Sang Hyeok Woo1,*, Sung-Keum Seo1,*, Yoonhwa Park1,2, Eun-Kyu Kim3, Min-Ki Seong4, Hyun-Ah Kim4, Jie-Young Song1, Sang-Gu Hwang1, Jin Kyung Lee5, Woo Chul Noh4, In-Chul Park1
1Divisionof Radiation Cancer Research, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Seoul, 01812, République de Corée
2Schoolof Life Science and Biotechnology, Korea University, Seongbuk-gu, Seoul, 02841, République de Corée
3Department of Surgery, Breast Cancer Center, Seoul National University Bundang Hospital, Seoul National University College of Medicine, Bundang-gu, Seongnam, 13620, République de Corée
4Départementde chirurgie, Korea Cancer Center Hospital, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Séoul, 01812, République de Corée
5KIRAMSRadiation Biobank, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Séoul, 01812, République de Corée
*Cesauteurs ont contribué à parts égales à ce travail
Correspondance à : In-Chul Park, courriel : [email protected]
Mots-clés : tamoxifène, cancer du sein, dichloroacétate, récepteur du facteur de croissance épidermique, pyruvate déshydrogénase kinase
Reçu : 18 mai 2016
Accepté : 22 juillet 2016
Publié en ligne : 01 août 2016
Résumé
La reprogrammation métabolique dans les cellules cancéreuses a récemment été reconnue comme une caractéristique essentielle de la néoplasie. Dans ce contexte, les altérations métaboliques représentent une cible thérapeutique attrayante, et des résultats encourageants avec des médicaments ciblant divers processus métaboliques ont été obtenus dans des études précliniques. Récemment, plusieurs études ont suggéré que le dichloroacétate (DCA), un inhibiteur spécifique de la pyruvate déshydrogénase kinase, pourrait être un médicament anticancéreux potentiel dans un grand nombre de tumeurs diverses. Cependant, le mécanisme précis n’est pas entièrement compris, ce qui est important pour l’utilisation du DCA dans le traitement du cancer. Dans la présente étude, nous avons constaté que le DCA sensibilisait les cellules cancéreuses du sein MCF7 à la mort cellulaire induite par le tamoxifène en diminuant l’expression du récepteur du facteur de croissance épidermique (EGFR). La régulation négative de l’EGFR a été causée par la dégradation de la protéine. En outre, la protéine kinase mitogène p38 a joué un rôle important dans la dégradation de l’EGFR induite par le DCA/tamoxifène. Enfin, le DCA a également favorisé une mort cellulaire comparable à celle induite par le tamoxifène dans les cellules MCF7 résistantes au tamoxifène, qui ont été établies par un traitement à long terme au tamoxifène. En résumé, nos résultats suggèrent que le DCA est un médicament potentiel intéressant qui sensibilise les cellules à la mort cellulaire induite par le tamoxifène et qui permet de surmonter la résistance au tamoxifène via la régulation négative de l’expression de l’EGFR dans les cellules cancéreuses du sein.
INTRODUCTION
Les cellules cancéreuses en prolifération ont des besoins métaboliques considérablement différents de ceux de la plupart des cellules normales différenciées. Par exemple, pour soutenir une croissance et une prolifération cellulaires rapides, les cellules cancéreuses modifient de manière différentielle le flux métabolique par rapport au tissu environnant afin de fournir suffisamment de bioénergie et d’intermédiaires biosynthétiques. Un phénomène bien connu observé dans la plupart des cellules cancéreuses est le passage à la glycolyse aérobie, indépendamment de l’apport en oxygène, appelé « effet Warburg », dans lequel le pyruvate est directement converti en acide lactique au lieu d’entrer dans le cycle de l’acide citrique [1]. Comme toutes les cellules cancéreuses dépendent de cette modification du métabolisme, ces voies modifiées représentent des cibles thérapeutiques intéressantes [2]. Des efforts ont été faits pour cibler la reprogrammation du métabolisme, seule ou en combinaison avec la chimiothérapie anticancéreuse, tant dans les études précliniques que cliniques [3]. Il est intéressant de noter que ce remodelage métabolique spécifique au cancer est inversé par le dichloroacétate (DCA), une petite molécule ciblant les mitochondries qui peut pénétrer dans la plupart des tissus après administration orale [4]. Elle inhibe spécifiquement la pyruvate déshydrogénase kinase (PDK), un membre de la famille des kinases, ce qui entraîne la réactivation de la pyruvate déshydrogénase (PDH), une enzyme clé qui déplace le flux de pyruvate dans la mitochondrie pour favoriser l’oxydation du glucose au lieu de la glycolyse [4]. Bien que le DCA ait récemment été évalué dans plusieurs essais précliniques sur le cancer [5], les réponses des cellules cancéreuses au traitement par le DCA, qui déterminent si le DCA apportera un avantage clinique dans le traitement du cancer, n’ont pas été entièrement élucidées.
Plus de 70 % des cancers du sein expriment le récepteur d’œstrogènes (ER) et dépendent des œstrogènes pour la croissance et la progression de la tumeur [6]. Ainsi, le traitement endocrinien doit être considéré comme complémentaire à la chirurgie chez la majorité des patientes, car il induit une rémission tumorale et apporte des avantages cliniques constants. Le tamoxifène, un médicament anti-œstrogène, est le traitement le plus couramment utilisé chez les patientes atteintes d’un cancer du sein ER-positif, tant au stade précoce qu’au stade avancé/métastatique [7]. En tant que traitement adjuvant du cancer du sein au stade précoce, le tamoxifène améliore la survie globale, et l’on pense que son utilisation généralisée a contribué de manière significative à la réduction de la mortalité par cancer du sein observée au cours de la dernière décennie [8]. Malgré les avantages évidents du traitement par le tamoxifène dans le cancer du sein, presque toutes les patientes atteintes d’une maladie métastatique et jusqu’à 25 % des patientes recevant un traitement adjuvant par le tamoxifène finissent par rechuter et mourir de la maladie [9, 10]. Les mécanismes biologiques qui sous-tendent la résistance intrinsèque (de novo) et acquise au tamoxifène revêtent donc une importance clinique considérable. Une meilleure compréhension de ces mécanismes pourrait suggérer de nouvelles stratégies pour surmonter la résistance au tamoxifène et améliorer le traitement du cancer du sein.
Dans la présente étude, nous avons démontré que l’expression de l’EGFR dans les cellules du cancer du sein était diminuée par le traitement au DCA. Une combinaison de DCA et de tamoxifène a permis de réduire davantage les niveaux d’EGFR. Nous avons montré que le DCA renforçait la cytotoxicité du tamoxifène sur les cellules cancéreuses du sein en inhibant l’expression de l’EGFR. En outre, le DCA a sensibilisé les cellules MCF7 résistantes au tamoxifène au tamoxifène par le biais de la régulation négative de l’EFGR. Ces résultats suggèrent l’utilisation potentielle du DCA dans le traitement du cancer du sein en atténuant la voie de signalisation de l’EGFR.
