Чон Лиел Рох a, *, Джин Янг Парк a, Ын Хе Ким a, Хе Джин Чан a, Минсу Квон b
a Отделение отоларингологии, Медицинский центр Асан, Медицинский колледж Университета Ульсан, Сеул, Республика Корея
b Отделение отоларингологии, Медицинская школа при больнице Национального университета Кёнсан, Чинджу, Республика Корея
Автор переписки: Тел: +82 2 3010 3965; факс: +82 2 489 2773. Адрес электронной почты: [email protected] (J.-L. Roh).
Получено: 9 сентября 2015 г.
Пересмотрено: исправленная форма 13 ноября 2015 г.
Принято: 14 ноября 2015 г
Аннотация
Дихлорацетат (ДХА), сиротский препарат, способствующий переходу от гликолиза к окислительному фосфорилированию, был повторно использован для лечения рака. В настоящем исследовании изучалось, может ли DCA преодолеть резистентность к цисплатину при раке головы и шеи (РГШ). Были использованы две устойчивые к цисплатину линии клеток РГШ (AMC-HN4R и -HN9R), их родительские линии и другие линии клеток РГШ человека. Эффект DCA, отдельно и в комбинации с цисплатином, оценивали путем измерения клеточного цикла, жизнеспособности, гибели, продукции реактивных форм кислорода (ROS), мембранного потенциала митохондрий (ΔΨm) и экспрессии белков в доклинических моделях опухолевых ксенотрансплантатов мыши. Увеличение гликолиза коррелировало со снижением чувствительности к цисплатину и уменьшалось под действием DCA. Устойчивые к цисплатину клетки сверхэкспрессировали киназу пируватдегидрогеназы 2 (PDK2). DCA индуцировал гибель клеток HNC путем снижения ΔΨm и стимулирования производства митохондриальной ROS. Этот эффект снижался антиоксидантом N-ацетил-л-цистеином или ингибированием апоптоза, опосредованного каспазой. Активация митохондриального окисления глюкозы под действием DCA в конечном итоге активировала нижележащие митохондриальные апоптотические сигналы, что привело к гибели химиорезистентных раковых клеток. Таким образом, DCA значительно сенсибилизировал устойчивые клетки HNC к цисплатину in vitro и in vivo. Высокий гликолиз и сверхэкспрессия PDK2 тесно связаны с устойчивостью клеток НЯК к цисплатину; последняя может быть преодолена с помощью DCA.
Ключевые слова: Рак головы и шеи, устойчивость к цисплатину, PDK2, дихлорацетат, ремоделирование митохондрий
Сокращения: РГШ, рак головы и шеи; ДХА, дихлорацетат; CDDP, цисплатин; OXPHOS, окислительное фосфорилирование; PDK2, киназа 2 пируватдегидрогеназы; PDHE1α, изоформа E1α пируватдегидрогеназы; ROS, реактивные виды кислорода; ΔΨm, мембранный потенциал митохондрий; NAC, N-ацетил-цистеин; DCF-DA, 2′,7′- дихлорфлуоресцеин диацетат; PARP, поли(АДФ-рибоза) полимераза; siRNA, короткая интерферирующая РНК; 18F-FDG, 18F-фтордезоксиглюкоза; PET, позитронно-эмиссионная томография; SUV, стандартизированное значение поглощения; MTV, метаболический объем опухоли; TUNEL, терминальная дезоксинуклеотидилтрансфераза-опосредованное мечение ник-конца ДУТФ.
© 2015 Elsevier Ireland Ltd. Все права защищены.
ВВЕДЕНИЕ
Рак головы и шеи (РГШ) является восьмым по распространенности раком во всем мире, ежегодно диагностируется более полумиллиона новых случаев [1]. Опухоли возникают в верхних отделах пищеварительного тракта, включая ротовую или носовую полость, глотку и гортань, и метастазируют в регионарные лимфатические узлы и отдаленные участки. Современный стандарт лечения ГНК включает мультимодальный подход, включающий хирургию, химиотерапию и радиотерапию, особенно при распространенном ГНК. Наряду с недавним интересом к стратегиям сохранения органов, нехирургические методы, такие как радиотерапия в сочетании с химиотерапией, все чаще используются в качестве первой линии терапии при НЯК [2]. В настоящее время системная химиотерапия является центральным компонентом нескольких лечебных подходов, включая комбинацию с окончательной лучевой терапией или индукционным лечением, превышающим резервирование только для паллиации [3]. Цисплатин, платиновое производное цис-диамминдихлорплатина (II) (CDDP), остается химиотерапевтическим препаратом первой линии в нехирургических методах лечения НЯК, несмотря на последние достижения в области таргетной терапии [4]. Однако за последние три десятилетия, несмотря на диагностические и терапевтические достижения, общая выживаемость при НЯК существенно не изменилась, что является результатом стойкой резистентности раковых клеток к терапии, включая цисплатин и облучение [2].
