Sang Hyeok Woo1,*, Sung-Keum Seo1,*, Yoonhwa Park1,2, Eun-Kyu Kim3, Min-Ki Seong4, Hyun-Ah Kim4, Jie-Young Song1, Sang-Gu Hwang1, Jin Kyung Lee5, Woo Chul Noh4, In-Chul Park1
1Divisiónde Investigación del Cáncer por Radiación, Instituto Coreano de Ciencias Radiológicas y Médicas, Nowon-gu, Seúl, 01812, República de Corea
2Escuelade Ciencias de la Vida y Biotecnología, Universidad de Corea, Seongbuk-gu, Seúl, 02841, República de Corea
3Departamento de Cirugía, Centro de Cáncer de Mama, Hospital Bundang de la Universidad Nacional de Seúl, Facultad de Medicina de la Universidad Nacional de Seúl, Bundang-gu, Seongnam, 13620, República de Corea
4Departamentode Cirugía, Hospital del Centro Oncológico de Corea, Instituto Coreano de Ciencias Radiológicas y Médicas, Nowon-gu, Seúl, 01812, República de Corea
5KIRAMSRadiation Biobank, Instituto Coreano de Ciencias Radiológicas y Médicas, Nowon-gu, Seúl, 01812, República de Corea
*Estosautores han contribuido a partes iguales a este trabajo
Correspondencia a: In-Chul Park, correo electrónico: [email protected]
Palabras clave: tamoxifeno, cáncer de mama, dicloroacetato, receptor del factor de crecimiento epidérmico, piruvato deshidrogenasa cinasa
Recibido: 18 de mayo de 2016
Aceptado: 22 de julio de 2016
Publicado en línea: 01 de agosto de 2016
Resumen
Recientemente se ha reconocido que la reprogramación metabólica en las células cancerosas es una característica esencial de la neoplasia. En este contexto, las alteraciones metabólicas representan una diana terapéutica atractiva, y en estudios preclínicos se han obtenido resultados alentadores con fármacos dirigidos a diversos procesos metabólicos. Recientemente, varios estudios han sugerido que el dicloroacetato (DCA), un inhibidor específico de la piruvato deshidrogenasa cinasa, puede ser un fármaco potencial contra el cáncer en un gran número de tumores diversos. Sin embargo, el mecanismo exacto no se entiende completamente, lo cual es importante para el uso de DCA en el tratamiento del cáncer. En el presente estudio, descubrimos que el DCA sensibilizaba las células de cáncer de mama MCF7 a la muerte celular inducida por tamoxifeno mediante la disminución de la expresión del receptor del factor de crecimiento epidérmico (EGFR). La regulación a la baja del EGFR se produjo por degradación de la proteína. Además, la proteína cinasa activada por mitógenos p38 desempeñó un papel importante en la degradación del EGFR inducida por DCA/tamoxifeno. Por último, el DCA también promovió la muerte celular inducida por tamoxifeno comparable en células MCF7 resistentes al tamoxifeno, que se establecieron mediante el tratamiento a largo plazo con tamoxifeno. En resumen, nuestros resultados sugieren que el DCA es un fármaco potencialmente atractivo que sensibiliza las células a la muerte celular inducida por tamoxifeno y supera la resistencia al tamoxifeno mediante la regulación a la baja de la expresión de EGFR en células de cáncer de mama.
INTRODUCCIÓN
Las células cancerosas en proliferación tienen unas necesidades metabólicas considerablemente diferentes a las de la mayoría de las células diferenciadas normales. Por ejemplo, para apoyar el rápido crecimiento y proliferación celular, las células cancerosas alteran diferencialmente el flujo metabólico en comparación con el tejido circundante para proporcionar suficiente bioenergética e intermediarios biosintéticos. Un fenómeno bien conocido que se observa en la mayoría de las células cancerosas es un cambio a la glucólisis aeróbica, independientemente del suministro de oxígeno, que se denomina «efecto Warburg», en el que el piruvato se convierte directamente en ácido láctico en lugar de entrar en el ciclo del ácido cítrico [1]. Como todas las células cancerosas dependen de este cambio en el metabolismo, estas vías alteradas representan dianas terapéuticas atractivas [2]. Tanto en estudios preclínicos como clínicos [3], se han realizado esfuerzos para atacar el metabolismo reprogramado solo o en combinación con quimioterapia contra el cáncer. Curiosamente, esta remodelación metabólica específica del cáncer es revertida por el dicloroacetato (DCA), una pequeña molécula dirigida a las mitocondrias que puede penetrar en la mayoría de los tejidos tras su administración oral [4]. Inhibe específicamente la piruvato deshidrogenasa cinasa (PDK), un miembro de la familia de las cinasas, lo que provoca la reactivación de la piruvato deshidrogenasa (PDH), una enzima clave que desplaza el flujo de piruvato hacia la mitocondria para promover la oxidación de la glucosa en lugar de la glucólisis [4]. Aunque el DCA se ha evaluado recientemente en varios ensayos preclínicos contra el cáncer [5], las respuestas de las células cancerosas al tratamiento con DCA, que determinan si el DCA proporcionará beneficios clínicos en el tratamiento del cáncer, no se han dilucidado completamente.
Más del 70% de los cánceres de mama expresan el receptor de estrógenos (RE) y dependen de los estrógenos para impulsar el crecimiento y la progresión del tumor [6]. Así pues, la terapia endocrina debe considerarse complementaria a la cirugía en la mayoría de las pacientes, ya que induce la remisión tumoral y proporciona beneficios clínicos constantes. El fármaco antiestrógeno tamoxifeno es el tratamiento más utilizado en pacientes con cáncer de mama RE-positivo, tanto en estadios tempranos como avanzados o metastásicos [7]. Como terapia adyuvante en el cáncer de mama precoz, el tamoxifeno mejora la supervivencia global, y se cree que su uso generalizado ha contribuido significativamente a la reducción de la mortalidad por cáncer de mama observada en la última década [8]. A pesar de los evidentes beneficios del tratamiento con tamoxifeno en el cáncer de mama, casi todas las pacientes con enfermedad metastásica y hasta el 25% de las pacientes que reciben tamoxifeno adyuvante acaban recayendo y muriendo a causa de la enfermedad [9, 10]. Por lo tanto, los mecanismos biológicos que subyacen a la resistencia intrínseca (de novo) y adquirida al tamoxifeno revisten una importancia clínica considerable. Una mejor comprensión de estos mecanismos puede sugerir nuevas estrategias para superar la resistencia al tamoxifeno y mejorar el tratamiento del cáncer de mama.