Résultats
L’inhibition de la PDK réduit l’EGFR et augmente la mort cellulaire induite par le tamoxifène dans les cellules de cancer du sein
Des études récentes ont montré que le ciblage de la PDK par un inhibiteur de la PDK, tel que le DCA, fait passer le métabolisme des cellules cancéreuses de la glycolyse à la phosphorylation oxydative en déphosphorylant la pyruvate déshydrogénase mitochondriale [4, 11]. Pour élucider la manière dont les voies de signalisation des facteurs de croissance et des kinases, conjointement avec la PDK, régulent l’effet Warburg dans le cancer du sein, nous avons examiné les niveaux d’expression de plusieurs récepteurs de facteurs de croissance dans les cellules MCF7 dont la PDK a été supprimée. Il est intéressant de noter que la déplétion de la PDK par un traitement à l’ARNsi a entraîné une diminution de l’EGFR, alors que les autres récepteurs de facteurs de croissance n’ont subi aucun effet ou des effets marginaux (figure 1A). Quatre isoenzymes PDK (PDK1, PDK2, PDK3, PDK4) ont été identifiés dans les tissus des mammifères [12]. Pour confirmer la régulation négative de l’EGFR par le traitement par siPDK, nous avons étudié l’expression de l’EGFR dans les cellules traitées avec un siRNA contre chaque isoforme de PDK. Chaque traitement par siRNA a seulement aboli l’expression du PDK ciblé, mais tous ont provoqué la régulation négative de l’EGFR, ce qui suggère que le PDK ne peut pas réguler l’expression de l’EGFR d’une manière isoforme-spécifique (Figure 1B). Conformément à ces résultats, le DCA a également réduit les niveaux d’EGFR de manière dose-dépendante (Figure 1C). Lorsque nous avons analysé la concentration de lactate dans les milieux de culture, nous n’avons constaté aucun changement significatif de la concentration de lactate par le traitement au tamoxifène/DCA ou à l’ARNsi contre l’EGFR, même si elle a été réduite par le traitement au DCA (figure supplémentaire S1). Nous suggérons donc que la régulation négative de l’EGFR n’est peut-être pas associée au changement métabolique induit par le DCA dans les cellules cancéreuses du sein. L’activation de la voie de signalisation de l’EGFR contribuant à la résistance au tamoxifène [13], nous avons examiné si la régulation négative de l’EGFR par l’inhibition de la PDK sensibilisait les cellules au tamoxifène. Comme le montre la figure 1D, le co-traitement avec le tamoxifène et le DCA a entraîné une réduction marquée de la viabilité cellulaire. Le traitement combiné pendant 72 heures a réduit la viabilité cellulaire à moins de 30 % de celle du témoin (figure 1E). Ensuite, la mort cellulaire dans les cellules co-traitées a été évaluée par coloration à l’Annexin V/PI. Quarante-huit heures après le traitement, l’association du tamoxifène et du DCA a induit une mort cellulaire de 30 %, contre 15 % ou 8 % pour le tamoxifène ou le DCA seuls, respectivement (figure 1F). Nous avons précédemment signalé que la mort cellulaire apoptotique dans les cellules de cancer du sein était causée par la perte du potentiel de la membrane mitochondriale (MMP) [14]. Nous avons donc vérifié si la perte du potentiel de la membrane mitochondriale était impliquée dans la mort cellulaire induite par le co-traitement, mais aucun changement significatif du potentiel de la membrane mitochondriale n’a été observé entre les cellules non traitées et celles traitées par le tamoxifène/DCA (figure supplémentaire S2).
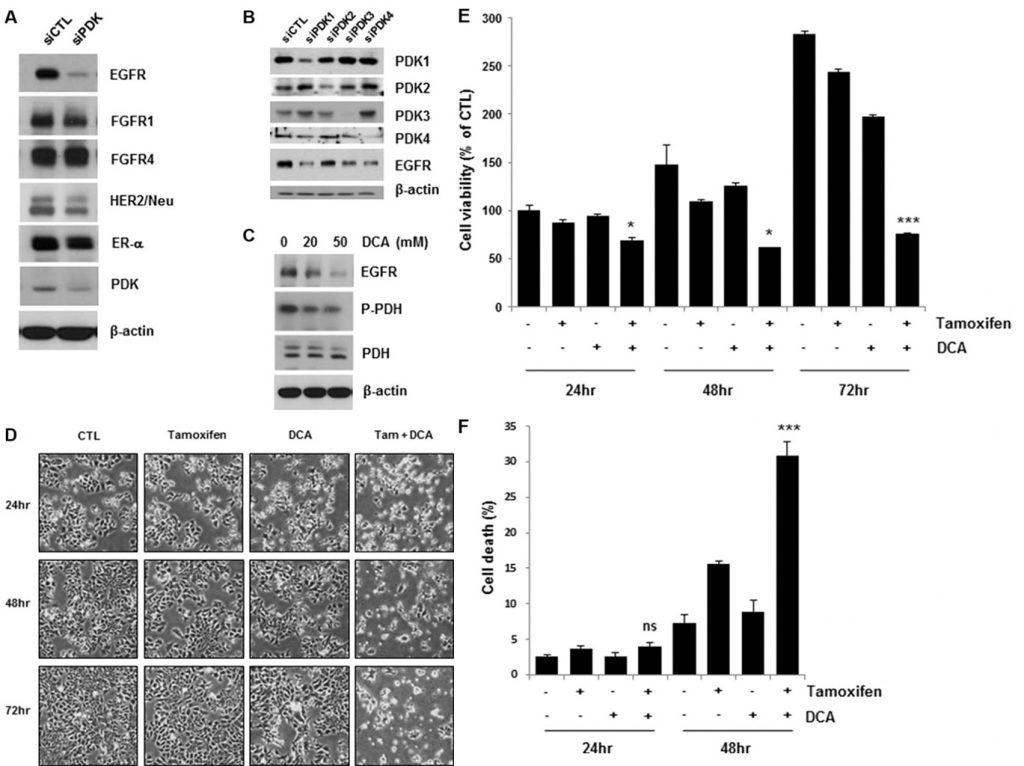
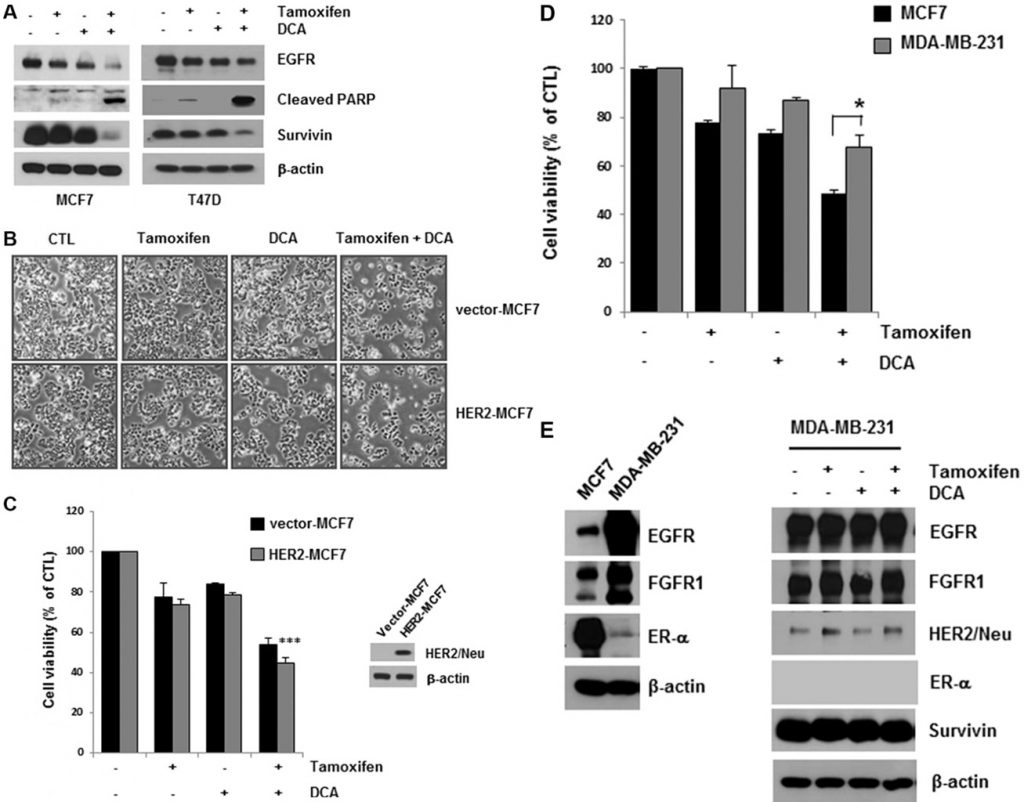
