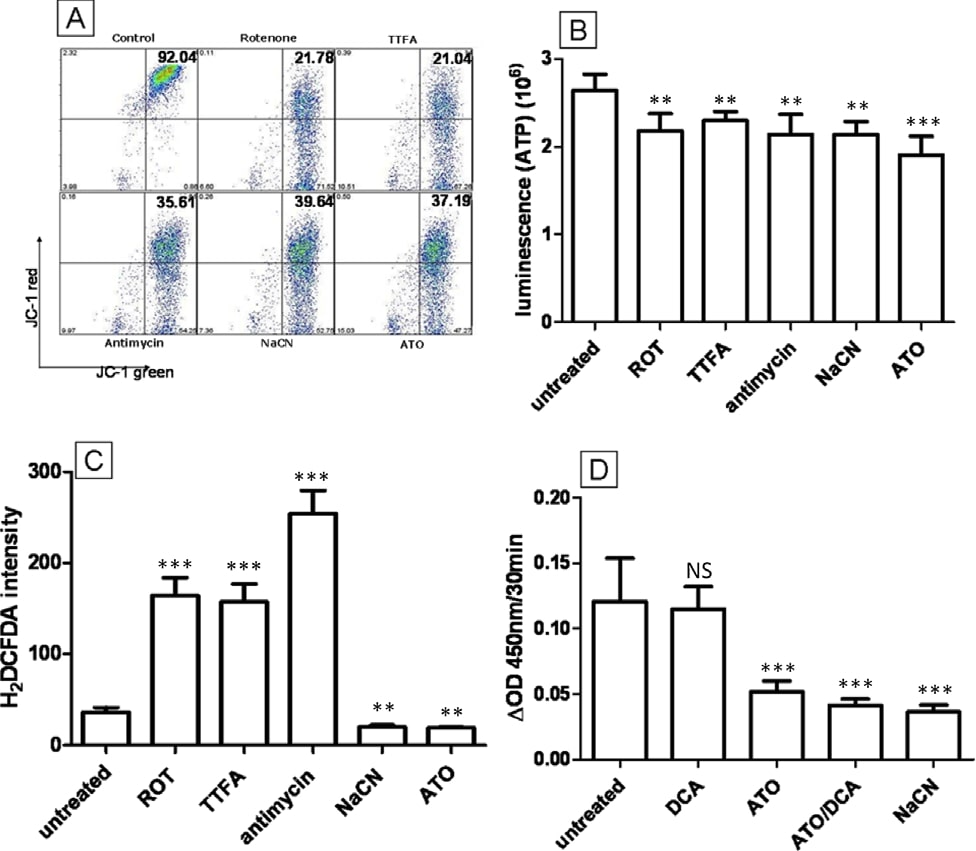
Inhibitoren van de mitochondriale overgangspoort blokkeerden gedeeltelijk de door ATO geïnduceerde caspase-activering, maar hadden geen effect op ROS, ATP en MMP
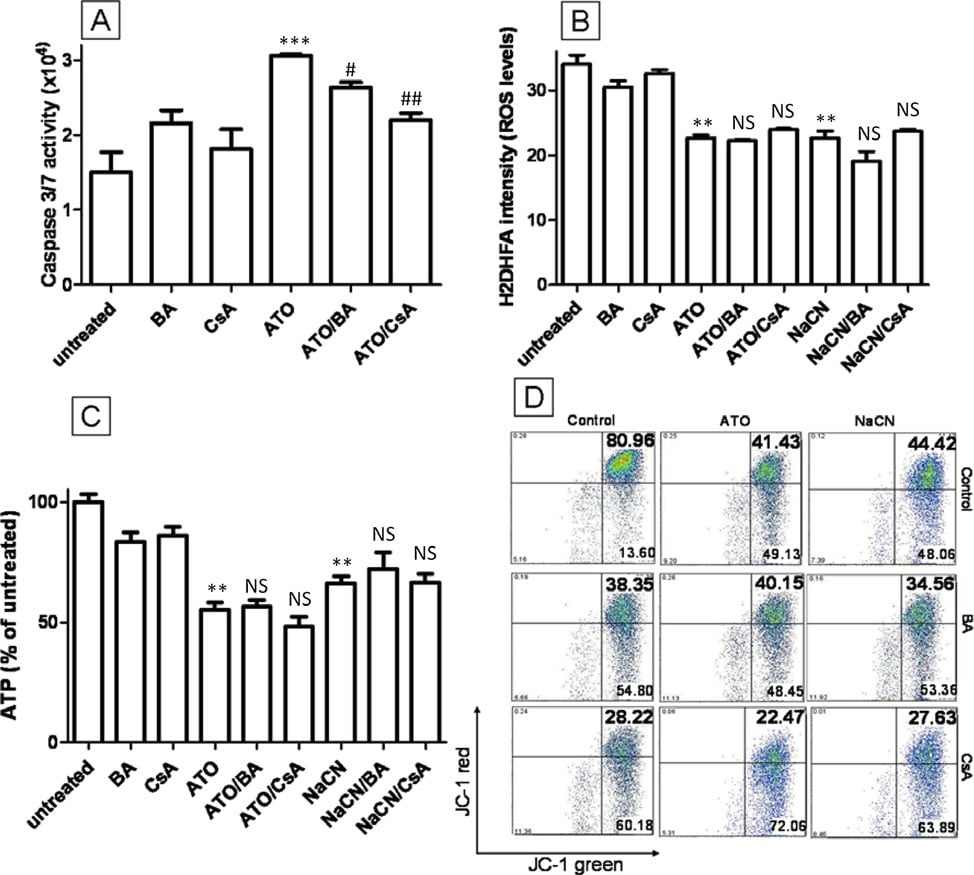
Een mechanisme dat eerder is beschreven als de sleutel tot ATO-cytotoxiciteit is de opening van de mitochondriale overgangspoort (MTP). ATO blijkt de opening van de MTP in geïsoleerde mitochondriën te induceren en kan de afgifte van cytochroom C verhogen [11] en nucleaire apoptose induceren in celvrije systemen [31]. De opening van MTP door ATO is voorgesteld om mitochondriale permeabilisatie te induceren, het MMP af te voeren en intracellulaire ROS te verhogen. Om te testen of de MTP belangrijk is voor de toxiciteit van ATO in T-47D cellen, werden bongkrekinezuur (BA) en cyclosporine A (CsA), beide krachtige MTP porieblokkers [32], gecombineerd met ATO om te zien of zij de door ATO geïnduceerde intracellulaire veranderingen kunnen remmen. De toevoeging van 5 μM CsA of 50 μM BA aan met ATO (5 μM) behandelde cellen had geen invloed op het vermogen van ATO om de ROS-productie na 12 uur behandeling te verlagen, de ATP-productie te verminderen of de MMP te depolariseren (P > 0,05) (figuur 6B, C en 6D). Soortgelijke gegevens zijn te zien voor NaCN- en CsA/BA-behandeling (P > 0,05) (figuur 6B, C en 6D). Om de activiteit van CsA en BA in onze experimentele omstandigheden te bevestigen, werd de activiteit van caspase 3/7, een apoptosemarker stroomafwaarts van MTP opening, geactiveerd door het vrijkomen van cytochroom C, beoordeeld na behandeling met hoge concentratie ATO (figuur 6A). ATO-behandeling (20 μM) verdubbelde de caspase 3/7-activiteit in vergelijking met onbehandelde cellen (2,1-voudig, figuur 6A) en dit effect werd verminderd door BA en CsA (respectievelijk 1,7-voudig en 1,4-voudig), hetgeen erop wijst dat CsA of BA de MTP-opening in T-47D-cellen kan blokkeren. Deze gegevens tonen duidelijk aan dat, hoewel ATO (20 μM) de opening van MTP kan induceren, dit mechanisme de veranderingen in ROS, MMP en ATP tijdens een behandeling met 5 μM ATO niet kan verklaren. Wij concluderen dat deze veranderingen waarschijnlijk te wijten zijn aan de remming van ETC-complex IV.
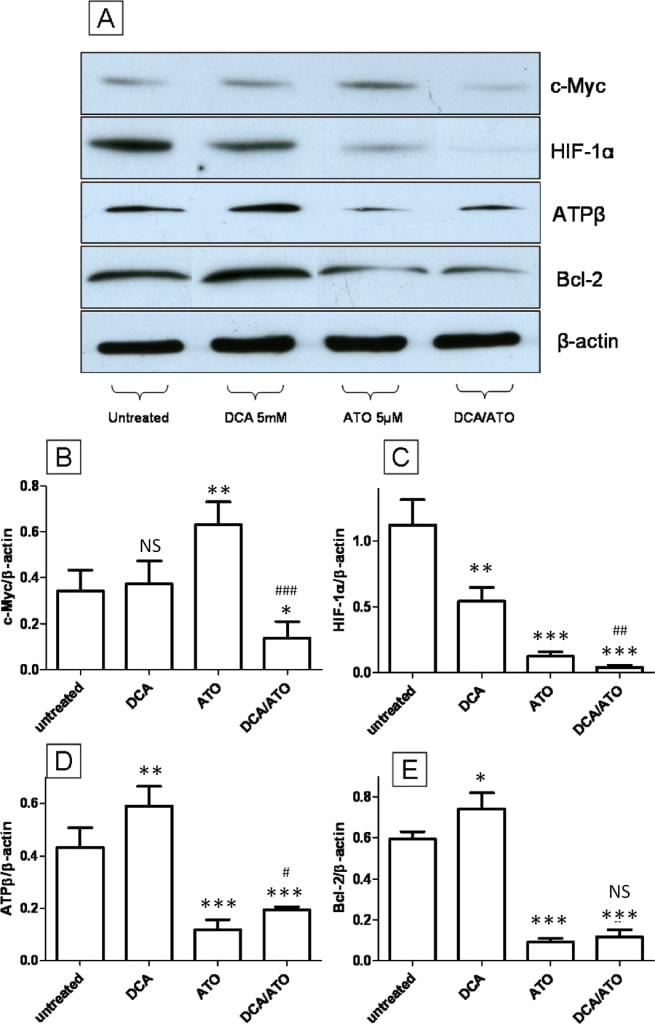