Метаболические изменения являются общей чертой раковых клеток: сдвиг в генерации клеточной энергии от митохондриального окислительного фосфорилирования к аэробному гликолизу обеспечивает биосинтетическое преимущество раковых клеток [5]. Увеличение гликолиза в раковых клетках, называемое эффектом Варбурга, обычно связано с фенотипическими изменениями, включая адаптацию к гипоксии и недостатку питательных веществ, устойчивость к окислительному стрессу и апоптотическим стимулам, а также повышенный синтез биомассы [6]. Дерегулированный метаболизм связан с устойчивостью к лечению при терапии рака [7]. В гликолитическом пути повышение уровня транспортера глюкозы (GLUT), гексокиназы (HK), пируваткиназы M2 (PKM2), киназы пируватдегидрогеназы (PDK), лактатдегидрогеназы-A (LDHA), синтеза жирных кислот (FASN) и глутаминазы, среди прочих, связано с устойчивостью к противораковым препаратам [7]. Имеются данные о том, что регулирование метаболизма раковых клеток может улучшить терапию и преодолеть устойчивость к химиотерапии или радиотерапии [8,9].
Дихлорацетат (ДХА), сиротский препарат для лечения молочнокислого ацидоза, сдвигает метаболизм раковых клеток с гликолиза на окислительное фосфорилирование [10]. DCA избирательно ингибирует PDK, который активируется во многих видах рака, что приводит к активации пируватдегидрогеназы (PDH), комплекса ферментов, которые превращают цитозольный пируват в митохондриальный ацетил-КоА. Ингибирование PDK с помощью малой интерферирующей РНК (siRNA) или лечение DCA изменяет биоэнергетику рака и восстанавливает митохондрий-зависимый апоптоз в раковых клетках [11]. DCA эффективен в лечении рака с агрессивным фенотипом in vitro и in vivo[12,13], а также преодолевает резистентность к сорафенибу в гепатоцеллюлярной карциноме путем активации митохондриального окислительного фосфорилирования [14]. Таким образом, DCA может быть подходящим противораковым препаратом для повышения эффективности химиотерапии и преодоления резистентности к химиотерапевтическим средствам при раке человека; однако, хотя DCA широко изучался при раке, он редко тестировался на клетках НЯК [15], особенно в условиях резистентности к химиотерапевтическим средствам. Необходимо дальнейшее изучение механизмов действия DCA и синергизма с традиционными химиотерапевтическими препаратами. Здесь мы показываем, что DCA смещает биоэнергетику НЯК в сторону митохондриального окисления глюкозы и приводит к накоплению в клетках митохондриальных реактивных форм кислорода (mROS), тем самым сенсибилизируя химиорезистентные клетки НЯК к цисплатину in vitro и in vivo.
Материалы и методы
Культура клеток и создание цисплатин-устойчивых клеток НГШ
Клетки рака головы и шеи человека (РГШ) AMC-HN2, -HN3, -HN4, -HN5, -HN9 и -HN10 выращивали в минимальной основной среде Орла (Life TechnologiesTM, Carlsbad, CA, USA); Клетки SNU-1041, -1066 и -1076 выращивали в среде Roswell Park Memorial Institute (Life TechnologiesTM ); а клетки UMSCC1 выращивали в модифицированной среде Дульбекко Eagle (Life TechnologiesTM ). Среда была дополнена 10% фетальной бычьей сывороткой. Все линии раковых клеток были подтверждены путем профилирования ДНК (короткие тандемные повторы, STR), предоставленной Корейским банком клеточных линий. Клетки инкубировали при 37 °C в увлажненной атмосфере, содержащей 5% CO2.
Устойчивые к цисплатину клетки AMC-HN4 и HN9 (HN4R и HN9R) были получены из родительских чувствительных к цисплатину клеток AMC-HN4 и HN9, соответственно, путем воздействия возрастающих концентраций цисплатина (цис-платина (II) диамин дихлорид [CDDP]; Sigma-Aldrich, Луис, МО, США) [16]. Устойчивость к цисплатину в созданных клеточных линиях оценивали с помощью анализа жизнеспособности клеток и сравнивали с родительскими клетками.
