Йонг Вон Чой1, Ин Кён Лим*
1 Кафедра биохимии и молекулярной биологии, Проект трансформации и восстановления клеток BK21, Медицинская школа Университета Аджоу, Сувон 443-721, Республика Корея
* Кафедра биохимии и молекулярной биологии, Медицинская школа Университета Аджоу, Сан 5 Вончон-дон, Ёнтонгу, Сувон 443-721, Республика Корея; тел: +82 31 21 219 5051; факс: +82 31 219 5059. Адрес электронной почты: [email protected] (И.К. Лим).
Получено: 29 октября 2013 г.
Принята: 20 января 2014 г.
Пересмотрена: в исправленном виде 8 января 2014 г.
Опубликована/доступна онлайн: 27 января 2014 г
Аннотация
Для изучения сенсибилизации метформин-цитотоксичности раковые клетки обрабатывали дихлорацетатом (DCA), ингибитором киназы пируватдегидрогеназы (PDK). Метформин-цитотоксичность в основном зависела от наличия глюкозы и восстановительной энергии, генерируемой пентозофосфатным путем, в то время как сопутствующее лечение DCA усиливало метформин-цитотоксичность путем перепрограммирования метаболизма глюкозы посредством ингибирования PDK и увеличения митохондриального дыхания. Несмотря на высокий уровень глюкозы и высокий уровень GSH, сопутствующее лечение DCA вызвало гибель клеток, а не их выживание. В заключение следует отметить, что DCA сенсибилизировал метформин-цитотоксичность путем перепрограммирования метаболизма глюкозы от аэробного гликолиза к митохондриальному окислению, о чем свидетельствуют измерения потребления глюкозы, высвобождения лактата и соотношения скорости потребления кислорода и скорости внеклеточного окисления.
Ключевые слова: Дихлорацетат метформина (ДХА), окислительный стресс, лишение глюкозы, содержание глутатиона
© 2014 Опубликовано Elsevier Ireland Ltd.
ВВЕДЕНИЕ
Все больше доказательств указывает на то, что метаболические нарушения раковых клеток, такие как аэробный гликолиз или глутаминовая зависимость, неизбежны для канцерогенеза не только как эпифеномен [13], [31], и огромное количество усилий было потрачено на поиск лекарственных мишеней и химических веществ-кандидатов на метаболизм рака [5], [30]. На основании эпидемиологических, доклинических и клинических данных метформин, бигуанид, используемый для лечения сахарного диабета, стал одним из наиболее привлекательных и перспективных препаратов-мишеней для метаболизма рака. Хотя механизм действия метформина до конца не выяснен, известно, что внутриклеточная функция метформина заключается в ингибировании комплекса I дыхательной цепи [8], [21]. В настоящее время многие данные свидетельствуют о том, что активация AMPK является главным узлом противоопухолевого действия метформина [3], [16],[23], поэтому LKB1-/- раковые клетки более устойчивы к метформин-индуцированной цитотоксичности в системе культур in vitro [22], [34]. Напротив, было показано, что активация AMPK защищает раковые клетки от энергетического стресса через регуляцию гомеостаза NADPH [12], поэтому LKB1-/-раковые клетки более чувствительны к фенформин-индуцированному метаболическому стрессу в мышиной модели рака легких [24]. В путаном контексте имеются сообщения о том, что противоопухолевые эффекты метформина зависят от концентрации глюкозы в культуральной среде [11], [17], [25]; лишение глюкозы значительно усиливает цитотоксичность метформина [17], тогда как активация AMPK, индуцированная метформином, неэффективна в условиях высокого содержания глюкозы (25 мМ) [25]. Таким образом, прежде чем использовать метформин в качестве противоракового средства, направленного на метаболизм глюкозы, необходимо выяснить биохимический механизм метформин-цитотоксичности.
Эффект Варбурга, часто наблюдаемый в раковых клетках, важен не только для получения энергии, но и для поддержания сниженного статуса в условиях враждебного микроокружения опухоли. Глюкоза является основным источником восстановительной силы, NADPH, через пентозофосфатный путь [1], [9], [22]. Действительно, 2-дезоксиглюкоза(2DG) опосредованная гибель раковых клеток зависит в основном от непереносимого окислительного стресса в дополнение к энергетическому кризису [15]. Исходя из концепции, что усиление гликолиза защищает раковые клетки от окислительного стресса, считается, что усиленный метформином гликолиз способен защитить клетки от митохондриального окислительного стресса, возникающего в результате ингибирования комплекса I дыхательной цепи [35]. Поэтому мы исследовали, может ли метформин-индуцированный митохондриальный стресс усиливаться окислительным стрессом, вызванным истощением GSH при недостатке глюкозы или обработкой H2O2 в условиях достаточного количества глюкозы. На основании сообщений о том, что ингибирование гликолиза 2-ДГ усиливает цитотоксическое действие метформина [2], [6], [14] и что дихлорацетат (ДХА) снижает гликолиз, активируя прируватдегидрогеназу (ПДГ) [32], наряду с окислительным метаболизмом и противоопухолевой активностью [20].
В настоящем исследовании мы изучили влияние DCA на метформин-цитотоксичность и регуляцию метаболизма глюкозы. В отличие от воздействия только метформина, комбинированное лечение раковых клеток метформином и DCA значительно усиливало активность PDH, но не гликолиза, таким образом восстанавливая митохондриальное дыхание в раковых клетках. Перепрограммирование метаболизма глюкозы было тесно связано с сильным окислительным стрессом. Таким образом, DCA-опосредованное перепрограммирование аэробного гликолиза на митохондриальное окисление усилило вызванный метформином митохондриальный и клеточный окислительно-восстановительный стресс, которого было достаточно, чтобы вызвать массовую гибель клеток, несмотря на высокий уровень глюкозы.
Материалы и методы
Клетки и реактивы
Клетки HeLa, MCF7 и MDA-MB-231 выращивали в среде DMEM (Gibco), содержащей глюкозу (0-25 мМ), дополненной 10% фетальной бычьей сывороткой и 100 Ед/мл гентамицина при 37 °C и 5%CO2. Метформин, дихлорацетат (DCA), л-бутионин-сульфоксимин (BSO), H2O2, моноэтиловый эфир глутатиона (GSH-MEE) и N-ацетил-цистеин (NAC) были получены от Sigma Chemical Co. (Сент-Луис, МО). Тролокс был получен от Biomol International, L.P., а 2′,7′-дихлордигидрофлуоресцеин диацетат (H2DCFDA), MitoSOX Red, пропидий йодид (PI) и Calcein AM были получены от Molecular Probes (Eugene, OR).