En el presente estudio, demostramos que la expresión de EGFR en células de cáncer de mama disminuía con el tratamiento con DCA. Una combinación de DCA y tamoxifeno redujo aún más los niveles de EGFR. Demostramos que el DCA aumentaba la citotoxicidad del tamoxifeno en las células de cáncer de mama mediante la inhibición de la expresión del EGFR. Además, el DCA sensibilizó al tamoxifeno a las células MCF7 resistentes al tamoxifeno a través de la regulación a la baja de EFGR. Estos resultados sugieren la posible utilización del DCA en el tratamiento del cáncer de mama mediante la atenuación de la vía de señalización del EGFR.
Resultados
La inhibición de la PDK regula a la baja el EGFR y aumenta la muerte celular inducida por tamoxifeno en células de cáncer de mama
Estudios recientes han demostrado que la inhibición de la PDK, como el DCA, desplaza el metabolismo de las células cancerosas de la glucólisis a la fosforilación oxidativa mediante la desfosforilación de la piruvato deshidrogenasa mitocondrial [4, 11]. Para dilucidar cómo las vías de señalización del factor de crecimiento y de la quinasa, junto con la PDK, regulan el efecto Warburg en el cáncer de mama, examinamos los niveles de expresión de varios receptores del factor de crecimiento en las células MCF7 eliminadas por PDK. Curiosamente, la depleción de PDK mediante tratamiento con siRNA redujo la regulación de EGFR, en contraste con efectos nulos o marginales sobre otros receptores de factores de crecimiento (Figura 1A). Se han identificado cuatro isoenzimas PDK (PDK1, PDK2, PDK3, PDK4) en tejidos de mamíferos [12]. Para confirmar la regulación a la baja de EGFR por el tratamiento con siPDK, exploramos la expresión de EGFR en las células tratadas con siRNA contra cada isoforma de PDK. Sin embargo, todas ellas provocaron la regulación a la baja del EGFR, lo que sugiere que es posible que la PDK no regule la expresión del EGFR de forma específica para cada isoforma (Figura 1B). En consonancia con estos resultados, el DCA también redujo los niveles de EGFR de forma dependiente de la dosis (Figura 1C). Cuando analizamos la concentración de lactato en los medios de cultivo, no hubo cambios significativos en la concentración de lactato por el tratamiento con tamoxifeno/DCA o siRNA contra EGFR, aunque disminuyó por el tratamiento con DCA (Figura Suplementaria S1). Por lo tanto, sugerimos que la regulación a la baja del EGFR puede no estar asociada con el cambio metabólico inducido por el DCA en las células de cáncer de mama. Dado que la activación de la vía de señalización del EGFR contribuye a la resistencia al tamoxifeno [13], examinamos si la regulación a la baja del EGFR mediante la inhibición de la PDK sensibilizaba las células al tamoxifeno. Como se muestra en la Figura 1D, el co-tratamiento con tamoxifeno y DCA condujo a una marcada reducción de la viabilidad celular. El tratamiento combinado durante 72 h redujo la viabilidad celular a menos del 30% de la del control (Figura 1E). A continuación, se evaluó la muerte celular en las células co-tratadas mediante tinción con Annexin V/PI. Cuarenta y ocho horas después del tratamiento, la combinación de tamoxifeno y DCA indujo un 30% de muerte celular, en comparación con el 15% o el 8% por tamoxifeno o DCA solos, respectivamente (Figura 1F). Anteriormente, informamos de que la muerte celular apoptótica en células de cáncer de mama estaba causada por la pérdida de potencial de membrana mitocondrial (MMP) [14]. Por lo tanto, hemos probado si la pérdida de MMP involucrados en la muerte celular inducida por el co-tratamiento, sin embargo, no hubo cambios significativos en MMP entre no tratados y tamoxifeno / DCA células tratadas (Suplementario Figura S2).