Анализ жизнеспособности клеток
Жизнеспособность клеток определяли методом исключения трипанового синего, МТТ и клоногенным анализом. Исключение трипанового синего проводили в клетках HNC, высеянных в количестве 1 x105 в 6-луночные планшеты. Клеткам позволяли достичь 60-70% конфлюентности и подвергали воздействию дихлорацетата (DCA; Sigma-Aldrich) в течение 72 ч. Затем клетки трипсинизировали, окрашивали 0,4% трипановым синим (Life TechnologiesTM ) и подсчитывали с помощью гемацитометра. МТТ-анализ проводился на клетках HNC, высеянных в 96-луночных планшетах в количестве 3-5 x103 клеток на лунку. Клетки инкубировали в течение ночи, затем подвергали воздействию DCA и цисплатина, отдельно или в комбинации, в течение 72 ч. Затем клетки подвергали воздействию тетразолиевого соединения 3-[4,5-диметил-2-тиазолил]-2,5-дифенил-2Н-тетразолия бромида (МТТ; Sigma-Aldrich) в течение 4 ч, после чего добавляли буфер для солюбилизации на 2 ч. Абсорбцию в каждой лунке измеряли при 570 нм с помощью микропланшетного ридера SpectraMax M2 (Molecular Devices, Sunnyvale, CA, США). Для клоногенного анализа клетки подвергали воздействию DCA или транспортного средства в течение 72 ч, а затем инкубировали в среде без лекарств в течение 7-10 дней. Лунки окрашивали 0,5% раствором кристаллического фиолетового и подсчитывали количество колоний. Все анализы проводили в трех экземплярах и повторяли три раза.
Цитотоксичность цисплатина оценивали по МТТ через 72 ч, и рассчитывали половину максимальной ингибирующей концентрации (IC50) для каждой клетки HNC. Взаимодействие двух препаратов считалось синергическим, если ингибирование роста, вызванное комбинацией препаратов, было больше, чем сумма ингибирований, вызванных одним из препаратов: индекс комбинации (CI) = 1, аддитивное взаимодействие; CI < 1, синергическое взаимодействие; и CI > 1, антагонистическое взаимодействие [17].
Измерение биоэнергетики и продукции ROS
Клетки HNC (1 x104 клеток на лунку) высевали в 24-луночные планшеты, на следующий день их промывали и инкубировали в бессывороточной среде DMEM, дополненной 100 нг/мл олигомицина (ингибитор АТФ-синтазы; Sigma-Aldrich) или без него, в течение 6 ч. Культуральная среда (50 мкл) была собрана из каждого планшета, и концентрация лактата в культуральной среде была измерена с помощью набора для определения лактата (Biovision, Mountain View, CA, США). Lac (о) и Lac (в) означают концентрацию лактата в среде после 6 ч инкубации в присутствии и отсутствии олигомицина, соответственно. Вклад гликолиза и окислительного фосфорилирования (OXPHOS) в клеточную биоэнергетику рассчитывали по следующей формуле: гликолиз (%) = Lac (c)/Lac (o) x 100; OXPHOS (%) = 100 — гликолиз (%) [14].
Клетки подвергали воздействию 15 или 30 мМ DCA в течение 24 ч, и генерацию ROS определяли в клетках, инкубированных с 10 мкМ 2′,7′-дихлорфлуоресцеин диацетата (DCF-DA) (Enzo Life Sciences, Farmingdale, NY, USA). Клетки инкубировали с 10 мкМ DCF-DA в течение 30 мин при 37 °C, дважды промывали PBS и анализировали на проточном цитометре FACScalibur. Перед воздействием 30 мМ DCA клетки предварительно обрабатывали 3 мМ N-ацетил-цистеина (NAC; Sigma-Aldrich) в течение 1 ч или 10 мкМ ингибитора супероксиддисмутазы (СОД) диэтил-дитиокарбамата (Sigma-Aldrich). Уровни ROS измерялись методом проточной цитометрии с использованием DCF-DA и представлены в виде изменения в разах по сравнению с контрольным (базальным) уровнем.