DCA en ATO hebben tegengestelde effecten op ATP synthase βsubuniten Bcl-2 expressie terwijl ze samenwerken bij het omlaag brengen van c-Myc en HIF-1α eiwitniveaus
Wij onderzochten het effect van 5 mM DCA en 5 μM ATO behandeling (12 uur) op de expressie c-Myc en HIF1α, twee belangrijke transcriptiefactoren waarvan bekend is dat ze het Warburg-effect [33] en de mitochondriale activiteit [34] reguleren, en op de expressie van de mitochondriale eiwitten ATP-synthase β-subeenheid (ATPβ) en Bcl-2. Immunoblotting voor c-Myc toonde geen verandering na behandeling met DCA, terwijl ATO de up-regulatie van c-Myc induceerde (figuur 7A en 7B), maar ATO in combinatie met DCA verlaagde het c-Myc-niveau in vergelijking met de onbehandelde controle. De HIF-1α-niveaus daarentegen vertoonden een neerwaartse regulering na behandeling met zowel DCA als ATO. De combinatie van ATO en DCA liet een verdere down-regulatie van HIF-1α zien in vergelijking met de met één middel behandelde en controlegroepen (figuur 7A en 7C). Bcl-2 is een lid van de BH3-eiwitfamilie die rechtstreeks bindt aan Bad en Bax en apoptose voorkomt [35]. Immunoblotting voor ATPβ wees uit dat DCA-behandeling ATPβ verhoogde, terwijl ATO een tegengesteld effect vertoonde en de ATPβ niveaus verlaagde (Figuur 7A en 7D), en ATO en DCA gecombineerde behandeling toonde een intermediaire respons vergeleken met ATO of DCA behandelde groepen. Immunoblotting voor het pro-overlevingseiwit Bcl-2 toonde een up-regulering na DCA, een down-regulering voor ATO-behandeling, en een intermediaire respons van de combinatie van de twee geneesmiddelen (figuur 7A en 7E). Deze resultaten komen ook sterk overeen met de ATP-niveaus en de ROS-productie, waar DCA en ATO tegengestelde effecten hadden (figuur 2C en 3B). De Bcl-2- en ATPβ-resultaten hangen rechtstreeks samen met het vermogen van DCA en ATO om de ROS-productie en de ATP-niveaus te beïnvloeden.
Bespreking
In de afgelopen tien jaar is ATO effectief gebruikt voor de behandeling van zowel nieuw gediagnosticeerde als teruggekeerde APL-patiënten, waarbij patiënten volledige remissie vertoonden na behandeling met ATO in een lage dosis [4]. Het anti-kanker vermogen van ATO is niet beperkt tot APL [36] en vele andere tumoren in diermodellen zijn gevoelig gebleken voor behandeling met ATO [37,38]. Echter, het gebrek aan informatie over de werkingsplaatsen van ATO-cytotoxiciteit in andere tumortypes dan APL heeft de toepassing ervan voor de behandeling van andere kankers tot voor kort beperkt [5].
