Sang Hyeok Woo1,*, Sung-Keum Seo1,*, Yoonhwa Park1,2, Eun-Kyu Kim3, Min-Ki Seong4, Hyun-Ah Kim4, Jie-Young Song1, Sang-Gu Hwang1, Jin Kyung Lee5, Woo Chul Noh4, In-Chul Park1
1Divisionof Radiation Cancer Research, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Seoul, 01812, Republic of Korea
2Schoolof Life Science and Biotechnology, Korea University, Seongbuk-gu, Seoul, 02841, Republic of Korea
3Department of Surgery, Breast Cancer Center, Seoul National University Bundang Hospital, Seoul National University College of Medicine, Bundang-gu, Seongnam, 13620, Republiek Korea
4Departmentof Surgery, Korea Cancer Center Hospital, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Seoul, 01812, Republiek Korea
5KIRAMSRadiation Biobank, Korea Institute of Radiological and Medical Sciences, Nowon-gu, Seoul, 01812, Republiek Korea
*Dezeauteurs hebben in gelijke mate aan dit werk bijgedragen
Correspondentie aan: In-Chul Park, e-mail: [email protected]
Trefwoorden: tamoxifen, borstkanker, dichlooracetaat, epidermale groeifactorreceptor, pyruvaatdehydrogenase kinase
Ontvangen: 18 mei 2016
Geaccepteerd: 22 juli 2016
Online gepubliceerd: 01 augustus 2016
Abstract
Metabole herprogrammering in kankercellen is onlangs erkend als een essentieel kenmerk van neoplasie. In deze context vormen metabole veranderingen een aantrekkelijk therapeutisch doelwit, en in preklinische studies zijn bemoedigende resultaten verkregen met geneesmiddelen gericht op verschillende metabole processen. Recentelijk hebben verschillende studies gesuggereerd dat dichlooracetaat (DCA), een specifieke pyruvaatdehydrogenase kinaseremmer, een potentieel antikankermedicijn kan zijn voor een groot aantal uiteenlopende tumoren. Het precieze mechanisme wordt echter niet volledig begrepen, wat belangrijk is voor het gebruik van DCA bij de behandeling van kanker. In deze studie vonden we dat DCA MCF7 borstkankercellen gevoelig maakte voor tamoxifen-geïnduceerde celdood door de expressie van de epidermale groeifactor receptor (EGFR) te verminderen. De downregulatie van EGFR werd veroorzaakt door afbraak van het eiwit. Voorts speelde p38 mitogen-activated protein kinase een belangrijke rol bij DCA/tamoxifen-geïnduceerde EGFR-degradatie. Tenslotte bevorderde DCA ook vergelijkbare tamoxifen-geïnduceerde celdood in tamoxifen-resistente MCF7-cellen, die tot stand waren gekomen door langdurige behandeling met tamoxifen. Samengevat suggereren onze resultaten dat DCA een aantrekkelijk potentieel geneesmiddel is dat cellen gevoelig maakt voor tamoxifen-geïnduceerde celdood en tamoxifen-resistentie overwint via downregulatie van EGFR-expressie in borstkankercellen.
INLEIDING
Prolifererende kankercellen hebben aanzienlijk andere metabolische behoeften dan de meeste normale gedifferentieerde cellen. Om snelle celgroei en proliferatie te ondersteunen, veranderen kankercellen bijvoorbeeld de metabole flux anders dan het omringende weefsel om voldoende bio-energetische en biosynthetische tussenproducten te leveren. Een bekend verschijnsel dat in de meeste kankercellen wordt waargenomen is een verschuiving naar aërobe glycolyse, ongeacht de zuurstoftoevoer, wat het “Warburg-effect” wordt genoemd, waarbij pyruvaat rechtstreeks wordt omgezet in melkzuur in plaats van in de citroenzuurcyclus [1]. Aangezien alle kankercellen afhankelijk zijn van deze verandering in het metabolisme, vormen deze veranderde pathways aantrekkelijke therapeutische doelwitten [2]. Zowel in preklinische als in klinische studies zijn pogingen gedaan om het geherprogrammeerde metabolisme alleen of in combinatie met kankerchemotherapie aan te pakken [3]. Interessant is dat deze kankerspecifieke metabolische remodellering wordt omgekeerd door dichlooracetaat (DCA), een mitochondria-gerichte kleine molecule die na orale toediening in de meeste weefsels kan doordringen [4]. Het remt specifiek pyruvaat dehydrogenase kinase (PDK), een lid van de kinase familie, wat leidt tot reactivering van pyruvaat dehydrogenase (PDH), een sleutelenzym dat de flux van pyruvaat naar de mitochondria verplaatst om glucose-oxidatie te bevorderen in plaats van glycolyse [4]. Hoewel DCA onlangs is geëvalueerd in verschillende preklinische kankeronderzoeken [5], zijn de reacties van kankercellen op de behandeling met DCA, die bepalen of DCA klinisch voordeel oplevert bij de behandeling van kanker, nog niet volledig opgehelderd.
Meer dan 70% van de borstkankers brengen de oestrogeenreceptor (ER) tot expressie en zijn afhankelijk van oestrogeen om de tumorgroei en -progressie aan te sturen [6]. Daarom moet endocriene therapie bij de meeste patiënten worden beschouwd als een aanvulling op chirurgie, omdat deze therapie tumorremissie induceert en consistente klinische voordelen biedt. Het anti-oestrogeen geneesmiddel tamoxifen is de meest gebruikte behandeling voor patiënten met ER-positieve borstkanker in zowel het vroege als het gevorderde/metastatische stadium [7]. Als adjuvante therapie bij vroege borstkanker verbetert tamoxifen de algemene overleving, en men denkt dat het wijdverbreide gebruik ervan aanzienlijk heeft bijgedragen tot de vermindering van de borstkankersterfte die de laatste tien jaar is waargenomen [8]. Ondanks de duidelijke voordelen van tamoxifenbehandeling bij borstkanker, hervallen bijna alle patiënten met uitgezaaide ziekte en maar liefst 25% van de patiënten die adjuvant tamoxifen krijgen uiteindelijk en sterven aan de ziekte [9, 10]. De biologische mechanismen die ten grondslag liggen aan intrinsieke (de novo) en verworven resistentie tegen tamoxifen zijn daarom van groot klinisch belang. Een beter begrip van deze mechanismen kan nieuwe strategieën voorstellen om tamoxifenresistentie te overwinnen en de behandeling van borstkanker te verbeteren.