L’association de DCA et de tamoxifène a permis de réduire davantage les taux d’EGFR dans les cellules MCF7 et T47D par rapport à l’association de DCA seul (figure 2A). La mort cellulaire induite par le co-traitement a été confirmée par la détection du clivage de PARP, un marqueur d’apoptose (figure 2A). La survivine est une molécule anti-apoptotique ainsi qu’une cible du RE [15]. Le co-traitement a également entraîné une diminution de la survivine, ce qui pourrait favoriser l’apoptose dans les cellules (figure 2A). Bien que le traitement au tamoxifène ait légèrement diminué les niveaux d’EGFR dans les cellules MCF7 et T47D, aucune augmentation significative de la mort cellulaire n’a été observée dans les cellules, ce qui suggère qu’un niveau critique d’EGFR est nécessaire pour la survie des cellules cancéreuses du sein (Figure 2A).
Des preuves provenant de lignées cellulaires ont montré que la surexpression des voies HER2 peut contribuer à la résistance acquise aux thérapies endocriniennes [13]. Pour déterminer si la surexpression de HER2 influence la cytotoxicité du tamoxifène et du DCA, nous avons examiné la viabilité cellulaire des cellules MCF7 surexprimant HER2 (HER2-MCF7) après traitement par tamoxifène et DCA. Les résultats ont montré que le tamoxifène et le DCA réduisaient considérablement la viabilité cellulaire, même dans les cellules HER2-MCF7 (figures 2B et 2C), ce qui suggère que le DCA pourrait renforcer la mort cellulaire induite par le tamoxifène dans les cellules cancéreuses du sein surexprimant HER2. Nous avons ensuite évalué les effets inhibiteurs de croissance du co-traitement sur la lignée cellulaire de cancer du sein triple négatif MDA-MB-231. Comme le montre la figure 2D, les cellules MDA-MB-231 étaient moins sensibles au tamoxifène et au DCA que les cellules MCF7. La régulation négative de l’EGFR étant observée dans les cellules ER-positives, nous avons examiné les effets du tamoxifène et du DCA sur les niveaux d’EGFR dans les cellules MDA-MB-231. L’EGFR était fortement exprimé dans les cellules MDA-MB-231 par rapport aux cellules MCF7, et les niveaux n’ont pas été significativement réduits par le tamoxifène et le DCA (Figure 2E). Ensuite, nous avons examiné la cytotoxicité du tamoxifène et du DCA dans la lignée de cellules épithéliales mammaires immortalisées non tumorigènes MCF10A. Il est intéressant de noter que l’expression de l’EGFR dans les cellules MCF10A était comparable à celle des cellules MDA-MB-231 et que ni la régulation négative de l’EGFR ni la mort cellulaire n’ont été observées dans les cellules MCF10A après traitement par le tamoxifène et le DCA (figure supplémentaire S3). Ces résultats indiquent que les effets anti-prolifératifs du tamoxifène et du DCA sur les cellules cancéreuses du sein dépendent de la régulation négative de l’EGFR.
Letraitement combiné de tamoxifène et de DCA induit une dégradation de l’EGFR médiée par p38 MAPK
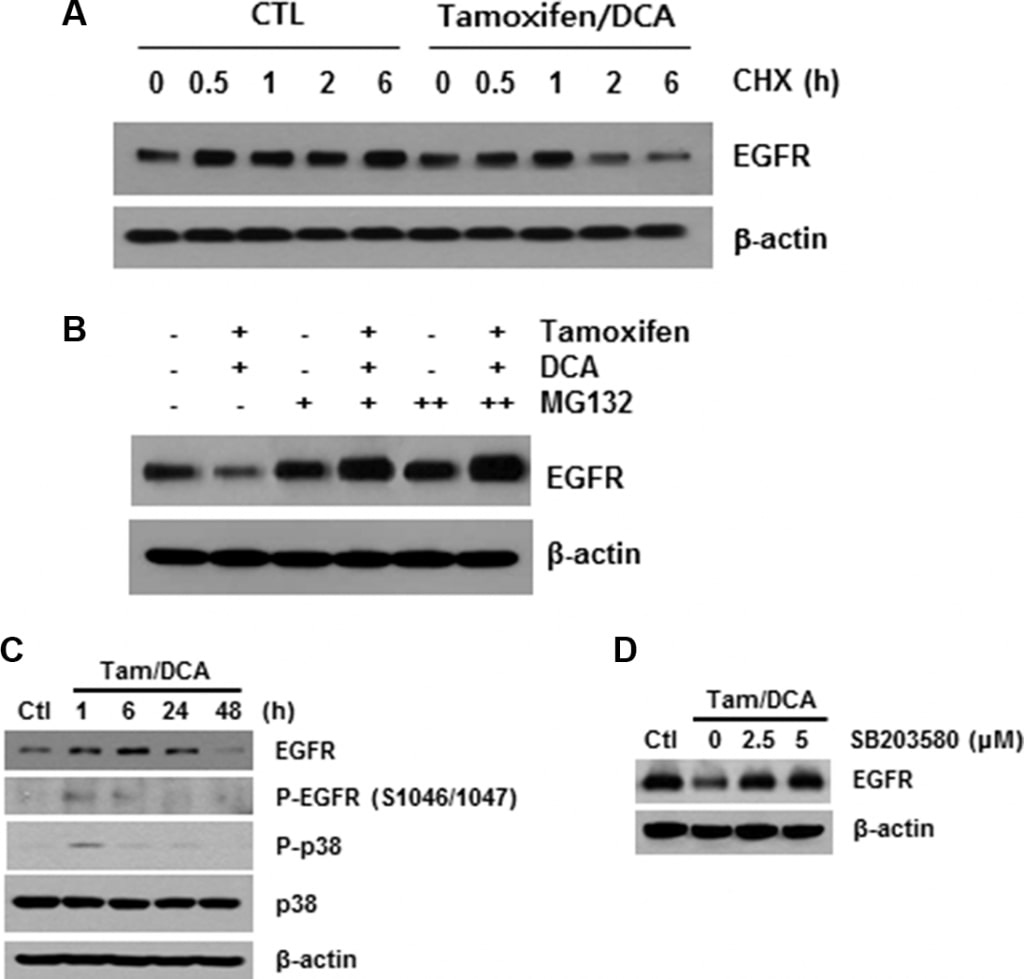
Comme décrit ci-dessus, la liaison au ligand provoque une autophosphorylation rapide, ce qui entraîne l’élimination de l’EGFR de la surface cellulaire par endocytose dans un compartiment endosomal précoce [16]. Par conséquent, nous avons ensuite étudié le rôle de la modification du récepteur dans la régulation négative de l’EGFR médiée par le tamoxifène/DCA. Après avoir bloqué la synthèse des protéines à l’aide de cycloheximide, nous avons constaté que la stabilité de l’EGFR était considérablement compromise dans les cellules traitées par le tamoxifène/DCA par rapport à celles du groupe témoin (figure 3A). Nous avons ensuite évalué les effets du MG132, un inhibiteur du protéasome, sur la dégradation de l’EGFR induite par le tamoxifène/DCA. Le traitement par le MG132 a rétabli l’expression de l’EGFR dans les cellules traitées par le tamoxifène/DCA de manière dose-dépendante (figure 3B).