In deze studie hebben wij aangetoond dat complex IV van de ETC een doelwit is voor ATO. Uit een panel van ETC-remmers werd de afname van ROS, de afname van de ATP-productie en de MMP-depolarisatie veroorzaakt door de behandeling van T-47D-cellen met een lage dosis ATO alleen gereproduceerd door cyanide, een goed gekarakteriseerde complex IV-remmer (figuur 5). Directe meting van de cytochroom-C-oxidase-activiteit in hele cellen die met ATO zijn behandeld, bevestigde het vermogen van ATO om deze enzymactiviteit rechtstreeks te remmen. Van het cytochroom C-oxidasecomplex van de ETC is bekend dat het dicht bij elkaar gelegen cysteïneresten bevat [39]en deze zullen waarschijnlijk reageren met ATO.
De meeste kankercellen hebben een hoger ROS-niveau dan normalecellen[40] en het ETC wordt beschouwd als de belangrijkste bron van intracellulaire ROS. De vermindering van ROS tot 60% door NaCN of ATO toonde aan dat complex IV verantwoordelijk is voor het merendeel van de ROS-productie in T-47D-cellen. De oxidatieve stress van verhoogde ROS-productie is beschreven als een tweesnijdend zwaard. Er is aangetoond dat een matige toename van ROS gepaard gaat met een verhoogd proliferatiepotentieel [41], verhoogde antioxidantenzymen zoals glutathiontransferase [42], superoxide dismutase en catalase [43] en verhoogde prosurvival eiwitten zoals Bcl-2 [42] en survivin [44]. Ondraaglijke hoeveelheden ROS leiden echter uiteindelijk tot celdood [45]. De afname van ROS na 12 uur behandeling met 5 μM ATO correleerde ook direct met een afname van de Bcl-2 niveaus (figuur 7E), wat suggereert dat T-47D cellen zich in een apoptose-resistente toestand bevinden als gevolg van verhoogde ROS niveaus. Voortdurende remming van complex IV van het ETC zal leiden tot depolarisatie van het MMP en remming van de ATP-productie. De celdood die het gevolg is van deze mitochondriale ontregeling zal aanleiding geven tot de verhoogde ROS die na 4 uur of bij een hogere ATO-concentratie (figuur 3C en 3D) worden waargenomen, zoals gerapporteerd door anderen [46] na ATO-behandeling. Verhoogde ROS is dus een gevolg, niet de oorzaak, van de door ATO veroorzaakte celdood.