In de huidige studie toonden wij aan dat de expressie van EGFR in borstkankercellen werd verminderd door behandeling met DCA. Een combinatie van DCA en tamoxifen verlaagde de EGFR niveaus verder. Wij toonden aan dat DCA de cytotoxiciteit van tamoxifen voor borstkankercellen verhoogde door de EGFR-expressie te remmen. Bovendien sensibiliseerde DCA de tamoxifen-resistente MCF7-cellen voor tamoxifen via EFGR-downregulatie. Deze resultaten suggereren het mogelijke gebruik van DCA bij de behandeling van borstkanker door het afremmen van de EGFR signaalroute.
Resultaten
Remming van PDK downreguleert EGFR en verhoogt tamoxifen-geïnduceerde celdood in borstkankercellen
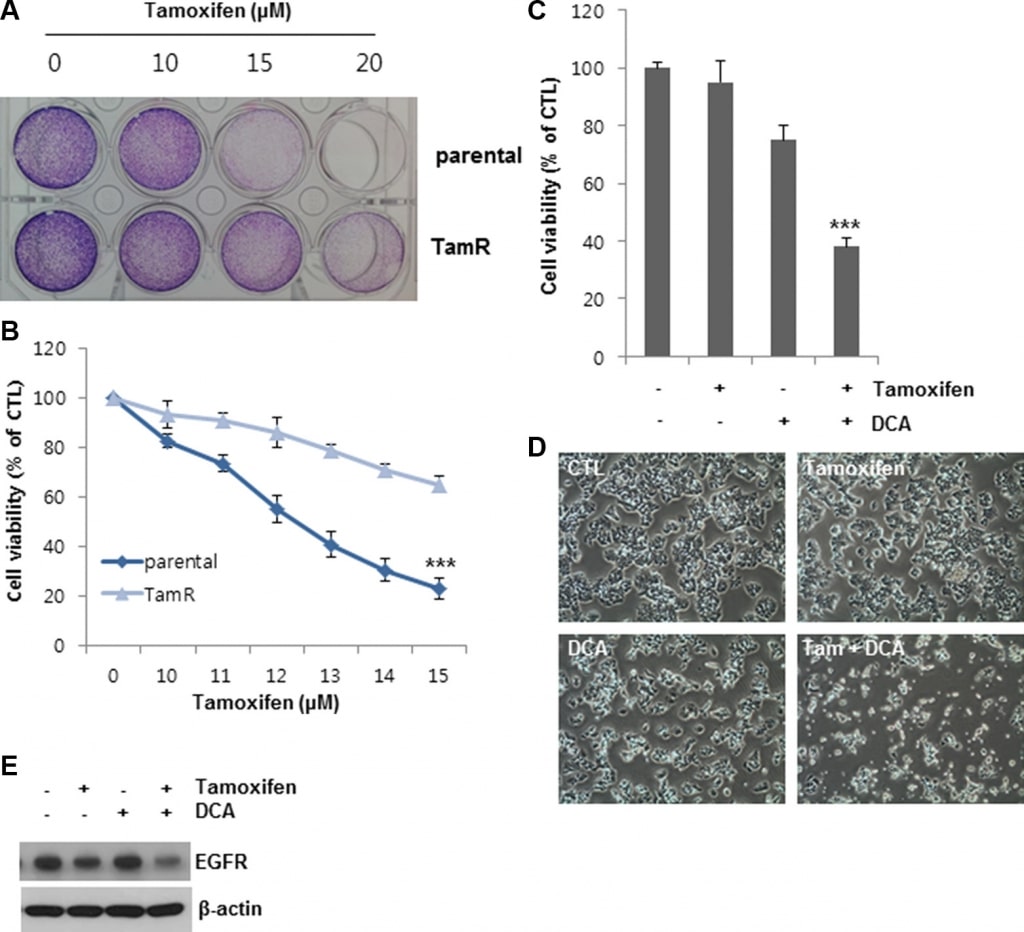
Recente studies hebben aangetoond dat het aanpakken van PDK door een PDK-remmer, zoals DCA, het metabolisme van de kankercel verandert van glycolyse naar oxidatieve fosforylering door mitochondriaal pyruvaatdehydrogenase te fosforyleren [4, 11]. Om na te gaan hoe groeifactor- en kinasesignaleringswegen in combinatie met PDK het Warburg-effect in borstkanker reguleren, onderzochten wij de expressieniveaus van verschillende groeifactorreceptoren in PDK-knockdown MCF7-cellen. Interessant is dat de depletie van PDK door siRNA-behandeling EGFR downreguleerde, in tegenstelling tot geen of marginale effecten op andere groeifactorreceptoren (figuur 1A). Vier PDK-isoenzymen (PDK1, PDK2, PDK3, PDK4) zijn geïdentificeerd in zoogdierweefsels [12]. Om de downregulatie van EGFR door siPDK-behandeling te bevestigen, onderzochten we de EGFR-expressie in de cellen behandeld met siRNA tegen elke isovorm van PDK. Elke siRNA-behandeling schafte alleen de expressie van de beoogde PDK af, maar ze veroorzaakten allemaal de downregulatie van EGFR, wat suggereert dat PDK de EGFR-expressie niet op een isovorm-specifieke manier reguleert (figuur 1B). Consistent met deze resultaten, DCA ook verminderd EGFR niveaus in een dosis-afhankelijke wijze (figuur 1C). Toen we de lactaatconcentratie in de kweekmedia analyseerden, was er geen significante verandering van de lactaatconcentratie door behandeling met tamoxifen/DCA of siRNA tegen EGFR, ook al werd deze verlaagd door behandeling met DCA (aanvullende figuur S1). Wij suggereren dus dat EGFR-downregulatie mogelijk niet geassocieerd is met de DCA-geïnduceerde metabolische verschuiving in borstkankercellen. Omdat de activering van de EGFR-signaleringsroute bijdraagt tot tamoxifenresistentie [13], onderzochten we of de downregulatie van EGFR door remming van PDK de cellen gevoelig maakte voor tamoxifen. Zoals getoond in figuur 1D, leidde co-behandeling met tamoxifen en DCA tot een duidelijke vermindering van de levensvatbaarheid van de cellen. De gecombineerde behandeling gedurende 72 uur verminderde de levensvatbaarheid van de cellen tot minder dan 30% van die van de controle (figuur 1E). Vervolgens werd de celdood in de medebehandelde cellen beoordeeld met behulp van Annexine V/PI-kleuring. Achtenveertig uur na de behandeling induceerde de combinatie van tamoxifen en DCA 30% celdood, vergeleken met 15% of 8% door tamoxifen of DCA alleen, respectievelijk (figuur 1F). Eerder rapporteerden wij dat apoptotische celdood in borstkankercellen werd veroorzaakt door het verlies van mitochondriaal membraanpotentieel (MMP) [14]. Wij hebben dus getest of verlies van MMP betrokken is bij celdood geïnduceerd door de medebehandeling, maar er was geen significante verandering in MMP tussen onbehandelde en tamoxifen/DCA-behandelde cellen (aanvullende figuur S2).
