Sang Hyeok Woo1,*, Sung-Keum Seo1,*, Yoonhwa Park1,2, Eun-Kyu Kim3, Min-Ki Seong4, Hyun-Ah Kim4, Jie-Young Song1, Sang-Gu Hwang1, Jin Kyung Lee5, Woo Chul Noh4, In-Chul Park1
1Divisionof Radiation Cancer Research, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Seoul, 01812, Republik Korea
2Schoolof Life Science and Biotechnology, Korea University, Seongbuk-gu, Seoul, 02841, Republik Korea
3Abteilungfür Chirurgie, Brustkrebszentrum, Seoul National University Bundang Hospital, Seoul National University College of Medicine, Bundang-gu, Seongnam, 13620, Republik Korea
4Abteilungfür Chirurgie, Korea Cancer Center Hospital, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Seoul, 01812, Republik Korea
5KIRAMSRadiation Biobank, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Seoul, 01812, Republik Korea
*DieseAutoren haben gleichermaßen zu dieser Arbeit beigetragen
Korrespondenz mit: In-Chul Park, E-Mail: [email protected]
Schlüsselwörter: Tamoxifen, Brustkrebs, Dichloracetat, epidermaler Wachstumsfaktor-Rezeptor, Pyruvat-Dehydrogenase-Kinase
Eingereicht: May 18, 2016
Accepted: July 22, 2016
Published online: August 01, 2016
Zusammenfassung
Die Reprogrammierung des Stoffwechsels in Krebszellen wurde kürzlich als ein wesentliches Merkmal der Neoplasie erkannt. In diesem Zusammenhang stellen Stoffwechselveränderungen ein attraktives therapeutisches Ziel dar, und in präklinischen Studien wurden ermutigende Ergebnisse mit Medikamenten erzielt, die auf verschiedene Stoffwechselprozesse abzielen. In jüngster Zeit haben mehrere Studien darauf hingewiesen, dass Dichloracetat (DCA), ein spezifischer Pyruvat-Dehydrogenase-Kinase-Inhibitor, ein potenzielles Krebsmedikament für eine große Zahl verschiedener Tumoren sein könnte. Der genaue Mechanismus ist jedoch noch nicht vollständig geklärt, was für den Einsatz von DCA in der Krebsbehandlung wichtig ist. In der vorliegenden Studie fanden wir heraus, dass DCA MCF7-Brustkrebszellen für den Tamoxifen-induzierten Zelltod sensibilisierte, indem es die Expression des epidermalen Wachstumsfaktorrezeptors (EGFR) verringerte. Die Herunterregulierung des EGFR wurde durch den Abbau des Proteins verursacht. Außerdem spielte die mitogen-aktivierte Proteinkinase p38 eine wichtige Rolle beim DCA/Tamoxifen-induzierten EGFR-Abbau. Schließlich förderte DCA auch einen vergleichbaren Tamoxifen-induzierten Zelltod in Tamoxifen-resistenten MCF7-Zellen, die durch Langzeitbehandlung mit Tamoxifen entstanden waren. Zusammenfassend deuten unsere Ergebnisse darauf hin, dass DCA ein attraktiver potenzieller Wirkstoff ist, der die Zellen für den Tamoxifen-induzierten Zelltod sensibilisiert und die Tamoxifen-Resistenz über die Herunterregulierung der EGFR-Expression in Brustkrebszellen überwindet.
EINFÜHRUNG
Proliferierende Krebszellen haben im Vergleich zu den meisten normalen, differenzierten Zellen ganz andere Stoffwechselbedürfnisse. Um ein schnelles Zellwachstum und eine rasche Vermehrung zu unterstützen, verändern Krebszellen beispielsweise den Stoffwechselfluss im Vergleich zum umgebenden Gewebe, um ausreichend Bioenergetik und biosynthetische Zwischenprodukte bereitzustellen. Ein bekanntes Phänomen, das bei den meisten Krebszellen zu beobachten ist, ist die Umstellung auf aerobe Glykolyse, unabhängig von der Sauerstoffzufuhr, was als „Warburg-Effekt“ bezeichnet wird, bei dem Pyruvat direkt in Milchsäure umgewandelt wird, anstatt in den Zitronensäurezyklus zu gelangen [1]. Da alle Krebszellen von dieser Veränderung des Stoffwechsels abhängig sind, stellen diese veränderten Stoffwechselwege attraktive therapeutische Ziele dar [2]. Sowohl in präklinischen als auch in klinischen Studien wurden Anstrengungen unternommen, den umprogrammierten Stoffwechsel allein oder in Kombination mit einer Krebschemotherapie anzugehen [3]. Interessanterweise wird diese krebsspezifische Umgestaltung des Stoffwechsels durch Dichloracetat (DCA) rückgängig gemacht, ein kleines Molekül, das auf die Mitochondrien abzielt und nach oraler Verabreichung in die meisten Gewebe eindringen kann [4]. Es hemmt spezifisch die Pyruvat-Dehydrogenase-Kinase (PDK), ein Mitglied der Kinase-Familie, was zu einer Reaktivierung der Pyruvat-Dehydrogenase (PDH) führt, einem Schlüsselenzym, das den Fluss von Pyruvat in die Mitochondrien verlagert, um die Glukoseoxidation statt der Glykolyse zu fördern [4]. Obwohl DCA kürzlich in mehreren präklinischen Krebsversuchen untersucht wurde [5]wurde, sind die Reaktionen der Krebszellen auf die DCA-Behandlung, die ausschlaggebend dafür sind, ob DCA einen klinischen Nutzen für die Krebsbehandlung bringt, noch nicht vollständig geklärt.
Mehr als 70 % der Brustkrebse exprimieren den Östrogenrezeptor (ER) und sind auf Östrogen angewiesen, um das Wachstum und Fortschreiten des Tumors zu fördern [6]. Daher sollte die endokrine Therapie bei der Mehrzahl der Patientinnen als Ergänzung zur Operation angesehen werden, da sie eine Tumorremission bewirkt und konsistente klinische Vorteile bietet. Das Anti-Östrogen-Medikament Tamoxifen ist die am häufigsten verwendete Behandlung für Patientinnen mit ER-positivem Brustkrebs sowohl im frühen als auch im fortgeschrittenen/metastasierten Stadium [7]. Als adjuvante Therapie bei Brustkrebs im Frühstadium verbessert Tamoxifen die Gesamtüberlebenszeit, und man geht davon aus, dass sein weit verbreiteter Einsatz wesentlich zu der in den letzten zehn Jahren beobachteten Verringerung der Brustkrebssterblichkeit beigetragen hat [8]. Trotz der offensichtlichen Vorteile einer Tamoxifen-Behandlung bei Brustkrebs erleiden fast alle Patientinnen mit metastasierter Erkrankung und bis zu 25 % der Patientinnen, die adjuvant mit Tamoxifen behandelt werden, schließlich einen Rückfall und sterben an der Krankheit [9, 10]. Die biologischen Mechanismen, die der intrinsischen (de novo) und erworbenen Resistenz gegen Tamoxifen zugrunde liegen, sind daher von erheblicher klinischer Bedeutung. Ein besseres Verständnis dieser Mechanismen könnte neue Strategien zur Überwindung der Tamoxifen-Resistenz und zur Verbesserung der Behandlung von Brustkrebs aufzeigen.