Het eerder voorgestelde mitochondriale doelwit voor ATO is het MTP. Zheng et al toonden aan dat ATO de opening van de MTP in geïsoleerde mitochondriën van de rattenlever induceert [11]wat leidde tot het voorstel dat MTP verantwoordelijk is voor het vrijkomen van ROS en de depolarisatie van de MMP tijdens arseenbehandeling, en de daaropvolgende apoptose [47]. Onze studie sluit MTP niet uit als een doelwit voor ATO. Onze gegevens wijzen erop dat remming van de MTP via CsA en BA de door hoge concentratie ATO geïnduceerde caspase 3/7 activering gedeeltelijk kan blokkeren, maar dat de MTP-blokkers er niet in slagen de depolarisatie van de MMP en de door lage concentratie ATO veroorzaakte verlaging van de ROS- en ATP-niveaus te remmen. Hoewel ATO dus de MTP kan openen, zijn er alternatieve doelen nodig om de werking van ATO bij lage concentraties te verklaren. Er is aangetoond dat remming van het thioredoxinesysteem optreedt in MCF-7-cellen na behandeling met 2-5 μM ATO en dat het sommige cellulaire effecten van ATO bij lage concentraties kan mediëren [13], maar dit mechanisme zou leiden tot verhoogde oxidatieve stress, en kan de verminderde ROS waargenomen in de huidige studie niet verklaren.
DCA, als een pyruvaat dehydrogenase kinase remmer, kan het glycolytische effect omkeren en heeft zowel in vitro als in vivo belangrijke anti-kanker eigenschappen aangetoond [17-23]. Sommige studies tonen verhoogde apoptose met DCA [17,18,20]. Onze studies bij borstkanker en studies van Stockwin et al wijzen er echter op dat DCA eerder cytostatisch dan cytotoxisch werkt bij een reeks cellijnen, behalve bij zeer hoge concentraties [22,48]. Michelakis meldde onlangs dat de dalserumniveaus van DCA 0,5 mM bedroegen bij glioblastomapatiënten die tweemaal daags 6,25 mg/kg oraal innamen [23]de klinisch relevante concentraties liggen dus waarschijnlijk tussen 0,5 en 5 mM. Er is aangetoond dat DCA een kleine maar significante toename van de activiteit van caspase 3/7 induceert, zelfs wanneer geen apoptose werd waargenomen, en er is voorgesteld dat DCA kankercellen kan sensibiliseren voor cytotoxische middelen [22]. De huidige studie bevestigde deze hypothese door aan te tonen dat DCA T-47D-cellen gevoelig maakte voor ATO-behandeling, zoals blijkt uit AV+/PI+ dubbele kleuring (figuur 1E en tekst). ATO werd geselecteerd als de agent voor het testen vanwege zijn anti-mitochondriale eigenschappen, zoals we voorgesteld dat door het omkeren van de glycolytische fenotype met DCA, en het sturen van meer pyruvaat in mitochondriale oxidatieve fosforylering, terwijl tegelijkertijd gericht op de mitochondriën met ATO, een coöperatief effect zou worden gezien met deze drug combinatie. Het principe van deze dual targeting-strategie wordt ondersteund door de resultaten van twee recente publicaties waaruit blijkt dat cellen met mitochondriale defecten (bv. rho (0) of na behandeling met niet-drugs mitochondriale remmers zoals rotenon) een grotere gevoeligheid vertonen voor de groeiremmende effecten van DCA [48,49]. Hoewel de werkzaamheid van deze tweeledige doelgerichte strategie nog in vivo moet worden aangetoond, werden de versterkte combinatie-effecten van ATO/DCA gezien op zowel groeiremming als apoptose bij geneesmiddelconcentraties in klinisch relevante bereiken. Sommige effecten aan de onderkant van het concentratiebereik waren niet groot (bv. 15% toename van apoptotische cellen), maar het effect in vivo gedurende weken in plaats van uren behandeling rechtvaardigt verder onderzoek.
Het is goed gedocumenteerd dat DCA het mitochondriaal gedrag kan veranderen, d.w.z. de MMP depolariseren [18,20] [18,20]. Dit fenomeen is echter niet goed verklaard. Wij constateerden dat DCA de ATP kan verhogen terwijl het de MMP depolariseert in T-47D cellen (figuur 2). Op basis van deze gegevens stelden wij voor dat DCA de mitochondriale functie verandert via complex V van de ETC, ATP synthase. ATP-synthase is de laatste stap van de ETC en gebruikt het door de ETC gegenereerde MMP om ATP te produceren. ATPb is gedownreguleerd in long-, hersen-, borst- en maagkanker [50]wat de hypergepolariseerde mitochondriale membraan kan verklaren die in veel kankercellen wordt aangetroffen [51]. Wij veronderstelden dat DCA de ATP synthase activiteit kan verhogen, hetzij via allosterische regulering vanwege zijn homologie met pyruvaat, hetzij via verhoging van ATP synthase eiwit niveaus na de omkering van het Warburg effect en verlaagde HIF-1α niveaus. Immunoblotting toonde duidelijk aan dat ATPβ is geüpreguleerd na behandeling met DCA (figuur 7D), wat suggereert dat DCA de ATP synthase activiteit kan verhogen, wat weer bijdraagt tot verhoogde ATP productie en uitputting van het MMP.