Анализ клеточного цикла и гибели клеток
Для анализа клеточного цикла клетки подвергали воздействию ДКА в течение 72 ч. Затем клетки трипсинизировали, фиксировали на ночь в ледяном этаноле и окрашивали в течение 30 мин йодистым пропидием (Sigma-Aldrich) при температуре 37 °С. Содержание клеточной ДНК измеряли с помощью проточного цитометра FACScalibur (BD Bioscience, Сан-Хосе, Калифорния, США). Для анализа клеточной гибели клетки культивировали с цисплатином и DCA, отдельно или в комбинации, или эквивалентным количеством ДМСО (транспортный контроль). Через 72 часа клетки собирали, промывали ледяным PBS и ресуспендировали в буфере для связывания. Клетки окрашивали аннексином V-FITC (флуоресцеин изотиоцианат) и йодистым пропидием с использованием набора для выявления апоптоза аннексином V-FITC (BD Biosciences, Franklin Lakes, NJ, USA), а затем анализировали методом проточной цитометрии. Все данные анализировали с помощью программного обеспечения Cell Quest (BD Biosciences). Статистическую значимость между различными группами лечения оценивали с помощью двуххвостового U-теста Манна-Уитни или t-теста Стьюдента.
Для анализа активности каспазы клетки HN4R и HN9R, посеянные в 96-луночный планшет, подвергали воздействию 100 мкл среды, содержащей цисплатин и DCA отдельно или в комбинации, в течение 72 ч, с или без 3 мМ NAC или 50 мкМ Z-VAD-fmk (R&D Systems, Minneapolis, MN, USA) перед воздействием 30 мМ DCA. Анализы проводили в трех лунках с использованием флуориметрического Homogeneous Caspase Assay (Roche Life Science, Базель, Швейцария). Добавляли рабочий раствор субстрата, и планшет инкубировали в темноте при 37 °C в течение 2-8 ч или при комнатной температуре в течение ночи. Поглощение в каждой лунке измеряли при длине волны возбуждения 485 нм и длине волны эмиссии 520 нм с помощью микропланшетного ридера SpectraMax M2.
Для измерения мембранного потенциала митохондрий (ΔΨm) клетки HN4R и HN9R, посеянные в 96-луночный планшет, подвергались воздействию 100 мкл среды, содержащей цисплатин и DCA отдельно или в комбинации в течение 36 ч. Клетки окрашивались 200 нМ тетраметилродамин этилового эфира (TMRE, Life Technologies TM ) в течение 20 мин и затем анализировались методом проточной цитометрии. Средняя интенсивность флуоресценции (MFI) каждой группы лечения была нормализована по отношению к контрольной группе.
Количественная обратная транскрипция-ПЦР в реальном времени
Общая клеточная РНК была выделена с помощью реагента для лизиса QIAzol и набора RNeasy Mini Kit (Qiagen, Valencia, CA, USA). кДНК была получена из очищенной РНК с помощью набора для обратной транскрипции QuantiTect (Qiagen) в соответствии с инструкциями производителя. КДНК киназы пируватдегидрогеназы 2 (PDK2) амплифицировали методом ПЦР с использованием следующих праймеров: 5′-ATGGCAGTCCTCCTCTCTGA-3′ (прямой) и 5′-CACCCACCCTCTTCCTAACA-3′ (обратный). Для β-актина (эндогенного контроля) использовались следующие праймеры: 5′-ACCCCCACTGAAAAAGATGA-3′ (прямой) и 5′-ATCTTCAAACCTCCATGATG-3′ (обратный). Количественная обратная транскрипция-ПЦР (qRT-PCR) в реальном времени проводилась с использованием SYBR Green (Qiagen) на системе 7900HT Fast Real-time PCR System (Applied Bioscience, Foster, CA, USA). Относительные уровни мРНК-мишеней нормировали на экспрессию β-актина.
siRNA
Для нокдауна PDK2 и PDHE1α клетки AMC-HN4-cisR и HN9-cisR высевали на 60 мм планшеты в среду без антибиотиков и через 18 ч трансфицировали 100 нмоль/л малой интерферирующей РНК (siRNA), нацеленной на гены PDK2 или PDHE1α человека, или скремблированной контрольной siRNA (Life Technologies TM ). Через 72 ч клетки подвергали воздействию DCA в течение еще 72 ч, а затем анализировали экспрессию белка. Нокдаун был подтвержден методом вестерн-блоттинга с использованием антител против PDK2 или PDHE1.