In der vorliegenden Studie konnten wir zeigen, dass die Expression von EGFR in Brustkrebszellen durch die Behandlung mit DCA verringert wurde. Eine Kombination aus DCA und Tamoxifen senkte die EGFR-Werte weiter. Wir konnten zeigen, dass DCA die Zytotoxizität von Tamoxifen auf Brustkrebszellen durch Hemmung der EGFR-Expression verstärkt. Darüber hinaus sensibilisierte DCA die tamoxifenresistenten MCF7-Zellen für Tamoxifen über eine EFGR-Downregulation. Diese Ergebnisse deuten auf eine mögliche Verwendung von DCA bei der Behandlung von Brustkrebs durch Abschwächung des EGFR-Signalwegs hin.
Ergebnisse
Die Hemmung von PDK regelt EGFR herunter und verstärkt den Tamoxifen-induzierten Zelltod in Brustkrebszellen
Jüngste Studien haben gezeigt, dass ein PDK-Inhibitor wie DCA den Stoffwechsel von Krebszellen von der Glykolyse zur oxidativen Phosphorylierung verlagert, indem er die mitochondriale Pyruvatdehydrogenase dephosphoryliert [4, 11]. Um zu klären, wie Wachstumsfaktor- und Kinase-Signalwege in Verbindung mit PDK den Warburg-Effekt bei Brustkrebs regulieren, untersuchten wir die Expressionsniveaus verschiedener Wachstumsfaktor-Rezeptoren in MCF7-Zellen mit PDK-Knockdown. Interessanterweise führte die Ausschaltung von PDK durch siRNA-Behandlung zu einer Herabregulierung von EGFR, während andere Wachstumsfaktorrezeptoren nicht oder nur geringfügig betroffen waren (Abbildung 1A). Vier PDK-Isoenzyme (PDK1, PDK2, PDK3, PDK4) sind in Säugetiergeweben identifiziert worden [12]. Um die Herunterregulierung des EGFR durch die siPDK-Behandlung zu bestätigen, untersuchten wir die EGFR-Expression in den Zellen, die mit siRNA gegen jede PDK-Isoform behandelt wurden. Jede siRNA-Behandlung hob nur die Expression der anvisierten PDK auf, aber alle verursachten die Herunterregulierung von EGFR, was darauf hindeutet, dass PDK die EGFR-Expression möglicherweise nicht isoformspezifisch reguliert (Abbildung 1B). In Übereinstimmung mit diesen Ergebnissen verringerte DCA die EGFR-Konzentration ebenfalls in einer dosisabhängigen Weise (Abbildung 1C). Bei der Analyse der Laktatkonzentration in den Kulturmedien zeigte sich keine signifikante Veränderung der Laktatkonzentration durch die Behandlung mit Tamoxifen/DCA oder siRNA gegen EGFR, obwohl sie durch die DCA-Behandlung verringert wurde (ergänzende Abbildung S1). Daher vermuten wir, dass die Herunterregulierung des EGFR nicht mit der DCA-induzierten Stoffwechselverschiebung in Brustkrebszellen verbunden ist. Da die Aktivierung des EGFR-Signalwegs zur Tamoxifen-Resistenz beiträgt [13]beiträgt, untersuchten wir, ob die Herunterregulierung des EGFR durch Hemmung von PDK die Zellen für Tamoxifen sensibilisierte. Wie in Abbildung 1D dargestellt, führte die gleichzeitige Behandlung mit Tamoxifen und DCA zu einer deutlichen Verringerung der Lebensfähigkeit der Zellen. Die kombinierte Behandlung über 72 Stunden reduzierte die Lebensfähigkeit der Zellen auf weniger als 30 % derjenigen der Kontrollgruppe (Abbildung 1E). Anschließend wurde der Zelltod in den mitbehandelten Zellen mittels Annexin V/PI-Färbung untersucht. Achtundvierzig Stunden nach der Behandlung führte die Kombination von Tamoxifen und DCA zu 30 % Zelltod, verglichen mit 15 % bzw. 8 % bei Tamoxifen oder DCA allein (Abbildung 1F). Zuvor hatten wir berichtet, dass der apoptotische Zelltod in Brustkrebszellen durch den Verlust des mitochondrialen Membranpotenzials (MMP) verursacht wird [14]. Daher testeten wir, ob der Verlust des MMP am Zelltod beteiligt ist, der durch die gleichzeitige Behandlung ausgelöst wird. Es gab jedoch keine signifikante Veränderung des MMP zwischen unbehandelten und mit Tamoxifen/DCA behandelten Zellen (ergänzende Abbildung S2).
