Sang Hyeok Woo1,*, Sung-Keum Seo1,*, Yoonhwa Park1,2, Eun-Kyu Kim3, Min-Ki Seong4, Hyun-Ah Kim4, Jie-Young Song1, Sang-Gu Hwang1, Jin Kyung Lee5, Woo Chul Noh4, In-Chul Park1
1Divisionedi ricerca sul cancro da radiazioni, Istituto coreano di scienze radiologiche e mediche, Nowon-gu, Seoul, 01812, Repubblica di Corea
2Scuoladi scienze della vita e biotecnologie, Università della Corea, Seongbuk-gu, Seoul, 02841, Repubblica di Corea
3Dipartimento di chirurgia, Centro per il cancro al seno, Ospedale Bundang dell’Università nazionale di Seoul, Collegio di medicina dell’Università nazionale di Seoul, Bundang-gu, Seongnam, 13620, Repubblica di Corea
4Dipartimentodi chirurgia, Korea Cancer Center Hospital, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Seoul, 01812, Repubblica di Corea
5KIRAMSRadiation Biobank, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Seoul, 01812, Repubblica di Corea
*Questiautori hanno contribuito in egual misura a questo lavoro
Corrispondenza a: In-Chul Park, e-mail: [email protected]
Parole chiave: tamoxifene, cancro al seno, dicloroacetato, recettore del fattore di crescita epidermico, piruvato deidrogenasi chinasi
Ricevuto: 18 maggio 2016
Accettato: 22 luglio 2016
Pubblicato online: 01 agosto 2016
Abstract
La riprogrammazione metabolica nelle cellule tumorali è stata recentemente riconosciuta come un tratto distintivo essenziale della neoplasia. In questo contesto, le alterazioni metaboliche rappresentano un interessante bersaglio terapeutico e negli studi preclinici sono stati ottenuti risultati incoraggianti con farmaci mirati a vari processi metabolici. Recentemente, diversi studi hanno suggerito che il dicloroacetato (DCA), un inibitore specifico della piruvato deidrogenasi chinasi, può essere un potenziale farmaco antitumorale in un gran numero di tumori diversi. Tuttavia, il meccanismo preciso non è completamente compreso, il che è importante per l’uso del DCA nel trattamento del cancro. Nel presente studio, abbiamo scoperto che il DCA sensibilizza le cellule di cancro al seno MCF7 alla morte cellulare indotta dal tamoxifene, diminuendo l’espressione del recettore del fattore di crescita epidermico (EGFR). La downregulation di EGFR è stata causata dalla degradazione della proteina. Inoltre, la p38 mitogeno-attivata proteina chinasi ha svolto un ruolo importante nella degradazione dell’EGFR indotta da DCA/tamoxifene. Infine, il DCA ha anche promosso la morte cellulare indotta dal tamoxifene in cellule MCF7 resistenti al tamoxifene, ottenute con un trattamento a lungo termine con tamoxifene. In sintesi, i nostri risultati suggeriscono che il DCA è un potenziale farmaco interessante che sensibilizza le cellule alla morte cellulare indotta dal tamoxifene e supera la resistenza al tamoxifene attraverso la downregolazione dell’espressione dell’EGFR nelle cellule di cancro al seno.
INTRODUZIONE
Le cellule tumorali in fase di proliferazione hanno esigenze metaboliche notevolmente diverse rispetto alla maggior parte delle cellule normali differenziate. Ad esempio, per sostenere la rapida crescita e proliferazione cellulare, le cellule tumorali alterano in modo differenziato il flusso metabolico rispetto al tessuto circostante, per fornire una quantità sufficiente di bioenergetici e intermedi biosintetici. Un fenomeno ben noto osservato nella maggior parte delle cellule tumorali è il passaggio alla glicolisi aerobica, indipendentemente dall’apporto di ossigeno, definito “effetto Warburg”, in cui il piruvato viene convertito direttamente in acido lattico invece di entrare nel ciclo dell’acido citrico [1]. Poiché tutte le cellule tumorali dipendono da questa alterazione del metabolismo, queste vie alterate rappresentano interessanti bersagli terapeutici [2]. Sono stati compiuti sforzi per colpire il metabolismo riprogrammato da solo o in combinazione con la chemioterapia antitumorale sia in studi preclinici che clinici [3]. È interessante notare che questo rimodellamento metabolico specifico del cancro viene invertito dal dicloroacetato (DCA), una piccola molecola mirata ai mitocondri che può penetrare nella maggior parte dei tessuti dopo la somministrazione orale [4]. Inibisce specificamente la piruvato deidrogenasi chinasi (PDK), un membro della famiglia delle chinasi, portando alla riattivazione della piruvato deidrogenasi (PDH), un enzima chiave che sposta il flusso di piruvato nei mitocondri per promuovere l’ossidazione del glucosio invece della glicolisi [4]. Sebbene il DCA sia stato recentemente valutato in diversi studi preclinici sul cancro [5], le risposte delle cellule tumorali al trattamento con DCA, che determinano se il DCA fornirà un beneficio clinico nel trattamento del cancro, non sono state completamente chiarite.
Oltre il 70% dei tumori al seno esprime il recettore degli estrogeni (ER) e dipende dagli estrogeni per guidare la crescita e la progressione del tumore [6]. Pertanto, la terapia endocrina dovrebbe essere considerata complementare alla chirurgia nella maggior parte delle pazienti, in quanto induce la remissione del tumore e fornisce benefici clinici costanti. Il farmaco anti-estrogeno tamoxifene è il trattamento più comunemente utilizzato per le pazienti con carcinoma mammario ER-positivo sia in fase iniziale che in fase avanzata/metastatica [7]. Come terapia adiuvante nel carcinoma mammario in fase iniziale, il tamoxifene migliora la sopravvivenza globale e si ritiene che il suo uso diffuso abbia contribuito in modo significativo alla riduzione della mortalità per cancro al seno osservata nell’ultimo decennio [8]. Nonostante gli evidenti benefici del trattamento con tamoxifene nel carcinoma mammario, quasi tutte le pazienti con malattia metastatica e ben il 25% delle pazienti sottoposte a tamoxifene adiuvante finiscono per avere una ricaduta e morire a causa della malattia [9, 10]. I meccanismi biologici alla base della resistenza intrinseca (de novo) e acquisita al tamoxifene sono quindi di notevole importanza clinica. Una migliore comprensione di questi meccanismi potrebbe suggerire nuove strategie per superare la resistenza al tamoxifene e migliorare il trattamento del carcinoma mammario.
Nel presente studio abbiamo dimostrato che l’espressione dell’EGFR nelle cellule di cancro al seno è stata ridotta dal trattamento con DCA. La combinazione di DCA e tamoxifene ha ulteriormente ridotto i livelli di EGFR. Abbiamo dimostrato che il DCA ha potenziato la citotossicità del tamoxifene sulle cellule di cancro al seno inibendo l’espressione dell’EGFR. Inoltre, il DCA ha sensibilizzato le cellule MCF7 resistenti al tamoxifene attraverso la downregulation dell’EFGR. Questi risultati suggeriscono il potenziale utilizzo del DCA nel trattamento del cancro al seno attraverso l’attenuazione della via di segnalazione dell’EGFR.