La phosphorylation de l’EGFR sur les résidus sérine et thréonine représente un mécanisme d’atténuation de l’activité de l’EGFR, et parmi eux, les sites de phosphorylation de la sérine 1046/1047 (Ser 1046/7) sont nécessaires à la désensibilisation de l’EGFR [17]. Il a récemment été signalé que la protéine kinase activée par des mitogènes (MAPK) p38 induit la phosphorylation de l’EGFR à Ser 1046/7, ce qui entraîne sa dégradation dans les cellules cancéreuses [18]. Par conséquent, nous avons ensuite examiné les effets du tamoxifène et du DCA sur la phosphorylation de p38 MAPK dans les cellules MCF7. p38 MAPK a été significativement phosphorylé en 1 h, et la phosphorylation a été maintenue pendant 24 h après le traitement au tamoxifène et au DCA (Figure 3C). De plus, la dégradation de l’EGFR induite par le co-traitement a été considérablement supprimée lorsque les cellules ont été prétraitées avec un inhibiteur spécifique de p38 MAPK, le SB203580 (Figure 3D), ce qui indique que l’activation de p38 MAPK joue un rôle dans la dérégulation de l’EGFR induite par le tamoxifène/DCA dans les cellules MCF7.
Lesinhibiteurs de l’EGFR augmentent la mort cellulaire induite par le tamoxifène
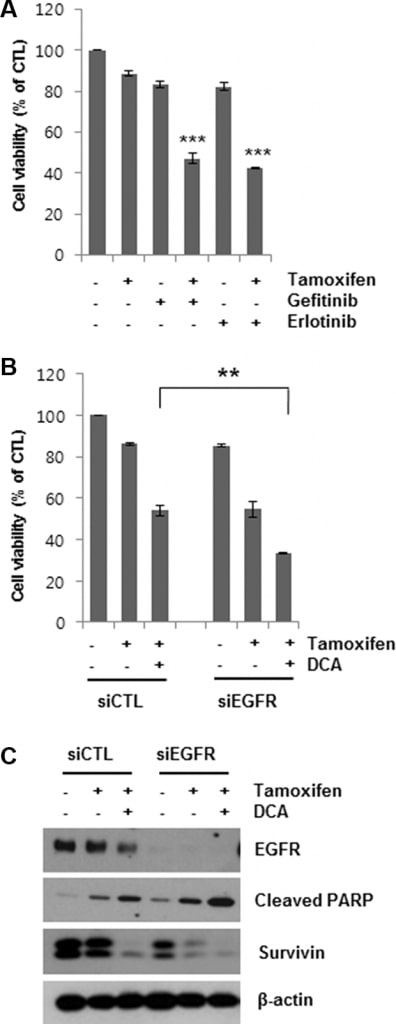
Ayant montré que la dégradation de l’EGFR médiée par le DCA pouvait augmenter la mort cellulaire induite par le tamoxifène dans les cellules MCF7, nous avons ensuite déterminé si l’inhibition de l’EGFR augmentait la mort cellulaire dans les cellules traitées par le tamoxifène. Le géfitinib et l’erlotinib sont des inhibiteurs sélectifs et réversibles de la liaison de la tyrosine kinase EGFR à l’ATP [19, 20]. Le co-traitement des cellules MCF7 avec 5 μM de géfitinib ou d’erlotinib pendant 48 h a nettement augmenté la mort cellulaire induite par le tamoxifène (figure 4A). De même, le knockdown de l’EGFR à l’aide du traitement siEGFR a augmenté la mort cellulaire induite par le tamoxifène (Figure 4B). La réduction de la viabilité cellulaire par le traitement combiné du siEGFR et du tamoxifène était comparable à celle obtenue par le DCA et le tamoxifène. En outre, le traitement par le siEGFR a permis de réduire davantage les niveaux d’EGFR dans les cellules traitées par le tamoxifène/DCA, ce qui a entraîné une augmentation de la mort cellulaire apoptotique ainsi qu’une régulation négative de la survivine par rapport au traitement par le si-contrôle (figure 4C). Ces résultats confirment la conclusion selon laquelle le DCA sensibilise les cellules cancéreuses du sein au tamoxifène par le biais de la régulation négative de l’EGFR.