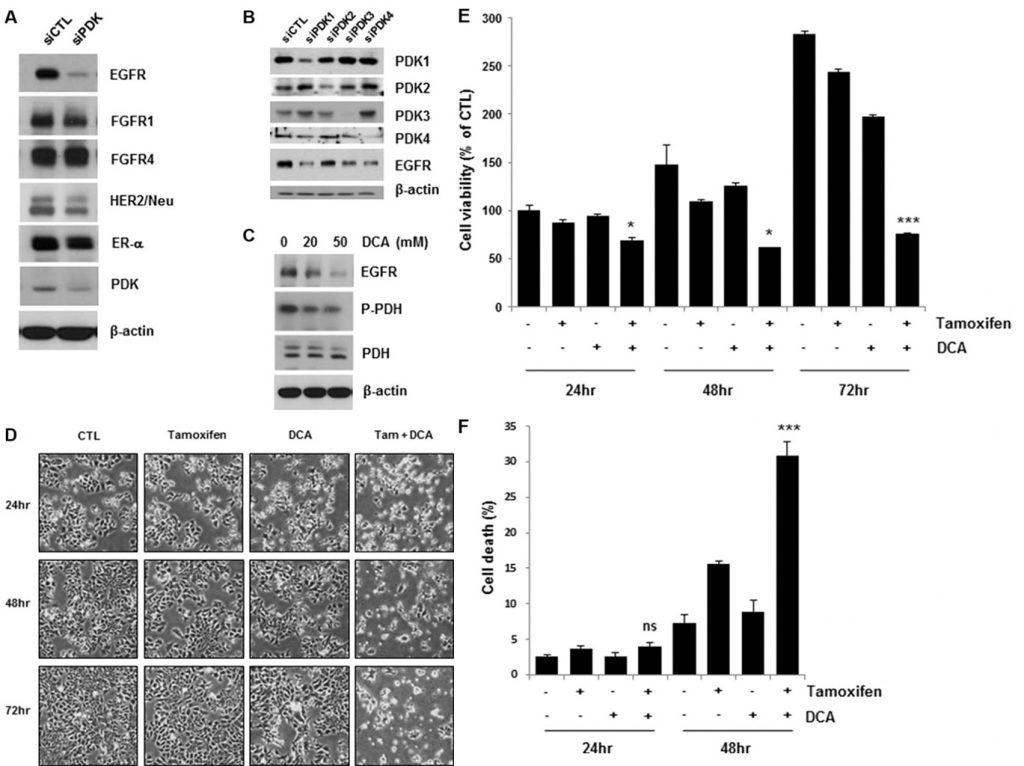
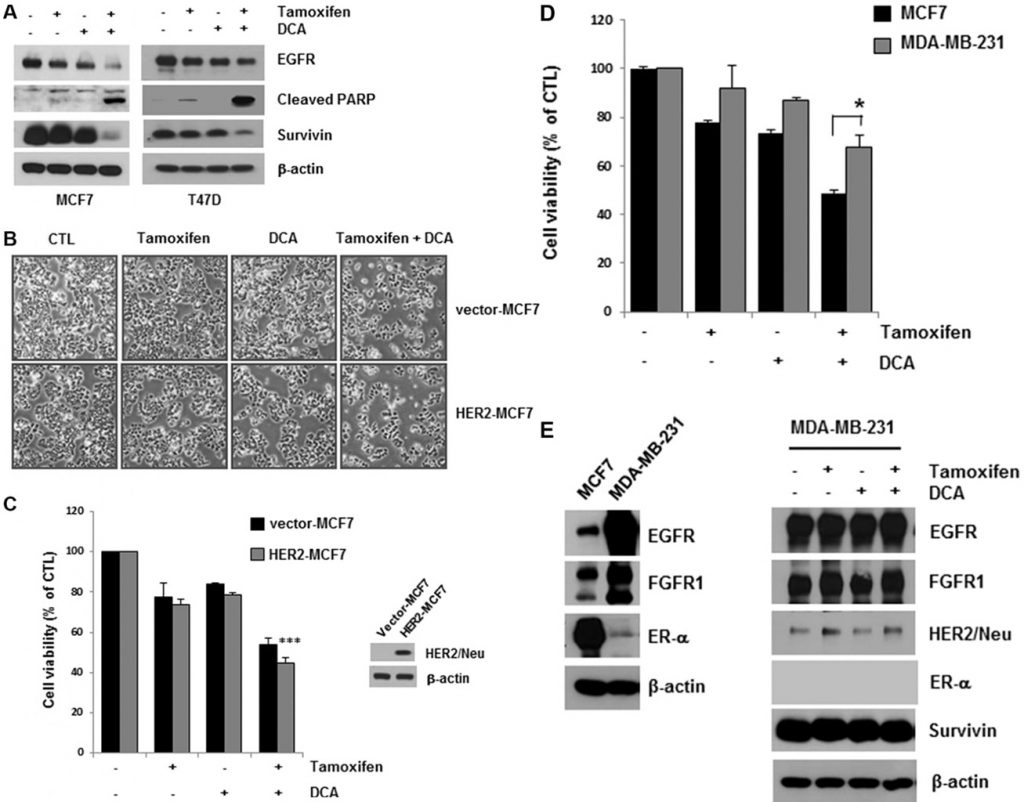
El DCA más el tamoxifeno redujeron aún más los niveles de EGFR en las células MCF7 y T47D en comparación con el DCA solo (Figura 2A). La muerte celular inducida por el tratamiento conjunto se confirmó mediante la detección de la escisión de PARP, un marcador de apoptosis (Figura 2A). La survivina es una molécula antiapoptótica y una diana del RE [15]. El tratamiento conjunto también redujo la survivina, que puede mediar en la apoptosis de las células (Figura 2A). Aunque el tratamiento con tamoxifeno disminuyó ligeramente los niveles de EGFR en las células MCF7 y T47D, no se observó un aumento significativo de la muerte celular en las células, lo que sugiere que se necesita un nivel crítico de EGFR para la supervivencia de las células de cáncer de mama (Figura 2A).
Las pruebas obtenidas en líneas celulares han demostrado que la sobreexpresión de las vías de HER2 puede contribuir a la resistencia adquirida a las terapias endocrinas [13]. Para determinar si la sobreexpresión de HER2 influye en la citotoxicidad del tamoxifeno y el DCA, examinamos la viabilidad celular en células MCF7 sobreexpresantes de HER2 (HER2-MCF7) tras el tratamiento con tamoxifeno y DCA. Los resultados mostraron que el tamoxifeno y el DCA redujeron significativamente la viabilidad celular incluso en las células HER2-MCF7 (Figura 2B y 2C), lo que sugiere que el DCA podría potenciar la muerte celular inducida por el tamoxifeno en las células de cáncer de mama sobreexpresoras de HER2. Evaluamos además los efectos inhibidores del crecimiento del tratamiento conjunto en la línea celular de cáncer de mama triple negativo MDA-MB-231. Como se muestra en la Figura 2D, las células MDA-MB-231 fueron menos sensibles al tamoxifeno y al DCA que las células MCF7. Dado que en las células ER-positivas se observó una regulación a la baja del EGFR, examinamos los efectos del tamoxifeno y el DCA sobre los niveles de EGFR en las células MDA-MB-231. En comparación con las células MCF7, las células MDA-MB-231 presentaban una elevada expresión de EGFR, cuyos niveles no disminuyeron significativamente con el tamoxifeno y el DCA (Figura 2E). A continuación, examinamos la citotoxicidad del tamoxifeno y el DCA en la línea celular epitelial mamaria inmortalizada no tumorigénica MCF10A. Curiosamente, la expresión de EGFR en las células MCF10A era comparable a la de las células MDA-MB-231 y no se observó ni regulación a la baja de EGFR ni muerte celular en las células MCF10A tras el tratamiento con tamoxifeno y DCA (Figura suplementaria S3). Estos resultados indican que los efectos antiproliferativos del tamoxifeno y el DCA en las células de cáncer de mama dependen de la regulación a la baja del EGFR.