Risultati
L’inibizione della PDK downregola l’EGFR e aumenta la morte cellulare indotta dal tamoxifene nelle cellule di carcinoma mammario
Studi recenti hanno dimostrato che l’inibizione della PDK da parte di un inibitore della PDK, come il DCA, sposta il metabolismo delle cellule tumorali dalla glicolisi alla fosforilazione ossidativa de-fosforilando la piruvato deidrogenasi mitocondriale [4, 11]. Per chiarire come le vie di segnalazione dei fattori di crescita e delle chinasi, insieme alla PDK, regolano l’effetto Warburg nel cancro al seno, abbiamo esaminato i livelli di espressione di diversi recettori dei fattori di crescita nelle cellule MCF7 knockdown della PDK. È interessante notare che la deplezione di PDK mediante trattamento con siRNA ha ridotto l’EGFR, mentre non ha avuto effetti o ha avuto effetti marginali sugli altri recettori dei fattori di crescita (Figura 1A). Nei tessuti dei mammiferi sono stati identificati quattro isoenzimi PDK (PDK1, PDK2, PDK3, PDK4) [12]. Per confermare la downregulation di EGFR con il trattamento siPDK, abbiamo analizzato l’espressione di EGFR nelle cellule trattate con siRNA contro ciascuna isoforma di PDK. Ogni trattamento con siRNA ha abolito solo l’espressione della PDK bersaglio, ma tutti hanno causato la downregulation di EGFR, suggerendo che PDK potrebbe non regolare l’espressione di EGFR in modo isoforma-specifico (Figura 1B). Coerentemente con questi risultati, anche il DCA ha ridotto i livelli di EGFR in modo dose-dipendente (Figura 1C). Quando abbiamo analizzato la concentrazione di lattato nei terreni di coltura, non c’è stata alcuna variazione significativa della concentrazione di lattato con il trattamento con tamoxifene/DCA o siRNA contro EGFR, anche se è stata ridotta dal trattamento con DCA (Figura S1 supplementare). Pertanto, suggeriamo che la downregulation dell’EGFR potrebbe non essere associata al cambiamento metabolico indotto dal DCA nelle cellule di cancro al seno. Poiché l’attivazione della via di segnalazione dell’EGFR contribuisce alla resistenza al tamoxifene [13], abbiamo esaminato se la downregulation dell’EGFR mediante l’inibizione della PDK sensibilizzasse le cellule al tamoxifene. Come mostrato nella Figura 1D, il co-trattamento con tamoxifene e DCA ha portato a una marcata riduzione della vitalità cellulare. Il trattamento combinato per 72 ore ha ridotto la vitalità cellulare a meno del 30% di quella del controllo (Figura 1E). Successivamente, la morte cellulare nelle cellule co-trattate è stata valutata utilizzando la colorazione con Annexin V/PI. Quarantotto ore dopo il trattamento, la combinazione di tamoxifene e DCA ha indotto una morte cellulare del 30%, rispetto al 15% o all’8% del tamoxifene o del DCA da soli, rispettivamente (Figura 1F). In precedenza, abbiamo riportato che la morte cellulare apoptotica nelle cellule di cancro al seno è causata dalla perdita del potenziale di membrana mitocondriale (MMP) [14]. Abbiamo quindi verificato se la perdita di MMP fosse coinvolta nella morte cellulare indotta dal co-trattamento; tuttavia, non vi è stata alcuna variazione significativa di MMP tra le cellule non trattate e quelle trattate con tamoxifene/DCA (Figura supplementare S2).
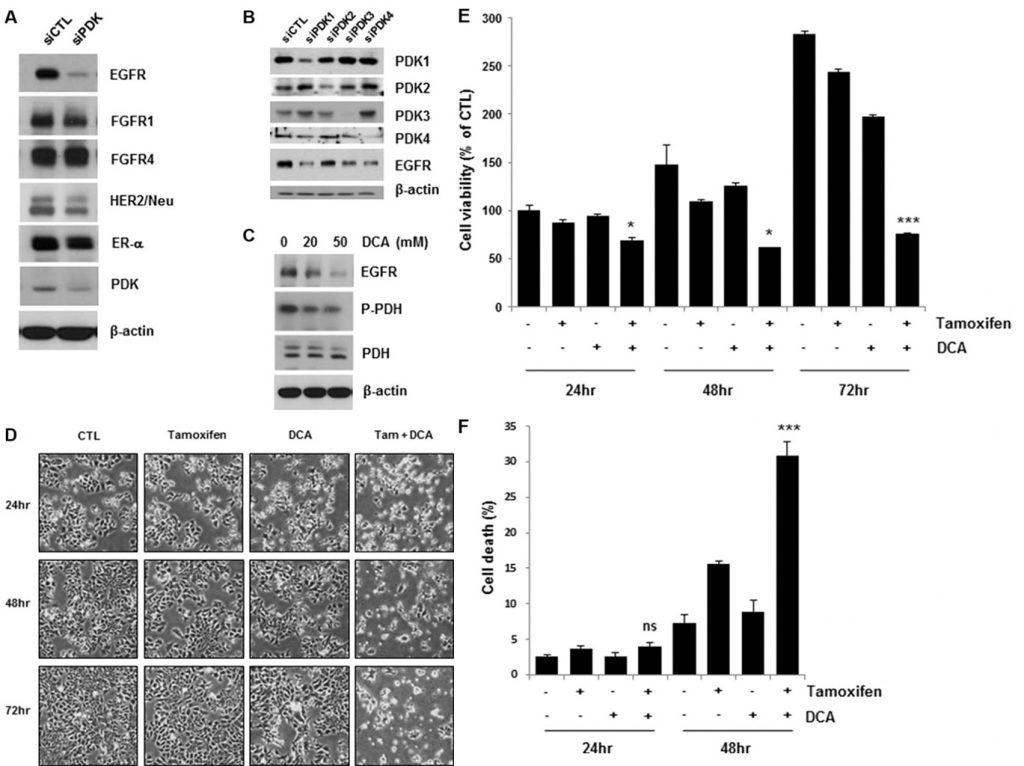
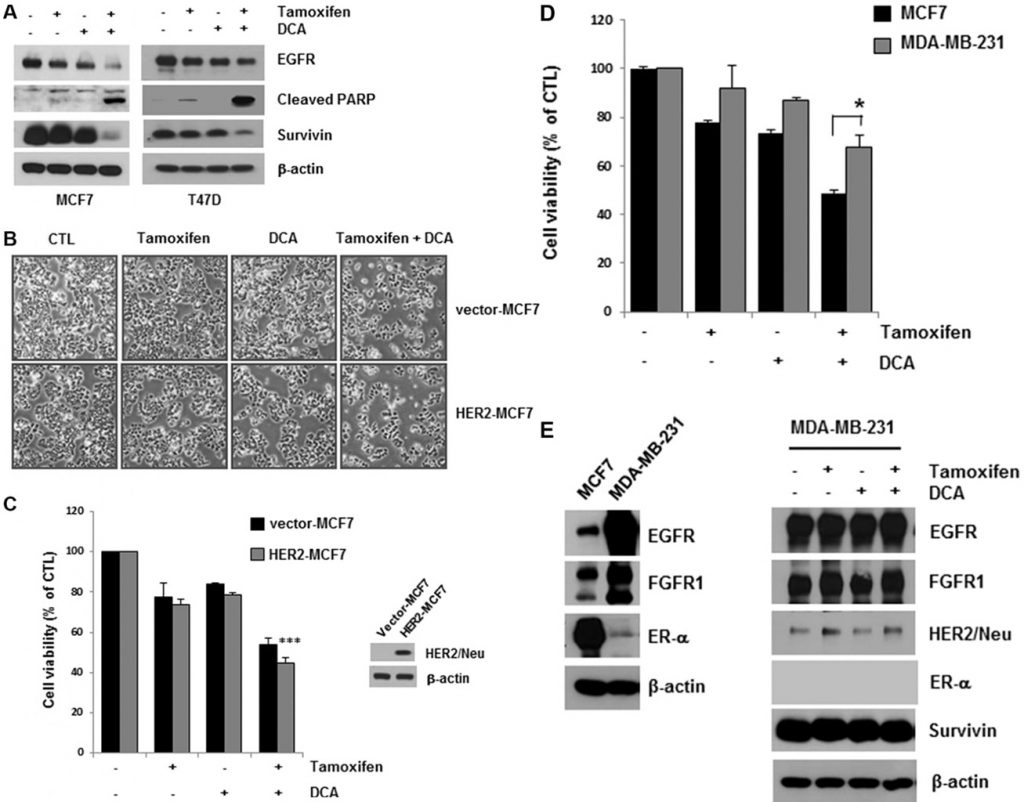