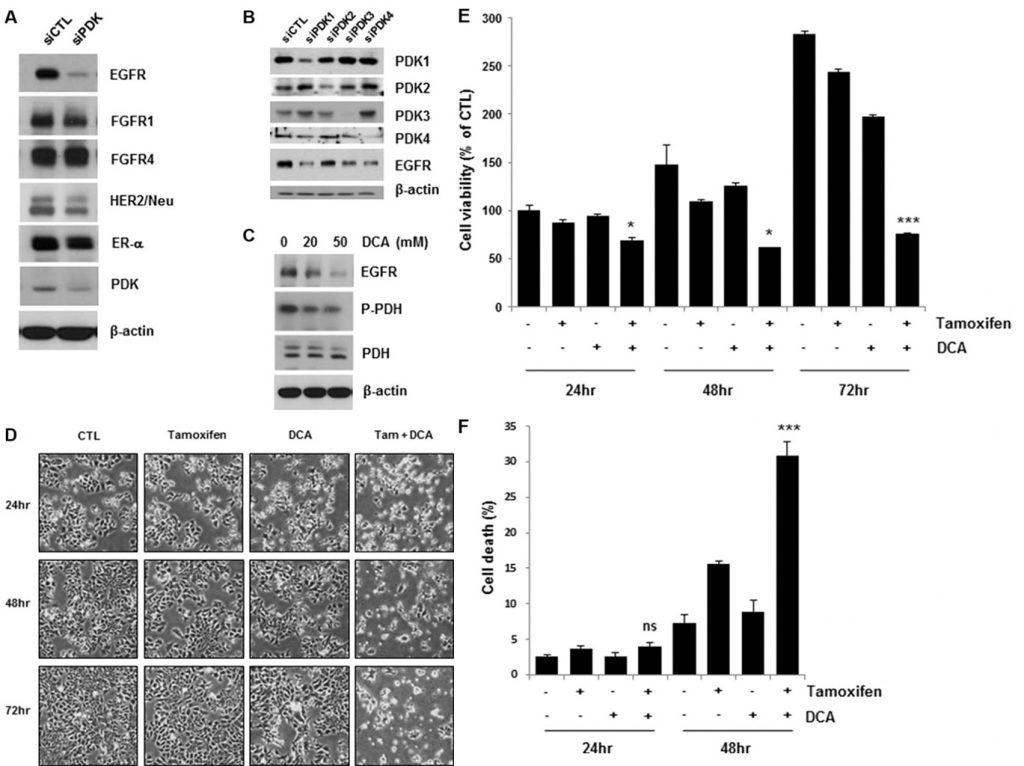
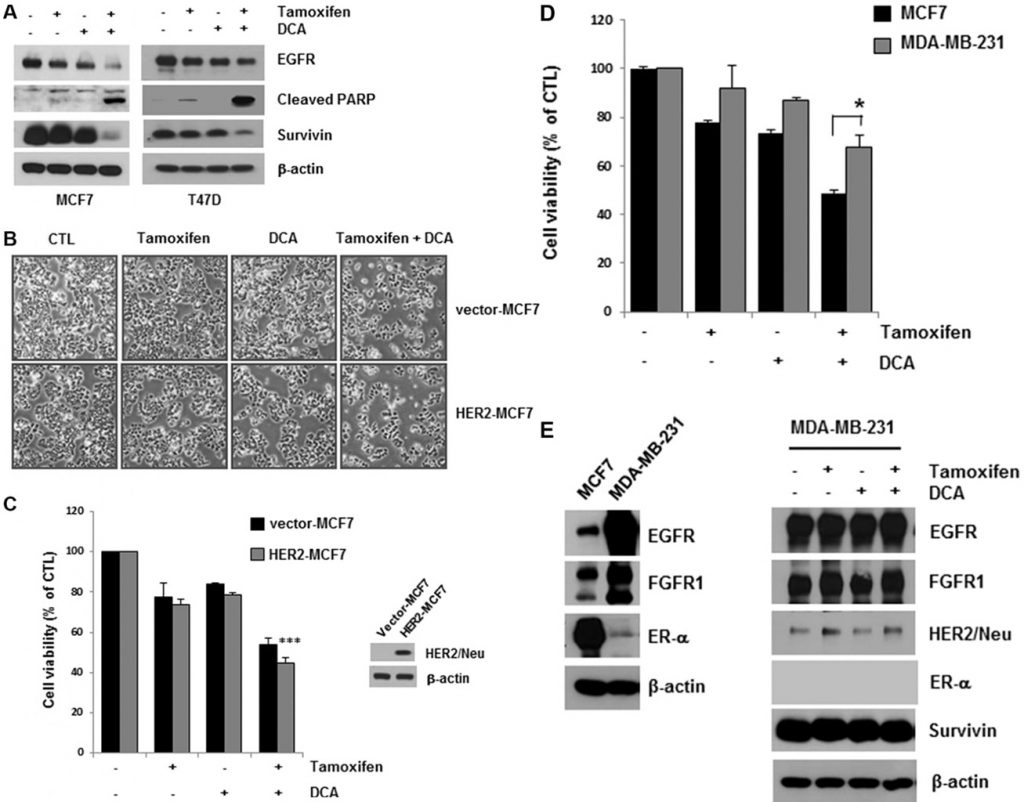
DCA plus tamoxifen verminderde de EGFR-niveaus in zowel MCF7- als T47D-cellen verder in vergelijking met die van DCA alleen (figuur 2A). De celdood geïnduceerd door de co-behandeling werd bevestigd door het detecteren van PARP-splitsing, een marker van apoptose (figuur 2A). Survivin is een anti-apoptotische molecule en een doelwit van de ER [15]. De medebehandeling downreguleerde ook survivin, dat apoptose in de cellen kan mediëren (figuur 2A). Hoewel tamoxifenbehandeling de EGFR-niveaus in MCF7- en T47D-cellen licht verminderde, werd geen significante toename van celdood in de cellen waargenomen, wat suggereert dat een kritisch niveau van EGFR nodig is voor de overleving van borstkankercellen (figuur 2A).