HIF-1α en c-Myc zijn twee belangrijke oncogene transcriptiefactoren waarvan bekend is dat zij het metabolisme in kankercellen regelen. HIF-1α kan de enzymen van de glycolytische route en melkzuurdehydrogenase opwaarts reguleren, waardoor het Warburg-effect in kankercellen wordt ondersteund [33]. Dit verhoogt niet alleen de ATP-productie, maar verhoogt ook de toevoer van precursoren zoals glucose-6-fosfaat en fructose-6-fosfaat voor de productie van nucleïnezuren via de pentosefosfaatroute [16]. c-Myc daarentegen speelt niet alleen een centrale rol in het bevorderen van de overgang van de G1 naar de S-fase van de celcyclus door de cyclines en hun kinasen en remmers te reguleren, maar verhoogt ook de eiwitcomponenten van de ETC zoals COXI-IV en helpt de mitochondriale activiteit te verhogen [52]. De combinatie van c-Myc en HIF-1α overexpressie is dus belangrijk voor het induceren van het Warburg effect en het verhogen van de mitochondriale activiteit, ter ondersteuning van de glutamine-TCA hub die essentieel is voor het anabolisme van aminozuren en vetzuren die nodig zijn voor de celdeling [16]. Deze metabole signatuur draagt bij tot carcinogenese en het kwaadaardige fenotype van vele tumoren [33,34]. Zowel ATO als DCA kunnen de HIF-1α niveaus aanzienlijk verlagen, wat wijst op een vermindering van het Warburg effect (figuur 7C). Terwijl DCA alleen geen effect had op de c-Myc niveaus, is het verrassend dat ATO de c-Myc niveaus aanzienlijk verhoogt (figuur 7B). Dit kan een positieve terugkoppeling zijn, waarbij de cellen proberen de ETC-activiteit te verhogen na de remming van complex IV. Opvallend is dat DCA het effect van ATO op c-Myc ongedaan maakte, en dat de combinatie van ATO en DCA de c-Myc-expressie sterk onderdrukte, hetgeen correleert met het vermogen van deze middelen om samen te werken bij het verminderen van de celproliferatie en het induceren van celdood (figuur 1).
De werkzaamheid van ATO tegen PML is toegeschreven aan verscheidene bijzondere kenmerken van deze maligniteit – differentiatie door inactivering van PML-RARα, lage antioxidantcapaciteit van PML-cellen ter bescherming tegen de verhoogde ROS-niveaus, en accumulatie van het geneesmiddel als gevolg van een verminderde osmoregulatie [5]. Er lopen veel proeven voor het gebruik van ATO bij een reeks vaste maligniteiten, hoewel er tot dusver weinig resultaten zijn gepubliceerd. De beschikbare resultaten suggereren dat ATO niet erg effectief is als enkelvoudig middel bij patiënten met gevorderde kanker van de alvleesklier, de lever of melanoom <a href=”#5″> [5]</a></sup>. Onderzoekers pleiten voor de evaluatie van ATO in combinatie met andere antikankergeneesmiddelen<sup><a href=”#53″> [53,54]</a></sup> met enkele fase I-proeven die zijn afgerond <sup><a href=”#55″>[55]</a></sup>. Naarmate ons begrip van de mechanismen van ATO blijft groeien, met name de effecten ervan via het kankermetabolisme, zullen we wellicht beter in staat zijn het kankerbestrijdende potentieel ervan in vivo te benutten in nieuwe combinaties van geneesmiddelen</p>
<h2>Conclusies</h2>
<p>Twee recente rapporten toonden aan dat DCA het meest effectief is tegen cellen met mitochondriale defecten <sup><a href=”#48″>[48,49].</a></sup> Dit rapport is het eerste dat aantoont dat het aanpakken van twee aspecten van het metabolisme – het omkeren van het Warburg-effect met DCA en het remmen van oxidatieve fosforylering met ATO – duidelijk een effectieve antikankerstrategie is in vitro tegen borstkankercellijnen. De identificatie van cytochroom C-oxidase als mitochondriaal doelwit voor ATO levert nieuwe mechanistische informatie op voor de toepassing van ATO bij de behandeling van andere tumortypes dan APL. Het vermogen van DCA om de ATP synthase b subunit expressie te verhogen, waardoor een ander wijdverbreid kanker metabolisch fenotype wordt omgekeerd, suggereert ook dat DCA relevant kan zijn voor een breed scala van tumortypes. Het vermogen van DCA om de cytotoxische effecten van andere chemotherapeutische middelen dan ATO in vitro en in vivo te versterken, verdient eveneens nader onderzoek. Het gebrek aan gevoeligheid van de niet-kankerachtige cellijn MCF-10A voor de geteste klinisch relevante concentraties ATO/DCA is ook bemoedigend, en suggereert dat deze behandelingsstrategie verder moet worden getest tegen vaste tumoren in vivo.</p>
<h2>Lijst van afkortingen</h2>
<p>APL: acute promyeloïde leukemie; ATO: arseentrioxide; ATPβ: ATP synthase β-subeenheid; AV: annexine V; BA: bongkrekinezuur; CFSE: carboxyfluoresceïne succinimidylester; CsA: cyclosporine A; DCA: dichlooracetaat; ETC: elektronentransportketen; H2DCFDA: 2′, 7′-dihydrochlorofluroresceïneacetaat; JC-1: 5,5′,6,6′-tetrachloor-1,1′, 3,3′-tetraethylbenzimidazol-carbocyaninejodide; MMP: mitochondriale membraanpotentiaal; MTP: mitochondriale overgangsporie; NaCN: natriumcyanide; PI: propidiumjodide; ROS: Reactive oxygen species; TTFA: thenolytrifluoroacetone.</p>
<h2>Bevestigingen</h2>
<p>Dit onderzoek werd ondersteund door een subsidie van de National Breast Cancer Foundation Australia (AB), en door NHMRC 366787 R.D. Wright Career Development Award (AB). PB wordt ondersteund door NHRMC en de Australian National University. RS werd ondersteund door een Australian National University Postgraduate beurs</p>
<h2>Auteursgegevens</h2>
<p>1 Molecular Genetics Group, Department of Translational Biosciences, John Curtin School of Medical Research, Building 131, Australian National University, P.O. Box 334, Canberra ACT 0200, AUSTRALIA. 2 Afdeling stralingsoncologie, Stanford School of Medicine, Stanford CA 94305 USA.</p>
<h2>Bijdragen van de auteurs</h2>
<p>RS ontwierp en voerde de experimenten uit, en stelde het manuscript op. PB nam deel aan de analyse van de gegevens en de voorbereiding van het manuscript. AB nam deel aan het ontwerp en de analyse van de gegevens, en bereidde het manuscript voor publicatie voor. Alle auteurs hebben het definitieve manuscript gelezen en goedgekeurd.</p>
<h2>Strijdige belangen</h2>
<p>De auteurs verklaren geen concurrerende belangen te hebben.<br></p>
<p>Ontvangen: 3 mei 2011 Aanvaard: 18 november 2011<br>Gepubliceerd: 18 november 2011</p>
<h2>Verwijzingen</h2>
<br><span id=”1″ class=”referencess blue-text”>1</span> Antman KH: Introduction: the history of arsenic trioxide in cancer therapy. Oncologist 2001, 6(Suppl 2):1-2.