DCA più tamoxifene ha ulteriormente ridotto i livelli di EGFR sia nelle cellule MCF7 che in quelle T47D rispetto a quelli del solo DCA (Figura 2A). La morte cellulare indotta dal co-trattamento è stata confermata dal rilevamento della scissione di PARP, un marcatore di apoptosi (Figura 2A). La Survivin è una molecola anti-apoptotica e un bersaglio dell’ER [15]. Il co-trattamento ha anche ridotto la survivin, che potrebbe mediare l’apoptosi nelle cellule (Figura 2A). Sebbene il trattamento con tamoxifene abbia ridotto leggermente i livelli di EGFR nelle cellule MCF7 e T47D, non è stato osservato un aumento significativo della morte cellulare nelle cellule, suggerendo che un livello critico di EGFR è necessario per la sopravvivenza delle cellule di carcinoma mammario (Figura 2A).
L’evidenza di linee cellulari ha dimostrato che la sovraespressione delle vie di HER2 può contribuire alla resistenza acquisita alle terapie endocrine [13]. Per determinare se la sovraespressione di HER2 influisce sulla citotossicità di tamoxifene e DCA, abbiamo esaminato la vitalità cellulare in cellule MCF7 iperesprimenti HER2 (HER2-MCF7) dopo il trattamento con tamoxifene e DCA. I risultati hanno mostrato che il tamoxifene e il DCA hanno ridotto significativamente la vitalità cellulare anche nelle cellule HER2-MCF7 (Figura 2B e 2C), suggerendo che il DCA potrebbe potenziare la morte cellulare indotta dal tamoxifene nelle cellule di carcinoma mammario HER2-sovrappressore. Abbiamo poi valutato gli effetti inibitori della crescita del co-trattamento sulla linea cellulare di cancro al seno triplo-negativo MDA-MB-231. Come mostrato nella Figura 2D, le cellule MDA-MB-231 sono risultate meno sensibili al tamoxifene e al DCA rispetto alle cellule MCF7. Poiché la downregulation di EGFR è stata osservata nelle cellule ER-positive, abbiamo esaminato gli effetti di tamoxifene e DCA sui livelli di EGFR nelle cellule MDA-MB-231. L’EGFR era altamente espresso nelle cellule MDA-MB-231 rispetto alle cellule MCF7 e i livelli non erano significativamente ridotti dal tamoxifene e dal DCA (Figura 2E). Successivamente, abbiamo esaminato la citotossicità di tamoxifene e DCA nella linea cellulare epiteliale mammaria immortalizzata non tumorale MCF10A. È interessante notare che l’espressione di EGFR nelle cellule MCF10A era paragonabile a quella delle cellule MDA-MB-231 e che nelle cellule MCF10A non è stata osservata né la downregulation di EGFR né la morte cellulare dopo il trattamento con tamoxifene e DCA (Figura S3 supplementare). Questi risultati indicano che gli effetti antiproliferativi del tamoxifene e del DCA nelle cellule di cancro al seno dipendono dalla downregulation dell’EGFR.
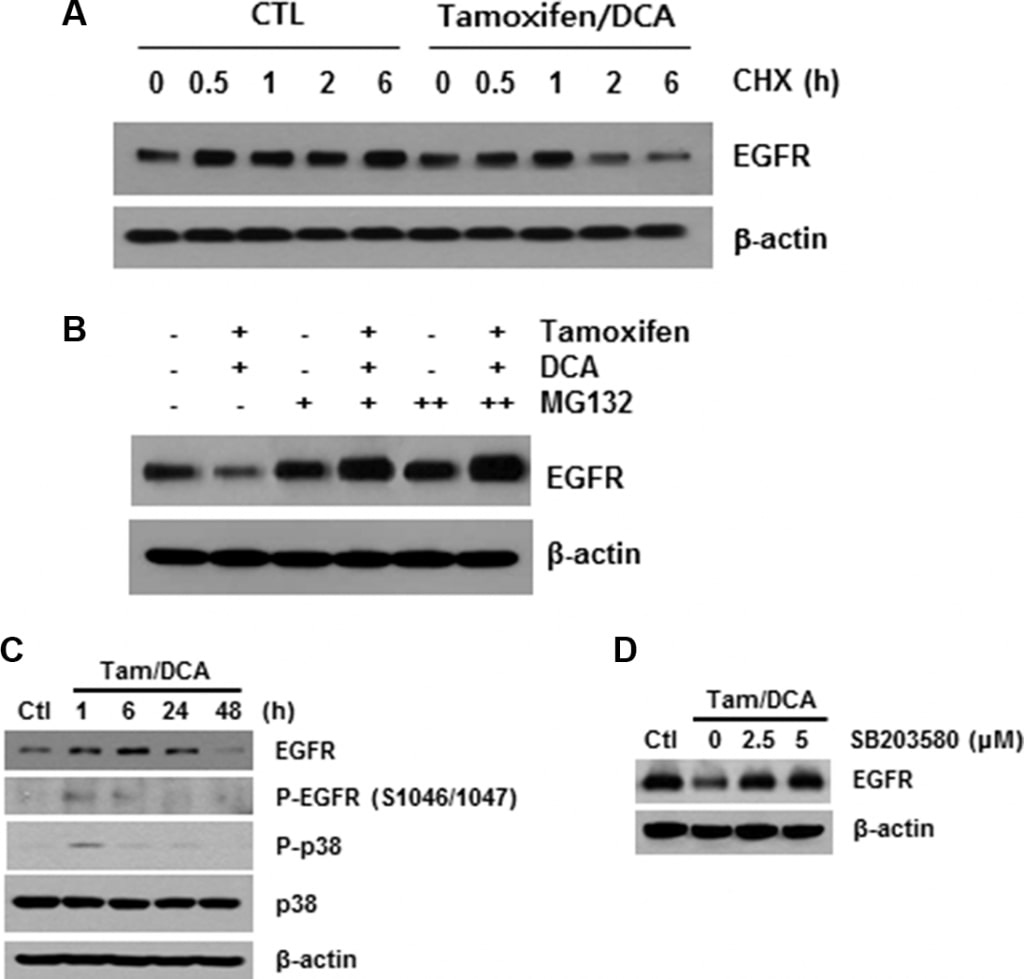
Il trattamento combinato di tamoxifene e DCA induce la degradazione dell’EGFR mediata da p38 MAPK
Come descritto in precedenza, il legame con il ligando provoca una rapida autofosforilazione, con conseguente rimozione dell’EGFR dalla superficie cellulare tramite endocitosi in un compartimento endosomiale precoce [16]. Pertanto, abbiamo studiato il ruolo della modificazione del recettore nella downregulation dell’EGFR mediata dal tamoxifene/DCA. Dopo aver bloccato la sintesi proteica con cicloeximide, abbiamo scoperto che la stabilità dell’EGFR era significativamente compromessa nelle cellule trattate con tamoxifene/DCA rispetto a quelle di controllo (Figura 3A). Abbiamo quindi valutato gli effetti di MG132, un inibitore del proteasoma, sulla degradazione di EGFR indotta da tamoxifene/DCA. Il trattamento con MG132 ha ripristinato l’espressione di EGFR nelle cellule trattate con tamoxifene/DCA in modo dose-dipendente (Figura 3B).