Uit bewijsmateriaal van cellijnen is gebleken dat overexpressie van HER2 pathways kan bijdragen tot verworven resistentie tegen endocriene therapieën [13]. Om te bepalen of HER2-overexpressie de cytotoxiciteit van tamoxifen en DCA beïnvloedt, onderzochten we de levensvatbaarheid van de cellen van HER2-overexpressie MCF7 (HER2-MCF7) na behandeling met tamoxifen en DCA. Uit de resultaten bleek dat tamoxifen en DCA de levensvatbaarheid van de cellen aanzienlijk verminderden, zelfs in HER2-MCF7-cellen (figuur 2B en 2C), wat suggereert dat DCA de tamoxifen-geïnduceerde celdood in HER2-overexpresserende borstkankercellen zou kunnen versterken. Wij evalueerden verder de groeiremmende effecten van de medebehandeling op de triple-negatieve borstkankercellijn MDA-MB-231. Zoals getoond in figuur 2D waren MDA-MB-231-cellen minder gevoelig voor tamoxifen en DCA dan MCF7-cellen. Omdat downregulatie van EGFR werd waargenomen in ER-positieve cellen, onderzochten we de effecten van tamoxifen en DCA op EGFR-niveaus in MDA-MB-231 cellen. EGFR kwam sterk tot expressie in MDA-MB-231 cellen in vergelijking met MCF7 cellen, en de niveaus werden niet significant verlaagd door tamoxifen en DCA (figuur 2E). Vervolgens onderzochten we de cytotoxiciteit van tamoxifen en DCA in de niet-tumorigene geïmmortaliseerde borstepitheelcellijn MCF10A. Interessant genoeg was de expressie van EGFR in MCF10A cellen vergelijkbaar met die van MDA-MB-231 cellen en werd noch EGFR-downregulatie noch celdood waargenomen in MCF10A cellen na behandeling met tamoxifen en DCA (aanvullende figuur S3). Deze resultaten wijzen erop dat de anti-proliferatieve effecten van tamoxifen en DCA in borstkankercellen afhankelijk zijn van EGFR-downregulatie.
De gecombineerde behandeling van tamoxifen en DCA induceert p38 MAPK-gemedieerde EGFR afbraak
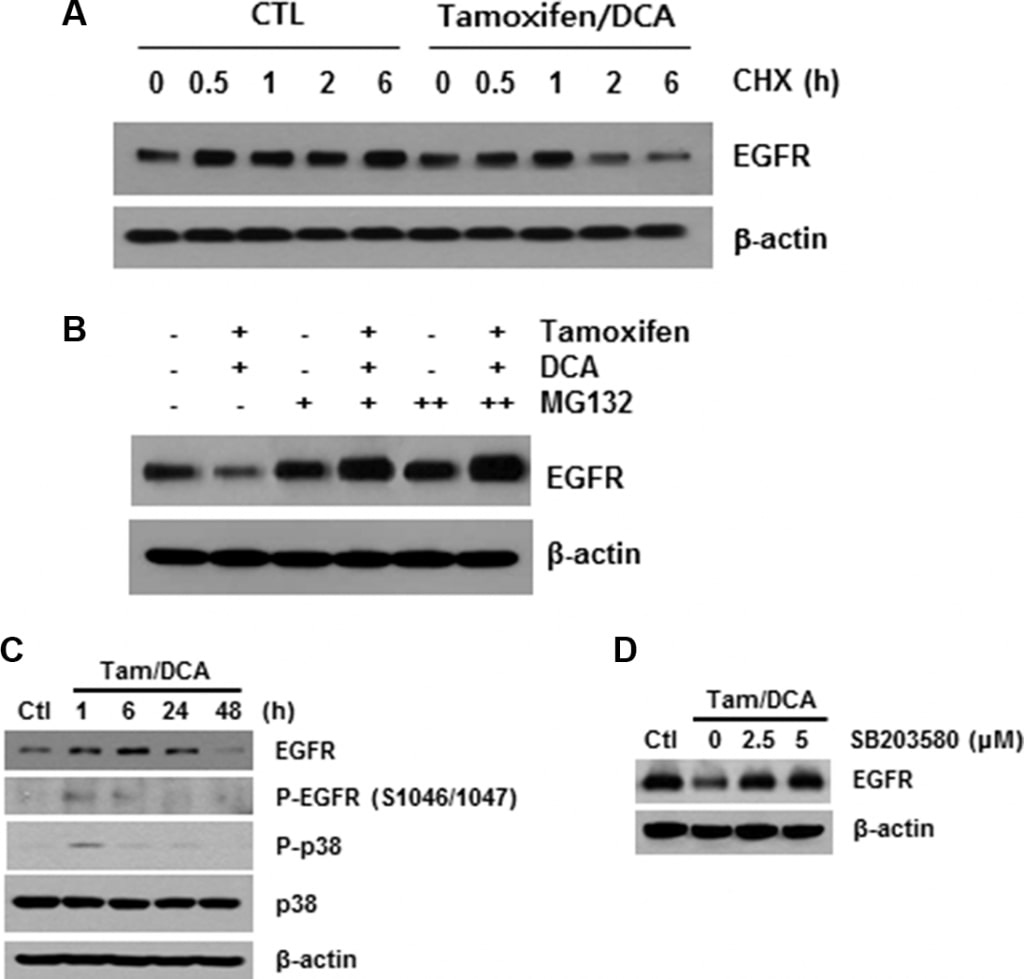
Zoals hierboven beschreven veroorzaakt ligand binding snelle autofosforylering, resulterend in de verwijdering van de EGFR van het celoppervlak via endocytose naar een vroeg endosomaal compartiment [16]. Daarom onderzochten we vervolgens de rol van receptormodificatie in tamoxifen/DCA-gemedieerde EGFR-downregulatie. Na het blokkeren van de eiwitsynthese met cycloheximide vonden we dat de stabiliteit van EGFR aanzienlijk was aangetast in met tamoxifen/DCA behandelde cellen, vergeleken met die van de controle (figuur 3A). Vervolgens evalueerden we de effecten van MG132, een proteasoomremmer, op tamoxifen/DCA-geïnduceerde EGFR-degradatie. Behandeling met MG132 herstelde de EGFR-expressie in met tamoxifen/DCA behandelde cellen op dosis-afhankelijke wijze (figuur 3B).
De fosforylering van EGFR op serine en threonine residuen vertegenwoordigt een mechanisme voor verzwakking van de EGFR activiteit, en onder hen zijn de serine 1046/1047 (Ser 1046/7) fosforyleringsplaatsen nodig voor EGFR desensitisatie [17]. Recent is gemeld dat p38 mitogen-activated protein kinase (MAPK) de fosforylering van EGFR op Ser 1046/7 induceert, wat leidt tot afbraak in kankercellen [18]. Daarom onderzochten we vervolgens de effecten van tamoxifen en DCA op de fosforylering van p38 MAPK in MCF7-cellen. p38 MAPK werd significant gefosforyleerd binnen 1 uur, en de fosforylering bleef gedurende 24 uur na behandeling met tamoxifen en DCA gehandhaafd (figuur 3C). Bovendien werd de degradatie van EGFR geïnduceerd door de co-behandeling significant onderdrukt wanneer de cellen werden voorbehandeld met een specifieke p38 MAPK-remmer, SB203580 (figuur 3D), wat aangeeft dat p38 MAPK-activatie een rol speelt in tamoxifen/DCA-geïnduceerde EGFR-downregulatie in MCF7-cellen.