Измерение жизнеспособности клеток и анализ колониеобразования в мягком агаре
Жизнеспособность клеток после различных обработок в течение указанного времени оценивали путем исключения красителя трипанового синего (Sigma-Aldrich). Для анализа 4 ×104 клеток высевали в 12-луночные планшеты и обрабатывали химическими веществами на следующий день. Клетки трипсинизировали и смешивали с 0,4% трипановым синим (1:1). Процент жизнеспособных клеток представляет собой количество неокрашенных клеток/количество всех клеток × 100. Гибель клеток также измеряли с помощью набора для определения высвобождения лактатдегидрогеназы(ЛДГ) в соответствии с протоколом производителя (Takara, Япония). Для колониеобразующего анализа на мягком агаре клетки HeLa (1,5 ×103) были помещены в 0,6% раствор агарозы и выложены на 1,0% слой агарозы перед обработкой различными концентрациями метформина, DCA или обоих в комбинации. Колонии размером более 125 мкм через 2 недели считались положительными.
Измерение уровня клеточных ROS
Внутриклеточные и митохондриальные ROS измеряли с помощью чувствительного к окислению флуоресцентного зонда дихлордигидрофлуресцин диацетата (H2DCF-DA) и MitoSOX Red (Invitrogen), соответственно. Клетки HeLa, обработанные метформином, DCA или обоими препаратами в течение указанного времени, инкубировали с DCF-DA (20 мкМ) или MitoSOX (5 мкМ) в течение 10 минут при 37 °C. Затем клетки промывали. Затем клетки дважды промывали фосфатным забуференным физраствором (PBS) и подвергали проточной цитометрии (BD FACSCanto II, BD Biosciences, San Jose, CA) для получения и анализа. Интенсивность флуоресценции определяли также с помощью флуоресцентной микроскопии.
Измерение уровня клеточного глутатиона
Для определения окислительно-восстановительного статуса внутриклеточного глутатиона, уровни восстановленного и окисленного глутатиона (GSH, GSSG) анализировали в соответствии с методом, описанным ранее [29]. Для определения общего глутатиона клетки собирали и суспендировали в 1% сульфосалициловой кислоте. После удаления агрегатов центрифугированием при 8000g в течение 10 мин супернатанты использовали для анализа. Реакцию начинали, добавляя супернатант (1 мкг) в 96-луночный микротитровальный планшет, содержащий 200 мкл аналитической смеси [125 мМ NaPO4, pH 7,5, 0,3 мМ NADPH, 6 мМ 5,50-дитиобис(2-нитробензойная кислота) (DNTB; Sigma, St. Louis, MO), 0,12 U глутатионредуктазы], и максимальную скорость восстановления DNTB на GSH получали путем измерения абсорбции при 405 нм. Количество общего глутатиона рассчитывали по калибровочной кривой с использованием стандартного GSH (Sigma). Для получения уровня GSSG клетки предварительно обрабатывали 2-винилпиридином, чтобы лишить их имеющегося GSH, а супернатант (50 мкг) обрабатывали по тому же протоколу, что и для общего глутатиона.
Оценка активности пируватдегидрогеназы (ПДГ)
Активность ПДГ измеряли с помощью набора PDH Enzyme Activity Microplate Assay Kit (Abcam, Cambridge, MA) в соответствии с протоколом. Клетки лизировали в буфере для образцов, поставляемом с набором, и общий клеточный белок в концентрации 5 мг/мл загружали в трех экземплярах на микропланшет, предварительно покрытый анти-PDH антителами, и инкубировали в течение 3 ч при комнатной температуре. После промывки образцов добавляли реакционную смесь, и поглощение продукта реакции связанного с репортерным красителем восстановления NAD+ до NADH измеряли при 450 нм в течение 50 мин с интервалом 0,5 мин с помощью микропланшетного спектрофотометра Eon (BioStack, Winooski, VT). Активность PDH рассчитывали по наклону кривой зависимости мОД от времени (мин) (ΔмОД/мин) и нормировали на контрольную группу.
Анализ потребления глюкозы и продукции лактата
Клетки Hela (1 ×104/мл), культивируемые в DMEM с 25 мМ глюкозы, обрабатывали метформином, DCA или обоими препаратами. Через 48 ч концентрацию глюкозы и лактата в среде измеряли с помощью многопараметрической биоаналитической системы YSI 7100. Для измерения исходной концентрации глюкозы и лактата использовали среду без клеточной культуры. Потребление глюкозы = (концентрация глюкозы в среде без культуры клеток) — (концентрация глюкозы в среде из обработанной группы). Выработка лактата = (концентрация лактата в среде из обработанной группы) — (концентрация лактата в среде без клеточной культуры).
Измерение скорости окислительного фосфорилирования и гликолиза
Скорость потребления кислорода (OCR) и скорость внеклеточного окисления (ECAR) измеряли с помощью анализатора внеклеточных потоков Seahorse Bioscience (North Billerica, MA) XF24 [33]. Так, клетки HeLa высевали в 24-луночные микропланшеты XF (1 ×104) и инкубировали при 37 °C в течение 24 до обработки метформином, DCA или обоими препаратами в течение 12 ч. Затем клетки промывали и уравновешивали в DMEM, дополненной GlutaMax-1 (200 мМ), глюкозой (25 мМ), хлоридом натрия (32 мМ) и феноловым красным без бикарбоната при 37 °C в течение 1 ч в инкубаторе безCO2. OCR (пмоль/мин) и ECAR (мпч/мин) измеряли 3 раза по 3 мин. каждый. После эксперимента подсчитывали количество клеток для нормализации.
Иммуноблот-анализ
Лизаты клеток (40 мкг), приготовленные в RIPA-буфере с ингибиторами фосфатаз, разделяли на SDS-PAGE перед переносом на PVDF-мембрану (Millipore Corp). Блотты гибридизировали с первичными антителами и визуализировали с помощью системы ECL (Amersham Biosciences, Бакингемшир, Великобритания). Антитела были приобретены: анти-p-PDH (S293) от Calbiochem, анти-PARP и анти-PDH от Abcam, анти-Caspase 3 от Cell signaling, анти-циклин D1, анти-циклин E и анти-β-актин от Santa Cruz Biotechnology.
Статистический анализ
Числовые данные были представлены как среднее ± SD независимых определений. Применялся независимый t-тестили ANOVA, множественные сравнения оценивались с помощью Tukey HSD. Значения P менее 0,05 считались значимыми.