La fosforilazione dell’EGFR su residui di serina e treonina rappresenta un meccanismo di attenuazione dell’attività dell’EGFR e, tra questi, i siti di fosforilazione della serina 1046/1047 (Ser 1046/7) sono necessari per la desensibilizzazione dell’EGFR [17]. Recentemente è stato riportato che la p38 mitogen-activated protein kinase (MAPK) induce la fosforilazione dell’EGFR a Ser 1046/7, con conseguente degradazione nelle cellule tumorali [18]. Pertanto, abbiamo esaminato gli effetti del tamoxifene e del DCA sulla fosforilazione di p38 MAPK nelle cellule MCF7. p38 MAPK è stata significativamente fosforilata entro 1 ora e la fosforilazione è stata mantenuta per 24 ore dopo il trattamento con tamoxifene e DCA (Figura 3C). Inoltre, la degradazione di EGFR indotta dal co-trattamento è stata significativamente soppressa quando le cellule sono state pretrattate con un inibitore specifico di p38 MAPK, SB203580 (Figura 3D), indicando che l’attivazione di p38 MAPK svolge un ruolo nella downregulation di EGFR indotta da tamoxifene/DCA nelle cellule MCF7.
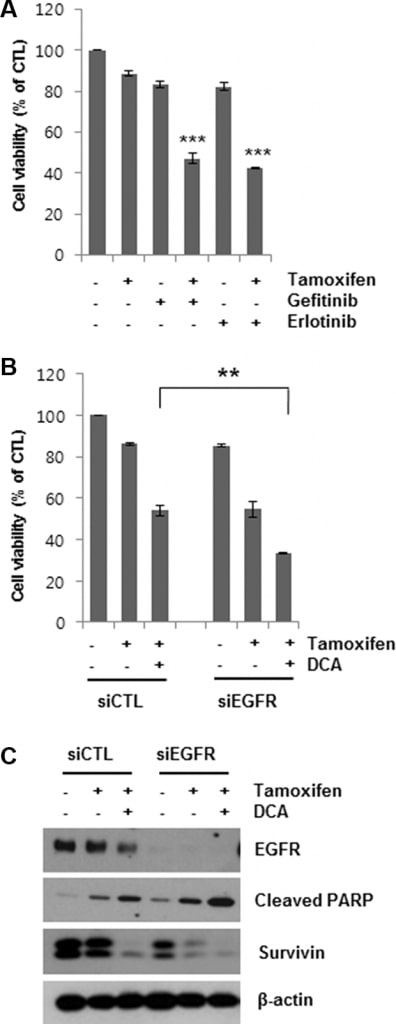
Gliinibitori dell’EGFR aumentano la morte cellulare indotta dal tamoxifene
Avendo dimostrato che la degradazione dell’EGFR mediata dal DCA può aumentare la morte cellulare indotta dal tamoxifene nelle cellule MCF7, abbiamo quindi determinato se l’inibizione dell’EGFR aumentasse la morte cellulare nelle cellule trattate con tamoxifene. Gefitinib ed erlotinib sono inibitori reversibili selettivi della tirosin-chinasi EGFR che si lega all’ATP [19, 20]. Il co-trattamento delle cellule MCF7 con 5 μM di gefitinib o erlotinib per 48 ore ha aumentato notevolmente la morte cellulare indotta dal tamoxifene (Figura 4A). Analogamente, il knockdown di EGFR mediante trattamento con siEGFR ha aumentato la morte cellulare indotta dal tamoxifene (Figura 4B). La riduzione della vitalità cellulare ottenuta con il trattamento combinato di siEGFR e tamoxifene è stata paragonabile a quella ottenuta con DCA e tamoxifene. Inoltre, il trattamento con siEGFR ha ulteriormente abbassato i livelli di EGFR nelle cellule trattate con tamoxifene/DCA, determinando un aumento della morte cellulare apoptotica e una downregulation della survivin rispetto al controllo con si (Figura 4C). Questi risultati supportano la conclusione che il DCA sensibilizza le cellule di cancro al seno al tamoxifene attraverso la downregulation di EGFR.
l’espressione di c-myc e Nanog è associata agli effetti citotossici del DCA nelle cellule MCF7 trattate con tamoxifene
È stato dimostrato che l’attivazione dell’EGFR nel cancro aumenta diversi fattori di trascrizione che possono influenzare il tipo e la durata della segnalazione dell’EGFR [21]. Tra questi, Nanog e c-myc hanno un ruolo pleiotropico nella tumorigenesi, compresa la resistenza alla terapia standard nel carcinoma mammario [22, 23]. Per approfondire le attività antitumorali mediate dalla downregulation dell’EGFR nelle cellule di carcinoma mammario, abbiamo esaminato l’espressione di c-myc e Nanog nelle cellule MCF7 dopo il cotrattamento con tamoxifene e DCA. I livelli di c-myc e Nanog sono diminuiti significativamente nelle cellule trattate con tamoxifene e DCA (Figura 5A). Analogamente, le espressioni di entrambe le proteine sono state soppresse dal tamoxifene in combinazione con gefitinib o erlotinib (Figura 5B), indicando che l’espressione di EGFR è necessaria per mantenere l’espressione di c-myc e Nanog nelle cellule MCF7 trattate con tamoxifene. Tuttavia, l’effetto di siEGFR sull’espressione di c-myc e Nanog è stato scarso o nullo, indicando che la downregulation di EGFR è necessaria ma non sufficiente per ridurre l’espressione delle proteine (Figura S4 supplementare). Per determinare se la ridotta espressione di queste due proteine sia coinvolta negli effetti citotossici del tamoxifene e del DCA nelle cellule MCF7, abbiamo testato gli effetti di siRNA contro c-myc e Nanog nelle cellule esposte a tamoxifene/DCA. L’abbattimento di c-myc e Nanog ha sensibilizzato significativamente le cellule al tamoxifene/DCA rispetto ai controlli (Figura 5C). Al contrario, la sovraespressione di FLAG-c-myc e FLAG-Nanog mediante trasfezione delle cellule con vettori FLAG-c-myc e FLAG-Nanog ha protetto significativamente le cellule dalla citotossicità indotta da tamoxifene/DCA (Figura 5D). Nel complesso, i nostri dati suggeriscono che la downregulation dell’EGFR da parte di una combinazione di tamoxifene e DCA può indurre la morte cellulare nelle cellule MCF7 in parte attraverso l’inibizione dell’espressione di c-myc e Nanog. È noto che i geni di auto-rinnovamento, come c-myc e Nanog, sono associati alle proprietà delle cellule staminali del cancro 24. Per verificare se il co-trattamento con tamoxifene e DCA inibisca le cellule simil-staminali del cancro al seno, abbiamo eseguito un’analisi di citometria a flusso per stimare la percentuale di sottopopolazione di cellule simil-staminali nelle cellule MCF7 in base all’espressione di CD44. Dopo il co-trattamento, la popolazione CD44-alta nelle cellule MCF7 si è ridotta dal 42,5% al 19,9% (Figura 5E). Questo risultato ha sollevato la possibilità che il DCA più il tamoxifene possano inibire la capacità delle cellule simil-cancro nelle cellule di cancro al seno.