<br><span id=”2″ class=”referencess blue-text”>2</span> Emadi A, Gore SD: Arsenic trioxide – An old drug rediscovered. Blood Rev 2010, 24:191-199.
<br><span id=”3″ class=”referencess blue-text”>3</span> Powell BL, Moser B, Stock W, Gallagher RE, Willman CL, Stone RM, Rowe JM, Coutre S, Feusner JH, Gregory J, et al: Arsenic trioxide verbetert event-free en overall survival voor volwassenen met acute promyelocytische leukemie: North American Leukemia Intergroup Study C9710. Bloed 2010, 116:3751-3757.
<br><span id=”4″ class=”referencess blue-text”>4</span> Wang ZY, Chen Z: Acute promyelocytic leukemia: from highly fatal to highly curable. Bloed 2008, 111:2505-2515.
<br><span id=”5″ class=”referencess blue-text”>5</span> Dilda PJ, Hogg PJ: Arsenical-based cancer drugs. Cancer Treat Rev 2007, 33:542-564.
<br><span id=”6″ class=”referencess blue-text”>6</span> Chen GQ, Zhu J, Shi XG, Ni JH, Zhong HJ, Si GY, Jin XL, Tang W, Li XS, Xong SM, et al: In vitro studies over cellulaire en moleculaire mechanismen van arseentrioxide (As2O3) bij de behandeling van acute promyelocytische leukemie: As2O3 induceert NB4-celapoptose met downregulatie van Bcl2-expressie en modulatie van PML-RAR alpha/PML-eiwitten. 1996, 88:1052-1061.
<br><span id=”7″ class=”referencess blue-text”>7</span> Kanzawa T, Kondo Y, Ito H, Kondo S, Germano I: Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res 2003, 63:2103-2108.
<br><span id=”8″ class=”referencess blue-text”>8</span> Nakagawa Y, Akao Y, Morikawa H, Hirata I, Katsu K, Naoe T, Ohishi N, Yagi K: Arsenic trioxide-induced apoptosis through oxidative stress in cells of colon cancer cell lines. Life Sciences 2002, 70:2253-2269.
<br><span id=”9″ class=”referencess blue-text”>9</span> Ralph SJ: Arsenic-based antineoplastic drugs and their mechanisms of action. Met Based Drugs 2008, 2008:260146.
<br><span id=”10″ class=”referencess blue-text”>10</span> Cai X, Shen YL, Zhu Q, Jia PM, Yu Y, Zhou L, Huang Y, Zhang JW, Xiong SM, Chen SJ, et al: Arsenic trioxide-induced apoptosis and differentiation are associated respectively with mitochondrial transmembrane potential collapse and retinoic acid signaling pathways in acute promyelocytic leukemia. Leukemie 2000, 14:262-270.
<br><span id=”11″ class=”referencess blue-text”>11</span> Zheng Y, Shi Y, Tian C, Jiang C, Jin H, Chen J, Almasan A, Tang H, Chen Q: Essential role of the voltage-dependent anion channel (VDAC) in mitochondrial permeability transition pore opening and cytochrome c release induced by arsenic trioxide. Oncogene 2004, 23:1239-1247.
<br><span id=”12″ class=”referencess blue-text”>12</span> Tian X, Ma X, Qiao D, Ma A, Yan F, Huang X: mCICR is vereist voor As2O3-geïnduceerde opening van de permeabiliteitsovergangsporie en afgifte van cytochroom c uit mitochondriën. Mol Cell Biochem 2005, 277:33-42.
<br><span id=”13″ class=”referencess blue-text”>13</span> Lu J, Chew EH, Holmgren A: Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc Natl Acad Sci USA 2007, 104:12288-12293.
<br><span id=”14″ class=”referencess blue-text”>14</span> Kim JW, Dang CV: Cancer’s molecular sweet tooth and the Warburg effect. Cancer Res 2006, 66:8927-8930.
<br><span id=”15″ class=”referencecess blue-text”>15</span> Warburg O, Wind F, Negelein E: Ueber den Stoffwechsel von Tumoren im körper. Klinische Wochenschrift 1926, 5:829-832.