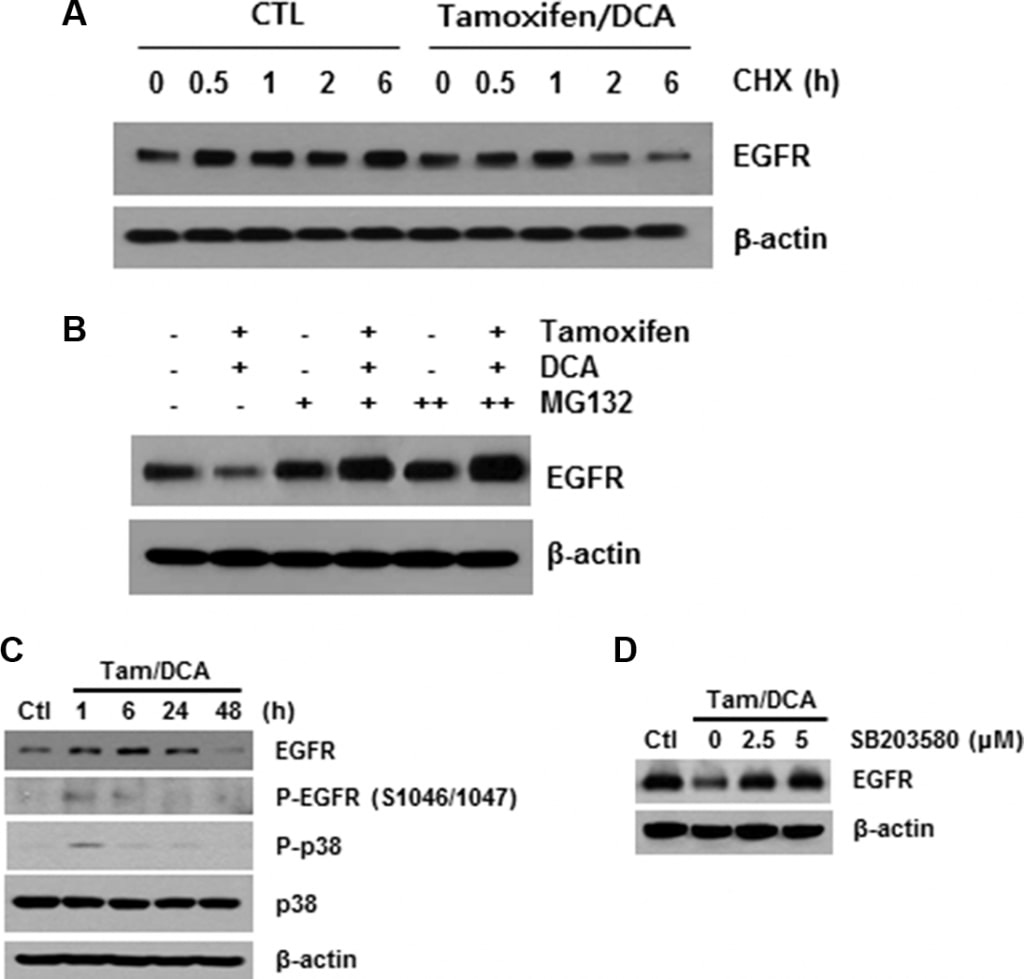
El tratamiento combinado de tamoxifeno y DCA induce la degradación del EGFR mediada por p38 MAPK
Como se ha descrito anteriormente, la unión del ligando provoca una rápida autofosforilación, lo que resulta en la eliminación del EGFR de la superficie celular mediante endocitosis a un compartimento endosomal temprano [16]. Por lo tanto, a continuación investigamos el papel de la modificación del receptor en la regulación a la baja del EGFR mediada por tamoxifeno/DCA. Tras bloquear la síntesis proteica con cicloheximida, observamos que la estabilidad del EGFR estaba significativamente comprometida en las células tratadas con tamoxifeno/DCA en comparación con el control (Figura 3A). A continuación, evaluamos los efectos de MG132, un inhibidor del proteasoma, sobre la degradación del EGFR inducida por tamoxifeno/DCA. El tratamiento con MG132 restauró la expresión de EGFR en las células tratadas con tamoxifeno/DCA de forma dosis-dependiente (Figura 3B).
La fosforilación del EGFR en residuos de serina y treonina representa un mecanismo de atenuación de la actividad del EGFR, y entre ellos, los sitios de fosforilación serina 1046/1047 (Ser 1046/7) son necesarios para la desensibilización del EGFR [17]. Recientemente se ha informado de que la proteína cinasa activada por mitógenos (MAPK) p38 induce la fosforilación del EGFR en Ser 1046/7, lo que provoca su degradación en las células cancerosas [18]. Por lo tanto, a continuación examinamos los efectos del tamoxifeno y el DCA sobre la fosforilación de la p38 MAPK en células MCF7. La p38 MAPK se fosforiló significativamente en 1 h, y la fosforilación se mantuvo durante 24 h tras el tratamiento con tamoxifeno y DCA (Figura 3C). Además, la degradación de EGFR inducida por el co-tratamiento se suprimió significativamente cuando las células se pretrataron con un inhibidor específico de p38 MAPK, SB203580 (Figura 3D), lo que indica que la activación de p38 MAPK desempeña un papel en la regulación a la baja de EGFR inducida por tamoxifeno/DCA en células MCF7.
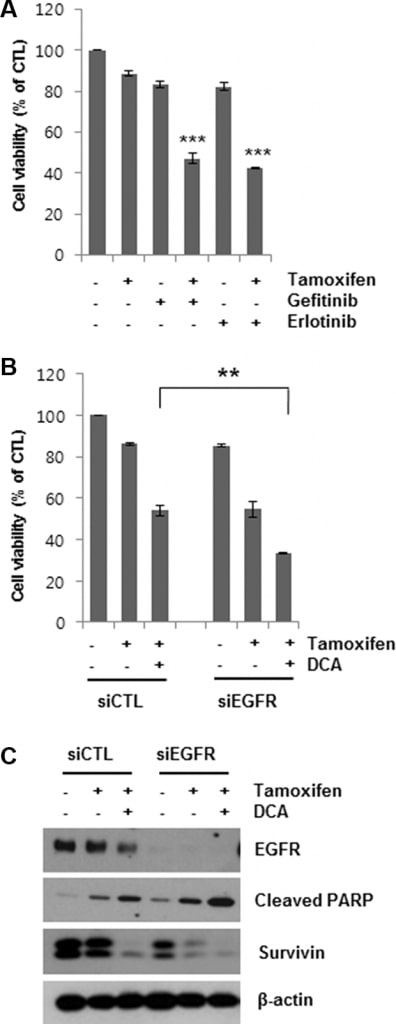
Los inhibidores de EGFR potencian la muerte celular inducida por tamoxifeno
Una vez demostrado que la degradación de EGFR mediada por DCA podía potenciar la muerte celular inducida por tamoxifeno en células MCF7, determinamos si la inhibición de EGFR potenciaba la muerte celular en células tratadas con tamoxifeno. Gefitinib y erlotinib son inhibidores selectivos reversibles de la unión de la tirosina quinasa EGFR al ATP [19, 20]. El co-tratamiento de las células MCF7 con 5 μM de gefitinib o erlotinib durante 48 h aumentó notablemente la muerte celular inducida por tamoxifeno (Figura 4A). Del mismo modo, el bloqueo del EGFR mediante el tratamiento con siEGFR aumentó la muerte celular inducida por tamoxifeno (Figura 4B). La reducción de la viabilidad celular por el tratamiento combinado de siEGFR y tamoxifeno fue comparable a la producida por DCA y tamoxifeno. Además, el tratamiento con siEGFR redujo aún más los niveles de EGFR en las células tratadas con tamoxifeno/DCA, lo que provocó un aumento de la muerte celular apoptótica junto con una regulación a la baja de la survivina en comparación con el control con siEGFR (Figura 4C). Estos resultados apoyan la conclusión de que el DCA sensibiliza las células de cáncer de mama al tamoxifeno a través de la regulación a la baja del EGFR.