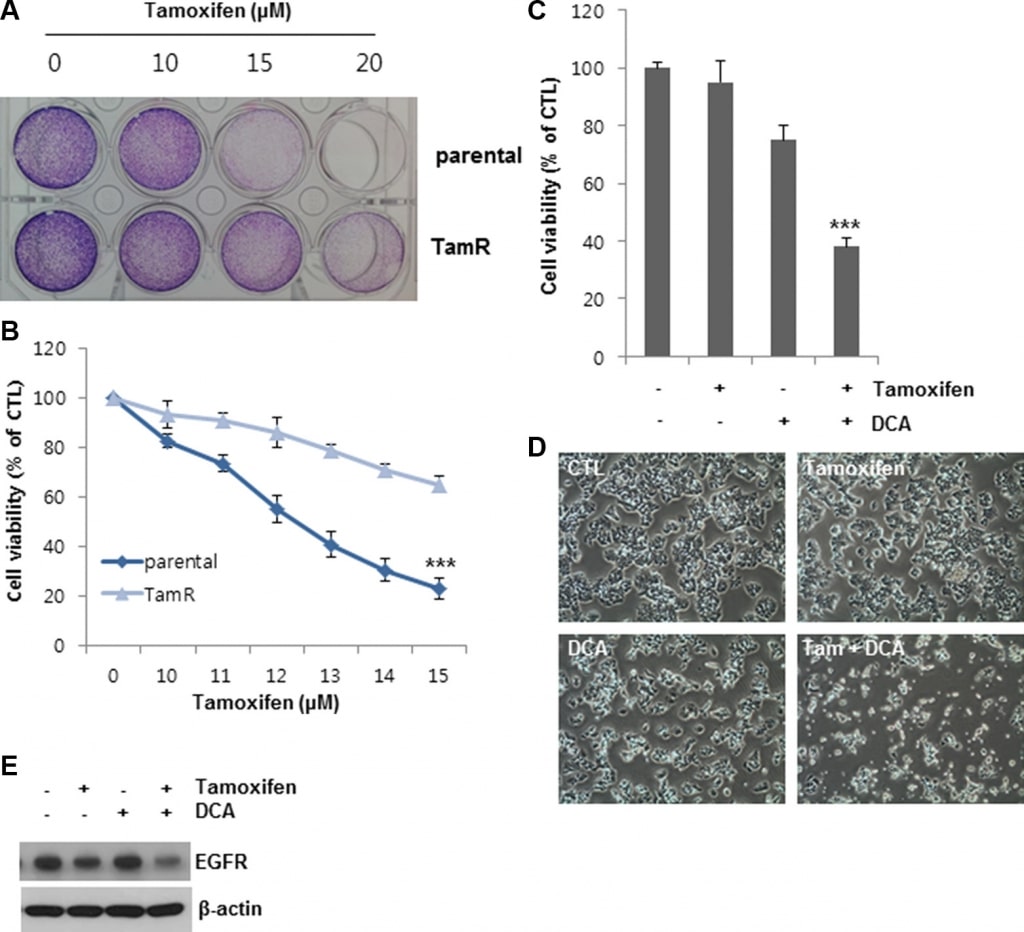
Il trattamento combinato con DCA e tamoxifene può superare la resistenza al tamoxifene nelle cellule di cancro al seno
Per confermare ulteriormente gli effetti osservati del DCA sulla morte cellulare indotta dal tamoxifene, abbiamo stabilito cellule MCF7 resistenti al tamoxifene (TamR) mediante trattamento con tamoxifene per un lungo periodo di tempo. Rispetto alle cellule non trattate, la vitalità cellulare delle cellule MCF7 e delle cellule MCF7 TamR era rispettivamente del 20 e del 60% dopo il trattamento con 13 μM di tamoxifene, indicando che le cellule MCF7 TamR erano meno sensibili alla stessa concentrazione di tamoxifene rispetto alle cellule MCF7 parentali (Figura 6A e 6B). Il DCA da solo ha inibito la crescita delle cellule MCF7 TamR di circa il 25% rispetto al controllo (Figura 6C e 6D). Al contrario, il co-trattamento di DCA con tamoxifene ha soppresso la crescita cellulare di oltre il 60% rispetto al controllo (Figura 6C e 6D). Anche i livelli di EGFR nelle cellule TamR sono stati ridotti dal co-trattamento (Figura 6E). Questi risultati suggeriscono che il DCA potrebbe superare la resistenza al tamoxifene nelle cellule di cancro al seno attraverso la downregulation dell’EGFR a livello proteico.
Discussione
Con l’aumento della comprensione del fenotipo metabolico delle cellule tumorali, l’individuazione delle differenze metaboliche tra cellule tumorali e normali è stata proposta come nuova strategia antitumorale. Nonostante l’aumento dei potenziali farmaci antitumorali che hanno come bersaglio i processi metabolici delle cellule tumorali, per colpire con successo il metabolismo del cancro sarà necessario chiarire come le cellule rispondono ai farmaci. Abbiamo dimostrato che il DCA inibisce non solo l’attività della PDK ma anche l’espressione dell’EGFR nelle cellule di cancro al seno. Il trattamento combinato di DCA e tamoxifene ha indotto la degradazione proteasoma-dipendente delle proteine EGFR nelle cellule di cancro al seno attraverso l’attivazione di p38 MAPK. I nostri risultati indicano che la downregulation dell’EGFR svolge un ruolo chiave nella morte cellulare apoptotica indotta dalla combinazione di DCA e tamoxifene. A nostra conoscenza, questo è il primo rapporto che dimostra che il DCA sensibilizza le cellule di cancro al seno alla morte cellulare indotta dal tamoxifene attraverso la downregulation dell’EGFR.