l’expression de c-myc et de Nanog est associée aux effets cytotoxiques du DCA dans les cellules MCF7 traitées au tamoxifène
Il a été démontré que l’activation de l’EGFR dans le cancer augmente divers facteurs de transcription qui peuvent affecter le type et la durée de la signalisation de l’EGFR [21]. Parmi eux, Nanog et c-myc jouent un rôle pléiotropique dans la tumorigenèse, y compris dans la résistance au traitement standard du carcinome mammaire [22, 23]. Afin d’étudier plus en détail les activités antitumorales médiées par la réduction de l’EGFR dans les cellules cancéreuses du sein, nous avons examiné l’expression de c-myc et de Nanog dans les cellules MCF7 après un co-traitement au tamoxifène et au DCA. Les niveaux de c-myc et de Nanog ont diminué de manière significative dans les cellules traitées par tamoxifène et DCA (Figure 5A). De même, les expressions des deux protéines ont été supprimées par le tamoxifène en association avec le gefitinib ou l’erlotinib (figure 5B), ce qui indique que l’expression de l’EGFR est nécessaire pour maintenir l’expression de c-myc et de Nanog dans les cellules MCF7 traitées par le tamoxifène. Néanmoins, le siEGFR n’a eu que peu ou pas d’effet sur l’expression de c-myc et Nanog, ce qui indique que la régulation négative de l’EGFR est nécessaire mais pas suffisante pour diminuer l’expression des protéines (figure supplémentaire S4). Pour déterminer si l’expression réduite de ces deux protéines est impliquée dans les effets cytotoxiques du tamoxifène et du DCA dans les cellules MCF7, nous avons testé les effets des siRNA contre c-myc et Nanog dans les cellules exposées au tamoxifène/DCA. Le knockdown de c-myc et Nanog a significativement sensibilisé les cellules au tamoxifène/DCA par rapport aux contrôles (Figure 5C). Inversement, la surexpression de FLAG-c-myc et FLAG-Nanog par transfection des cellules avec les vecteurs FLAG-c-myc et FLAG-Nanog a significativement protégé les cellules de la cytotoxicité induite par le tamoxifène/DCA (Figure 5D). L’ensemble de nos données suggère que la régulation négative de l’EGFR par une combinaison de tamoxifène et de DCA peut induire la mort cellulaire des cellules MCF7 en partie par l’inhibition de l’expression de c-myc et Nanog. Il est connu que les gènes d’auto-renouvellement, tels que c-myc et Nanog, sont associés aux propriétés des cellules souches cancéreuses 24. Pour vérifier si le co-traitement par le tamoxifène et le DCA inhibe les cellules semblables aux cellules souches du cancer du sein, nous avons effectué une analyse par cytométrie en flux pour estimer la proportion de la sous-population de cellules semblables aux cellules souches dans les cellules MCF7 sur la base de l’expression de CD44. Après le co-traitement, la population à forte expression CD44 dans les cellules MCF7 a été réduite de 42,5 % à 19,9 % (figure 5E). Cette découverte a soulevé la possibilité que le DCA et le tamoxifène puissent inhiber la capacité des cellules semblables aux cellules souches cancéreuses dans les cellules du cancer du sein.

Letraitement combiné de DCA et de tamoxifène peut surmonter la résistance au tamoxifène dans les cellules de cancer du sein
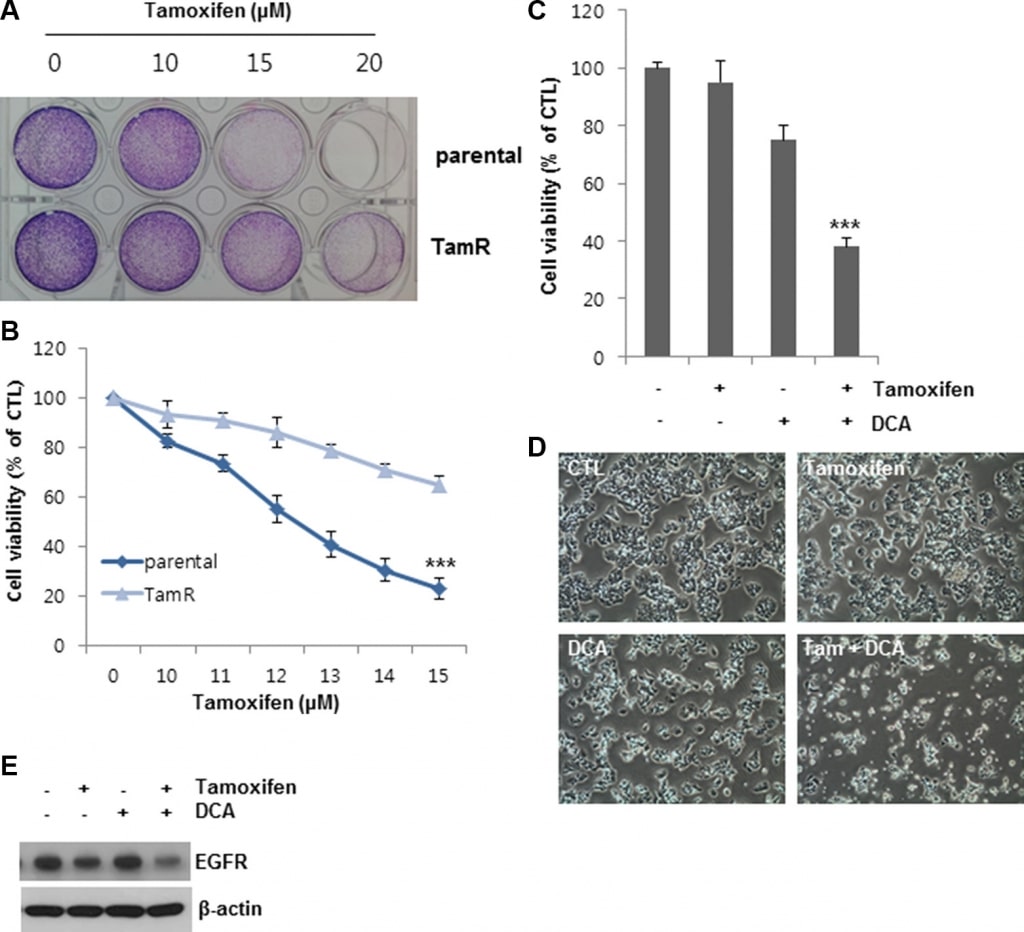
Pour confirmer davantage les effets observés du DCA sur la mort cellulaire induite par le tamoxifène, nous avons établi des cellules MCF7 résistantes au tamoxifène (TamR) par un traitement au tamoxifène sur une longue période. Par rapport aux cellules non traitées, la viabilité cellulaire des cellules MCF7 et TamR MCF7 était de 20 et 60 %, respectivement, après un traitement par 13 μM de tamoxifène, ce qui indique que les cellules TamR MCF7 étaient moins sensibles à la même concentration de tamoxifène que les cellules parentales MCF7 (figures 6A et 6B). Le DCA seul a inhibé la croissance des cellules TamR MCF7 d’environ 25 % par rapport au témoin (Figure 6C et 6D). En revanche, le co-traitement du DCA avec le tamoxifène a supprimé la croissance cellulaire de plus de 60 % par rapport au contrôle (Figure 6C et 6D). Les niveaux d’EGFR dans les cellules TamR ont également été réduits par le co-traitement (figure 6E). Ces résultats suggèrent que le DCA pourrait surmonter la résistance au tamoxifène des cellules cancéreuses du sein par la régulation négative de l’EGFR au niveau des protéines.
Discussion
La compréhension du phénotype métabolique des cellules tumorales s’étant accrue, le ciblage des différences métaboliques entre les cellules tumorales et normales a été proposé comme une nouvelle stratégie anticancéreuse. Malgré l’augmentation du nombre de médicaments anticancéreux potentiels qui ciblent les processus métaboliques des cellules cancéreuses, il sera nécessaire d’élucider la façon dont les cellules répondent aux médicaments pour réussir à cibler le métabolisme du cancer. Nous avons démontré ici que le DCA inhibe non seulement l’activité de la PDK mais aussi l’expression de l’EGFR dans les cellules cancéreuses du sein. Le traitement combiné de DCA et de tamoxifène a induit une dégradation des protéines EGFR dépendante du protéasome dans les cellules cancéreuses du sein via l’activation de p38 MAPK. Nos résultats indiquent que la régulation négative de l’EGFR joue un rôle clé dans la mort cellulaire apoptotique induite par une combinaison de DCA et de tamoxifène. À notre connaissance, il s’agit du premier rapport montrant que le DCA sensibilise les cellules cancéreuses du sein à la mort cellulaire induite par le tamoxifène via la régulation négative de l’EGFR.