la expresión de c-myc y Nanog está asociada a los efectos citotóxicos del DCA en células MCF7 tratadas con tamoxifeno
Se ha demostrado que la activación del EGFR en el cáncer aumenta varios factores de transcripción que pueden afectar al tipo y la duración de la señalización del EGFR [21]. Entre ellos, Nanog y c-myc tienen un papel pleiotrópico en la tumorigénesis, incluyendo la resistencia a la terapia estándar en el carcinoma de mama [22, 23]. Para investigar más a fondo las actividades antitumorales mediadas por la regulación a la baja del EGFR en células de cáncer de mama, examinamos la expresión de c-myc y Nanog en células MCF7 tras el cotratamiento con tamoxifeno y DCA. Los niveles de c-myc y Nanog disminuyeron significativamente en las células cotratadas con tamoxifeno y DCA (Figura 5A). Del mismo modo, las expresiones de ambas proteínas fueron suprimidas por el tamoxifeno en combinación con gefitinib o erlotinib (Figura 5B), lo que indica que la expresión de EGFR es necesaria para mantener la expresión de c-myc y Nanog en las células MCF7 tratadas con tamoxifeno. Sin embargo, el efecto del siEGFR sobre la expresión de c-myc y Nanog fue escaso o nulo, lo que indica que la regulación a la baja del EGFR es necesaria pero no suficiente para disminuir la expresión de las proteínas (Figura suplementaria S4). Para determinar si la expresión reducida de estas dos proteínas está implicada en los efectos citotóxicos del tamoxifeno y el DCA en las células MCF7, probamos los efectos de siRNAs contra c-myc y Nanog en células expuestas a tamoxifeno/DCA. El knockdown de c-myc y Nanog sensibilizó significativamente las células al tamoxifeno/DCA en comparación con los controles (Figura 5C). Por el contrario, la sobreexpresión de FLAG-c-myc y FLAG-Nanog mediante transfección de células con vectores FLAG-c-myc y FLAG-Nanog protegió significativamente a las células de la citotoxicidad inducida por tamoxifeno/DCA (Figura 5D). En conjunto, nuestros datos sugieren que la regulación a la baja del EGFR mediante una combinación de tamoxifeno y DCA puede inducir la muerte celular en las células MCF7 parcialmente a través de la inhibición de la expresión de c-myc y Nanog. Se sabe que los genes de autorrenovación, como c-myc y Nanog, están asociados a las propiedades de las células madre cancerosas 24. Para comprobar si el tratamiento conjunto con tamoxifeno y DCA inhibe las células similares a las células madre del cáncer de mama, realizamos un análisis de citometría de flujo para estimar la proporción de la subpoblación de células similares a las células madre en las células MCF7 basándonos en la expresión de CD44. Tras el tratamiento conjunto, la población de células MCF7 con expresión alta de CD44 se redujo del 42,5% al 19,9% (Figura 5E). Este hallazgo planteó la posibilidad de que el DCA más el tamoxifeno pudieran inhibir la capacidad de las células madre cancerosas en las células de cáncer de mama.

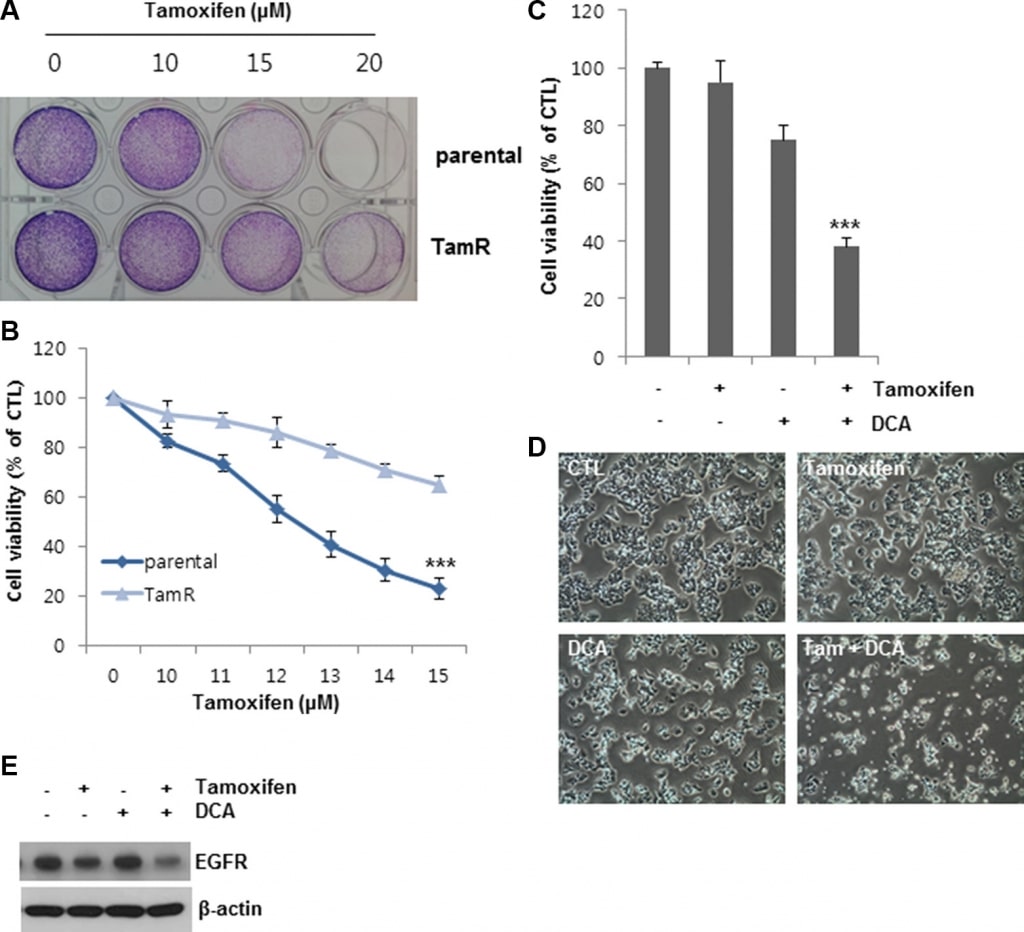
El tratamiento combinado con DCA y tamoxifeno puede superar la resistencia al tamoxifeno en células de cáncer de mama
Para confirmar aún más los efectos observados del DCA sobre la muerte celular inducida por tamoxifeno, establecimos células MCF7 resistentes al tamoxifeno (TamR) mediante tratamiento con tamoxifeno durante un largo periodo de tiempo. En comparación con las células no tratadas, la viabilidad celular de las células MCF7 y TamR MCF7 fue del 20 y 60%, respectivamente, tras el tratamiento con 13 μM de tamoxifeno, lo que indica que las células TamR MCF7 fueron menos sensibles a la misma concentración de tamoxifeno que las células parentales MCF7 (Figura 6A y 6B). El DCA por sí solo inhibió el crecimiento de las células MCF7 TamR en aproximadamente un 25% del del control (Figura 6C y 6D). Por el contrario, el tratamiento conjunto de DCA y tamoxifeno suprimió el crecimiento celular en más de un 60% en comparación con el control (Figura 6C y 6D). Los niveles de EGFR en las células TamR también se redujeron con el tratamiento conjunto (Figura 6E). Estos resultados sugieren que el DCA podría superar la resistencia al tamoxifeno en células de cáncer de mama mediante la regulación a la baja del EGFR a nivel proteico.