Nelle cellule di carcinoma mammario ER-positive che hanno sviluppato resistenza endocrina, l’espressione di ER può essere direttamente soppressa da una maggiore segnalazione dei recettori dei fattori di crescita dovuta alla sovraespressione di EGFR e HER2, che successivamente attivano MAPK e inibiscono la trascrizione di ER [25]. Inoltre, la resistenza al tamoxifene nelle cellule di cancro al seno è stata correlata alla sovraespressione di EGFR e ad alti livelli di chinasi extracellulare attivata fosforilata 1/2 [2]. Pertanto, la strategia di combinare un inibitore dell’EGFR con un agente endocrino è sufficientemente interessante da meritare ulteriori studi per la terapia del cancro al seno. In questo studio abbiamo scoperto che la downregulation di EGFR nelle cellule di carcinoma mammario trattate con DCA aumenta la sensibilità delle cellule al tamoxifene. La downregulation dell’EGFR mediante il co-trattamento del DCA con il tamoxifene è stata confermata in diverse linee cellulari di cancro al seno ER-positive, tra cui MCF7, T47D e BT474 (Figura 2A e non mostrato). Oltre alla linea cellulare di carcinoma mammario HER2-amplificata BT474, anche le cellule HER2-MCF7 hanno mostrato una downregulation di EGFR da parte di DCA/tamoxifene, suggerendo che il trattamento combinato sarà applicabile al carcinoma mammario HER2-positivo. Purtroppo, una linea cellulare di carcinoma mammario triplo-negativo, MDA-MB-231, che notoriamente sovraesprime l’EGFR [26], ha mostrato un fenotipo refrattario e nessun cambiamento evidente nei livelli di EGFR in risposta al co-trattamento. Una possibilità è che l’amplificazione dell’EGFR nelle cellule MDA-MB-231 ne sovverta la downregulation con il co-trattamento del DCA con il tamoxifene. L’altra spiegazione è che la via o le vie di segnalazione che mediano la downregulation dell’EGFR siano bloccate da un meccanismo sconosciuto nelle cellule MDA-MB-231. Saranno necessarie ulteriori indagini per chiarire il meccanismo alla base del mantenimento dell’espressione dell’EGFR in risposta al co-trattamento di DCA e tamoxifene nelle cellule di carcinoma mammario ER-negative.
Sono note quattro isoforme di PDK, ciascuna attiva in risposta a diverse condizioni intracellulari ed extracellulari. La PDK1 è attivata dall’ipossia [27]; la PDK2 è attivata dai prodotti PDH acetil CoA e NADH [28]; la PDK3 è attivata dall’ATP [29] e la PDK4 è regolata trascrizionalmente da segnali ormonali [30]. La PDK2 possiede la maggiore attività di fosforilazione del complesso piruvato deidrogenasi, seguita da PDK4, PDK1 e PDK3 [12]. La PDK2 è più suscettibile all’inibizione da parte del DCA a causa della sua espressione ubiquitaria [31]. Sebbene abbiamo rilevato l’espressione di quattro isoforme nelle cellule MCF7 (Figura 1C), le differenze funzionali tra le isoforme PDK nel tumore al seno rimangono ancora elusive. Sarà interessante indagare ulteriormente se le vie di segnalazione che sopprimono una particolare isoforma di PDK portano alla downregulation di EGFR. È noto che PDK funziona nei mitocondri e diversi studi hanno dimostrato che anche EGFR trasloca nei mitocondri dopo la stimolazione con EGF [32, 33]. Inoltre, è stato suggerito che l’interazione di EGFR con PDK nella matrice mitocondriale svolge un ruolo importante nella crescita tumorale indotta da EGFR nel glioblastoma multiforme [34]. In questo caso, abbiamo riscontrato che il blocco dell’attività di PDK mediante un inibitore o il silenziamento ha ridotto i livelli cellulari totali di EGFR nelle cellule di cancro al seno trattate con tamoxifene. Sono necessari ulteriori studi per determinare se solo l’EGFR associato ai mitocondri è soggetto a degradazione dal co-trattamento con DCA e tamoxifene nelle cellule di cancro al seno. In generale, l’esposizione delle cellule all’EGF provoca una rapida autofosforilazione, compresa la tirosina (Tyr) 1045, che fornisce un sito di aggancio per l’ubiquitina ligasi c-Cbl, determinando così l’ubiquitinazione dell’EGFR e la sua rimozione tramite endocitosi dalla superficie cellulare in un compartimento endosomiale precoce [35]. Tuttavia, il cotrattamento con DCA e tamoxifene non ha avuto alcun effetto sulla fosforilazione dell’EGFR a Tyr 1045 (dati non mostrati). Abbiamo invece osservato la fosforilazione di EGFR a Ser 1046/7 da parte di DCA e tamoxifene, bloccata da un inibitore specifico di p38 MAPK, SB203580. È stato suggerito che p38 MAPK svolga un ruolo centrale sia nell’internalizzazione che nella degradazione di EGFR [18]; tuttavia, sono necessarie ulteriori indagini per chiarire come la fosforilazione di EGFR a residui di serina causi la sua downregulation e per identificare il mediatore a monte dell’attivazione di p38 MAPK dopo il trattamento con DCA/tamoxifene.
Nanog e c-myc sono stati identificati come fattori di trascrizione e il loro ruolo nel mantenimento dell’autorinnovamento nelle cellule staminali embrionali è stato stabilito in studi precedenti [22, 36]. Un meccanismo recentemente proposto di resistenza intrinseca alla terapia endocrina prevede l’esistenza di un sottogruppo specializzato di cellule tumorali chiamato cellule iniziatrici del tumore (TIC) [37]. Le TIC hanno la capacità di auto-rinnovarsi e di generare nuovi tumori che consistono interamente di tipi di cellule clonalmente derivate presenti nel tumore parentale. Abbiamo dimostrato che il DCA ha ridotto l’espressione di c-myc e Nanog nelle cellule MCF7 trattate con tamoxifene. Inoltre, l’espressione di queste proteine è stata correlata agli effetti citotossici del trattamento combinato di DCA e tamoxifene. Pertanto, il DCA può essere sfruttato per espandere il potenziale terapeutico del tamoxifene sopprimendo lo sviluppo della resistenza intrinseca nella terapia del cancro al seno.
In conclusione, i nostri risultati hanno rivelato che il DCA sensibilizza le cellule di cancro al seno ER-positive al tamoxifene diminuendo i livelli di EGFR. Il trattamento con DCA e tamoxifene ha inibito l’espressione dei geni di auto-rinnovamento e la sopravvivenza delle cellule MCF7 resistenti al tamoxifene. Pertanto, proponiamo che il DCA possa essere un agente terapeutico efficace per il trattamento del cancro al seno resistente al tamoxifene. Inoltre, le strategie di combinazione con il DCA possono essere utili per potenziare l’efficacia terapeutica di altri chemioterapici citotossici o terapie mirate. Ulteriori esperimenti, compresi studi su animali e studi clinici, dovrebbero essere condotti in futuro.