Результаты
Цитотоксичность метформина восстанавливается при наличии глюкозы
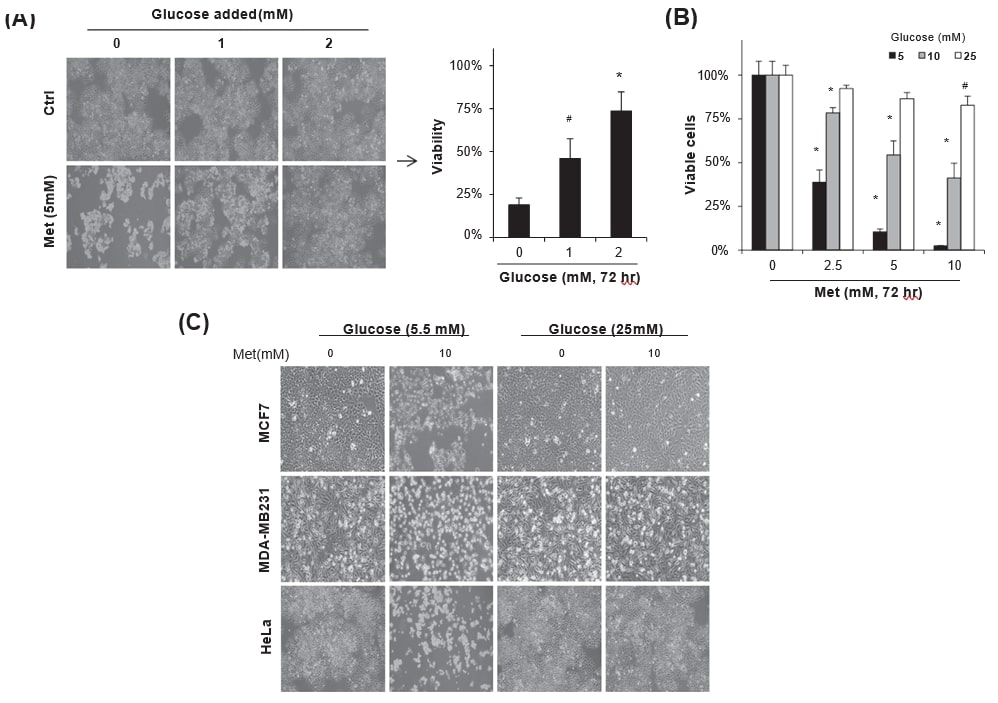
Для понимания биохимических путей, регулирующих метформин-индуцированную гибель раковых клеток, клетки HeLa, содержащиеся в DMEM с 5,5 мМ глюкозы, обрабатывали метформином (5 мМ) с ежедневным добавлением глюкозы (0-2 мМ) в течение 72 часов. Добавление глюкозы значительно повысило жизнеспособность клеток, обработанных метформином (рис. 1A, #p < 0,05 и *p < 0,001 по сравнению с отсутствием добавления) в хорошем соответствии с предыдущими отчетами [11], [17], [25]. Когда клетки HeLa, содержащиеся в среде, содержащей 5-25 мМ глюкозы, инкубировали с 0-10 мМ метформина в течение 72 ч, метформин-индуцированная гибель клеток зависела от концентрации глюкозы в культуральной среде. Особенно высокая концентрация глюкозы (25 мМ) значительно защищала метформин-цитотоксичность независимо от концентрации метформина (рис. 1B). Гибель клеток, индуцированная 10 мМ метформина, была защищена 25 мМ глюкозы в культуральной среде не только в клетках HeLa, но и в клетках рака молочной железы MCF7 и MDA-MB231, тогда как 5,5 мМ глюкозы не смогли защитить клетки от токсического эффекта (рис. 1С), наблюдаемого под фазово-контрастным микроскопом. Результаты показывают, что баланс между концентрациями метформина и глюкозы в культуральной среде важен для регуляции метформин-цитотоксичности.
Метформин-цитотоксичность регулируется GSH-опосредованными окислительно-восстановительными изменениями
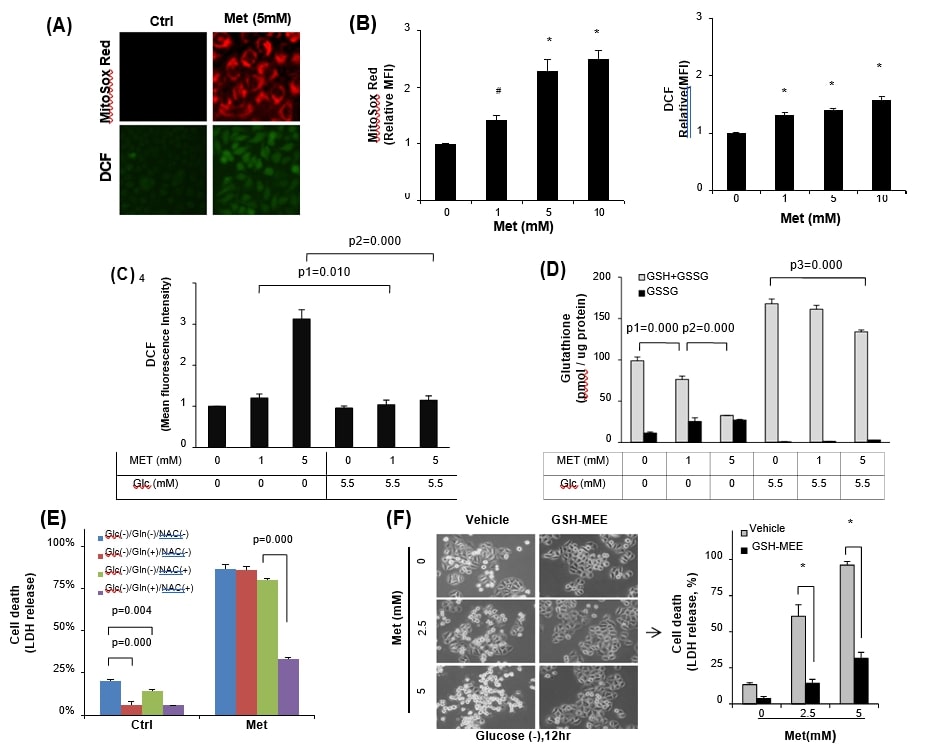
Поскольку метформин повышает уровень митохондриальной ROS через ингибирование комплекса I дыхательной цепи [8], [21], [36], мы исследовали, достаточно ли силен индуцированный метформином митохондриальный окислительный стресс, чтобы вызвать гибель раковых клеток в условиях недостатка глюкозы. Клетки HeLa, содержащиеся в глюкозосодержащей среде, обрабатывали 5 мМ метформина, а затем оценивали уровни митохондриального супероксида и общего клеточного ROS с помощью MitoSox Red и DCFDA, соответственно (рис. 2A). Действительно, метформин увеличивал уровни митохондриальной и общей клеточной ROS в зависимости от концентрации метоформина, однако флуоресценция MitoSox Red была в основном выше, чем уровень общей клеточной ROS в клетках HeLa (рис. 2B). Как и ожидалось, индуцированный метформином уровень клеточной ROS был значительно выше в условиях без глюкозы, чем в среде, содержащей глюкозу (рис. 2C). Эти данные убедительно подтверждают сообщение [15] о том, что лишение глюкозы снижает клеточный пул глутатиона (GSH + GSSG). Поэтому, когда измеряли пул глутатиона, снижение GSH относительно GSSG было более значительным в условиях отсутствия глюкозы (p = 0,000) в ответ на метформин, однако общее содержание глутатиона сохранялось без