Debate
A medida que se ha ido conociendo mejor el fenotipo metabólico de las células tumorales, se ha propuesto como nueva estrategia contra el cáncer atacar las diferencias metabólicas entre las células tumorales y las normales. A pesar del aumento de posibles fármacos contra el cáncer dirigidos a los procesos metabólicos de las células cancerosas, será necesario dilucidar cómo responden las células a los fármacos para poder atacar con éxito el metabolismo del cáncer. En este trabajo demostramos que el DCA inhibe no sólo la actividad PDK, sino también la expresión de EGFR en células de cáncer de mama. El tratamiento combinado de DCA y tamoxifeno indujo la degradación dependiente del proteasoma de las proteínas EGFR en células de cáncer de mama a través de la activación de p38 MAPK. Nuestros resultados indican que la regulación a la baja del EGFR desempeña un papel clave en la muerte celular apoptótica inducida por la combinación de DCA y tamoxifeno. Hasta donde sabemos, éste es el primer informe que demuestra que el DCA sensibiliza las células de cáncer de mama a la muerte celular inducida por tamoxifeno a través de la regulación a la baja del EGFR.
En las células de cáncer de mama RE-positivas que han desarrollado resistencia endocrina, la expresión del RE puede estar directamente suprimida por una mayor señalización del receptor del factor de crecimiento debido a la sobreexpresión de EGFR y HER2, que posteriormente activan la MAPK e inhiben la transcripción del RE [25]. Además, la resistencia al tamoxifeno en células de cáncer de mama se ha relacionado con la sobreexpresión de EGFR y altos niveles de quinasa activada extracelular fosforilada 1/2 [2]. Por lo tanto, la estrategia de combinar un inhibidor del EGFR con un agente endocrino es lo suficientemente interesante como para justificar un estudio más profundo para la terapia del cáncer de mama. En este estudio, descubrimos que la regulación a la baja del EGFR en células de cáncer de mama tratadas con DCA aumentaba la sensibilidad de las células al tamoxifeno. La regulación a la baja del EGFR mediante el tratamiento conjunto de DCA y tamoxifeno se ha confirmado en varias líneas celulares de cáncer de mama ER-positivo, incluidas MCF7, T47D y BT474 (Figura 2A y no mostrada). Además de la línea celular BT474 de cáncer de mama HER2-amplificado, las células HER2-MCF7 también mostraron una regulación a la baja del EGFR por DCA/tamoxifeno, lo que sugiere que el tratamiento combinado será aplicable al cáncer de mama HER2-positivo. Desafortunadamente, una línea celular de cáncer de mama triple negativo, MDA-MB-231, que se sabe que sobreexpresa EGFR [26], mostró un fenotipo refractario, así como ningún cambio obvio en los niveles de EGFR en respuesta al co-tratamiento. Una posibilidad es que la amplificación del EGFR en las células MDA-MB-231 anule su regulación a la baja mediante el tratamiento conjunto de DCA y tamoxifeno. La otra explicación es que la(s) vía(s) de señalización que media(n) en la regulación a la baja del EGFR esté(n) bloqueada(s) por un mecanismo desconocido en las células MDA-MB-231. Serán necesarias más investigaciones para dilucidar el mecanismo subyacente del mantenimiento de la expresión del EGFR en respuesta al tratamiento conjunto de DCA y tamoxifeno en células de cáncer de mama con RE negativo.
Se conocen cuatro isoformas de PDK, cada una activa en respuesta a diferentes condiciones intracelulares y extracelulares. La PDK1 se activa por hipoxia [27]; la PDK2 se activa por los productos de la PDH acetil CoA y NADH [28]; la PDK3 se activa por ATP [29]; y la PDK4 se regula transcripcionalmente por señales hormonales [30]. La PDK2 posee la mayor actividad de fosforilación del complejo piruvato deshidrogenasa, seguida de la PDK4, la PDK1 y la PDK3 [12]. PDK2 es más susceptible a la inhibición por DCA debido a su expresión ubicua [31]. Aunque detectamos la expresión de cuatro isoformas en las células MCF7 (Figura 1C), las diferencias funcionales entre las isoformas PDK en el cáncer de mama siguen siendo difíciles de determinar. Será interesante seguir investigando si las vías de señalización que suprimen una isoforma concreta de PDK conducen a la regulación a la baja del EGFR. Se sabe que la PDK funciona en la mitocondria, y varios estudios han demostrado que el EGFR también se transloca a la mitocondria tras la estimulación por EGF [32, 33]. Además, se ha sugerido que la interacción de EGFR con PDK en la matriz mitocondrial desempeña un papel importante en el crecimiento tumoral inducido por EGFR en el glioblastoma multiforme [34]. Aquí, encontramos que el bloqueo de la actividad de PDK por un inhibidor o silenciamiento disminuyó los niveles celulares totales de EGFR en células de cáncer de mama tratadas con tamoxifeno. Se necesitan más estudios para determinar si sólo el EGFR asociado a mitocondrias está sujeto a degradación por el co-tratamiento con DCA y tamoxifeno en células de cáncer de mama. En general, la exposición de las células al EGF causa una rápida autofosforilación, incluyendo la tirosina (Tyr) 1045, que proporciona un sitio de acoplamiento para la ubiquitina ligasa c-Cbl, lo que resulta en la ubiquitinación del EGFR y la eliminación del EGFR vía endocitosis desde la superficie celular a un compartimento endosomal temprano [35]. Sin embargo, el tratamiento conjunto con DCA y tamoxifeno no tuvo ningún efecto sobre la fosforilación del EGFR en Tyr 1045 (datos no mostrados). En cambio, observamos la fosforilación de EGFR en Ser 1046/7 por DCA y tamoxifeno, que fue bloqueada por un inhibidor específico de p38 MAPK, SB203580. Se ha sugerido que la p38 MAPK juega un papel fundamental tanto en la internalización como en la degradación del EGFR [18]; sin embargo, se requiere más investigación para dilucidar cómo la fosforilación del EGFR en residuos de serina causa su regulación a la baja y para identificar el mediador de la activación de la p38 MAPK tras el tratamiento con DCA/tamoxifeno.