Materiali e metodi
Coltura cellulare e reagenti
Le cellule di carcinoma mammario umano MCF7, T47D e MDA-MB-231 sono state acquistate dall’American Type Culture Collection (Rockville, MD, USA) e sono state coltivate nel terreno di crescita raccomandato (Invitrogen, Carlsbad, CA, USA). Le cellule MCF7 iperesprimenti HER2 (HER2-MCF7) e le cellule vettoriali di controllo (vector-MCF7) sono state gentilmente fornite dal Dr. Incheol Shin (Hanyang University, Seoul, Corea). Le cellule MCF7 resistenti al tamoxifene (TamR) sono state sviluppate coltivando le cellule MCF7 in presenza di 10 μM di tamoxifene per più di 6 mesi. Come controllo, le cellule parentali sono state coltivate per lo stesso periodo di tempo in terreni normali. Dopo l’instaurazione della resistenza, le cellule sono state sottoposte a passage per non più di 3 mesi. Per gli esperimenti che prevedevano il trattamento con tamoxifene, le cellule sono state coltivate di routine in terreno Dulbecco’s modified Eagle’s medium privo di fenolo e con il 10% di siero fetale bovino scremato dal carbone subito prima dei trattamenti. Gli anticorpi contro EGFR, fosfo-EGFR (S1046/1047), FGFR1, FGFR4, PDK1, PDH, survivin, PARP clivato, p38, fosfo-p38 e Nanog sono stati acquistati da Cell Signaling Technology (Danvers, MA, USA). Gli anticorpi contro HER2/Neu, ER-α e c-myc, i siRNA diretti contro EGFR, Nanog, PDK4 e c-myc e i siRNA di controllo negativo (scrambled) sono stati acquistati da Santa Cruz Biotechnology (Dallas, TX, USA). Gli anticorpi contro PDK2 e PDK3 sono stati acquistati da Thermo Fisher Scientific (Waltham, MA, USA). L’anticorpo β-actina, l’anticorpo FLAG, il tamoxifene, il DCA, la cicloeximide e l’MG132 sono stati acquistati da Sigma-Aldrich (St. Louis, MO, USA). L’anticorpo fosfo-PDH (S293) e SB203580 sono stati acquistati da BD Biosciences Pharmingen (San Diego, CA, USA), mentre gefitinib ed erlotinib sono stati ottenuti da Selleck Chemicals (London, ON, Canada).
Trasfezioni e trattamenti
Le cellule sono state trasfettate con il siRNA target (50 nM) utilizzando Lipofectamine RNAiMAX (Invitrogen) come descritto dal produttore. Le cellule sono state trasfettate con 1 μg di FLAG-c-myc pcDNA 3.1 e FLAG-Nanog-pcDNA 3.1 utilizzando Lipofectamine 2000 come descritto dal produttore. Dopo 6 ore, le cellule sono state trattate con tamoxifene e/o DCA per 24-48 ore e poi analizzate come descritto altrove.
Misurazione della vitalità cellulare
La vitalità cellulare è stata determinata misurando la conversione mitocondriale del 3-(4,5-dimetiltiazolil2)-2,5-difeniltetrazolio bromuro (MTT) in un prodotto colorato. Le cellule sono state trattate come indicato e il terreno è stato scambiato con terreno privo di siero contenente 1 mM di MTT. Dopo 2 ore di incubazione a 37°C, le cellule sono state solubilizzate in DMSO. La quantità di formazan, la forma convertita di MTT, è stata determinata misurando l’assorbanza a 595 nm. Valutazione dell’apoptosi
L’apoptosi è stata determinata mediante analisi di fluorescence-activated cell sorting utilizzando un kit di apoptosi Annexin V-FITC (BioVision, Milpitas, CA, USA) come indicato dal produttore. In breve, dopo il trattamento, le cellule sono state trattate con tripsina e poi risospese in un tampone di legame (10 mM HEPES/NaOH, pH 7,4, 140 mM NaCl, 2,5 mM CaCl2 ) con Annexin V-FITC e ioduro di propidio. Dopo un’incubazione di 15 minuti, la fluorescenza cellulare è stata analizzata mediante citometria a flusso. La morte cellulare è stata misurata come percentuale di cellule nella popolazione positiva all’Annexina V e al PI.
Western blotting
Le cellule sono state raccolte e lisate in tampone RIPA (50 mM Tris-HCl pH 7,5, 150 mM NaCl, 1% Nonidet P40, 0,5% sodio desossicolato e 0,1% SDS) integrato con un cocktail di inibitori della proteasi/fosfatasi (Roche, Mannheim, Germania). Quantità uguali di proteine (20-50 μg) sono state separate mediante SDS-PAGE e trasferite su una membrana di nitrocellulosa. Le membrane sono state bloccate incubando con latte scremato al 5% in soluzione salina Trisbuffered per 1 ora e sono state poi incubate per una notte con gli anticorpi primari appropriati. Le membrane sono state incubate con l’anticorpo secondario coniugato con HRP per 1 ora. Le proteine immunoreattive sono state visualizzate con reagenti a chemiluminescenza potenziata (Amersham Biosciences, Little Chalfont, UK).
Rilevazione della popolazione cellulare CD44 positiva
Le cellule sono state colorate con anticorpi alla diluizione 1∶100 in PBS per 15 minuti. Gli anticorpi utilizzati sono stati i seguenti: anticorpi di controllo isotipico FITC-CD44 e IgG di topo coniugate con FITC sono stati ottenuti da BD Biosciences Pharmingen. Le cellule marcate sono state analizzate in citometria a flusso. Le popolazioni di cellule positive al CD44 sono state determinate dall’intensità del FITC.
Analisi statistica
Tutti i dati presentati sono rappresentativi di almeno due esperimenti separati. I confronti tra i gruppi sono stati analizzati utilizzando il t-test di Student. Gli asterischi (***p < 0,001, **p < 0,01, *p < 0,05) indicano la significatività statistica.
Riconoscimenti E Finanziamenti
Questa ricerca è stata sostenuta dal Basic Science Research Program attraverso la National Research Foundation of Korea (NRF) finanziata dal Ministero della Scienza, dell’ICT e della Pianificazione Futura (No. 1711031812, 1711023318 e 1711031800).
Conflitti di interesse
Gli autori non dichiarano conflitti di interesse.
RIFERIMENTI
1 1. Vander Heiden MG, Cantley LC, Thompson CB. Capire l’effetto Warburg: i requisiti metabolici della proliferazione cellulare. Science. 2009; 324:1029-33.
2ZhaoY, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013; 4:e532.
3 Vander Heiden MG. Puntare sul metabolismo del cancro: si apre una finestra terapeutica. Nat Rev Drug Discov. 2011; 10:671-84.
4 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, et al. Un asse dei canali mitocondriali-K+ è soppresso nel cancro e la sua normalizzazione promuove l’apoptosi e inibisce la crescita del cancro. Cancer Cell. 2007; 11: 37-51.
5 Kankotia S, Stacpoole PW. Dicloroacetato e cancro: una nuova casa per un farmaco orfano? Biochim Biophys Acta. 2014; 1846:617-29.
6 Osborne CK. Il tamoxifene nel trattamento del cancro al seno. N Engl J Med. 1998 Nov 26; 339:1609-18.
7 Johnston SR. Nuove strategie nel carcinoma mammario positivo ai recettori degli estrogeni. Clin Cancer Res. 2010 Apr 1; 16:1979-87. doi: 10.1158/1078-0432.CCR-09-1823.
8 Peto R, Boreham J, Clarke M, Davies C, Beral V. I decessi per cancro al seno nel Regno Unito e negli Stati Uniti sono diminuiti del 25% nel 2000 all’età di 20-69 anni. Lancet. 2000; 355:1822.