<br><span id=”16″ class=”referencess blue-text”>16</span> DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB: Beyond aerobic glycolysis: Transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci 2007, 104:19345-19350.
<br><span id=”17″ class=”referencess blue-text”>17</span> Wong JY, Huggins GS, Debidda M, Munshi NC, De Vivo I: Dichlooracetaat induceert apoptose in endometriumkankercellen. Gynecol Oncol 2008, 109:394-402.
<br><span id=”18″ class=”referencess blue-text”>18</span> Cao W, Yacoub S, Shiverick KT, Namiki K, Sakai Y, Porvasnik S, Urbanek C, Rosser CJ: Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostaat 2008, 68:1223-1231.
<br><span id=”19″ class=”referencess blue-text”>19</span> Papandreou I, Goliasova T, Denko NC: Antikankermedicijnen die het metabolisme aanpakken: is dichlooracetaat het nieuwe paradigma? Int J Cancer 2011, 128:1001-1008.
<br><span id=”20″ class=”referencess blue-text”>20</span> Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, et al: A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11:37-51.
<br><span id=”21″ class=”referencess blue-text”>21</span> Chen Y, Cairns R, Papandreou I, Koong A, Denko NC: Oxygen consumption can regulate the growth of tumors, a new perspective on the Warburg effect. PLoS One 2009, 4:e7033.
<br><span id=”22″ class=”referencess blue-text”>22</span> Sun RC, Fadia M, Dahlstrom JE, Parish CR, Board PG, Blackburn AC: Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo. Breast Cancer Res Treat 2010, 120:253-260.
<br><span id=”23″ class=”referencess blue-text”>23</span> Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, et al: Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010, 2:31ra34.
<br><span id=”24″ class=”referencess blue-text”>24</span> Chrzanowska-Lightowlers ZMA, Turnbull DM, Lightowlers RN: A microtiter plate assay for cytochrome c oxidase in permeabilized whole cells. Analytische biochemie 1993, 214:45-49.
<br><span id=”25″ class=”referencess blue-text”>25</span> Theodoratos A, Tu WJ, Cappello J, Blackburn AC, Matthaei K, Board PG: Phenylalanine-induced leucopenia in genetic and dichloroacetic acid generated deficiency of glutathione transferase Zeta. Biochem Pharmacol 2009, 77:1358-1363.
<br><span id=”26″ class=”referencess blue-text”>26</span> Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, et al: Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL): II. Klinische werkzaamheid en farmacokinetiek bij hervallen patiënten. Bloed 1997, 89:3354-3360.
<br><span id=”27″ class=”referencess blue-text”>27</span> Maeda H, Hori S, Nishitoh H, Ichijo H, Ogawa O, Kakehi Y, Kakizuka A: Tumor Growth Inhibition by Arsenic Trioxide (As2O3) in the Orthotopic Metastasis Model of Androgen-independent Prostate Cancer. Cancer Res 2001, 61: 5432-5440.
<br><span id=”28″ class=”referencess blue-text”>28</span> Samikkannu T, Chen CH, Yih LH, Wang AS, Lin SY, Chen TC, Jan KY: Reactive oxygen species are involved in arsenic trioxide inhibition of pyruvate dehydrogenase activity. Chem Res Toxicol 2003, 16:409-414.
<br><span id=”29″ class=”referencess blue-text”>29</span> Campbell MK, Farrell SO: Biochemie. 4 edition. Thomson learning, Inc.; 2003.
<br><span id=”30″ class=”referencess blue-text”>30</span> Liu Y, Fiskum G, Schubert D: Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem 2002, 80:780-787.
<br><span id=”31″ class=”referencess blue-text”>31</span> Larochette N, Decaudin D, Jacotot E, Brenner C, Marzo I, Susin SA, Zamzami N, Xie Z, Reed J, Kroemer G: Arsenite induceert apoptose via een direct effect op de mitochondrial permeability transition pore. Exp Cell Res 1999, 249:413-421.
<br><span id=”32″ class=”referencess blue-text”>32</span> Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G: Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 1998, 281:2027-2031.
<br><span id=”33″ class=”referencess blue-text”>33</span> Semenza GL: Regeling van het kankercelmetabolisme door hypoxie-induceerbare factor 1. Semin Cancer Biol 2009, 19:12-16.
<br><span id=”34″ class=”referencecess blue-text”>34</span> Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, Li F: The c-Myc target gene network. Seminars in Cancer Biology 2006, 16:253-264. Sun et al. Molecular Cancer 2011, 10:142 http://www.molecular-cancer.com/content/10/1/142 Pagina 14 van 15
<br><span id=”35″ class=”referencess blue-text”>35</span> Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X: Prevention of apoptosis by Bcl-2: Release of cytochrome c from mitochondria blocked. Science 1997, 275:1129-1132.
<br><span id=”36″ class=”referencess blue-text”>36</span> Carney DA: Arsenic trioxide mechanisms of action-looking beyond acute promyelocytic leukemia. Leuk Lymfoom 2008, 49:1846-1851.
<br><span id=”37″ class=”referencess blue-text”>37</span> Kito M, Matsumoto K, Wada N, Sera K, Futatsugawa S, Naoe T, Nozawa Y, Akao Y: Antitumor effect van arseentrioxide in murine xenograft model. Cancer Sci 2003, 94:1010-1014.