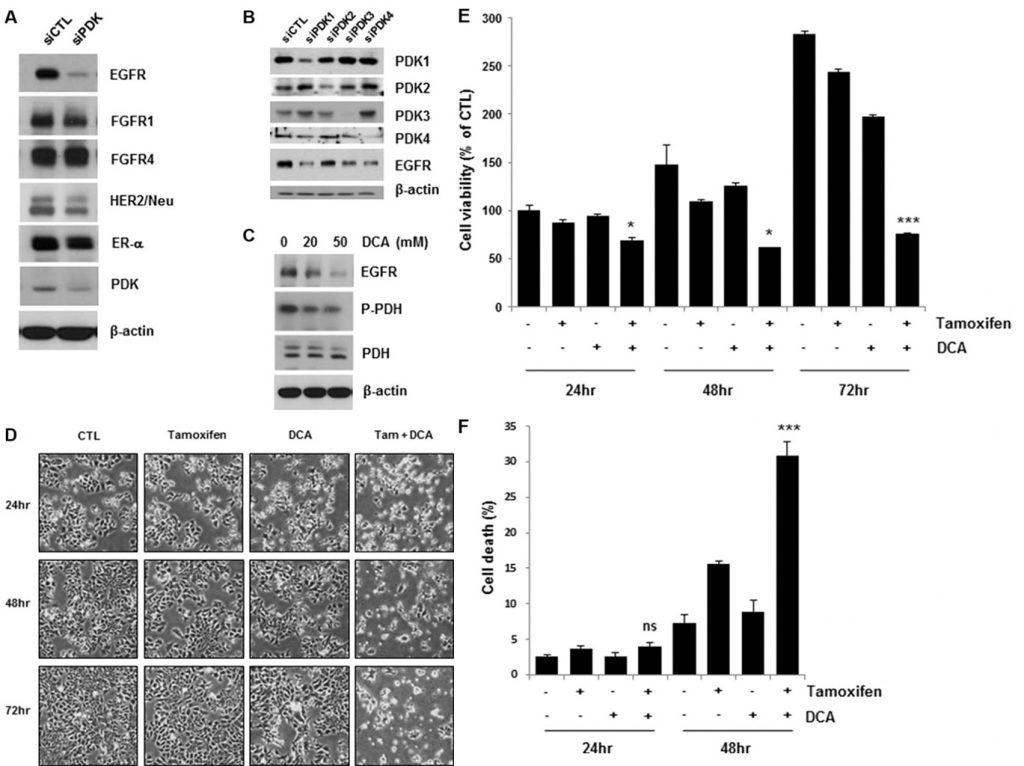
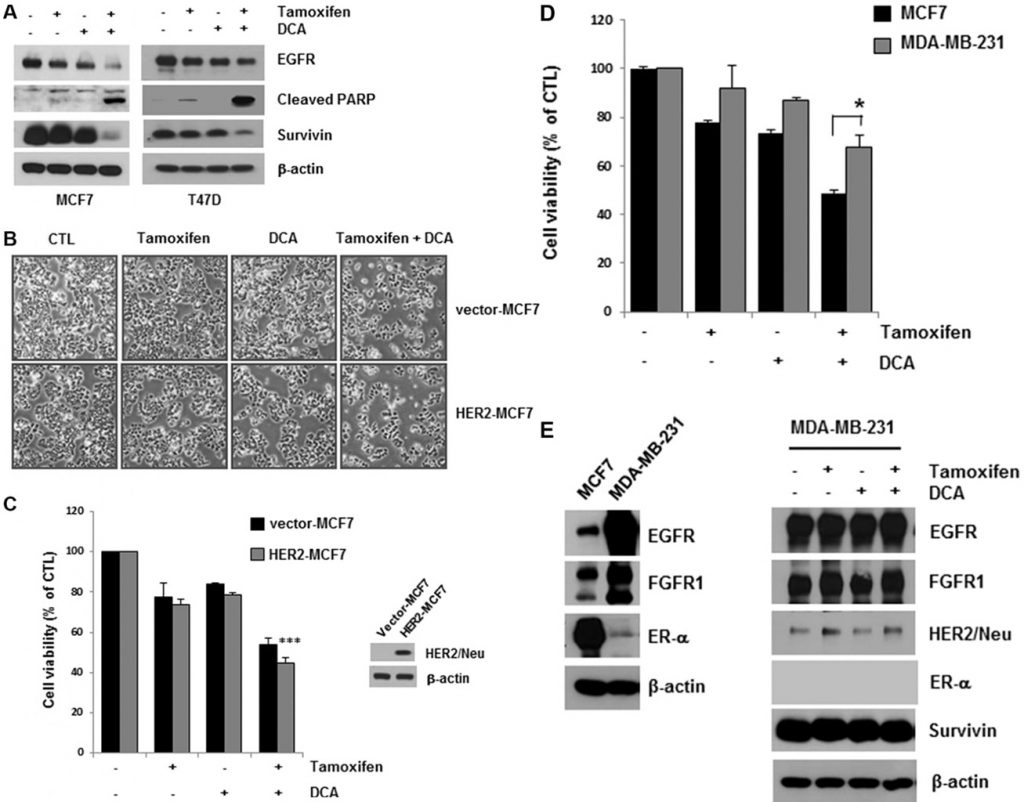
DCA plus Tamoxifen verringerte den EGFR-Spiegel sowohl in MCF7- als auch in T47D-Zellen im Vergleich zu DCA allein (Abbildung 2A). Der durch die gleichzeitige Behandlung ausgelöste Zelltod wurde durch den Nachweis der PARP-Spaltung, einem Marker für Apoptose, bestätigt (Abbildung 2A). Survivin ist sowohl ein anti-apoptotisches Molekül als auch ein Ziel des ER [15]. Durch die gleichzeitige Behandlung wurde auch Survivin herunterreguliert, das möglicherweise die Apoptose in den Zellen vermittelt (Abbildung 2A). Obwohl die Behandlung mit Tamoxifen die EGFR-Konzentration in MCF7- und T47D-Zellen leicht verringerte, wurde kein signifikanter Anstieg des Zelltods in den Zellen beobachtet, was darauf hindeutet, dass ein kritisches EGFR-Niveau für das Überleben von Brustkrebszellen erforderlich ist (Abbildung 2A).
Zelllinien haben gezeigt, dass eine Überexpression der HER2-Signalwege zur erworbenen Resistenz gegen endokrine Therapien beitragen kann [13]. Um festzustellen, ob die HER2-Überexpression die Zytotoxizität von Tamoxifen und DCA beeinflusst, untersuchten wir die Zelllebensfähigkeit von HER2-überexprimierenden MCF7-Zellen (HER2-MCF7) nach Behandlung mit Tamoxifen und DCA. Die Ergebnisse zeigten, dass Tamoxifen und DCA die Zelllebensfähigkeit auch in HER2-MCF7-Zellen deutlich verringerten (Abbildung 2B und 2C), was darauf hindeutet, dass DCA den Tamoxifen-induzierten Zelltod in HER2-überexprimierenden Brustkrebszellen verstärken könnte. Des Weiteren untersuchten wir die wachstumshemmende Wirkung der Co-Behandlung auf die dreifach negative Brustkrebszelllinie MDA-MB-231. Wie in Abbildung 2D dargestellt, reagierten MDA-MB-231-Zellen weniger empfindlich auf Tamoxifen und DCA als MCF7-Zellen. Da eine Herabregulierung des EGFR in ER-positiven Zellen beobachtet wurde, untersuchten wir die Auswirkungen von Tamoxifen und DCA auf die EGFR-Werte in MDA-MB-231-Zellen. EGFR wurde in MDA-MB-231-Zellen im Vergleich zu MCF7-Zellen stark exprimiert, und die Werte wurden durch Tamoxifen und DCA nicht signifikant verringert (Abbildung 2E). Als nächstes untersuchten wir die Zytotoxizität von Tamoxifen und DCA in der nicht-tumorigenen immortalisierten Brustepithelzelllinie MCF10A. Interessanterweise war die Expression von EGFR in MCF10A-Zellen vergleichbar mit der von MDA-MB-231-Zellen, und weder eine EGFR-Downregulation noch ein Zelltod wurden in MCF10A-Zellen nach der Behandlung mit Tamoxifen und DCA beobachtet (ergänzende Abbildung S3). Diese Ergebnisse deuten darauf hin, dass die proliferationshemmenden Wirkungen von Tamoxifen und DCA in Brustkrebszellen von der EGFR-Downregulation abhängig sind.
Die kombinierte Behandlung von Tamoxifen und DCA induziert den p38 MAPK-vermittelten EGFR-Abbau
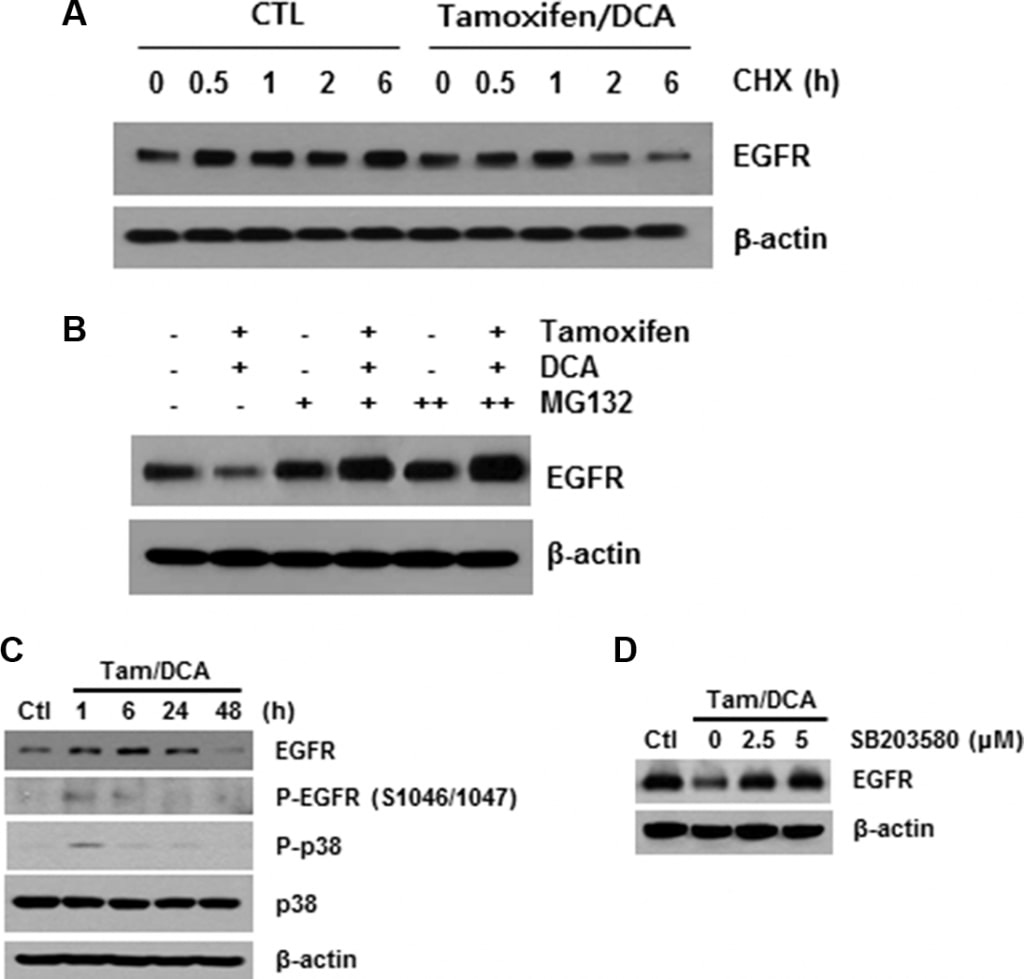
Wie oben beschrieben, führt die Ligandenbindung zu einer raschen Autophosphorylierung, die zur Entfernung des EGFR von der Zelloberfläche durch Endozytose in ein frühes endosomales Kompartiment führt [16]. Daher untersuchten wir als nächstes die Rolle der Rezeptormodifikation bei der Tamoxifen/DCA-vermittelten EGFR-Downregulation. Nach der Blockierung der Proteinsynthese mit Cycloheximid stellten wir fest, dass die Stabilität des EGFR in mit Tamoxifen/DCA behandelten Zellen im Vergleich zur Kontrolle deutlich beeinträchtigt war (Abbildung 3A). Anschließend untersuchten wir die Auswirkungen von MG132, einem Proteasom-Inhibitor, auf den Tamoxifen/DCA-induzierten EGFR-Abbau. Die Behandlung mit MG132 stellte die EGFR-Expression in den mit Tamoxifen/DCA behandelten Zellen dosisabhängig wieder her (Abbildung 3B).