Nanog y c-myc han sido identificados como factores de transcripción, y su papel en el mantenimiento de la autorrenovación en células madre embrionarias ha sido establecido en estudios previos [22, 36]. Un mecanismo recientemente propuesto de resistencia intrínseca a la terapia endocrina postula la existencia de un subconjunto especializado de células cancerosas denominadas células iniciadoras de tumores (TIC) [37]. Las TIC tienen la capacidad de autorrenovarse y generar nuevos tumores que consisten enteramente en tipos celulares derivados clonalmente presentes en el tumor parental. Aquí demostramos que el DCA disminuía la expresión de c-myc y Nanog en células MCF7 tratadas con tamoxifeno. Además, la expresión de estas proteínas estaba relacionada con los efectos citotóxicos del tratamiento combinado de DCA y tamoxifeno. Por lo tanto, el DCA puede aprovecharse para ampliar el potencial terapéutico del tamoxifeno suprimiendo el desarrollo de resistencia intrínseca en la terapia del cáncer de mama.
En conclusión, nuestros resultados revelaron que el DCA sensibilizó las células de cáncer de mama ER-positivas al tamoxifeno mediante la disminución de los niveles de EGFR. El tratamiento con DCA y tamoxifeno inhibió la expresión de genes de auto-renovación y la supervivencia de las células MCF7 resistentes al tamoxifeno. Por lo tanto, proponemos que el DCA puede ser un agente terapéutico eficaz para tratar el cáncer de mama resistente al tamoxifeno. Además, las estrategias de combinación con DCA pueden ser útiles para mejorar la eficacia del tratamiento de otras quimioterapias citotóxicas o terapias dirigidas. En el futuro deberán llevarse a cabo más experimentos, incluidos estudios con animales y ensayos clínicos.
Materiales y métodos
Cultivo celular y reactivos
Las células humanas de cáncer de mama MCF7, T47D y MDA-MB-231 se adquirieron en la American Type Culture Collection (Rockville, MD, EE.UU.) y se cultivaron en el medio de crecimiento recomendado (Invitrogen, Carlsbad, CA, EE.UU.). Las células MCF7 con sobreexpresión de HER2 (HER2-MCF7) y las células de vector de control (vector-MCF7) fueron proporcionadas amablemente por el Dr. Incheol Shin (Universidad de Hanyang, Seúl, Corea). Las células MCF7 resistentes al tamoxifeno (TamR) se desarrollaron cultivando células MCF7 en presencia de 10 μM de tamoxifeno durante más de 6 meses. Como control, se cultivaron células parentales durante el mismo tiempo en medios normales. Tras el establecimiento de la resistencia, las células se pasificaron durante no más de 3 meses. En los experimentos con tratamientos con tamoxifeno, las células se cultivaron de forma rutinaria en medio Eagle modificado sin rojo fenol de Dulbecco más un 10% de suero fetal bovino desprovisto de carbón justo antes de los tratamientos. Los anticuerpos contra EGFR, fosfo-EGFR (S1046/1047), FGFR1, FGFR4, PDK1, PDH, survivina, PARP escindida, p38, fosfo-p38 y Nanog se adquirieron a Cell Signaling Technology (Danvers, MA, EE.UU.). Los anticuerpos contra HER2/Neu, ER-α y c-myc, los ARNsi dirigidos contra EGFR, Nanog, PDK4 y c-myc y los ARNsi de control negativo (revueltos) se adquirieron de Santa Cruz Biotechnology (Dallas, TX, EE.UU.). Los anticuerpos contra PDK2 y PDK3 se adquirieron a Thermo Fisher Scientific (Waltham, MA, EE.UU.). El anticuerpo β-actina, el anticuerpo FLAG, el tamoxifeno, el DCA, la cicloheximida y la MG132 se adquirieron a Sigma-Aldrich (St. Louis, MO, EE.UU.). El anticuerpo fosfo-PDH (S293) y el SB203580 procedían de BD Biosciences Pharmingen (San Diego, CA, EE.UU.), y el gefitinib y el erlotinib se obtuvieron de Selleck Chemicals (London, ON, Canadá).
Transfecciones y tratamientos
Las células se transfectaron con el ARNsi diana (50 nM) utilizando Lipofectamine RNAiMAX (Invitrogen) según lo descrito por el fabricante. Las células se transfectaron con 1 μg de FLAG-c-myc-ADNc 3.1 y FLAG-Nanog-ADNc 3.1 utilizando Lipofectamine 2000 tal y como describe el fabricante. Después de 6 h, las células se trataron con tamoxifeno y/o DCA durante 24-48 h y luego se analizaron como se describe en otro lugar.
Medición de la viabilidad celular
La viabilidad celular se determinó midiendo la conversión mitocondrial del bromuro de 3-(4,5-dimetiltiazolil2)-2,5-difeniltetrazolio (MTT) en un producto coloreado. Las células se trataron como se indica y el medio se cambió por medio libre de suero que contenía 1 mM de MTT. Tras 2 h de incubación a 37 °C, las células se solubilizaron en DMSO. La cantidad de formazán, la forma convertida del MTT, se determinó midiendo la absorbancia a 595 nm. Evaluación de la apoptosis
La apoptosis se determinó mediante un análisis de clasificación celular activado por fluorescencia utilizando un kit de apoptosis Annexin V-FITC (BioVision, Milpitas, CA, EE.UU.) siguiendo las instrucciones del fabricante. Brevemente, tras el tratamiento, las células se trataron con tripsina y luego se resuspendieron en tampón de unión (10 mM HEPES/NaOH, pH 7,4, 140 mM NaCl, 2,5 mM CaCl2 ) incluyendo Annexin V-FITC y yoduro de propidio. Tras 15 min de incubación, se analizó la fluorescencia celular mediante citometría de flujo. La muerte celular se midió como el porcentaje de células en la población Annexin V y PI positiva.
Western blotting
Se cosecharon las células y se lisaron en tampón RIPA (50 mM Tris-HCl pH 7,5, 150 mM NaCl, 1% Nonidet P40, 0,5% desoxicolato sódico y 0,1% SDS) suplementado con un cóctel inhibidor de proteasa/fosfatasa (Roche, Mannheim, Alemania). Se separaron cantidades iguales de proteínas (20-50 μg) mediante SDS-PAGE y se transfirieron a una membrana de nitrocelulosa. Las membranas se bloquearon incubándolas con leche desnatada al 5% en solución salina tamponada con Trisbuffer durante 1 h y, a continuación, se incubaron durante la noche con los anticuerpos primarios apropiados. Las membranas se incubaron con anticuerpos secundarios conjugados con HRP durante 1 h. Las proteínas inmunorreactivas se visualizaron utilizando reactivos de quimioluminiscencia mejorada (Amersham Biosciences, Little Chalfont, Reino Unido).
Detección de población celular CD44 positiva
Las células se tiñeron con anticuerpos a una dilución de 1∶100 en PBS durante 15 minutos. Los anticuerpos utilizados fueron los siguientes: FITC-CD44 y anticuerpos de control de isotipo IgG de ratón conjugados con FITC se obtuvieron de BD Biosciences Pharmingen. Las células marcadas se analizaron mediante citometría de flujo. Las poblaciones de células CD44 positivas se determinaron por la intensidad de FITC.