Dans les cellules cancéreuses du sein ER-positives qui ont développé une résistance endocrinienne, l’expression du RE peut être directement supprimée par une signalisation accrue des récepteurs des facteurs de croissance due à la surexpression de l’EGFR et de HER2, qui activent ensuite MAPK et inhibent la transcription du RE [25]. En outre, la résistance au tamoxifène dans les cellules cancéreuses du sein a été liée à la surexpression de l’EGFR et à des niveaux élevés de kinase 1/2 activée extracellulaire phosphorylée [2]. Ainsi, la stratégie consistant à combiner un inhibiteur de l’EGFR avec un agent endocrinien est suffisamment intéressante pour justifier une étude plus approfondie du traitement du cancer du sein. Dans cette étude, nous avons constaté que la régulation négative de l’EGFR dans les cellules de cancer du sein traitées par le DCA augmentait la sensibilité des cellules au tamoxifène. La régulation négative de l’EGFR par le co-traitement du DCA avec le tamoxifène a été confirmée dans plusieurs lignées cellulaires de cancer du sein ER-positif, notamment MCF7, T47D et BT474 (figure 2A et non montré). En plus de la lignée cellulaire de cancer du sein BT474 HER2-amplifiée, les cellules HER2-MCF7 ont également montré une régulation négative de l’EGFR par le DCA/tamoxifène, ce qui suggère que le traitement combiné sera applicable au cancer du sein HER2-positif. Malheureusement, une lignée cellulaire de cancer du sein triple négatif, MDA-MB-231, qui est connue pour surexprimer l’EGFR [26], a montré un phénotype réfractaire ainsi qu’aucun changement évident des niveaux d’EGFR en réponse au co-traitement. L’une des possibilités est que l’amplification de l’EGFR dans les cellules MDA-MB-231 écrase sa régulation négative par le co-traitement du DCA avec le tamoxifène. L’autre explication est que la ou les voies de signalisation qui interviennent dans la régulation négative de l’EGFR sont bloquées par un mécanisme inconnu dans les cellules MDA-MB-231. Des recherches plus approfondies seront nécessaires pour élucider le mécanisme sous-jacent du maintien de l’expression de l’EGFR en réponse au co-traitement par le DCA et le tamoxifène dans les cellules cancéreuses mammaires ER-négatives.
On connaît quatre isoformes de PDK, chacune étant active en réponse à différentes conditions intracellulaires et extracellulaires. La PDK1 est activée par l’hypoxie [27]; la PDK2 est activée par les produits de la PDH, l’acétyl CoA et le NADH [28]; la PDK3 est activée par l’ATP [29]; et la PDK4 est régulée transcriptionnellement par des signaux hormonaux [30]. PDK2 possède la plus grande activité de phosphorylation du complexe pyruvate déshydrogénase, suivie de PDK4, PDK1 et PDK3 [12]. PDK2 est le plus susceptible d’être inhibé par le DCA en raison de son expression ubiquitaire [31]. Bien que nous ayons détecté l’expression de quatre isoformes dans les cellules MCF7 (Figure 1C), les différences fonctionnelles entre les isoformes de PDK dans le cancer du sein restent encore insaisissables. Il sera intéressant d’étudier plus avant si les voies de signalisation supprimant une isoforme particulière de PDK entraînent une régulation négative de l’EGFR. La PDK est connue pour fonctionner dans les mitochondries, et plusieurs études ont démontré que l’EGFR se déplace également vers les mitochondries après une stimulation par l’EGF [32, 33]. En outre, il a été suggéré que l’interaction entre l’EGFR et la PDK dans la matrice mitochondriale joue un rôle important dans la croissance tumorale induite par l’EGFR dans le glioblastome multiforme [34]. Ici, nous avons constaté que le blocage de l’activité de la PDK par un inhibiteur ou un silencieux diminuait les niveaux cellulaires totaux d’EGFR dans les cellules de cancer du sein traitées au tamoxifène. D’autres études sont nécessaires pour déterminer si seul l’EGFR associé aux mitochondries est soumis à une dégradation par le co-traitement avec le DCA et le tamoxifène dans les cellules cancéreuses du sein. En général, l’exposition des cellules à l’EGF provoque une autophosphorylation rapide, notamment de la tyrosine (Tyr) 1045, qui fournit un site d’accueil pour l’ubiquitine ligase c-Cbl, ce qui entraîne l’ubiquitination de l’EGFR et son élimination par endocytose de la surface cellulaire vers un compartiment endosomal précoce [35]. Cependant, le co-traitement avec le DCA et le tamoxifène n’a eu aucun effet sur la phosphorylation de l’EGFR sur Tyr 1045 (données non présentées). Au contraire, nous avons observé la phosphorylation de l’EGFR à Ser 1046/7 par le DCA et le tamoxifène, qui a été bloquée par un inhibiteur spécifique de p38 MAPK, le SB203580. Il a été suggéré que p38 MAPK joue un rôle central à la fois dans l’internalisation et la dégradation de l’EGFR [18]; cependant, des recherches supplémentaires sont nécessaires pour élucider comment la phosphorylation de l’EGFR au niveau des résidus sérine entraîne sa régulation négative et pour identifier le médiateur en amont de l’activation de p38 MAPK après un traitement au DCA/tamoxifène.
Nanog et c-myc ont été identifiés comme des facteurs de transcription, et leur rôle dans le maintien de l’autorenouvellement des cellules souches embryonnaires a été établi dans des études antérieures [22, 36]. Un mécanisme récemment proposé de résistance intrinsèque à la thérapie endocrinienne postule l’existence d’un sous-ensemble spécialisé de cellules cancéreuses appelé cellules initiatrices de tumeurs (TIC) [37]. Les TIC ont la capacité de s’auto-renouveler et de générer de nouvelles tumeurs entièrement constituées de types cellulaires dérivés de clones présents dans la tumeur parentale. Ici, nous avons démontré que le DCA diminuait l’expression de c-myc et de Nanog dans les cellules MCF7 traitées au tamoxifène. De plus, l’expression de ces protéines était liée aux effets cytotoxiques du traitement combiné de DCA et de tamoxifène. Par conséquent, le DCA peut être exploité pour étendre le potentiel thérapeutique du tamoxifène en supprimant le développement de la résistance intrinsèque dans le traitement du cancer du sein.