Die Phosphorylierung des EGFR an Serin- und Threoninresten stellt einen Mechanismus zur Abschwächung der EGFR-Aktivität dar, wobei die Phosphorylierungsstellen an Serin 1046/1047 (Ser 1046/7) für die Desensibilisierung des EGFR erforderlich sind [17]. Kürzlich wurde berichtet, dass die mitogen-aktivierte Proteinkinase (MAPK) p38 die Phosphorylierung des EGFR an Ser 1046/7 induziert, was zu seinem Abbau in Krebszellen führt [18]. Daher untersuchten wir als Nächstes die Auswirkungen von Tamoxifen und DCA auf die Phosphorylierung von p38 MAPK in MCF7-Zellen. p38 MAPK wurde innerhalb von 1 Stunde signifikant phosphoryliert, und die Phosphorylierung blieb nach der Behandlung mit Tamoxifen und DCA 24 Stunden lang erhalten (Abbildung 3C). Darüber hinaus wurde der durch die gleichzeitige Behandlung induzierte Abbau von EGFR signifikant unterdrückt, wenn die Zellen mit einem spezifischen p38-MAPK-Inhibitor, SB203580, vorbehandelt wurden (Abbildung 3D), was darauf hindeutet, dass die p38-MAPK-Aktivierung eine Rolle bei der Tamoxifen/DCA-induzierten EGFR-Downregulation in MCF7-Zellen spielt.
EGFR-Inhibitoren verstärken den Tamoxifen-induzierten Zelltod
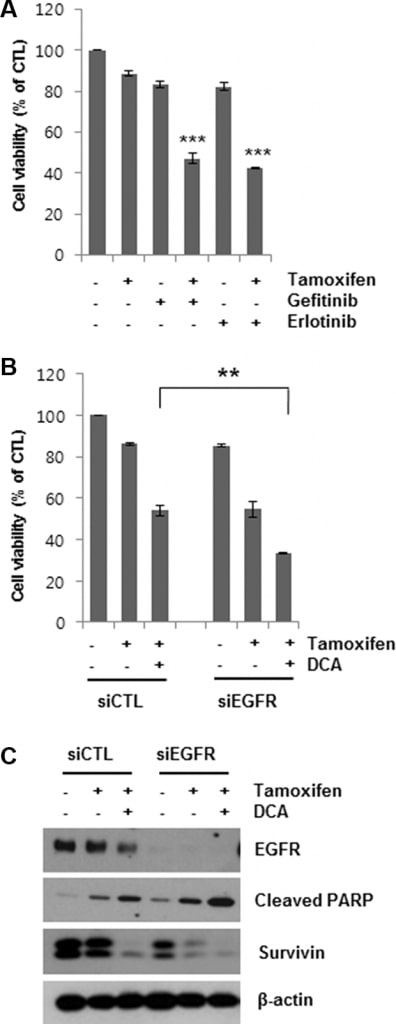
Nachdem wir gezeigt haben, dass der DCA-vermittelte EGFR-Abbau den Tamoxifen-induzierten Zelltod in MCF7-Zellen verstärken kann, haben wir anschließend untersucht, ob eine EGFR-Hemmung den Zelltod in Tamoxifen-behandelten Zellen verstärkt. Gefitinib und Erlotinib sind selektive reversible Inhibitoren der EGFR-Tyrosinkinase, die an ATP [19, 20]. Die gleichzeitige Behandlung von MCF7-Zellen mit 5 μM Gefitinib oder Erlotinib für 48 Stunden erhöhte den Tamoxifen-induzierten Zelltod deutlich (Abbildung 4A). In ähnlicher Weise verstärkte die Ausschaltung des EGFR durch die Behandlung mit siEGFR den Tamoxifen-induzierten Zelltod (Abbildung 4B). Die Verringerung der Zelllebensfähigkeit durch die kombinierte Behandlung von siEGFR und Tamoxifen war vergleichbar mit der durch DCA und Tamoxifen. Darüber hinaus senkte die siEGFR-Behandlung die EGFR-Spiegel in den mit Tamoxifen/DCA behandelten Zellen weiter, was zu einem erhöhten apoptotischen Zelltod zusammen mit einer Herabregulierung von Survivin im Vergleich zur si-Kontrolle führte (Abbildung 4C). Diese Ergebnisse stützen die Schlussfolgerung, dass DCA Brustkrebszellen über die Herunterregulierung des EGFR für Tamoxifen sensibilisiert.