Análisis estadístico
Todos los datos presentados son representativos de al menos dos experimentos distintos. Las comparaciones entre grupos se analizaron mediante la prueba t de Student. Los asteriscos (***p < 0,001, **p < 0,01, *p < 0,05) indican significación estadística.
Agradecimientos y financiación
Esta investigación fue apoyada por el Programa de Investigación Científica Básica a través de la Fundación Nacional de Investigación de Corea (NRF) financiado por el Ministerio de Ciencia, TIC y Planificación del Futuro (No. 1711031812, 1711023318 y 1711031800).
Conflictos de intereses
Los autores declaran no tener ningún conflicto de intereses.
REFERENCIAS
1 1. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009; 324:1029-33.
2ZhaoY, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013; 4:e532.
3 Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011; 10:671-84.
4 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11: 37-51.
5 Kankotia S, Stacpoole PW. Dicloroacetato y cáncer: ¿nuevo hogar para un medicamento huérfano? Biochim Biophys Acta. 2014; 1846:617-29.
6 Osborne CK. Tamoxifeno en el tratamiento del cáncer de mama. N Engl J Med. 1998 Nov 26; 339:1609-18.
7 Johnston SR. New strategies in estrogen receptor-positive breast cancer. Clin Cancer Res. 2010 Apr 1; 16:1979-87. doi: 10.1158/1078-0432.CCR-09-1823.
8 Peto R, Boreham J, Clarke M, Davies C, Beral V. UK and USA breast cancer deaths down 25% in year 2000 at ages 20-69 years. Lancet. 2000; 355:1822.
9 Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005; 365:1687-717.
10 Early Breast Cancer Trialists’ Collaborative Group (EBCTCG), Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, Dowsett M, Ingle J, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011; 378:771-84.
11 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013; 73:7277-89.
12 Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: An old metabolic gatekeeper regulated by new pathways and pharmacological agents. Int J Cancer. 2016; 138:809-17.
13 Osborne CK, Schiff R. Mecanismos de resistencia endocrina en el cáncer de mama. Annu Rev Med. 2011; 62:233-47.
14YunSM, Woo SH, Oh ST, Hong SE, Choe TB, Ye SK, Kim EK, Seong MK, Kim HA, Noh WC, Lee JK, Jin HO, Lee YH, et al. Melatonin enhances arsenic trioxide-induced cell death via sustained upregulation of Redd1 expression in breast cancer cells. Mol Cell Endocrinol. 2016; 422:64-73.
15 Zhu J, Lu X, Hua KQ, Sun H, Yu YH, Feng YJ. Oestrogen receptor α mediates 17β-estradiol enhancement of ovarian cancer cell motility through up-regulation of survivin expression. Arch Gynecol Obstet. 2012; 286: 729-37.
16 Arteaga CL. Dependencia del receptor del factor de crecimiento epidérmico en tumores humanos: ¿algo más que expresión? Oncologist. 2002; 7:31-9.
17 Sorkin A, Goh LK. Endocitosis and intracellular trafficking of ErbBs. Exp Cell Res. 2009; 315:683-96.
18 Adachi S, Shimizu M, Shirakami Y, Yamauchi J, Natsume H, Matsushima-Nishiwaki R, To S, Weinstein IB, Moriwaki H, Kozawa O. (-)-Epigallocatechin gallate downregulates EGF receptor via phosphorylation at Ser1046/1047 by p38 MAPK in colon cancer cells. Carcinogenesis. 2009; 30:1544-52.
19CiardielloF, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G. Antitumor effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin Cancer Res. 2000; 6:2053-63.
20 Pollack VA, Savage DM, Baker DA, Tsaparikos KE, Sloan DE, Moyer JD, Barbacci EG, Pustilnik LR, Smolarek TA, Davis JA, Vaidya MP, Arnold LD, Doty JL, et al. Inhibición de la fosforilación de tirosina asociada al receptor del factor de crecimiento epidérmico en carcinomas humanos con CP-358.774: dinámica de la inhibición del receptor in situ y efectos antitumorales en ratones atímicos. J Pharmacol Exp Ther. 1999; 291:739-48.
21 Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006; 366:2-16.
22 Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014; 135:2741-8.
23 Chen Y, Olopade OI. MYC in breast tumor progression (MYC en la progresión del tumor de mama). Expert Rev Anticancer Ther. 2008; 8: 1689-98.
24 Hou ZJ, Luo X, Zhang W, Peng F, Cui B, Wu SJ, Zheng FM, Xu J, Xu LZ, Long ZJ, Wang XT, Li GH, Wan XY, et al. Flubendazole, FDA-approved anthelmintic, targets breast cancer stem-like cells. Oncotarget. 2015; 6:6326-40. doi:10.18632/oncotarget.3436.
25 Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Activation of mitogenactivated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res. 2006; 66:3903-11.
26 Biswas DK, Cruz AP, Gansberger E, Pardee AB. Epidermal growth factor-induced nuclear factor kappa B activation: A major pathway of cell-cycle progression in estrogenreceptor negative breast cancer cells. Proc Natl Acad Sci U S A. 2000; 97:8542-7.
27 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006; 3:177-85.
28 Hiromasa Y, Hu L, Roche TE. Efectos inducidos por ligando en la piruvato deshidrogenasa cinasa isoforma 2. J Biol Chem. 2006; 281:12568-79.
29 Kato M, Chuang JL, Tso SC, Wynn RM, Chuang DT. Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. EMBO J. 2005; 24:1763-74.
30 Kwon HS, Huang B, Unterman TG, Harris RA. Protein kinase B-alpha inhibits human pyruvate dehydrogenase kinase-4 gene induction by dexamethasone through inactivation of FOXO transcription factors. Diabetes. 2004; 53:899-910.
31 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998; 329 :191-6.
32 Dasari VR, Velpula KK, Alapati K, Gujrati M, Tsung AJ. Cord blood stem cells inhibit epidermal growth factor receptor translocation to mitochondria in glioblastoma. PLoS One. 2012; 7:e31884.
33 Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, Hüttemann M, Douglas R, Haddad G, Parsons SJ. Epidermal growth factor receptor translocation to the mitochondria: regulation and effect. J Biol Chem. 2009; 284:36592-604.
34 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013; 73:7277-89.
35 Massie C, Mills IG. El papel en desarrollo de receptores y adaptadores. Nat Rev Cancer. 2006; 6:403-9.
36 Chappell J, Dalton S. Roles de MYC en el establecimiento y mantenimiento de la pluripotencia. Cold Spring Harb Perspect Med. 2013;3:a014381.
37 Wei W, Lewis MT. Identifying and targeting tumor-initiating cells in the treatment of breast cancer. Endocr Relat Cancer. 2015; 22:R135-55.