EGFR-remmers versterken de tamoxifen-geïnduceerde celdood
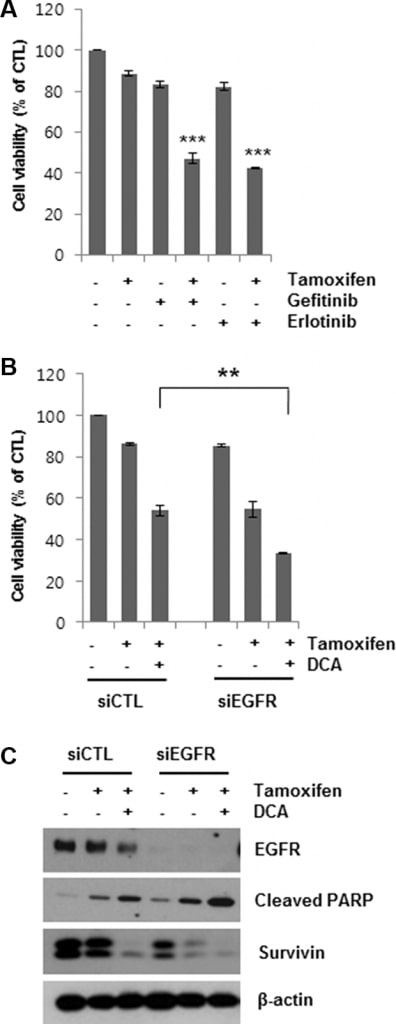
Na te hebben aangetoond dat DCA-gemedieerde EGFR-degradatie de tamoxifen-geïnduceerde celdood in MCF7-cellen kon versterken, hebben we vervolgens bepaald of EGFR-remming de celdood in met tamoxifen behandelde cellen verbeterde. Gefitinib en erlotinib zijn selectieve omkeerbare remmers van EGFR tyrosine kinase binding aan ATP [19, 20]. Medebehandeling van MCF7-cellen met 5 μM gefitinib of erlotinib gedurende 48 uur verhoogde de tamoxifen-geïnduceerde celdood aanzienlijk (figuur 4A). Ook het uitschakelen van EGFR met siEGFR versterkte de tamoxifen-geïnduceerde celdood (figuur 4B). De vermindering van de levensvatbaarheid van de cellen door de gecombineerde behandeling van siEGFR en tamoxifen was vergelijkbaar met die van DCA en tamoxifen. Bovendien verlaagde siEGFR-behandeling verder de EGFR-niveaus in met tamoxifen/DCA behandelde cellen, wat resulteerde in een verhoogde apoptotische celdood samen met downregulatie van survivin in vergelijking met die van de si-controle (figuur 4C). Deze resultaten ondersteunen de conclusie dat DCA borstkankercellen gevoelig maakt voor tamoxifen via downregulatie van EGFR.
c-myc en Nanog expressie zijn geassocieerd met de cytotoxische effecten van DCA in met tamoxifen behandelde MCF7 cellen
EGFR activatie in kanker blijkt verschillende transcriptiefactoren te verhogen die het type en de duur van EGFR signalering kunnen beïnvloeden [21]. Onder hen hebben Nanog en c-myc een pleiotrope rol in tumorigenese, met inbegrip van resistentie tegen standaardtherapie bij borstcarcinoom [22, 23]. Om de anti-tumoractiviteiten gemedieerd door EGFR-downregulatie in borstkankercellen verder te onderzoeken, onderzochten wij de expressie van c-myc en Nanog in MCF7-cellen na co-behandeling met tamoxifen en DCA. De niveaus van c-myc en Nanog werden significant verlaagd in de cellen die behandeld werden met tamoxifen en DCA (figuur 5A). Ook werden de expressies van beide eiwitten onderdrukt door tamoxifen in combinatie met gefitinib of erlotinib (figuur 5B), wat erop wijst dat EGFR-expressie nodig is om de expressie van c-myc en Nanog in met tamoxifen behandelde MCF7-cellen te handhaven. Toch was er weinig of geen effect van siEGFR op de expressie van c-myc en Nanog, wat erop wijst dat EGFR-downregulatie noodzakelijk maar niet voldoende is om de expressie van de eiwitten te verminderen (aanvullende figuur S4). Om te bepalen of de verminderde expressie van deze twee eiwitten betrokken is bij de cytotoxische effecten van tamoxifen en DCA in MCF7-cellen, testten we de effecten van siRNA’s tegen c-myc en Nanog in cellen die blootgesteld waren aan tamoxifen/DCA. Knockdown van c-myc en Nanog maakte de cellen aanzienlijk gevoeliger voor tamoxifen/DCA in vergelijking met de controles (figuur 5C). Omgekeerd beschermde overexpressie van FLAG-c-myc en FLAG-Nanog door transfectie van cellen met FLAG-c-myc en FLAG-Nanog vectoren de cellen aanzienlijk tegen tamoxifen/DCA-geïnduceerde cytotoxiciteit (figuur 5D). Al met al suggereren onze gegevens dat EGFR-downregulatie door een combinatie van tamoxifen en DCA celdood in MCF7-cellen kan induceren, gedeeltelijk door remming van c-myc- en Nanog-expressie. Het is bekend dat zelfvernieuwende genen, zoals c-myc en Nanog geassocieerd zijn met kankerstamachtige celeigenschappen 24. Om te testen of co-behandeling met tamoxifen en DCA borstkankerstamcellen remt, voerden we flowcytometrie-analyse uit om het aandeel van stamcel-subpopulatie in MCF7-cellen te schatten op basis van de expressie CD44. Na de co-behandeling werd de CD44-hoge populatie in MCF7-cellen verminderd van 42,5% tot 19,9% (figuur 5E). Deze bevinding deed de mogelijkheid rijzen dat DCA plus tamoxifen het vermogen van kankerstamcellen in borstkankercellen zou kunnen remmen.

De gecombineerde behandeling met DCA en tamoxifen kan tamoxifenresistentie in borstkankercellen overwinnen
Om de waargenomen effecten van DCA op tamoxifen-geïnduceerde celdood verder te bevestigen, stelden we tamoxifenresistente (TamR) MCF7-cellen op door tamoxifenbehandeling over een lange periode. Vergeleken met de onbehandelde cellen was de levensvatbaarheid van de MCF7 en TamR MCF7-cellen respectievelijk 20 en 60% na behandeling met 13 uM tamoxifen, wat aangeeft dat de TamR MCF7-cellen minder gevoelig waren voor dezelfde concentratie tamoxifen dan de MCF7-oudercellen (figuur 6A en 6B). DCA alleen remde de groei van TamR MCF7-cellen met ongeveer 25% van die van de controle (figuur 6C en 6D). Daarentegen onderdrukte co-behandeling van DCA met tamoxifen de celgroei met meer dan 60% ten opzichte van de controle (figuur 6C en 6D). De EGFR-niveaus in TamR-cellen werden ook verlaagd door de medebehandeling (figuur 6E). Deze resultaten suggereren dat DCA tamoxifen-resistentie in borstkankercellen kan overwinnen door EGFR-downregulatie op eiwitniveau.