die Expression von c-myc und Nanog ist mit den zytotoxischen Wirkungen von DCA in Tamoxifen-behandelten MCF7-Zellen verbunden
Es hat sich gezeigt, dass die EGFR-Aktivierung bei Krebs verschiedene Transkriptionsfaktoren erhöht, die die Art und Dauer der EGFR-Signalübertragung beeinflussen können [21]. Unter ihnen spielen Nanog und c-myc eine pleiotrope Rolle bei der Tumorentstehung, einschließlich der Resistenz gegenüber Standardtherapien bei Brustkrebs [22, 23]. Um die Anti-Tumor-Aktivitäten, die durch die Herunterregulierung des EGFR in Brustkrebszellen vermittelt werden, weiter zu untersuchen, untersuchten wir die Expression von c-myc und Nanog in MCF7-Zellen nach gemeinsamer Behandlung mit Tamoxifen und DCA. Die Spiegel von c-myc und Nanog waren in den mit Tamoxifen und DCA kotreatierten Zellen signifikant verringert (Abbildung 5A). In ähnlicher Weise wurde die Expression beider Proteine durch Tamoxifen in Kombination mit Gefitinib oder Erlotinib unterdrückt (Abbildung 5B), was darauf hindeutet, dass die EGFR-Expression erforderlich ist, um die Expression von c-myc und Nanog in mit Tamoxifen behandelten MCF7-Zellen aufrechtzuerhalten. Die Wirkung von siEGFR auf die Expression von c-myc und Nanog war jedoch gering oder gar nicht vorhanden, was darauf hindeutet, dass die Herunterregulierung des EGFR zwar notwendig, aber nicht ausreichend ist, um die Expression der Proteine zu verringern (ergänzende Abbildung S4). Um festzustellen, ob die verringerte Expression dieser beiden Proteine an den zytotoxischen Wirkungen von Tamoxifen und DCA in MCF7-Zellen beteiligt ist, testeten wir die Wirkung von siRNAs gegen c-myc und Nanog in Zellen, die Tamoxifen/DCA ausgesetzt waren. Die Ausschaltung von c-myc und Nanog sensibilisierte die Zellen im Vergleich zu den Kontrollen signifikant für Tamoxifen/DCA (Abbildung 5C). Umgekehrt schützte die Überexpression von FLAG-c-myc und FLAG-Nanog durch Transfektion der Zellen mit FLAG-c-myc- und FLAG-Nanog-Vektoren die Zellen signifikant vor Tamoxifen/DCA-induzierter Zytotoxizität (Abbildung 5D). Insgesamt deuten unsere Daten darauf hin, dass die Herunterregulierung des EGFR durch eine Kombination von Tamoxifen und DCA den Zelltod in MCF7-Zellen teilweise durch die Hemmung der c-myc- und Nanog-Expression auslösen kann. Es ist bekannt, dass Selbsterneuerungsgene wie c-myc und Nanog mit krebsstammähnlichen Zelleigenschaften verbunden sind 24. Um zu testen, ob die gleichzeitige Behandlung mit Tamoxifen und DCA die stammähnlichen Zellen von Brustkrebs hemmt, führten wir eine Durchflusszytometrie-Analyse durch, um den Anteil der stammähnlichen Zellsubpopulation in MCF7-Zellen anhand der CD44-Expression zu bestimmen. Nach der gleichzeitigen Behandlung wurde die CD44-reiche Population in MCF7-Zellen von 42,5% auf 19,9% reduziert (Abbildung 5E). Dieser Befund deutet darauf hin, dass DCA plus Tamoxifen die Fähigkeit von Brustkrebszellen, Krebsstammzellen zu bilden, hemmen könnte.

Die kombinierte Behandlung mit DCA und Tamoxifen kann die Tamoxifen-Resistenz in Brustkrebszellen überwinden
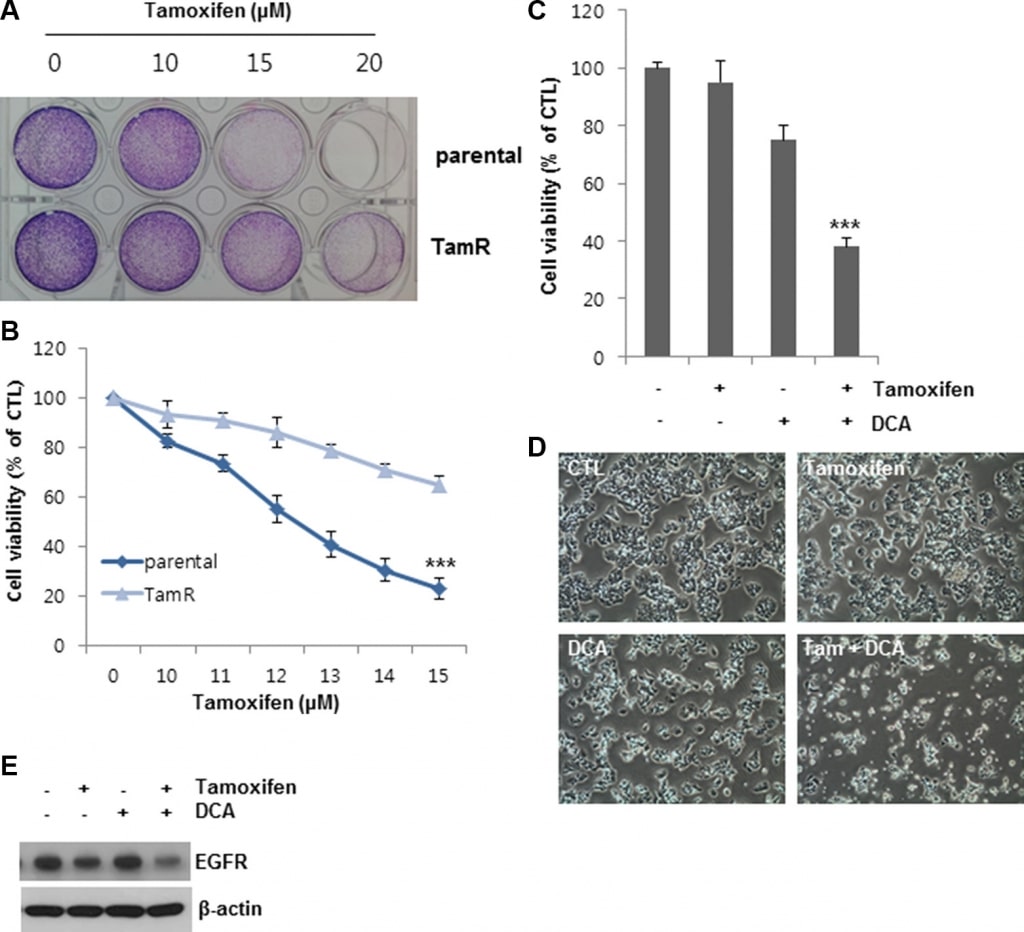
Um die beobachteten Auswirkungen von DCA auf den Tamoxifen-induzierten Zelltod weiter zu bestätigen, haben wir tamoxifenresistente (TamR) MCF7-Zellen durch Tamoxifen-Behandlung über einen langen Zeitraum etabliert. Im Vergleich zu den unbehandelten Zellen lag die Zelllebensfähigkeit der MCF7- und TamR-MCF7-Zellen nach der Behandlung mit 13 μM Tamoxifen bei 20 bzw. 60 %, was darauf hindeutet, dass die TamR-MCF7-Zellen weniger empfindlich auf dieselbe Tamoxifenkonzentration reagieren als die MCF7-Elternzellen (Abbildung 6A und 6B). DCA allein hemmte das Wachstum der TamR-MCF7-Zellen um etwa 25 % des Wachstums der Kontrollgruppe (Abbildung 6C und 6D). Im Gegensatz dazu unterdrückte die gleichzeitige Behandlung von DCA mit Tamoxifen das Zellwachstum um mehr als 60 % im Vergleich zur Kontrolle (Abbildung 6C und 6D). Die EGFR-Spiegel in TamR-Zellen wurden durch die gleichzeitige Behandlung ebenfalls herunterreguliert (Abbildung 6E). Diese Ergebnisse deuten darauf hin, dass DCA die Tamoxifen-Resistenz in Brustkrebszellen durch eine Herunterregulierung des EGFR auf Proteinebene überwinden kann.