Иммуноблоттинг
Клетки лизировали при 4 °C в буфере для радиоиммунопреципитации (RIPA) (Life TechnologiesTM ). Иммуноблоттинг проводили в соответствии со стандартными процедурами. Вкратце, 50 мкг белка растворяли додецилсульфат натрия-полиакриламидным гель-электрофорезом (SDS-PAGE) на 10-12% гелях, переносили на нитроцеллюлозные или поливинилидендифторидные мембраны и зондировали первичными и вторичными антителами. Были использованы следующие первичные антитела: p53 (DO1) (Santa Cruz Biotechnology, Santa Cruz, CA, США); гексокиназа 2 (HK2), p21 WAF1/CIP1, PUMA, расщепленная поли(ADP-рибоза) полимераза (PARP), фосфо-p53-Ser15 и расщепленная каспаза-3 (Cell Signaling Technology, Danvers, MA, США); PDK2 (Abcam, Cambridge, MA, USA); и PDHE1α и фосфо-PDHE1α (ser293) (Calbiochem, Billerica, MA, USA). β-Актин (Sigma-Aldrich) использовался в качестве контроля нагрузки. Все антитела разбавляли от 1:250 до 1:5000.
Конфокальная микроскопия
ΔΨm визуализировали в живых раковых клетках с помощью TMRM (Life TechnologiesTM ), чувствительного к митохондриям и напряжению красителя на основе родамина (красная флуоресценция). Митохондриальный супероксид (mROS, Life TechnologiesTM ), образующийся во время клеточного дыхания, измерялся с помощью MitoSOX (красная флуоресценция). Изображения флуоресценции TMRM или mitoSOX были получены с помощью двухфотонного конфокального микроскопа Zeiss LSM 510 (Carl Zeiss AG, Хайденхайм, Германия), и средняя флуоресценция была оценена количественно (произвольные единицы флуоресценции, AFU) в раковых клетках в культуре с помощью программного обеспечения Zen imaging (Carl Zeiss AG). Получение изображений и анализ проводились двумя учеными, которые не имели представления об источнике образцов клеток.
Позитронно-эмиссионная томография (ПЭТ)
Визуализация поглощения глюкозы проводилась in vivo у голых мышей при трансплантации клеток HN4R или HN9R с использованием газового наркоза изофлураном (20% кислорода) и 18 F-фтордезоксиглюкозы (18 F-ФДГ) в качестве радиотрассера. ПЭТ-визуализация проводилась с помощью микроПЭТ FOCUS 120 (Concorde Microsystem Inc., Knoxville, TN, USA). Мыши голодали в течение ночи, затем им внутривенно вводили 0,15 мКи. Через 1 ч проводилось ПЭТ-сканирование с 18 F-ФДГ во всем теле. Изображения получения данных ПЭТ показаны с помощью псевдоцветной карты, где красный цвет указывает на высокое поглощение 18 F-ФДГ.
Максимальное и среднее стандартизированное значение поглощения (SUVmax и SUVmean) использовались для определения активности 18 F-ФДГ-ПЭТ. SUV анализировали по уравнению SUV = A/(ID/BW), где A — активность в ткани с поправкой на распад (в мКи/мл), ID — введенная доза ФДГ (в мКи), BW — масса тела мыши (в граммах). Сферические или эллиптические области интереса (ROIs) располагались над очагами поражения, видимыми на ПЭТ-изображениях. SUVmax и SUVmean рассчитывались путем автоматического построения ROI над наиболее интенсивным срезом опухолей, видимых на ПЭТ-изображениях. MTV рассчитывались на основе данных ПЭТ с коррекцией затухания с помощью коммерческого пакета программного обеспечения (INFINITT PACS; INFINITT Healthcare Co., Ltd). данные ПЭТ с 18 F-ФДГ поступали на рабочую станцию в формате DICOM, и значения интенсивности автоматически преобразовывались в SUVs. Для расчетов MTV контурные границы опухоли определялись с использованием фиксированного SUV 2,0, а объем опухоли затем очерчивался с помощью изоконтура SUV 2,0. ПЭТ-изображения были получены у мышей с имплантацией опухоли на 21-й день после начала лечения. Средние значения SUVmax и MTV опухоли сравнивались между различными группами лечения.