Discussie
Naarmate het inzicht in het metabole fenotype van tumorcellen is gegroeid, is het aanpakken van de metabole verschillen tussen tumor- en normale cellen voorgesteld als een nieuwe strategie tegen kanker. Ondanks de toename van potentiële geneesmiddelen tegen kanker die gericht zijn op metabole processen van kankercellen, moet worden opgehelderd hoe cellen op de geneesmiddelen reageren om het kankermetabolisme met succes aan te pakken. Hier is aangetoond dat DCA niet alleen de PDK activiteit maar ook de EGFR expressie in borstkankercellen remt. De gecombineerde behandeling van DCA en tamoxifen induceerde proteasoom-afhankelijke afbraak van EGFR eiwitten in borstkankercellen via p38 MAPK activering. Onze resultaten wijzen erop dat EGFR-downregulatie een sleutelrol speelt in de apoptotische celdood geïnduceerd door een combinatie van DCA en tamoxifen. Voor zover wij weten is dit het eerste rapport dat aantoont dat DCA borstkankercellen gevoelig maakt voor tamoxifen-geïnduceerde celdood via EGFR-downregulatie.
In ER-positieve borstkankercellen die endocriene resistentie hebben ontwikkeld, kan ER-expressie rechtstreeks worden onderdrukt door versterkte groeifactorreceptorsignalering als gevolg van overexpressie van EGFR en HER2, die vervolgens MAPK activeren en ER-transcriptie remmen [25]. Bovendien is resistentie tegen tamoxifen in borstkankercellen in verband gebracht met overexpressie van EGFR en hoge niveaus van gefosforyleerd extracellulair geactiveerd kinase 1/2 [2]. De strategie van het combineren van een EGFR-remmer met een endocrien middel is dus van voldoende belang om verder onderzoek naar de behandeling van borstkanker te rechtvaardigen. In deze studie vonden we dat downregulatie van EGFR in met DCA behandelde borstkankercellen de gevoeligheid van de cellen voor tamoxifen verhoogde. EGFR-downregulatie door gelijktijdige behandeling van DCA met tamoxifen is bevestigd in verschillende ER-positieve borstkankercellijnen, waaronder MCF7, T47D en BT474 (figuur 2A en niet getoond). Naast de HER2-geamplificeerde borstkankercellijn BT474 vertoonden HER2-MCF7-cellen ook EGFR-downregulatie door DCA/tamoxifen, wat suggereert dat de combinatiebehandeling toepasbaar is op HER2-positieve borstkanker. Helaas vertoonde een triple-negatieve borstkankercellijn, MDA-MB-231, waarvan bekend is dat zij EGFR overexpresseert [26], een refractair fenotype en geen duidelijke verandering in EGFR-niveaus in reactie op de medebehandeling. Eén mogelijkheid is dat de amplificatie van EGFR in MDA-MB-231 cellen de downregulatie ervan door de medebehandeling van DCA met tamoxifen tenietdoet. De andere verklaring is dat de signaalweg(en) die voor de EGFR-downregulatie zorgen, door een onbekend mechanisme in MDA-MB-231 cellen worden geblokkeerd. Verder onderzoek zal nodig zijn om het onderliggende mechanisme op te helderen voor het behoud van EGFR-expressie in reactie op de co-behandeling van DCA en tamoxifen in ER-negatieve borstkankercellen.
Er zijn vier isovormen van PDK bekend, die elk actief zijn in reactie op verschillende intracellulaire en extracellulaire omstandigheden. PDK1 wordt geactiveerd door hypoxie [27]; PDK2 wordt geactiveerd door de PDH-producten acetyl CoA en NADH [28]; PDK3 wordt geactiveerd door ATP [29]; en PDK4 wordt transcriptioneel gereguleerd door hormonale signalen [ 30]. PDK2 bezit de grootste activiteit om het pyruvaat dehydrogenase complex te fosforyleren, gevolgd door PDK4, PDK1 en PDK3 [12]. PDK2 is het meest gevoelig voor remming door DCA vanwege zijn alomtegenwoordige expressie [31]. Hoewel wij de expressie van vier isovormen in MCF7-cellen ontdekten (figuur 1C), zijn de functionele verschillen tussen PDK-isovormen in borstkanker nog steeds onduidelijk. Verder onderzoek of signaalwegen die bepaalde isovormen van PDK onderdrukken leiden tot EGFR-downregulatie zal interessant zijn. Van PDK is bekend dat het in de mitochondria functioneert, en verschillende studies hebben aangetoond dat EGFR ook naar de mitochondria transloceert na EGF-stimulatie [32, 33]. Voorts is gesuggereerd dat de interactie van EGFR met PDK in de mitochondriale matrix een belangrijke rol speelt bij EGFR-geïnduceerde tumorgroei in glioblastoma multiforme [ 34]. Hier vonden we dat de blokkade van PDK activiteit door een remmer of silencing de totale cellulaire EGFR niveaus in met tamoxifen behandelde borstkankercellen verminderde. Verdere studies zijn nodig om te bepalen of alleen mitochondriaal-geassocieerd EGFR onderhevig is aan degradatie door co-behandeling met DCA en tamoxifen in borstkankercellen. In het algemeen veroorzaakt blootstelling van cellen aan EGF snelle autofosforylering, met inbegrip van tyrosine (Tyr) 1045, die een docking site biedt voor het ubiquitin-ligase c-Cbl, wat resulteert in de ubiquitinatie van de EGFR en verwijdering van de EGFR via endocytose van het celoppervlak naar een vroeg endosomaal compartiment [35]. Cotreatment met DCA en tamoxifen had echter geen effect op de fosforylering van EGFR op Tyr 1045 (data niet getoond). In plaats daarvan zagen we de fosforylering van EGFR op Ser 1046/7 door DCA en tamoxifen, die werd geblokkeerd door een specifieke p38 MAPK-remmer, SB203580. Er is gesuggereerd dat p38 MAPK een centrale rol speelt in zowel de internalisatie als de degradatie van EGFR [18]; verder onderzoek is echter nodig om op te helderen hoe EGFR-fosforylering op serineresten de downregulatie ervan veroorzaakt en om de upstream mediator van p38 MAPK activering na DCA/tamoxifen behandeling te identificeren.