значительного увеличения GSSG в среде, содержащей глюкозу (5,5 мМ) (рис. 2D), что указывает на то, что глюкоза в культуральной среде играет роль важного источника генерации GSH. Для изучения регуляции метформин-цитотоксичности окислительно-восстановительным стрессом при недостатке глюкозы использовали антиоксиданты. Обработка клеток HeLa 10 мМ глутамином или N-ацетил-цистеином (NAC) значительно снижала гибель клеток, наблюдаемую в среде без глюкозы без добавления метформина (рис. 2E, p = 0,000 и p = 0,004 по сравнению с отсутствием обработки, соответственно), однако эта же обработка не уменьшала метформин (5 мМ) цитотоксичность в среде без глюкозы. С другой стороны, комбинированная обработка глутамина с NAC могла защитить клетки HeLa от цитотоксичности метформина (5 мМ) (рис. 2E, p = 0,000). Чтобы непосредственно подтвердить, играет ли восстановленный глутатион роль в ингибировании метформин-цитотоксичности, была использована проницаемая для клеток форма GSH (GSH-MEE). Добавление GSH-MEE значительно снизило гибель клеток HeLa по сравнению с контролем с использованием транспортного средства даже в условиях отсутствия глюкозы (рис. 2F). Эти данные свидетельствуют о синергетическом эффекте лишения глюкозы в цитотоксичности метформина через окислительный стресс путем истощения GSH.
Усиление окислительного стресса усугубляет метформин-цитотоксичность, несмотря на высокий уровень глюкозы
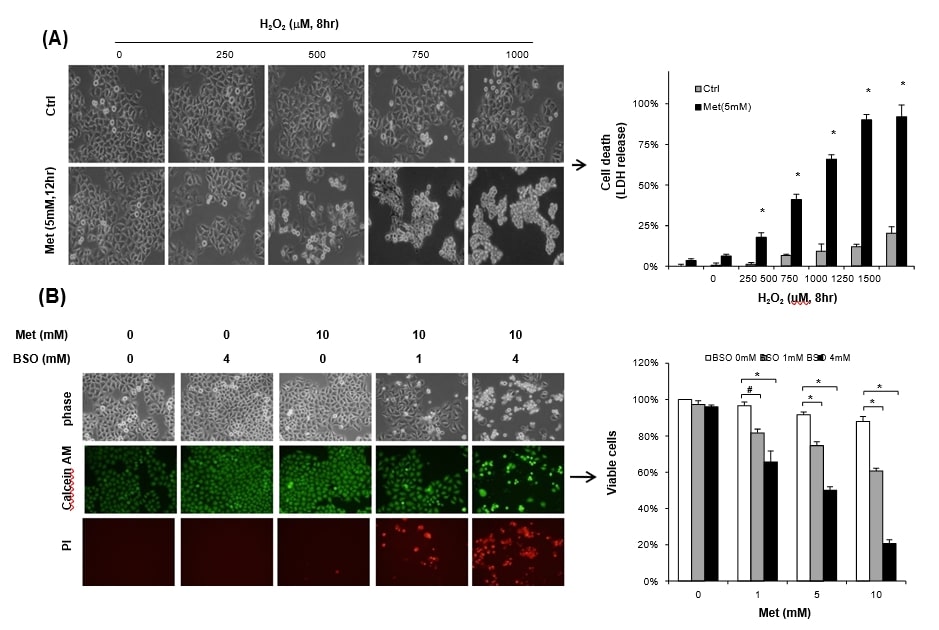
Чтобы проверить, становятся ли клетки, подверженные воздействию метформина, более уязвимыми к стрессу, клетки HeLa, содержащиеся на 25 мМ глюкозы, обрабатывали 5 мМ метформина вместе с H2O2 в течение 12 часов. Однократная обработка H2O2 или 5 мМ метформина не вызвала значительной гибели клеток, однако совместная обработка метформина и H2O2 (более 500 мкМ) заметно увеличила гибель клеток, измеренную по высвобождению LDH (рис. 3A). Не только экзогенный H2O2, но и истощение пула глутатиона с помощью BSO, ингибитора синтеза GSH, также приводило к значительному уровню метформин-цитотоксичности. Инкубация с метформином (10 мМ) или 4 мМ BSO в течение 48 ч не привела к значительным признакам гибели клеток в условиях высокого содержания глюкозы (25 мМ) при оценке окрашивания Calcein AM (рис. 3B левая панель). Однако комбинированное лечение метформином и BSO значительно увеличивало гибель клеток, окрашенных йодистым пропидием, при мониторинге методом исключения трипанового синего. BSO (1 мМ) значительно сенсибилизировал цитотоксичность 1 мМ метформина в условиях высокого содержания глюкозы (25 мМ) (рис. 3В правая панель). Наши результаты хорошо подтверждаются более ранними исследованиями, согласно которым GSH является наиболее многочисленным и важным биологическим антиоксидантом против окислительного стресса в раковых клетках [7], [10].