Доклинические исследования
Все процедуры исследования на животных проводились в соответствии с протоколами, утвержденными Комитетом по уходу и использованию животных нашего учреждения. Шестинедельные атимичные самцы мышей BALB/c (nu/nu) были приобретены у Central Lab Animal Inc. (Сеул, Республика Корея). Клетки AMC-HN4R или HN9R (5 x 10 6 ) вводили подкожно во фланг. Объем опухоли и вес тела измеряли каждые 3 дня. Опухоли измеряли с помощью штангенциркуля, а объем рассчитывали как (длина х ширина2 )/2. Лечение начиналось, когда клеточные имплантаты становились пальпируемыми узелками (= день 0). Мыши были рандомизированы на четыре группы лечения: транспортное средство, DCA, цисплатин и DCA плюс цисплатин.
Мышей лечили питьевой водой с добавлением 0,5 г/л DCA, или внутривенным введением 5 мг/кг цисплатина один раз в неделю, или комбинацией DCA и цисплатина по одинаковым графикам. Мышей приносили в жертву на 24-й день, опухоли выделяли и анализировали с помощью иммуноблотинга и in situ терминальной дезоксинуклеотидилтрансферазы-опосредованного мечения никкеля ДУТФ (TUNEL) (EMD Millipore, Billerica, MA, USA). Количество апоптотических телец подсчитывали вслепую в десяти случайно выбранных полях высокой мощности. Статистическую значимость между различными группами лечения оценивали с помощью двуххвостового U-теста Манна-Уитни или t-теста Стьюдента.
Результаты
Увеличение гликолиза в клетках HNC связано с резистентностью к цисплатину и обратимо под действием DCA
Все клеточные линии, использованные в нашем исследовании, были человеческими HNC. Цитотоксический эффект DCA оценивался в клетках HNC с помощью исключения трипанового синего, окрашивания кристаллическим фиолетовым и МТТ. DCA, протестированный в концентрации до 30 мМ в течение 72 ч, заметно подавлял рост клеток HNC (МТТ-тест, рис. 1А). Биоэнергетика клеток НЯК изменялась под действием ДКА: клеточный гликолиз значительно