<br><span id=”38″ class=”referencess blue-text”>38</span> Xu HY, Yang YL, Liu SM, Bi L, Chen SX: Effect of arsenic trioxide on human hepatocarcinoma in nude mice. World J Gastroenterol 2004, 10:3677-3679.
<br><span id=”39″ class=”referencess blue-text”>39</span> Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, ShinzawaItoh K, Nakashima R, Yaono R, Yoshikawa S: The whole structure of the 13- subunit oxidized cytochrome c oxidase at 2.8 A. Science 1996, 272:1136-1144.
<br><span id=”40″ class=”referencess blue-text”>40</span> Pelicano H, Carney D, Huang P: ROS stress in cancer cells and therapeutic implications. Drug Resist Updat 2004, 7:97-110.
<br><span id=”41″ class=”referencess blue-text”>41</span> Kamata H, Hirata H: Redox regulation of cellular signalling. Cellular Signalling 1999, 11:1-14.
<br><span id=”42″ class=”referencess blue-text”>42</span> Hayes JD, McLellan LI: Glutathion and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radic Res 1999, 31:273-300.
<br><span id=”43″ class=”referencess blue-text”>43</span> Nordberg J, Arner ESJ: Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radical Biology and Medicine 2001, 31:1287-1312.
<br><span id=”44″ class=”referencess blue-text”>44</span> Yao LL, Wang YG, Cai WJ, Yao T, Zhu YC: Survivin mediates the anti-apoptotic effect of δ-opioid receptor stimulation in cardiomyocytes. Journal of Cell Science 2007, 120:895-907.
<br><span id=”45″ class=”referencess blue-text”>45</span> Martindale JL, Holbrook NJ: Cellulaire respons op oxidatieve stress: Signalen voor zelfmoord en overleving. J Cell Physiol 2002, 192:1-15.
<br><span id=”46″ class=”referencess blue-text”>46</span> Perkins C, Kim CN, Fang G, Bhalla KN: Arsenic induces apoptosis of multidrug-resistant human myeloid leukemia cells that express Bcr-Abl or overexpress MDR, MRP, Bcl-2, or Bcl-x(L). Bloed 2000, 95:1014-1022.
<br><span id=”47″ class=”referencess blue-text”>47</span> Ma XD, Qiao DF, Tian XM, Yan F, Ma AD: Mechanism of opening of mitochondrial permeability transition pore induced by arsenic trioxide. Ai Zheng 2006, 25:17-21.
<br><span id=”48″ class=”referencess blue-text”>48</span> Stockwin LH, Yu SX, Borgel S, Hancock C, Wolfe TL, Phillips LR, Hollingshead MG, Newton DL: Natriumdichlooracetaat (DCA) richt zich selectief op cellen met defecten in de mitochondriale ETC. Int J Cancer 2010.
<br><span id=”49″ class=”referencess blue-text”>49</span> Sanchez-Arago M, Chamorro M, Cuezva JM: Selection of cancer cells with repressed mitochondria triggers colon cancer progression. Carcinogenesis 2010, 31:567-576.
<br><span id=”50″ class=”referencess blue-text”>50</span> Isidoro A, Martinez M, Fernandez PL, Ortega AD, Santamaria G, Chamorro M, Reed JC, Cuezva JM: Alteration of the bioenergetic phenotype of mitochondria is a hallmark of breast, gastric, lung and oesophageal cancer. Biochemical Journal 2004, 378:17-20.
<br><span id=”51″ class=”referencess blue-text”>51</span> Susin SA, Zamzami N, Kroemer G: Mitochondria as regulators of apoptosis: Twijfel niet meer. Biochimica et Biophysica Acta – Bioenergetica 1998, 1366:151-165.
<br><span id=”52″ class=”referencess blue-text”>52</span> Shim H, Dolde C, Lewis BC, Wu CS, Dang G, Jungmann RA, Dalla-Favera R, Dang CV: c-Myc transactivation of LDH-A: Implications for tumor metabolism and growth. Proc Natl Acad Sci USA 1997, 94:6658-6663.
<br><span id=”53″ class=”referencess blue-text”>53</span> Kim KB, Bedikian AY, Camacho LH, Papadopoulos NE, McCullough C: A phase II trial of arsenic trioxide in patients with metastatic melanoma. Cancer 2005, 104:1687-1692.
<br><span id=”54″ class=”referencess blue-text”>54</span> Subbarayan PR, Lima M, Ardalan B: Arsenic trioxide/ascorbic acid therapy in patients with refractory metastatic colorectal carcinoma: a clinical experience. Acta Oncol 2007, 46:557-561.
<br><span id=”55″ class=”referencess blue-text”>55</span> Ardalan B, Subbarayan PR, Ramos Y, Gonzalez M, Fernandez A, Mezentsev D, Reis I, Duncan R, Podolsky L, Lee K, et al: A phase I study of 5-fluorouracil/leucovorin and arsenic trioxide for patients with refractory/relapsed colorectal carcinoma. Clin Cancer Res 2010, 16:3019-3027.
<p></p>
<p>Gerelateerde inhoud:</p>
<figure class=”wp-block-embed is-type-wp-embed is-provider-dca-guide wp-block-embed-dca-guide”><div class=”wp-block-embed__wrapper”>
</div></figuur>
<figure class=”wp-block-embed is-type-wp-embed is-provider-dca-guide wp-block-embed-dca-guide”><div class=”wp-block-embed__wrapper”>
</div></figuur>