Дихлорацетат усиливает митохондриальное дыхание через активацию пируватдегидрогеназы
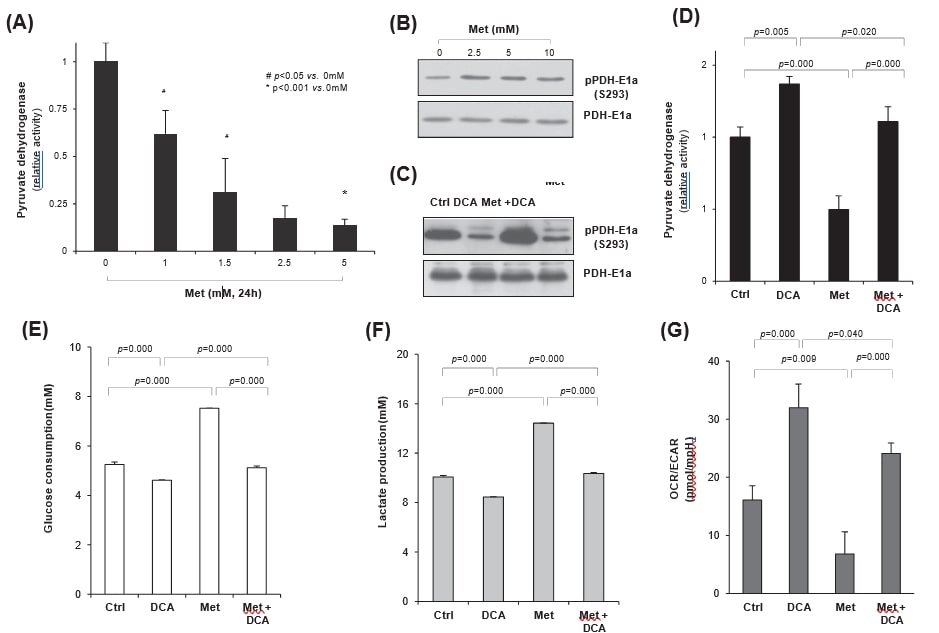
Поскольку PDH определяет судьбу метаболизма глюкозы либо в митохондриях, либо в цитозоле, весьма вероятно, что усиление активности PDH может увеличить митохондриальное дыхание, а не аэробный гликолиз в раковых клетках. Поэтому мы исследовали, регулирует ли метформин-индуцированный гликолиз DCA, ингибитор киназы PDH (PDK). Когда клетки HeLa обрабатывали метформином, активность PDH значительно снижалась в дозозависимой манере (рис. 4A). Лечение метформином явно увеличивало фосфорилирование PDH-E1a, что указывает на активацию PDK в отличие от ингибирования PDH (рис. 4B). С другой стороны, обработка DCA значительно снижала метформин-индуцированное фосфорилирование PDH-E1a на остатке S293 независимо от обработки метформином, что свидетельствует об увеличении или поддержании активности PDH под действием DCA (рис. 4C). Действительно, DCA значительно повышал активность PDH (p = 0,005), в отличие от значительного ингибирования метформином (p = 0,000), по сравнению с контролем. Более того, совместное применение ДКА и метформина значительно восстанавливало подавленную метформином активность PDH (p = 0,000) до контрольного уровня (рис. 4D); ДКА антагонизировал эффект метформина на активность PDH. Для оценки регуляции метаболических изменений под действием DCA или метформина измеряли скорость потребления глюкозы и продукции лактата в культуральной среде; обработка DCA значительно снижала потребление глюкозы (p = 0,000), в отличие от значительного увеличения под действием метформина (p = 0,000, рис. 4E). Однако при совместном применении метформина и DCA изменение было незначительным (p = 0,125 по сравнению с контролем), что еще раз подтверждает антагонистическую связь между DCA и метформином. Более того, высвобождение лактата (рис. 4F) под действием как ДКА, так и метформина выявило ту же картину, что и скорость потребления глюкозы (рис. 4E), что свидетельствует об изменении метаболизма глюкозы с аэробного гликолиза, индуцированного метформином, на митохондриальное окисление при совместном лечении ДКА. Когда скорость потребления кислорода (OCR) и скорость внеклеточного окисления (ECAR) были измерены анализатором XF24, отношение OCR/ECAR значительно увеличилось при лечении DCA (p = 0,000), что отражает увеличение митохондриального дыхания, но уменьшилось при лечении метформином (p = 0,009) по сравнению с контролем. Действительно, совместное лечение метформином и ДКА значительно восстановило митохондриальное дыхание по сравнению с лечением только метформином (p = 0,000), хотя оно было немного ниже, чем при лечении только ДКА (p = 0,04). В целом, совместное лечение метформином и DCA частично перепрограммировало метаболизм глюкозы с аэробного гликолиза на митохондриальное окисление (рис. 4G).
Дихлорацетат усиливает окислительный стресс и цитотоксичность метформина в условиях повышенного содержания глюкозы
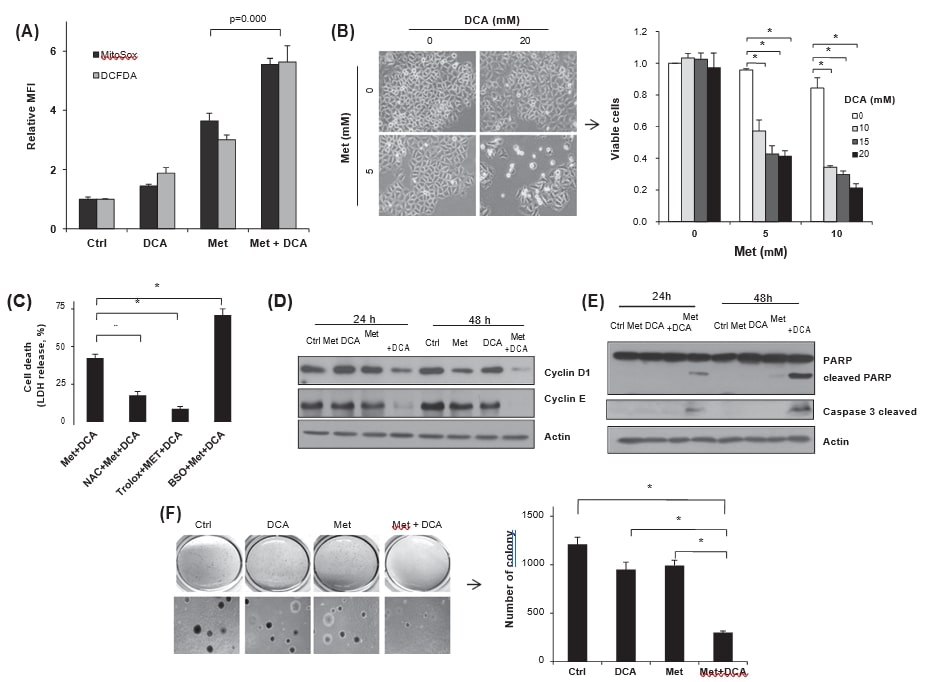
Для изучения влияния ДКА-опосредованного метаболического перепрограммирования на уровень ROS в клетках были проведены FACS-анализы с использованием MitoSox Red и DCFDA. Совместная обработка клеток HeLa метформином и DCA повышала уровень супероксида митохондрий и общего клеточного ROS по сравнению с каждой из обработок (рис. 5A), что свидетельствует о том, что метаболическое перепрограммирование может усугублять окислительный стресс под действием DCA. Чтобы оценить, усиливает ли усиленная ДКА генерация ROS цитотоксичность метформина или нет, была подсчитана жизнеспособность клеток после обработки метформином, ДКА или обоими препаратами. В отличие от незначительного уровня гибели клеток только от ДКА (∼20 мМ), совместная обработка ДКА и метформина заметно снижала жизнеспособность, несмотря на высокий уровень глюкозы (рис. 5B). Чтобы оценить влияние повышенного уровня ROS на гибель клеток, антиоксиданты, NAC или тролокс, или прооксидант, BSO, были предварительно обработаны перед совместным применением метформина и DCA. Действительно, антиоксиданты значительно снижали высвобождение ЛДГ в среду, тогда как при предварительной обработке BSO оно увеличивалось (рис.5C), что указывает на значительную регуляцию метформин-цитотоксичности через модуляцию окислительного стресса DCA. Такой синергетический эффект цитотоксичности наблюдался и в других раковых клетках, MDA-MB-231, EJ и MCF7 (данные не показаны). Более того, совместное лечение метформином и DCA значительно ингибировало синтез циклина D1 и циклина E наряду с расщеплением PARP (рис. 5D и E), что указывает на снижение пролиферации клеток и индукцию апоптоза. Наконец, совместное лечение значительно снизило якорно-независимый рост клеток HeLa по сравнению с одиночным лечением и контролем (рис. 5F).