9 Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effetti della chemioterapia e della terapia ormonale per il cancro al seno precoce sulla recidiva e sulla sopravvivenza a 15 anni: una panoramica degli studi randomizzati. Lancet. 2005; 365:1687-717.
10 Early Breast Cancer Trialists’ Collaborative Group (EBCTCG), Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, Dowsett M, Ingle J, et al. Rilevanza dei recettori ormonali del tumore al seno e di altri fattori sull’efficacia del tamoxifene adiuvante: meta-analisi a livello di paziente di studi randomizzati. Lancet. 2011; 378:771-84.
11 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. La combinazione di PDK1 ed EGFR innesca la regressione del glioblastoma invertendo l’effetto Warburg. Cancer Res. 2013; 73:7277-89.
12 Saunier E, Benelli C, Bortoli S. Il complesso della piruvato deidrogenasi nel cancro: Un vecchio gatekeeper metabolico regolato da nuove vie e agenti farmacologici. Int J Cancer. 2016; 138:809-17.
13 Osborne CK, Schiff R. Meccanismi di resistenza endocrina nel cancro al seno. Annu Rev Med. 2011; 62:233-47.
14YunSM, Woo SH, Oh ST, Hong SE, Choe TB, Ye SK, Kim EK, Seong MK, Kim HA, Noh WC, Lee JK, Jin HO, Lee YH, et al. La melatonina aumenta la morte cellulare indotta dal triossido di arsenico attraverso una sostenuta upregulation dell’espressione di Redd1 nelle cellule di cancro al seno. Mol Cell Endocrinol. 2016; 422:64-73.
15 Zhu J, Lu X, Hua KQ, Sun H, Yu YH, Feng YJ. Il recettore α degli estrogeni media il potenziamento della motilità delle cellule di cancro ovarico da parte del 17β-estradiolo attraverso l’up-regolazione dell’espressione di survivin. Arch Gynecol Obstet. 2012; 286: 729-37.
16 Arteaga CL. Dipendenza dal recettore del fattore di crescita epidermico nei tumori umani: più di una semplice espressione? Oncologo. 2002; 7:31-9.
17 Sorkin A, Goh LK. Endocitosi e traffico intracellulare degli ErbB. Exp Cell Res. 2009; 315:683-96.
18 Adachi S, Shimizu M, Shirakami Y, Yamauchi J, Natsume H, Matsushima-Nishiwaki R, To S, Weinstein IB, Moriwaki H, Kozawa O. (-)-Epigallocatechina gallato downregola il recettore EGF attraverso la fosforilazione a Ser1046/1047 da parte di p38 MAPK in cellule di cancro del colon. Carcinogenesi. 2009; 30:1544-52.
19CiardielloF, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G. Effetto antitumorale e potenziamento dell’attività dei farmaci citotossici in cellule tumorali umane da parte di ZD-1839 (Iressa), un inibitore tirosin-chinasico selettivo del recettore del fattore di crescita epidermico. Clin Cancer Res. 2000; 6:2053-63.
20 Pollack VA, Savage DM, Baker DA, Tsaparikos KE, Sloan DE, Moyer JD, Barbacci EG, Pustilnik LR, Smolarek TA, Davis JA, Vaidya MP, Arnold LD, Doty JL, et al. Inibizione della fosforilazione della tirosina associata al recettore del fattore di crescita epidermico in carcinomi umani con CP-358,774: dinamica dell’inibizione del recettore in situ ed effetti antitumorali in topi atimici. J Pharmacol Exp Ther. 1999; 291:739-48.
21 Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. La segnalazione del recettore del fattore di crescita epidermico (EGFR) nel cancro. Gene. 2006; 366:2-16.
22 Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Ruolo emergente di nanog nella tumorigenesi e nelle cellule staminali del cancro. Int J Cancer. 2014; 135:2741-8.
23 Chen Y, Olopade OI. MYC nella progressione del tumore al seno. Expert Rev Anticancer Ther. 2008; 8: 1689-98.
24 Hou ZJ, Luo X, Zhang W, Peng F, Cui B, Wu SJ, Zheng FM, Xu J, Xu LZ, Long ZJ, Wang XT, Li GH, Wan XY, et al. Il flubendazolo, antielmintico approvato dalla FDA, ha come bersaglio le cellule staminali del cancro al seno. Oncotarget. 2015; 6:6326-40. doi:10.18632/oncotarget.3436.
25 Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. L’attivazione della proteina chinasi mitogena in cellule di cancro al seno positive al recettore degli estrogeni in vitro induce un fenotipo molecolare in vivo di tumori al seno umani negativi al recettore degli estrogeni. Cancer Res. 2006; 66:3903-11.
26 Biswas DK, Cruz AP, Gansberger E, Pardee AB. Attivazione del fattore di crescita epidermico indotta dal fattore nucleare kappa B: Una via principale della progressione del ciclo cellulare nelle cellule di cancro al seno negative ai recettori per gli estrogeni. Proc Natl Acad Sci U S A. 2000; 97:8542-7.
27 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. Espressione mediata da HIF1 della piruvato deidrogenasi chinasi: un interruttore metabolico necessario per l’adattamento cellulare all’ipossia. Cell Metab. 2006; 3:177-85.
28 Hiromasa Y, Hu L, Roche TE. Effetti indotti dai ligandi sulla piruvato deidrogenasi chinasi isoforma 2. J Biol Chem. 2006; 281:12568-79.
29 Kato M, Chuang JL, Tso SC, Wynn RM, Chuang DT. Struttura cristallina della piruvato deidrogenasi chinasi 3 legata al dominio lipoilico 2 del complesso piruvato deidrogenasi umano. EMBO J. 2005; 24:1763-74.
30 Kwon HS, Huang B, Unterman TG, Harris RA. La proteina chinasi B-alfa inibisce l’induzione del gene della piruvato deidrogenasi chinasi-4 umana da parte del desametasone attraverso l’inattivazione dei fattori di trascrizione FOXO. Diabete. 2004; 53:899-910.
31 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Prove dell’esistenza di una regolazione tessuto-specifica del complesso della piruvato deidrogenasi nei mammiferi. Biochem J. 1998; 329 :191-6.
32 Dasari VR, Velpula KK, Alapati K, Gujrati M, Tsung AJ. Le cellule staminali del sangue cordonale inibiscono la traslocazione del recettore del fattore di crescita epidermico nei mitocondri nel glioblastoma. PLoS One. 2012; 7:e31884.
33 Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, Hüttemann M, Douglas R, Haddad G, Parsons SJ. Traslocazione del recettore del fattore di crescita epidermico nei mitocondri: regolazione ed effetto. J Biol Chem. 2009; 284:36592-604.
34 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Il bersaglio combinato di PDK1 ed EGFR innesca la regressione del glioblastoma invertendo l’effetto Warburg. Cancer Res. 2013; 73:7277-89.
35 Massie C, Mills IG. Il ruolo in evoluzione di recettori e adattatori. Nat Rev Cancer. 2006; 6:403-9.
36 Chappell J, Dalton S. Ruolo di MYC nella creazione e nel mantenimento della pluripotenza. Cold Spring Harb Perspect Med. 2013;3:a014381.
37 Wei W, Lewis MT. Identificare e colpire le cellule che iniziano il tumore nel trattamento del cancro al seno. Endocr Relat Cancer. 2015; 22:R135-55.