Nanog en c-myc zijn geïdentificeerd als transcriptiefactoren, en hun rol bij de instandhouding van zelfvernieuwing in embryonale stamcellen is in eerdere studies vastgesteld [22, 36]. Een recent voorgesteld mechanisme van intrinsieke resistentie tegen endocriene therapie stelt het bestaan van een gespecialiseerde subset van kankercellen, tumor-initiërende cellen (TIC’s) genaamd [ 37]. TIC’s hebben het vermogen om zichzelf te vernieuwen en nieuwe tumoren te genereren die volledig bestaan uit klonaal afgeleide celtypes die aanwezig zijn in de ouderlijke tumor. Hier toonden we aan dat DCA de expressie van c-myc en Nanog verminderde in met tamoxifen behandelde MCF7-cellen. Bovendien was de expressie van deze eiwitten gerelateerd aan de cytotoxische effecten van de gecombineerde behandeling van DCA en tamoxifen. Daarom kan DCA worden gebruikt om het therapeutisch potentieel van tamoxifen uit te breiden door de ontwikkeling van intrinsieke resistentie bij borstkankertherapie te onderdrukken.
Concluderend bleek uit onze resultaten dat DCA ER-positieve borstkankercellen gevoelig maakte voor tamoxifen door de EGFR-niveaus te verlagen. Behandeling met DCA en tamoxifen remde de expressie van zelfvernieuwende genen en de overleving van tamoxifen-resistente MCF7-cellen. Daarom stellen wij voor dat DCA een effectief therapeutisch middel kan zijn voor de behandeling van tamoxifen-resistente borstkanker. Bovendien kunnen combinatiestrategieën met DCA nuttig zijn om de doeltreffendheid van andere cytotoxische chemotherapieën of gerichte therapieën te verbeteren. Verdere experimenten, waaronder dierstudies en klinische proeven, moeten in de toekomst worden uitgevoerd.
Materialen en methoden
Celcultuur en reagentia
MCF7, T47D en MDA-MB-231 menselijke borstkankercellen werden gekocht van de American Type Culture Collection (Rockville, MD, USA) en werden gekweekt in het aanbevolen groeimedium (Invitrogen, Carlsbad, CA, USA). HER2-overexpressie MCF7 (HER2-MCF7) en controlevectorcellen (vector-MCF7) werden vriendelijk ter beschikking gesteld door Dr. Incheol Shin (Hanyang University, Seoul, Korea). Tamoxifen-resistente (TamR) MCF7-cellen werden ontwikkeld door het kweken van MCF7-cellen in de aanwezigheid van 10 uM tamoxifen gedurende meer dan 6 maanden. Als controle werden de oudercellen gedurende dezelfde tijd in gewone media gekweekt. Na de vaststelling van resistentie werden de cellen niet langer dan 3 maanden gepasseerd. Voor experimenten met tamoxifenbehandelingen werden de cellen routinematig gekweekt in fenolroodvrij Dulbecco’s gemodificeerd Eagle’s medium plus 10% met houtskool gestript foetaal runderserum vlak voor de behandelingen. Antilichamen tegen EGFR, fosfo-EGFR (S1046/1047), FGFR1, FGFR4, PDK1, PDH, survivin, gespleten PARP, p38, fosfo-p38, en Nanog werden verkregen van Cell Signaling Technology (Danvers, MA, USA). Antilichamen tegen HER2/Neu, ER-α en c-myc, siRNA’s gericht tegen EGFR, Nanog, PDK4 en c-myc en de negatieve controle (gescrambelde) siRNA’s werden verkregen van Santa Cruz Biotechnology (Dallas, TX, VS). Antilichamen tegen PDK2 en PDK3 werden verkregen van Thermo Fisher Scientific (Waltham, MA, USA). Het β-actine antilichaam, het FLAG antilichaam, tamoxifen, DCA, cycloheximide en MG132 werden gekocht bij Sigma-Aldrich (St. Louis, MO, USA). Het fosfo-PDH (S293) antilichaam en SB203580 waren afkomstig van BD Biosciences Pharmingen (San Diego, CA, USA), en gefitinib en erlotinib werden verkregen van Selleck Chemicals (London, ON, Canada).
Transfecties en behandelingen
Cellen werden getransfecteerd met het doelsiRNA (50 nM) met behulp van Lipofectamine RNAiMAX (Invitrogen) zoals beschreven door de fabrikant. Cellen werden getransfecteerd met 1 ug FLAG-c-myc pcDNA 3.1 en FLAG-Nanog-pcDNA 3.1 met behulp van Lipofectamine 2000 zoals beschreven door de fabrikant. Na 6 uur werden de cellen gedurende 24-48 uur behandeld met tamoxifen en/of DCA en vervolgens geanalyseerd zoals elders beschreven.
Meting van de levensvatbaarheid van de cellen
De levensvatbaarheid van de cellen werd bepaald door de mitochondriale omzetting van 3-(4,5-dimethylthiazolyl2)-2,5-difenyltetrazoliumbromide (MTT) in een gekleurd product te meten. De cellen werden behandeld zoals aangegeven, en het medium werd vervangen door serumvrij medium met 1 mM MTT. Na 2 uur incubatie bij 37°C werden de cellen opgelost in DMSO. De hoeveelheid formazan, de omgezette vorm van MTT, werd bepaald door de absorptie bij 595 nm te meten. Evaluatie van apoptose
Apoptose werd bepaald door fluorescentie-geactiveerde celsorteringsanalyse met behulp van een Annexin V-FITC apoptosekit (BioVision, Milpitas, CA, USA) volgens de aanwijzingen van de fabrikant. Kort gezegd werden de cellen na behandeling behandeld met trypsine en vervolgens geresuspendeerd in bindingsbuffer (10 mM HEPES/NaOH, pH 7,4, 140 mM NaCl, 2,5 mM CaCl2 ) met Annexin V-FITC en propidiumjodide. Na incubatie gedurende 15 min. werd de celfluorescentie geanalyseerd met flowcytometrie. Celdood werd gemeten als het percentage cellen in de Annexine V- en PI-positieve populatie.
Western blotting
De cellen werden geoogst en gelyseerd in RIPA-buffer (50 mM Tris-HCl pH 7,5, 150 mM NaCl, 1% Nonidet P40, 0,5% natriumdeoxycholaat en 0,1% SDS) aangevuld met een protease/fosfatase-inhibitor cocktail (Roche, Mannheim, Duitsland). Gelijke hoeveelheden eiwitten (20-50 μg) werden gescheiden door SDS-PAGE en overgebracht op een nitrocellulosemembraan. De membranen werden geblokkeerd door incubatie met 5% magere melk in Trisbuffered Saline gedurende 1 uur en werden vervolgens een nacht geïncubeerd met de juiste primaire antilichamen. Membranen werden geïncubeerd met HRP-geconjugeerd secundair antilichaam gedurende 1 uur. Immunoreactieve eiwitten werden gevisualiseerd met behulp van versterkte chemiluminiscentiereagentia (Amersham Biosciences, Little Chalfont, UK).