En conclusion, nos résultats ont révélé que le DCA sensibilisait les cellules cancéreuses du sein ER-positives au tamoxifène en diminuant les niveaux d’EGFR. Le traitement au DCA et au tamoxifène a inhibé l’expression des gènes d’auto-renouvellement et la survie des cellules MCF7 résistantes au tamoxifène. Par conséquent, nous proposons que le DCA puisse être un agent thérapeutique efficace pour traiter le cancer du sein résistant au tamoxifène. En outre, les stratégies d’association avec le DCA peuvent être utiles pour améliorer l’efficacité du traitement d’autres chimiothérapies cytotoxiques ou de thérapies ciblées. D’autres expériences, notamment des études animales et des essais cliniques, devraient être menées à l’avenir.
Matériel et méthodes
Culture cellulaire et réactifs
Les cellules de cancer du sein humain MCF7, T47D et MDA-MB-231 ont été achetées auprès de l’American Type Culture Collection (Rockville, MD, USA) et ont été cultivées dans le milieu de croissance recommandé (Invitrogen, Carlsbad, CA, USA). Les cellules MCF7 surexprimant HER2 (HER2-MCF7) et les cellules du vecteur de contrôle (vector-MCF7) ont été aimablement fournies par le Dr Incheol Shin (Université Hanyang, Séoul, Corée). Les cellules MCF7 résistantes au tamoxifène (TamR) ont été développées en cultivant des cellules MCF7 en présence de 10 μM de tamoxifène pendant plus de 6 mois. À titre de contrôle, les cellules parentales ont été cultivées pendant la même durée dans un milieu ordinaire. Après l’établissement de la résistance, les cellules ont été passées pendant 3 mois au maximum. Pour les expériences impliquant des traitements au tamoxifène, les cellules ont été systématiquement cultivées dans un milieu Dulbecco’s modified Eagle’s exempt de rouge de phénol et contenant 10 % de sérum fœtal bovin dépouillé de charbon de bois juste avant les traitements. Les anticorps contre EGFR, phospho-EGFR (S1046/1047), FGFR1, FGFR4, PDK1, PDH, survivine, PARP clivé, p38, phospho-p38 et Nanog ont été acquis auprès de Cell Signaling Technology (Danvers, MA, USA). Les anticorps contre HER2/Neu, ER-α et c-myc, les siRNA ciblant EGFR, Nanog, PDK4 et c-myc et les siRNA de contrôle négatif (brouillés) ont été acquis auprès de Santa Cruz Biotechnology (Dallas, TX, USA). Les anticorps contre PDK2 et PDK3 ont été acquis auprès de Thermo Fisher Scientific (Waltham, MA, USA). L’anticorps β-actine, l’anticorps FLAG, le tamoxifène, le DCA, le cycloheximide et le MG132 ont été achetés auprès de Sigma-Aldrich (St. Louis, MO, USA). L’anticorps phospho-PDH (S293) et le SB203580 ont été fournis par BD Biosciences Pharmingen (San Diego, CA, USA), et le gefitinib et l’erlotinib ont été obtenus auprès de Selleck Chemicals (London, ON, Canada).
Transfections et traitements
Les cellules ont été transfectées avec le siRNA cible (50 nM) en utilisant Lipofectamine RNAiMAX (Invitrogen) comme décrit par le fabricant. Les cellules ont été transfectées avec 1 μg de FLAG-c-myc pcDNA 3.1 et FLAG-Nanog-pcDNA 3.1 en utilisant la Lipofectamine 2000 comme décrit par le fabricant. Après 6 h, les cellules ont été traitées avec du tamoxifène et/ou du DCA pendant 24-48 h, puis analysées comme décrit ailleurs.
Mesure de la viabilité cellulaire
La viabilité cellulaire a été déterminée en mesurant la conversion mitochondriale du bromure de 3-(4,5-diméthylthiazolyl2)-2,5-diphényltétrazolium (MTT) en un produit coloré. Les cellules ont été traitées comme indiqué, et le milieu a été remplacé par un milieu sans sérum contenant 1 mM de MTT. Après 2 h d’incubation à 37°C, les cellules ont été solubilisées dans du DMSO. La quantité de formazan, la forme convertie du MTT, a été déterminée en mesurant l’absorbance à 595 nm. Évaluation de l’apoptose
L’apoptose a été déterminée par une analyse de tri cellulaire activé par fluorescence à l’aide d’un kit d’apoptose Annexin V-FITC (BioVision, Milpitas, CA, USA) selon les instructions du fabricant. En bref, après le traitement, les cellules ont été traitées avec de la trypsine puis remises en suspension dans un tampon de liaison (10 mM HEPES/NaOH, pH 7,4, 140 mM NaCl, 2,5 mM CaCl2) comprenant de l’Annexin V-FITC et de l’iodure de propidium. Après une incubation de 15 min, la fluorescence des cellules a été analysée par cytométrie de flux. La mort cellulaire a été mesurée comme le pourcentage de cellules dans la population positive à l’Annexine V et au PI.
Western blotting
Les cellules ont été récoltées et lysées dans un tampon RIPA (50 mM Tris-HCl pH 7,5, 150 mM NaCl, 1% Nonidet P40, 0,5% de désoxycholate de sodium et 0,1% de SDS) complété par un cocktail d’inhibiteurs de protéase/phosphatase (Roche, Mannheim, Allemagne). Des quantités égales de protéines (20-50 μg) ont été séparées par SDS-PAGE et transférées sur une membrane de nitrocellulose. Les membranes ont été bloquées par incubation avec du lait écrémé à 5 % dans une solution saline trisbuffée pendant 1 h, puis ont été incubées pendant la nuit avec les anticorps primaires appropriés. Les membranes ont été incubées avec un anticorps secondaire conjugué au HRP pendant 1 h. Les protéines immunoréactives ont été visualisées à l’aide de réactifs de chimiluminescence améliorée (Amersham Biosciences, Little Chalfont, UK).
Détection de la population cellulaire positive CD44
Les cellules ont été colorées avec des anticorps à une dilution de 1∶100 dans du PBS pendant 15 minutes. Les anticorps utilisés étaient les suivants : les anticorps de contrôle de l’isotype IgG de souris FITC-CD44 et FITC-conjugués ont été obtenus auprès de BD Biosciences Pharmingen. Les cellules marquées ont été analysées par cytométrie en flux. Les populations de cellules positives CD44 ont été déterminées par l’intensité du FITC.
Analyse statistique
Toutes les données présentées sont représentatives d’au moins deux expériences distinctes. Les comparaisons entre les groupes ont été analysées à l’aide du test t de Student. Les astérisques (***p < 0,001, **p < 0,01, *p < 0,05) indiquent la signification statistique.
Remerciements et financement
Cette recherche a été soutenue par le programme de recherche scientifique de base de la Fondation nationale de la recherche de Corée (NRF), financé par le ministère des Sciences, des TIC et de la Planification future (n° 1711031812, 1711023318 et 1711031800).
Conflits d’intérêts
Les auteurs ne déclarent aucun conflit d’intérêts.
RÉFÉRENCES
1 1. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect : the metabolic requirements of cell proliferation. Science. 2009 ; 324:1029-33.
2ZhaoY, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013 ; 4:e532.
3 Vander Heiden MG. Cibler le métabolisme du cancer : une fenêtre thérapeutique s’ouvre. Nat Rev Drug Discov. 2011 ; 10:671-84.