уменьшался, а окислительное фосфорилирование увеличивалось (рис. 1В). Изменение биоэнергетики варьировалось между линиями клеток НЯК и было значительно связано с чувствительностью к цисплатину: клетки НЯК с высоким гликолизом демонстрировали устойчивость к цисплатину (R2 = 0,859, P < 0,001; рис. 1С ). Кроме того, при воздействии DCA раковые клетки с наибольшим изменением биоэнергетики, скорее всего, становились более апоптотичными (R2 = 0,769, P = 0,002; рис. 1D ). Это позволяет предположить, что DCA вызывает специфический для раковых клеток апоптоз путем снижения гликолиза в клетках HNC.
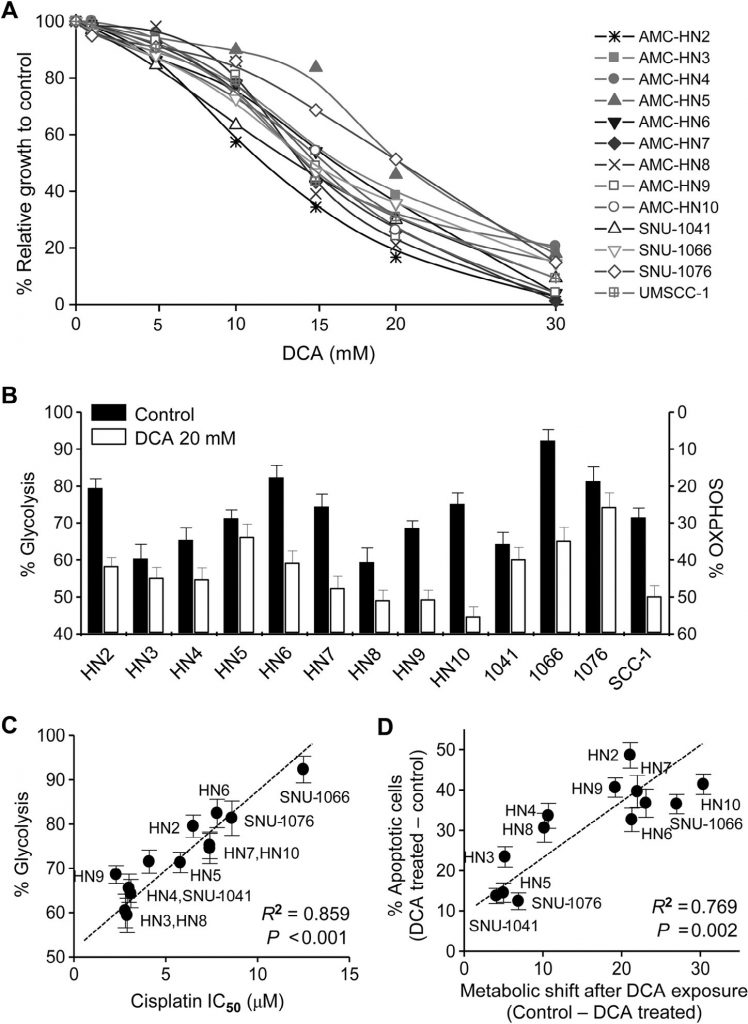
Экспрессия PDK2 связана с устойчивостью к цисплатину в HNC
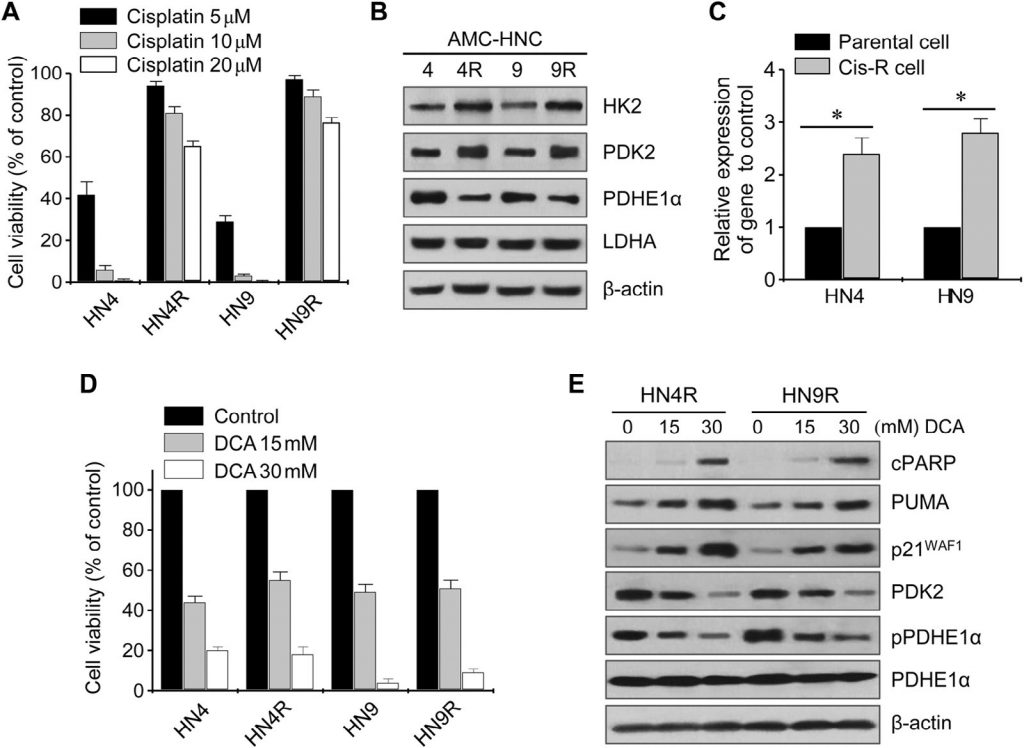
Цитотоксическое действие цисплатина было проверено на культивируемых клеточных линиях HN4-cisR и HN9-cisR и родительских раковых клетках (рис. 2А). Устойчивые к цисплатину клетки HN4-cisR и HN9-cisR показали 12-кратное и 18-кратное увеличение IC50, соответственно, по сравнению с соответствующими родительскими линиями. Вестерн-блот анализ показал, что HK2 и PDK2 были высоко экспрессированы в клетках HN4-cisR и HN9-cisR по сравнению с соответствующими родительскими клетками, в то время как экспрессия PDHE1α была низкой в клетках, устойчивых к цисплатину (рис. 2B). Уровень экспрессии мРНК PDK2 также был выше в резистентных клетках, чем в чувствительных (P < 0,01) (рис. 2C). DCA подавлял рост цисплатин-резистентных клеток HNC в той же степени, что и цисплатин-чувствительных клеток HNC (рис. 2D). Вестерн-блот анализ показал, что DCA значительно снизил уровень PDK2 и фосфо-PDHE1α (pPDHE1α), но повысил уровень проапоптотических белков, включая расщепленный PARP (cPARP) и PUMA, а также p21 в HN4-cisR и HN9-cisR (рис. 2E). Это говорит о том, что DCA может эффективно подавлять рост цисплатин-резистентных клеток HNC, а также чувствительных к цисплатину клеток HNC.