Обсуждение
Мы представили доказательства того, что перепрограммирование метаболизма глюкозы путем обработки клеток HeLa меформином и дихлорацетатом усиливало гибель клеток за счет повышения уровня ROS путем увеличения митохондриального окисления, а не аэробного гликолиза (рис. 6). DCA был использован в качестве агента, способствующего окислительному фосфорилированию вместо гликолиза и предназначенного для перепрограммирования метаболизма глюкозы, опосредованного метформином. DCA активирует PDH через ингибирование PDK, таким образом, пируват может быть преобразован в ацетил-КоА вместо лактата. В качестве биохимического механизма сенсибилизации раковых клеток к цитотоксичности метформина, вызванной недостатком глюкозы, мы продемонстрировали, что окислительный стресс, индуцированный метформином, усиливался в условиях недостатка глюкозы (рис. 2C). Действительно, метформин потреблял глутатиона гораздо больше, чем в базовом режиме (рис. 2D), тогда как в присутствии глюкозы (5,5 мМ) этот показатель был гораздо меньше. Хотя предварительная обработка клеток HeLa NAC значительно блокировала гибель клеток в условиях отсутствия глюкозы, добавление цистеина в NAC без глутамина было недостаточным для поддержания уровня глутатиона, чтобы противостоять метформин-индуцированному окислительному стрессу (рис. 2E). Это явление может быть подтверждено исследованием, согласно которому глутамин важен для восстановительного карбоксилирования в опухолевых клетках с дефектными митохондриями [19]. Поэтому добавление NAC к глютамину значительно снижало метформин-цитотоксичность в условиях отсутствия глюкозы (рис. 2E).
В условиях высокого содержания глюкозы (25 мМ) 10 мМ метформина проявляли лишь слабый туморостатический эффект, но не приводили к значительной гибели клеток (рис. 3B). Напротив, 2,5 мМ метформина вызывали значительную гибель клеток при концентрации глюкозы 5,5 мМ (рис. 1В), что указывает на глюкозу как важный источник восстановительной силы. Поэтому добавление 1-2 мМ глюкозы было достаточным для блокирования метформин-цитотоксичности при обычном уровне глюкозы (рис. 1A), предполагая, что метформин-цитотоксичность может быть недостаточной, чтобы вызвать гибель клеток при физиологическом состоянии глюкозы. Однако лечение метформином значительно увеличивало митохондриальную ROS в условиях отсутствия лимитирования глюкозы. До сих пор не было сообщений, посвященных генерации митохондриальной ROS в контексте метформин-цитотоксичности. Здесь мы наблюдали, что 1 мМ метформина уже значительно увеличивал митохондриальный супероксид наряду с клеточной ROS (рис. 2B), что индуцировало набухание митохондрий в раковых клетках, причем набухание не было необратимым (дополнительные рис. S1A и S1B). Тем не менее, увеличение митохондриального ROS, по-видимому, недостаточно для увеличения общего клеточного ROS и окислительного повреждения. Раковые клетки, подвергшиеся воздействию метформина, пытаются минимизировать окислительное повреждение для поддержания жизнеспособности клеток путем индукции аэробного гликолиза и выработки NADPH. Эта идея частично подтверждается тем фактом, что истощение восстановительной способности с помощью BSO или экзогенного H2O2 усиливало метформин-цитотоксичность, несмотря на высокое содержание глюкозы (рис. 3). Раковые клетки могут противостоять окислительному стрессу посредством аэробного гликолиза, а уровень GSH может поддерживаться в основном за счет NADPH, образующегося в пентозофосфатном пути [12].
Противоопухолевая активность DCA была показана in vitro [4] и in vivo на мышиных моделях [27], [28], а также у пациентов с глиобластомой [18], однако значение IC50 DCA относительно высоко (>17 мМ, 48 ч), чтобы вызвать гибель клеток. Более того, в протестированных линиях раковых клеток нет специфичности опухолевых клеток [26], а DCA избирательно активен против клеток с дефектами митохондрий, таких как клетки rho(0) или клетки HCT116 p53 null с дефицитом комплекса IV. Мы также наблюдали, что 20 мМ DCA сам по себе не вызывал эффективной гибели клеток, однако в сочетании с метформином противоопухолевая активность DCA синергировала с 10 мМ (рис. 5B и F).
Когда клетки обрабатывали метформином, активность PDH значительно ингибировалась через фосфолитирование остатка Ser293 (рис. 4C) без существенного изменения экспрессии мРНК PDK (данные не показаны), что указывает на посттранскрипционную модификацию. Хотя мы не смогли определить механизм ингибирования метформином активности PDH, снижение активности PDK под действием DCA усилило вызванный метформином окислительный стресс за счет подавления фосфорилирования PDH (рис. 4), что указывает на ингибирование активности PDK под действием DCA, но увеличение окислительного фосфорилирования наряду с редокс-стрессом. В соответствии с вышеизложенными результатами, DCA частично восстанавливал ингибированное метформином митохондриальное дыхание (рис. 4F) и значительно усиливал генерацию митохондриальной ROS, индуцированную метформином (рис. 5A). Защитная роль антиоксидантов против комбинированного воздействия метформина и DCA позволила нам сделать вывод, что DCA может потенцировать цитотоксичность метформина через перепрограммирование и усиление окислительного стресса (рис. 5C).