Diskussion
In dem Maße, in dem das Verständnis des metabolischen Phänotyps von Tumorzellen gewachsen ist, wurde die gezielte Beeinflussung der metabolischen Unterschiede zwischen Tumor- und normalen Zellen als neue Krebsbekämpfungsstrategie vorgeschlagen. Trotz der zunehmenden Zahl potenzieller Krebsmedikamente, die auf die Stoffwechselprozesse von Krebszellen abzielen, muss geklärt werden, wie die Zellen auf die Medikamente reagieren, um den Krebsstoffwechsel erfolgreich zu beeinflussen. Hier konnten wir zeigen, dass DCA nicht nur die PDK-Aktivität, sondern auch die EGFR-Expression in Brustkrebszellen hemmt. Die kombinierte Behandlung mit DCA und Tamoxifen induzierte den proteasomabhängigen Abbau von EGFR-Proteinen in Brustkrebszellen über die Aktivierung von p38 MAPK. Unsere Ergebnisse deuten darauf hin, dass die Herunterregulierung des EGFR eine Schlüsselrolle beim apoptotischen Zelltod spielt, der durch eine Kombination von DCA und Tamoxifen ausgelöst wird. Unseres Wissens nach ist dies der erste Bericht, der zeigt, dass DCA Brustkrebszellen für den Tamoxifen-induzierten Zelltod durch EGFR-Downregulation sensibilisiert.
In ER-positiven Brustkrebszellen, die eine endokrine Resistenz entwickelt haben, kann die ER-Expression direkt durch eine verstärkte Wachstumsfaktor-Rezeptor-Signalisierung aufgrund einer Überexpression von EGFR und HER2 unterdrückt werden, die anschließend MAPK aktivieren und die ER-Transkription hemmen [25]. Darüber hinaus wurde die Tamoxifen-Resistenz von Brustkrebszellen mit der Überexpression von EGFR und hohen Konzentrationen von phosphorylierter extrazellulärer aktivierter Kinase 1/2 in Verbindung gebracht [2]. Daher ist die Strategie, einen EGFR-Inhibitor mit einem endokrinen Wirkstoff zu kombinieren, von ausreichendem Interesse, um weitere Studien zur Brustkrebstherapie zu rechtfertigen. In dieser Studie haben wir festgestellt, dass die Herunterregulierung des EGFR in DCA-behandelten Brustkrebszellen die Empfindlichkeit der Zellen gegenüber Tamoxifen erhöht. Die Herabregulierung des EGFR durch die gleichzeitige Behandlung von DCA und Tamoxifen wurde in mehreren ER-positiven Brustkrebszelllinien, darunter MCF7, T47D und BT474, bestätigt (Abbildung 2A und nicht gezeigt). Neben der HER2-amplifizierten Brustkrebszelllinie BT474 zeigten auch die HER2-MCF7-Zellen eine EGFR-Downregulation durch DCA/Tamoxifen, was darauf hindeutet, dass die Kombinationsbehandlung auch bei HER2-positivem Brustkrebs anwendbar sein wird. Leider zeigte die dreifach negative Brustkrebszelllinie MDA-MB-231, von der bekannt ist, dass sie den EGFR überexprimiert [26], einen refraktären Phänotyp und keine offensichtliche Veränderung der EGFR-Werte als Reaktion auf die Co-Behandlung. Eine Möglichkeit ist, dass die Amplifikation des EGFR in MDA-MB-231-Zellen dessen Herunterregulierung durch die gleichzeitige Behandlung von DCA und Tamoxifen überwindet. Die andere Erklärung ist, dass der/die Signalweg(e), der/die die EGFR-Herabregulierung vermittelt/vermitteln, durch einen unbekannten Mechanismus in MDA-MB-231-Zellen blockiert wird/werden. Weitere Untersuchungen sind erforderlich, um den zugrunde liegenden Mechanismus für die Aufrechterhaltung der EGFR-Expression als Reaktion auf die gleichzeitige Behandlung mit DCA und Tamoxifen in ER-negativen Brustkrebszellen zu klären.
Es sind vier Isoformen der PDK bekannt, die jeweils auf unterschiedliche intra- und extrazelluläre Bedingungen reagieren. PDK1 wird durch Hypoxie aktiviert [27]; PDK2 wird durch die PDH-Produkte Acetyl-CoA und NADH aktiviert [28]; PDK3 wird durch ATP aktiviert [29]; und PDK4 wird durch hormonelle Signale transkriptionell reguliert [30]. PDK2 besitzt die größte Aktivität bei der Phosphorylierung des Pyruvatdehydrogenase-Komplexes, gefolgt von PDK4, PDK1 und PDK3 [12]. PDK2 ist aufgrund seiner ubiquitären Expression am anfälligsten für eine Hemmung durch DCA [31]. Obwohl wir die Expression von vier Isoformen in MCF7-Zellen nachweisen konnten (Abbildung 1C), sind die funktionellen Unterschiede zwischen den PDK-Isoformen bei Brustkrebs nach wie vor nicht klar. Es wäre interessant, weiter zu untersuchen, ob Signalwege, die eine bestimmte PDK-Isoform unterdrücken, zur Herunterregulierung des EGFR führen. Es ist bekannt, dass PDK in den Mitochondrien wirkt, und mehrere Studien haben gezeigt, dass EGFR nach EGF-Stimulation ebenfalls in die Mitochondrien verlagert wird [32, 33]. Darüber hinaus wurde vermutet, dass die Interaktion von EGFR mit PDK in der mitochondrialen Matrix eine wichtige Rolle beim EGFR-induzierten Tumorwachstum beim Glioblastoma multiforme spielt [34]. Wir haben hier festgestellt, dass die Blockade der PDK-Aktivität durch einen Inhibitor oder durch Silencing die zellulären EGFR-Gesamtwerte in mit Tamoxifen behandelten Brustkrebszellen verringert. Weitere Studien sind erforderlich, um festzustellen, ob nur der mitochondrial-assoziierte EGFR durch die gleichzeitige Behandlung mit DCA und Tamoxifen in Brustkrebszellen abgebaut wird. Im Allgemeinen führt die Exposition von Zellen gegenüber EGF zu einer schnellen Autophosphorylierung, einschließlich Tyrosin (Tyr) 1045, das eine Andockstelle für die Ubiquitin-Ligase c-Cbl darstellt, was zur Ubiquitinierung des EGFR und zur Entfernung des EGFR durch Endozytose von der Zelloberfläche in ein frühes endosomales Kompartiment führt [35]. Die gleichzeitige Behandlung mit DCA und Tamoxifen hatte jedoch keinen Einfluss auf die Phosphorylierung des EGFR an Tyr 1045 (Daten nicht gezeigt). Stattdessen beobachteten wir die Phosphorylierung von EGFR an Ser 1046/7 durch DCA und Tamoxifen, die durch einen spezifischen p38 MAPK-Inhibitor, SB203580, blockiert wurde. Es wurde vermutet, dass p38 MAPK sowohl bei der Internalisierung als auch beim Abbau von EGFR eine zentrale Rolle spielt [18]; es sind jedoch weitere Untersuchungen erforderlich, um zu klären, wie die EGFR-Phosphorylierung an Serinresten zu seiner Downregulation führt, und um den vorgeschalteten Mediator der p38 MAPK-Aktivierung nach DCA/Tamoxifen-Behandlung zu identifizieren.
Nanog und c-myc sind als Transkriptionsfaktoren identifiziert worden, und ihre Rolle bei der Aufrechterhaltung der Selbsterneuerung in embryonalen Stammzellen wurde in früheren Studien nachgewiesen [22, 36]. Ein kürzlich vorgeschlagener Mechanismus der intrinsischen Resistenz gegen eine endokrine Therapie geht von der Existenz einer spezialisierten Untergruppe von Krebszellen aus, die als tumorinitierende Zellen (TICs) bezeichnet werden [37]. TICs haben die Fähigkeit, sich selbst zu erneuern und neue Tumore zu erzeugen, die ausschließlich aus klonal abgeleiteten Zelltypen des Elterntumors bestehen. Hier konnten wir zeigen, dass DCA die Expression von c-myc und Nanog in mit Tamoxifen behandelten MCF7-Zellen verringert. Außerdem stand die Expression dieser Proteine im Zusammenhang mit den zytotoxischen Wirkungen der kombinierten Behandlung von DCA und Tamoxifen. Daher kann DCA genutzt werden, um das therapeutische Potenzial von Tamoxifen zu erweitern, indem die Entwicklung einer intrinsischen Resistenz bei der Brustkrebstherapie unterdrückt wird.