Detectie van CD44 positieve celpopulatie
Cellen werden gedurende 15 minuten gekleurd met antilichamen in een verdunning van 1∶100 in PBS. De gebruikte antilichamen waren als volgt: FITC-CD44 en FITC-geconjugeerde isotypecontrole-muis-IgG-antilichamen werden verkregen van BD Biosciences Pharmingen. Gelabelde cellen werden geanalyseerd op flowcytometrie. Populaties van CD44-positieve cellen werden bepaald aan de hand van de intensiteit van FITC.
Statistische analyse
Alle gepresenteerde gegevens zijn representatief voor ten minste twee afzonderlijke experimenten. Vergelijkingen tussen groepen werden geanalyseerd met de Student’s t-test. Sterretjes (***p < 0,001, **p < 0,01, *p < 0,05) wijzen op statistische significantie.
Dankbetuigingen en financiering
Dit onderzoek werd ondersteund door Basic Science Research Program via de National Research Foundation van Korea (NRF), gefinancierd door het Ministerie van Wetenschap, ICT & Toekomstplanning (nr. 1711031812, 1711023318 en 1711031800).
Belangenverstrengeling
De auteurs verklaren geen belangenconflicten te hebben.
VERWIJZINGEN
1 1. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029-33.
2ZhaoY, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013; 4:e532.
3 Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011; 10:671-84.
4 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11: 37-51.
5 Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug? Biochim Biophys Acta. 2014; 1846:617-29.
6 Osborne CK. Tamoxifen bij de behandeling van borstkanker. N Engl J Med. 1998 Nov 26; 339:1609-18.
7 Johnston SR. Nieuwe strategieën bij oestrogeenreceptor-positieve borstkanker. Clin Cancer Res. 2010 Apr 1; 16:1979-87. doi: 10.1158/1078-0432.CCR-09-1823.
8 Peto R, Boreham J, Clarke M, Davies C, Beral V. UK and USA breast cancer deaths down 25% in year 2000 at ages 20-69 years. Lancet. 2000; 355:1822.
9 Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005; 365:1687-717.
10 Early Breast Cancer Trialists’ Collaborative Group (EBCTCG), Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, Dowsett M, Ingle J, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011; 378:771-84.
11 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013; 73:7277-89.
12 Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer. 2016; 138:809-17.
13 Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med. 2011; 62:233-47.
14YunSM, Woo SH, Oh ST, Hong SE, Choe TB, Ye SK, Kim EK, Seong MK, Kim HA, Noh WC, Lee JK, Jin HO, Lee YH, et al. Melatonin enhances arsenic trioxide-induced cell death via sustained upregulation of Redd1 expression in breast cancer cells. Mol Cell Endocrinol. 2016; 422:64-73.
15 Zhu J, Lu X, Hua KQ, Sun H, Yu YH, Feng YJ. Oestrogen receptor α mediates 17β-estradiol enhancement of ovarian cancer cell motility through up-regulation of survivin expression. Arch Gynecol Obstet. 2012; 286: 729-37.
16 Arteaga CL. Epidermal growth factor receptor dependence in human tumors: more than just expression? Oncologist. 2002; 7:31-9.
17 Sorkin A, Goh LK. Endocytosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009; 315:683-96.
18 Adachi S, Shimizu M, Shirakami Y, Yamauchi J, Natsume H, Matsushima-Nishiwaki R, To S, Weinstein IB, Moriwaki H, Kozawa O. (-)-Epigallocatechin gallate downregulates EGF receptor via phosphorylation at Ser1046/1047 by p38 MAPK in colon cancer cells. Carcinogenesis. 2009; 30:1544-52.
19CiardielloF, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000; 6:2053-63.
20 Pollack VA, Savage DM, Baker DA, Tsaparikos KE, Sloan DE, Moyer JD, Barbacci EG, Pustilnik LR, Smolarek TA, Davis JA, Vaidya MP, Arnold LD, Doty JL, et al. Inhibition of epidermal growth factor receptor associated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor inhibition in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther. 1999; 291:739-48.
21 Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signalering in kanker. Gene. 2006; 366:2-16.
22 Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014; 135:2741-8.
23 Chen Y, Olopade OI. MYC in borsttumorprogressie. Expert Rev Anticancer Ther. 2008; 8: 1689-98.
24 Hou ZJ, Luo X, Zhang W, Peng F, Cui B, Wu SJ, Zheng FM, Xu J, Xu LZ, Long ZJ, Wang XT, Li GH, Wan XY, et al. Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells. Oncotarget. 2015; 6:6326-40. doi:10.18632/oncotarget.3436.
25 Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Activation of mitogenactivated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induceert een in vivo moleculair fenotype van estrogen receptor alpha-negative human breast tumors. Cancer Res. 2006; 66:3903-11.
26 Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: A major pathway of cell-cycle progression in estrogenreceptor negative breast cancer cells. Proc Natl Acad Sci U S A. 2000; 97:8542-7.
27 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006; 3:177-85.
28 Hiromasa Y, Hu L, Roche TE. Ligand-induced effects on pyruvate dehydrogenase kinase isoform 2. J Biol Chem. 2006; 281:12568-79.
29 Kato M, Chuang JL, Tso SC, Wynn RM, Chuang DT. Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. EMBO J. 2005; 24:1763-74.
30 Kwon HS, Huang B, Unterman TG, Harris RA. Protein kinase B-alpha inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes. 2004; 53:899-910.
31 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998; 329 :191-6.
32 Dasari VR, Velpula KK, Alapati K, Gujrati M, Tsung AJ. Stamcellen uit navelstrengbloed remmen translocatie epidermale groeifactorreceptor naar mitochondriën in glioblastoom. PLoS One. 2012; 7:e31884.
33 Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, Hüttemann M, Douglas R, Haddad G, Parsons SJ. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem. 2009; 284:36592-604.
34 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Gecombineerde targeting van PDK1 en EGFR triggert regressie van glioblastoom door omkering van het Warburg-effect. Cancer Res. 2013; 73:7277-89.
35 Massie C, Mills IG. De ontwikkelende rol van receptoren en adaptoren. Nat Rev Cancer. 2006; 6:403-9.
36 Chappell J, Dalton S. Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harb Perspect Med. 2013;3:a014381.
37 Wei W, Lewis MT. Identifying and targeting tumor-initiating cells in the treatment of breast cancer. Endocr Relat Cancer. 2015; 22:R135-55.