Таким образом, цитотоксичность, вызванная метформином, усиливалась при недостатке глюкозы. Даже в условиях высокого содержания глюкозы дополнительный ROS стресс, вызванный истощением GSH или экзогенным H2O2, усиливал метформин-цитотоксичность наряду с увеличением митохондриального дыхания. Мы пришли к выводу, что DCA-опосредованное перепрограммирование метаболизма глюкозы с аэробного гликолиза на окислительное дыхание усугубляет вызванный метформином окислительный стресс и сенсибилизирует метформин-цитотоксичность в раковых клетках (рис. 6).

Конфликт интересов
Никаких конкурирующих финансовых интересов, связанных с данной работой, нет.
Благодарности
Данная работа была поддержана грантом Национальной научно-исследовательской программы по борьбе с раком Министерства здравоохранения и социального обеспечения Республики Корея (131280).
Приложение A. Дополнительный материал
Дополнительные данные, связанные с этой статьей, можно найти в онлайновой версии на сайте http://dx.doi.org/10.1016/j.canlet.2014.01.015.
ССЫЛКИ
1 I.M. Ahmad, N. Aykin-Burns, J.E. Sim, S.A. Walsh, R. Higashikubo, G.R. Buettner, S. Venkataraman, M.A. Mackey, S.W. Flanagan, L.W. Oberley, D.R. Spitz Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells J. Biol. Chem., 280 (2005), pp. 4254-42632 И. Бен Сахра, К. Лоран, С. Джулиано, Ф. Ларбре, Г. Понцио, П. Гунон, Й. Ле Маршан-Брюстель, С. Джорджетти-Перальди, М. Кормон, К. Бертолотто, М. Декерт, П. Обергер, Ж.Ф. Танти, Ф. Бост Направленный метаболизм раковых клеток: комбинация метформина и 2-дезоксиглюкозы вызывает р53-зависимый апоптоз в клетках рака предстательной железы Cancer Res., 70 (2010), pp. 2465-2475
3 И. Бен Сахра, Й. Ле Маршан-Брюстель, Ж.Ф. Танти, Ф. Бост Метформин в терапии рака: новые перспективы для старого противодиабетического препарата? Mol. Cancer Ther., 9 (2010), pp. 1092-1099
4 С. Бонне, С.Л. Арчер, Ж. Аллалунис-Тернер, А. Хароми, К. Болье, Р. Томпсон, К.Т. Ли, Г.Д. Лопащук, Л. Путтагунта, С. Бонне, Г. Гарри, К. Хашимото, К.Дж. Портер, М.А. Андраде, Б. Тебо, Э.Д. Микелакис Ось митохондрий-К+ каналов подавляется при раке, и ее нормализация способствует апоптозу и подавляет рост рака Cancer Cell, 11 (2007), pp. 37-51
5 J.R. Cantor, D.M. Sabatini Метаболизм раковых клеток: одна отличительная черта, много лиц Cancer Discov., 2 (2012), pp. 881-898
6 J.H. Cheong, E.S. Park, J. Liang, J.B. Dennison, D. Tsavachidou, C. Nguyen-Charles, K. Wa Cheng, H. Hall, D. Zhang, Y. Lu, M. Ravoori, V. Kundra, J. Ajani, J.S. Lee, W. Ki Hong, G.B. Mills Двойное ингибирование энергетического пути опухоли 2-дезоксиглюкозой и метформином эффективно против широкого спектра доклинических моделей рака Mol. Cancer Ther., 10 (2011), pp. 2350-2362
7 E.P. Clark, E.R. Epp, J.E. Biaglow, M. Morse-Gaudio, E. Zachgo Glutathione depletion, radiosensitization, and misonidazole potentiation in hypoxic Chinese hamster ovary cells by buthionine sulfoximine Radiat. Res., 98 (1984), pp. 370-380
8 М.Я. Эль-Мир, В. Ногейра, Э. Фонтен, Н. Аверет, М. Ригуле, X. Леверве Диметилбигуанид ингибирует клеточное дыхание через косвенный эффект, направленный на комплекс дыхательной цепи I J. Biol. Chem., 275 (2000), pp. 223-228
9 Р.Б. Хаманака, Н.С. Чандель Биология клетки. Эффект Варбурга и окислительно-восстановительный баланс Наука, 334 (2011), с. 1219-1220
10 Т. Ишимото, О. Нагано, Т. Яе, М. Тамада, Т. Мотохара, Х. Осима, М. Осима, Т. Икеда, Р. Асаба, Х. Яги, Т. Масуко, Т. Симидзу, Т. Исикава, К. Каи, Э. Такахаси, Й. Имамура, Й. Baba, M. Ohmura, M. Suematsu, H. Baba, H. Saya Вариант CD44 регулирует редокс-статус в раковых клетках путем стабилизации субъединицы xCT системы xc(-) и тем самым способствует росту опухоли Cancer Cell, 19 (2011), pp. 387-400
11 S. Javeshghani, M. Zakikhani, S. Austin, M. Bazile, M.J. Blouin, I. Topisirovic, J. St-Pierre, M.N. Pollak Источник углерода и экспрессия myc влияют на антипролиферативное действие метформина Cancer Res., 72 (2012), pp. 6257-6267
12 S.M. Jeon, N.S. Chandel, N. Hay AMPK регулирует гомеостаз NADPH для содействия выживанию опухолевых клеток при энергетическом стрессе Nature, 485 (2012), pp. 661-665
13 G. Kroemer, J. Pouyssegur Метаболизм опухолевых клеток: ахиллесова пята рака Cancer Cell, 13 (2008), pp. 472-482
14 M.A. Lea, J. Chacko, S. Bolikal, J.Y. Hong, R. Chung, A. Ortega, C. Desbordes Добавление 2-дезоксиглюкозы усиливает ингибирование роста, но обращает вспять закисление в клетках рака толстой кишки, обработанных фенформином Противораковые исследования, 31 (2011), с. 421-426
15 X. Линь, Ф. Чжан, К.М. Брэдбери, А. Каушал, Л. Ли, Д.Р. Спитц, Р.Л. Афт, Д. Гиус 2-Дезокси-d-глюкоза-индуцированная цитотоксичность и радиосенсибилизация в опухолевых клетках опосредуется через нарушения в тиоловом обмене Cancer Res., 63 (2003), pp. 3413-3417