4 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibit cancer growth. Cancer Cell. 2007 ; 11 : 37-51.
5 Kankotia S, Stacpoole PW. Dichloroacétate et cancer : une nouvelle maison pour un médicament orphelin ? Biochim Biophys Acta. 2014 ; 1846:617-29.
6 Osborne CK. Le tamoxifène dans le traitement du cancer du sein. N Engl J Med. 1998 Nov 26 ; 339:1609-18.
7 Johnston SR. Nouvelles stratégies dans le cancer du sein à récepteurs d’œstrogènes positifs. Clin Cancer Res. 1 avril 2010 ; 16:1979-87. doi : 10.1158/1078-0432.CCR-09-1823.
8 Peto R, Boreham J, Clarke M, Davies C, Beral V. Les décès par cancer du sein au Royaume-Uni et aux États-Unis ont diminué de 25 % en 2000 chez les 20-69 ans. Lancet. 2000 ; 355:1822.
9 Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival : an overview of the randomised trials. Lancet. 2005 ; 365:1687-717.
10 Early Breast Cancer Trialists’ Collaborative Group (EBCTCG), Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, Dowsett M, Ingle J, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen : patient-level meta-analysis of randomised trials. Lancet. 2011 ; 378:771-84.
11 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Le ciblage combiné de PDK1 et EGFR déclenche la régression du glioblastome en inversant l’effet Warburg. Cancer Res. 2013 ; 73:7277-89.
12 Saunier E, Benelli C, Bortoli S. Le complexe pyruvate déshydrogénase dans le cancer : Un ancien portier métabolique régulé par de nouvelles voies et de nouveaux agents pharmacologiques. Int J Cancer. 2016 ; 138:809-17.
13 Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011 ; 62:233-47.
14YunSM, Woo SH, Oh ST, Hong SE, Choe TB, Ye SK, Kim EK, Seong MK, Kim HA, Noh WC, Lee JK, Jin HO, Lee YH, et al. La mélatonine améliore la mort cellulaire induite par le trioxyde d’arsenic via une régulation à la hausse soutenue de l’expression de Redd1 dans les cellules du cancer du sein. Mol Cell Endocrinol. 2016 ; 422:64-73.
15 Zhu J, Lu X, Hua KQ, Sun H, Yu YH, Feng YJ. Le récepteur α des œstrogènes médiatise l’amélioration par le 17β-estradiol de la motilité des cellules cancéreuses ovariennes par la régulation à la hausse de l’expression de la survivine. Arch Gynecol Obstet. 2012 ; 286 : 729-37.
16 Arteaga CL. Dépendance du récepteur du facteur de croissance épidermique dans les tumeurs humaines : plus qu’une simple expression ? Oncologue. 2002 ; 7:31-9.
17 Sorkin A, Goh LK. Endocytose et trafic intracellulaire des ErbBs. Exp Cell Res. 2009 ; 315:683-96.
18 Adachi S, Shimizu M, Shirakami Y, Yamauchi J, Natsume H, Matsushima-Nishiwaki R, To S, Weinstein IB, Moriwaki H, Kozawa O. (-)-Epigallocatechin gallate downregulates EGF receptor via phosphorylation at Ser1046/1047 by p38 MAPK in colon cancer cells. Carcinogenèse. 2009 ; 30:1544-52.
19CiardielloF, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000 ; 6:2053-63.
20 Pollack VA, Savage DM, Baker DA, Tsaparikos KE, Sloan DE, Moyer JD, Barbacci EG, Pustilnik LR, Smolarek TA, Davis JA, Vaidya MP, Arnold LD, Doty JL, et al. Inhibition of epidermal growth factor receptorassociated tyrosine phosphorylation in human carcinomas with CP-358,774 : dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther. 1999 ; 291:739-48.
21 Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. La signalisation du récepteur du facteur de croissance épidermique (EGFR) dans le cancer. Gene. 2006 ; 366:2-16.
22 Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014 ; 135:2741-8.
23 Chen Y, Olopade OI. MYC dans la progression des tumeurs du sein. Expert Rev Anticancer Ther. 2008 ; 8 : 1689-98.
24 Hou ZJ, Luo X, Zhang W, Peng F, Cui B, Wu SJ, Zheng FM, Xu J, Xu LZ, Long ZJ, Wang XT, Li GH, Wan XY, et al. Le flubendazole, un anthelminthique approuvé par la FDA, cible les cellules souches du cancer du sein. Oncotarget. 2015 ; 6:6326-40. doi:10.18632/oncotarget.3436.
25 Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. L’activation de la protéine kinase activée par des agents mitogènes dans les cellules cancéreuses mammaires à récepteur d’œstrogènes alpha positif in vitro induit un phénotype moléculaire in vivo de tumeurs mammaires humaines à récepteur d’œstrogènes alpha négatif. Cancer Res. 2006 ; 66:3903-11.
26 Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation : A major pathway of cell-cycle progression in estrogenreceptor negative breast cancer cells. Proc Natl Acad Sci U S A. 2000 ; 97:8542-7.
27 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF1-mediated expression of pyruvate dehydrogenase kinase : a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006 ; 3:177-85.
28 Hiromasa Y, Hu L, Roche TE. Ligand-induced effects on pyruvate dehydrogenase kinase isoform 2. J Biol Chem. 2006 ; 281:12568-79.
29 Kato M, Chuang JL, Tso SC, Wynn RM, Chuang DT. Structure cristalline de la pyruvate déshydrogénase kinase 3 liée au domaine lipoyle 2 du complexe pyruvate déshydrogénase humain. EMBO J. 2005 ; 24:1763-74.
30 Kwon HS, Huang B, Unterman TG, Harris RA. La protéine kinase B-alpha inhibe l’induction du gène de la pyruvate déshydrogénase kinase-4 humaine par la dexaméthasone par l’inactivation des facteurs de transcription FOXO. Diabetes. 2004 ; 53:899-910.
31 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998 ; 329 :191-6.
32 Dasari VR, Velpula KK, Alapati K, Gujrati M, Tsung AJ. Les cellules souches du sang de cordon inhibent la translocation du récepteur du facteur de croissance épidermique vers les mitochondries dans le glioblastome. PLoS One. 2012 ; 7:e31884.
33 Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, Hüttemann M, Douglas R, Haddad G, Parsons SJ. Translocation du récepteur du facteur de croissance épidermique vers la mitochondrie : régulation et effet. J Biol Chem. 2009 ; 284:36592-604.
34 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Le ciblage combiné de PDK1 et EGFR déclenche la régression du glioblastome en inversant l’effet Warburg. Cancer Res. 2013 ; 73:7277-89.
35 Massie C, Mills IG. Le rôle en développement des récepteurs et des adaptateurs. Nat Rev Cancer. 2006 ; 6:403-9.
36 Chappell J, Dalton S. Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harb Perspect Med. 2013;3:a014381.
37 Wei W, Lewis MT. Identifier et cibler les cellules initiatrices de tumeurs dans le traitement du cancer du sein. Endocr Relat Cancer. 2015 ; 22:R135-55.