Unsere Ergebnisse zeigen, dass DCA ER-positive Brustkrebszellen für Tamoxifen sensibilisiert, indem es die EGFR-Spiegel senkt. Die Behandlung mit DCA und Tamoxifen hemmte die Expression von Selbsterneuerungsgenen und das Überleben von tamoxifenresistenten MCF7-Zellen. Daher schlagen wir vor, dass DCA ein wirksames Therapeutikum zur Behandlung von tamoxifenresistentem Brustkrebs sein könnte. Darüber hinaus könnten Kombinationsstrategien mit DCA nützlich sein, um die Wirksamkeit anderer zytotoxischer Chemotherapien oder gezielter Therapien zu erhöhen. Weitere Experimente, einschließlich Tierstudien und klinischer Versuche, sollten in Zukunft durchgeführt werden.
Materialien und Methoden
Zellkultur und Reagenzien
Die menschlichen Brustkrebszellen MCF7, T47D und MDA-MB-231 wurden von der American Type Culture Collection (Rockville, MD, USA) erworben und in dem empfohlenen Wachstumsmedium (Invitrogen, Carlsbad, CA, USA) gezüchtet. HER2-überexprimierende MCF7- (HER2-MCF7) und Kontrollvektorzellen (Vektor-MCF7) wurden freundlicherweise von Dr. Incheol Shin (Hanyang University, Seoul, Korea) zur Verfügung gestellt. Tamoxifen-resistente (TamR) MCF7-Zellen wurden durch Kultivierung von MCF7-Zellen in Gegenwart von 10 μM Tamoxifen für mehr als 6 Monate entwickelt. Als Kontrolle wurden die elterlichen Zellen für die gleiche Zeit in normalen Medien kultiviert. Nach der Etablierung der Resistenz wurden die Zellen nicht länger als 3 Monate lang passagiert. Bei Experimenten mit Tamoxifen-Behandlungen wurden die Zellen routinemäßig in phenolrotfreiem Dulbecco’s Modified Eagle’s Medium plus 10 % fötales Rinderserum, das von Holzkohle befreit wurde, kurz vor den Behandlungen kultiviert. Antikörper gegen EGFR, phospho-EGFR (S1046/1047), FGFR1, FGFR4, PDK1, PDH, survivin, gespaltenes PARP, p38, phospho-p38 und Nanog wurden von Cell Signaling Technology (Danvers, MA, USA) erworben. Antikörper gegen HER2/Neu, ER-α und c-myc, siRNAs, die auf EGFR, Nanog, PDK4 und c-myc abzielen, sowie die siRNAs der Negativkontrolle (Scrambled) wurden von Santa Cruz Biotechnology (Dallas, TX, USA) erworben. Antikörper gegen PDK2 und PDK3 wurden von Thermo Fisher Scientific (Waltham, MA, USA) erworben. Der β-Actin-Antikörper, der FLAG-Antikörper, Tamoxifen, DCA, Cycloheximid und MG132 wurden von Sigma-Aldrich (St. Louis, MO, USA) erworben. Der Phospho-PDH (S293)-Antikörper und SB203580 stammen von BD Biosciences Pharmingen (San Diego, CA, USA), und Gefitinib und Erlotinib wurden von Selleck Chemicals (London, ON, Kanada) bezogen.
Transfektionen und Behandlungen
Die Zellen wurden mit der Ziel-siRNA (50 nM) unter Verwendung von Lipofectamine RNAiMAX (Invitrogen) wie vom Hersteller beschrieben transfiziert. Die Zellen wurden mit 1 μg FLAG-c-myc pcDNA 3.1 und FLAG-Nanog-pcDNA 3.1 unter Verwendung von Lipofectamine 2000 wie vom Hersteller beschrieben transfiziert. Nach 6 Stunden wurden die Zellen 24-48 Stunden lang mit Tamoxifen und/oder DCA behandelt und dann wie an anderer Stelle beschrieben analysiert.
Messung der Lebensfähigkeit der Zellen
Die Lebensfähigkeit der Zellen wurde durch Messung der mitochondrialen Umwandlung von 3-(4,5-Dimethylthiazolyl2)-2,5-Diphenyltetrazoliumbromid (MTT) in ein farbiges Produkt bestimmt. Die Zellen wurden wie angegeben behandelt, und das Medium wurde gegen serumfreies Medium mit 1 mM MTT ausgetauscht. Nach 2 Stunden Inkubation bei 37 °C wurden die Zellen in DMSO gelöst. Die Menge an Formazan, der umgewandelten Form von MTT, wurde durch Messung der Absorption bei 595 nm bestimmt. Bewertung der Apoptose Die
Apoptose wurde durch fluoreszenzaktivierte Zellsortierungsanalyse unter Verwendung eines Annexin V-FITC-Apoptose-Kits (BioVision, Milpitas, CA, USA) gemäß den Anweisungen des Herstellers bestimmt. Nach der Behandlung wurden die Zellen mit Trypsin behandelt und anschließend in Bindungspuffer (10 mM HEPES/NaOH, pH 7,4, 140 mM NaCl, 2,5 mM CaCl2 ) mit Annexin V-FITC und Propidiumiodid resuspendiert. Nach 15-minütiger Inkubation wurde die Zellfluoreszenz durchflusszytometrisch analysiert. Der Zelltod wurde als Prozentsatz der Zellen in der Annexin V- und PI-positiven Population gemessen.
Western Blotting
Die Zellen wurden geerntet und in RIPA-Puffer (50 mM Tris-HCl pH 7,5, 150 mM NaCl, 1 % Nonidet P40, 0,5 % Natriumdeoxycholat und 0,1 % SDS) lysiert, der mit einem Protease/Phosphatase-Inhibitor-Cocktail (Roche, Mannheim, Deutschland) ergänzt wurde. Gleiche Mengen an Proteinen (20-50 μg) wurden durch SDS-PAGE aufgetrennt und auf eine Nitrocellulosemembran übertragen. Die Membranen wurden durch 1-stündige Inkubation mit 5 % Magermilch in trisgepufferter Kochsalzlösung blockiert und dann über Nacht mit den entsprechenden Primärantikörpern inkubiert. Die Membranen wurden 1 Stunde lang mit HRP-konjugierten sekundären Antikörpern inkubiert. Die immunreaktiven Proteine wurden mit verstärkten Chemilumineszenz-Reagenzien (Amersham Biosciences, Little Chalfont, UK) sichtbar gemacht.
Nachweis der CD44-positiven Zellpopulation
Die Zellen wurden 15 Minuten lang mit Antikörpern in einer Verdünnung von 1∶100 in PBS gefärbt. Es wurden folgende Antikörper verwendet: FITC-CD44 und FITC-konjugierte Maus-IgG-Isotyp-Kontrollantikörper von BD Biosciences Pharmingen. Die markierten Zellen wurden durchflusszytometrisch analysiert. Die Populationen der CD44-positiven Zellen wurden anhand der FITC-Intensität bestimmt.
Statistische Analyse
Alle dargestellten Daten sind repräsentativ für mindestens zwei separate Experimente. Die Vergleiche zwischen den Gruppen wurden mit dem Student’s t-Test ausgewertet. Sternchen (***p < 0,001, **p < 0,01, *p < 0,05) zeigen statistische Signifikanz an.