16 L. Luo, W. Huang, R. Tao, N. Hu, Z.X. Xiao, Z. Luo ATM и LKB1 зависимая активация AMPK сенсибилизирует раковые клетки к этопозид-индуцированному апоптозу Cancer Lett., 328 (2013), pp. 114-119
17 J.A. Menendez, C. Oliveras-Ferraros, S. Cufi, B. Corominas-Faja, J. Joven, B. Martin-Castillo, A. Vazquez-Martin Метформин является синтетически летальным при отмене глюкозы в раковых клетках Cell Cycle, 11 (2012), pp. 2782-2792
18 E.D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, C. Maguire, T.L. Gammer, J.R. Mackey, D. Fulton, B. Abdulkarim, M.S. McMurtry, K.C. Petruk Metabolic modulation of glioblastoma with dichloroacetate Sci. Translat. Med., 2 (2010), p. 31ra34
19 A.R. Mullen, W.W. Wheaton, E.S. Jin, P.H. Chen, L.B. Sullivan, T. Cheng, Y. Yang, W.M. Linehan, N.S. Chandel, R.J. DeBerardinis Reductive carboxylation supports growth in tumour cells with defective mitochondria Nature, 481 (2012), pp. 385-388
20 M.R. Niewisch, Z. Kuci, H. Wolburg, M. Sautter, L. Krampen, B. Deubzer, R. Handgretinger, G. Bruchelt Влияние дихлорацетата (DCA) на производство лактата и потребление кислорода в клетках нейробластомы: является ли DCA подходящим препаратом для лечения нейробластомы? Клетка. Физиол. биохим: Int. J. Exp. Cell. Physiol., Biochem., Pharmacol., 29 (2012), pp. 373-380
21 M.R. Owen, E. Doran, A.P. Halestrap Доказательства того, что метформин оказывает противодиабетическое действие через ингибирование комплекса 1 митохондриальной дыхательной цепи Biochem. J., 348 (Pt 3) (2000), pp. 607-614
22 C. Riganti, E. Gazzano, M. Polimeni, E. Aldieri, D. Ghigo The pentose phosphate pathway: an antioxidant defense and a crossroad in tumor cell fate Free Radical Biol. Med., 53 (2012), pp. 421-436
23 G.Z. Rocha, M.M. Dias, E.R. Ropelle, F. Osorio-Costa, F.A. Rossato, A.E. Vercesi, M.J. Saad, J.B. Carvalheira Metformin amplifies chemotherapy-induced AMPK activation and antitumoral growth Clin. Cancer Res..: Official J. Am. Assoc. Cancer Res., 17 (2011), pp. 3993-4005
24 D.B. Shackelford, E. Abt, L. Gerken, D.S. Vasquez, A. Seki, M. Leblanc, L. Wei, M.C. Fishbein, J. Czernin, P.S. Mischel, R.J. Shaw LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin Cancer Cell, 23 (2013), pp. 143-158
25 J. Sinnett-Smith, K. Kisfalvi, R. Kui, E. RozengurtМетформин ингибирует mTORC1 активации, синтеза ДНК и пролиферации в клетках рака поджелудочной железы: зависимость от концентрации глюкозы и роль AMPK Биохим. Biophys. Res. Commun., 430 (2013), pp. 352-357
26 L.H. Stockwin, S.X. Yu, S. Borgel, C. Hancock, T.L. Wolfe, L.R. Phillips, M.G. Hollingshead, D.L. Newton Sodium dichloroacetate selectively target cells with defects in the mitochondrial ETC Int. J. Cancer J. Int. Cancer, 127 (2010), pp. 2510-2519
27 R.C. Sun, M. Fadia, J.E. Dahlstrom, C.R. Parish, P.G. Board, A.C. Blackburn Обращение гликолитического фенотипа дихлорацетатом подавляет рост метастатических клеток рака молочной железы in vitro и in vivo Breast Cancer Res. Treat., 120 (2010), pp. 253-260
28 G. Sutendra, P. Dromparis, A. Kinnaird, T.H. Stenson, A. Haromy, J.M. Parker, M.S. McMurtry, E.D. Michelakis iMitochondrial activation by inhibition of PDKII suppres HIF1a signaling and angiogenesis in cancer Онкоген, 32 (2013), с. 1638-1650
29 Ф. Тиетце Энзимный метод количественного определения нанограммовых количеств общего и окисленного глутатиона: применение к крови млекопитающих и другим тканям m8 Anal. Биохим., 27 (1969), с. 502-522
30 M.G. Vander Heiden Targeting cancer metabolism: a therapeutic window opens Nat. Rev. Drug Discovery, 10 (2011), pp. 671-684
31 P.S. Ward, C.B. Thompson Метаболическое перепрограммирование: отличительная черта рака, которую не предвидел даже Варбург Cancer Cell, 21 (2012), pp. 297-308
32 S. Whitehouse, R.H. Cooper, P.J. Randle Механизм активации пируватдегидрогеназы дихлорацетатом и другими галогенированными карбоновыми кислотами Biochem. J., 141 (1974), pp. 761-774
33 M. Wu, A. Neilson, A.L. Swift, R. Moran, J. Tamagnine, D. Parslow, S. Armistead, K. Lemire, J. Orrell, J. Teich, S. Chomicz, D.A. Ferrick Многопараметрический метаболический анализ выявляет тесную связь между ослаблением биоэнергетической функции митохондрий и усилением зависимости от гликолиза в опухолевых клетках человека Am. J. Physiol. Физиол. клетки, 292 (2007), стр. C125-136
34 X. Xiao, Q. He, C. Lu, K.D. Werle, R.X. Zhao, J. Chen, B.C. Davis, R. Cui, J. Liang, Z.X. Xu Метформин ухудшает рост печеночной киназы B1-интактных клеток рака шейки матки Gynecol. Oncol., 127 (2012), pp. 249-255
35 Y. Zhuang, W.K. Miskimins Метформин вызывает как каспазозависимую, так и поли(ADP-рибоза) полимеразозависимую клеточную смерть в клетках рака молочной железы Mol. Cancer Res..: MCR, 9 (2011), pp. 603-615
36 J.W. Zmijewski, E. Lorne, X. Zhao, Y. Tsuruta, Y. Sha, G. Liu, G.P. Siegal, E. Abraham Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury Am. J. Respir. Crit. Care Med., 178 (2008), pp. 168-179