Danksagung UND Finanzierung
Diese Forschung wurde vom Basic Science Research Program durch die National Research Foundation of Korea (NRF) unterstützt, die vom Ministerium für Wissenschaft, IKT und Zukunftsplanung finanziert wird (Nr. 1711031812, 1711023318 und 1711031800).
Interessenkonflikte
Die Autoren erklären, dass keine Interessenkonflikte bestehen.
REFERENZEN
1 1. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029-33.
2ZhaoY, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013; 4:e532.
3Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011; 10:671-84.
4 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Krebszelle. 2007; 11: 37-51.
5 Kankotia S, Stacpoole PW. Dichloracetat und Krebs: ein neues Zuhause für ein Arzneimittel für seltene Krankheiten? Biochim Biophys Acta. 2014; 1846:617-29.
6 Osborne CK. Tamoxifen in der Behandlung von Brustkrebs. N Engl J Med. 1998 Nov 26; 339:1609-18.
7 Johnston SR. Neue Strategien bei Östrogenrezeptor-positivem Brustkrebs. Clin Cancer Res. 2010 Apr 1; 16:1979-87. doi: 10.1158/1078-0432.CCR-09-1823.
8 Peto R, Boreham J, Clarke M, Davies C, Beral V. UK und USA: 25 % weniger Brustkrebs-Todesfälle im Jahr 2000 im Alter von 20-69 Jahren. Lancet. 2000; 355:1822.
9 Early Breast Cancer Trialists‘ Collaborative Group (EBCTCG). Auswirkungen von Chemo- und Hormontherapie bei Brustkrebs im Frühstadium auf das Wiederauftreten und die 15-Jahres-Überlebensrate: ein Überblick über die randomisierten Studien. Lancet. 2005; 365:1687-717.
10 Early Breast Cancer Trialists‘ Collaborative Group (EBCTCG), Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, Dowsett M, Ingle J, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011; 378:771-84.
11 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013; 73:7277-89.
12 Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer. 2016; 138:809-17.
13 Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011; 62:233-47.
14YunSM, Woo SH, Oh ST, Hong SE, Choe TB, Ye SK, Kim EK, Seong MK, Kim HA, Noh WC, Lee JK, Jin HO, Lee YH, et al. Melatonin steigert den Arsentrioxid-induzierten Zelltod über eine anhaltende Hochregulierung der Redd1-Expression in Brustkrebszellen. Mol Cell Endocrinol. 2016; 422:64-73.
15 Zhu J, Lu X, Hua KQ, Sun H, Yu YH, Feng YJ. Oestrogen receptor α mediates 17β-estradiol enhancement of ovarian cancer cell motility through up-regulation of survivin expression. Arch Gynecol Obstet. 2012; 286: 729-37.
16 Arteaga CL. Die Abhängigkeit vom epidermalen Wachstumsfaktor-Rezeptor in menschlichen Tumoren: mehr als nur Expression? Oncologist. 2002; 7:31-9.
17 Sorkin A, Goh LK. Endozytose und intrazellulärer Transport von ErbBs. Exp Cell Res. 2009; 315:683-96.
18 Adachi S, Shimizu M, Shirakami Y, Yamauchi J, Natsume H, Matsushima-Nishiwaki R, To S, Weinstein IB, Moriwaki H, Kozawa O. (-)-Epigallocatechingallat regelt den EGF-Rezeptor über Phosphorylierung an Ser1046/1047 durch p38 MAPK in Dickdarmkrebszellen herunter. Carcinogenesis. 2009; 30:1544-52.
19CiardielloF, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G. Antitumorwirkung und Potenzierung der Aktivität zytotoxischer Arzneimittel in menschlichen Krebszellen durch ZD-1839 (Iressa), einen selektiven Tyrosinkinase-Inhibitor für den epidermalen Wachstumsfaktor-Rezeptor. Clin Cancer Res. 2000; 6:2053-63.
20 Pollack VA, Savage DM, Baker DA, Tsaparikos KE, Sloan DE, Moyer JD, Barbacci EG, Pustilnik LR, Smolarek TA, Davis JA, Vaidya MP, Arnold LD, Doty JL, et al. Inhibition of epidermal growth factor receptorassociated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther. 1999; 291:739-48.
21 Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Die Signalübertragung des epidermalen Wachstumsfaktor-Rezeptors (EGFR) bei Krebs. Gene. 2006; 366:2-16.
22 Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014; 135:2741-8.
23 Chen Y, Olopade OI. MYC in der Brusttumorprogression. Expert Rev Anticancer Ther. 2008; 8: 1689-98.
24 Hou ZJ, Luo X, Zhang W, Peng F, Cui B, Wu SJ, Zheng FM, Xu J, Xu LZ, Long ZJ, Wang XT, Li GH, Wan XY, et al. Flubendazole, FDA-zugelassenes Anthelminthikum, zielt auf stammähnliche Zellen von Brustkrebs. Oncotarget. 2015; 6:6326-40. doi:10.18632/oncotarget.3436.
25 Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Activation of mitogenactivated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res. 2006; 66:3903-11.
26 Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: Ein wichtiger Weg der Zellzyklusprogression in Östrogenrezeptor-negativen Brustkrebszellen. Proc Natl Acad Sci U S A. 2000; 97:8542-7.
27 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF1-vermittelte Expression der Pyruvat-Dehydrogenase-Kinase: ein metabolischer Schalter, der für die zelluläre Anpassung an Hypoxie erforderlich ist. Cell Metab. 2006; 3:177-85.
28 Hiromasa Y, Hu L, Roche TE. Liganden-induzierte Effekte auf die Pyruvat-Dehydrogenase-Kinase-Isoform 2. J Biol Chem. 2006; 281:12568-79.
29 Kato M, Chuang JL, Tso SC, Wynn RM, Chuang DT. Kristallstruktur der Pyruvat-Dehydrogenase-Kinase 3, gebunden an die Lipoyl-Domäne 2 des menschlichen Pyruvat-Dehydrogenase-Komplexes. EMBO J. 2005; 24:1763-74.
30 Kwon HS, Huang B, Unterman TG, Harris RA. Die Proteinkinase B-alpha hemmt die Induktion des menschlichen Pyruvat-Dehydrogenase-Kinase-4-Gens durch Dexamethason durch Inaktivierung der FOXO-Transkriptionsfaktoren. Diabetes. 2004; 53:899-910.
31 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Beweise für die Existenz einer gewebespezifischen Regulierung des Pyruvatdehydrogenase-Komplexes bei Säugetieren. Biochem J. 1998; 329 :191-6.
32 Dasari VR, Velpula KK, Alapati K, Gujrati M, Tsung AJ. Stammzellen aus Nabelschnurblut hemmen die Translokation des epidermalen Wachstumsfaktor-Rezeptors in Mitochondrien bei Glioblastom. PLoS One. 2012; 7:e31884.
33 Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, Hüttemann M, Douglas R, Haddad G, Parsons SJ. Die Translokation des epidermalen Wachstumsfaktor-Rezeptors in die Mitochondrien: Regulierung und Wirkung. J Biol Chem. 2009; 284:36592-604.
34 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013; 73:7277-89.
35 Massie C, Mills IG. Die sich entwickelnde Rolle von Rezeptoren und Adaptoren. Nat Rev Cancer. 2006; 6:403-9.
36 Chappell J, Dalton S. Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harb Perspect Med. 2013;3:a014381.
37 Wei W, Lewis MT. Identifying and targeting tumor-initiating cells in the treatment of breast cancer. Endocr Relat Cancer. 2015; 22:R135-55.