DCA индуцирует накопление ROS в клетках HNC
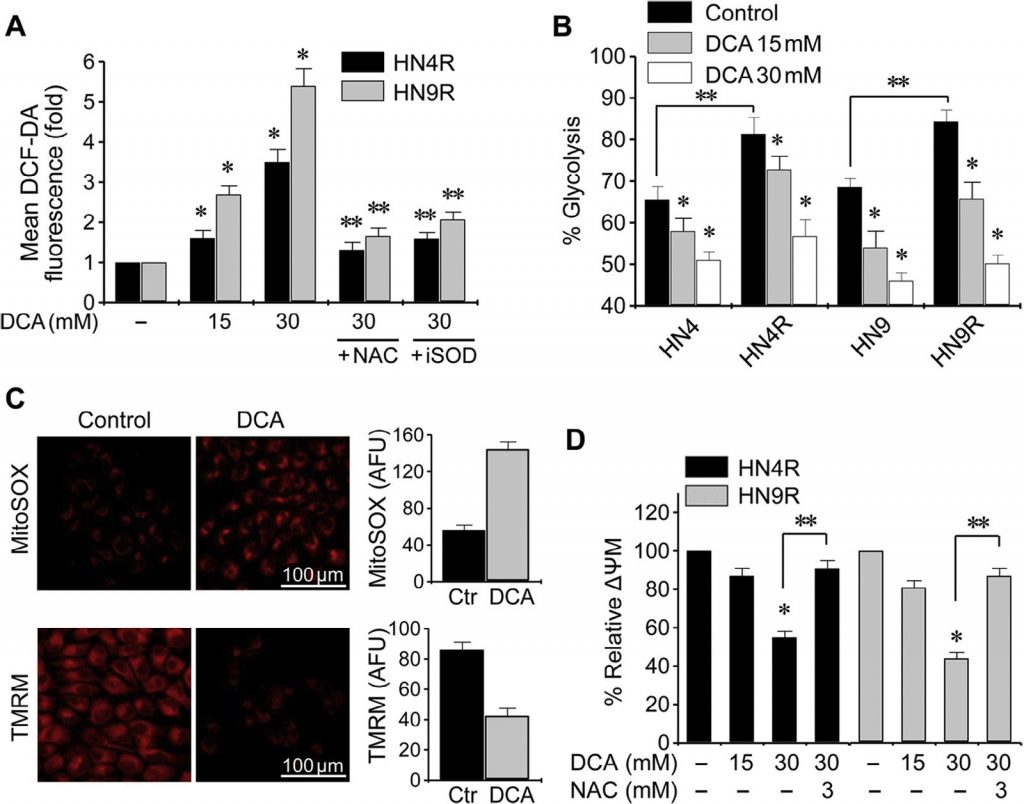
Изменение клеточной ROS под воздействием DCA оценивалось методом проточной цитометрии с использованием редокс-чувствительного флуоресцентного зонда DCF-DA. Воздействие DCA вызвало значительное повышение уровня ROS в клетках HNC (P < 0,01), которое блокировалось совместным воздействием NAC или ингибитором SOD диэтил-дитиокарбаматом (рис. 3A). Гликолиз значительно увеличился в цисплатин-устойчивых клетках HNC по сравнению с родительскими клетками (P < 0,01), но DCA вызвал значительное снижение гликолиза и увеличение окислительного фосфорилирования как в чувствительных к цисплатину, так и в цисплатин-устойчивых клетках HNC (P < 0,01) (рис. 3B). Мембранный потенциал митохондрий (ΔΨm) измеряли с помощью TMRM, чувствительного к напряжению митохондрий красителя на основе родамина, а митохондриальную ROS (mROS, супероксид митохондрий) измеряли с помощью MitoSOX red. В клетках HNC, обработанных DCA, наблюдалось снижение ΔΨm и увеличение mROS (P < 0,01) (рис. 3C). Изменение ΔΨm в цисплатин-устойчивых клетках HNC при воздействии DCA блокировалось предварительной обработкой NAC (рис. 3Д). Это говорит о том, что DCA может вызывать накопление ROS в клетках HNC путем активации окислительного фосфорилирования.
ДКА способствует остановке клеточного цикла и апоптозу в клетках HNC
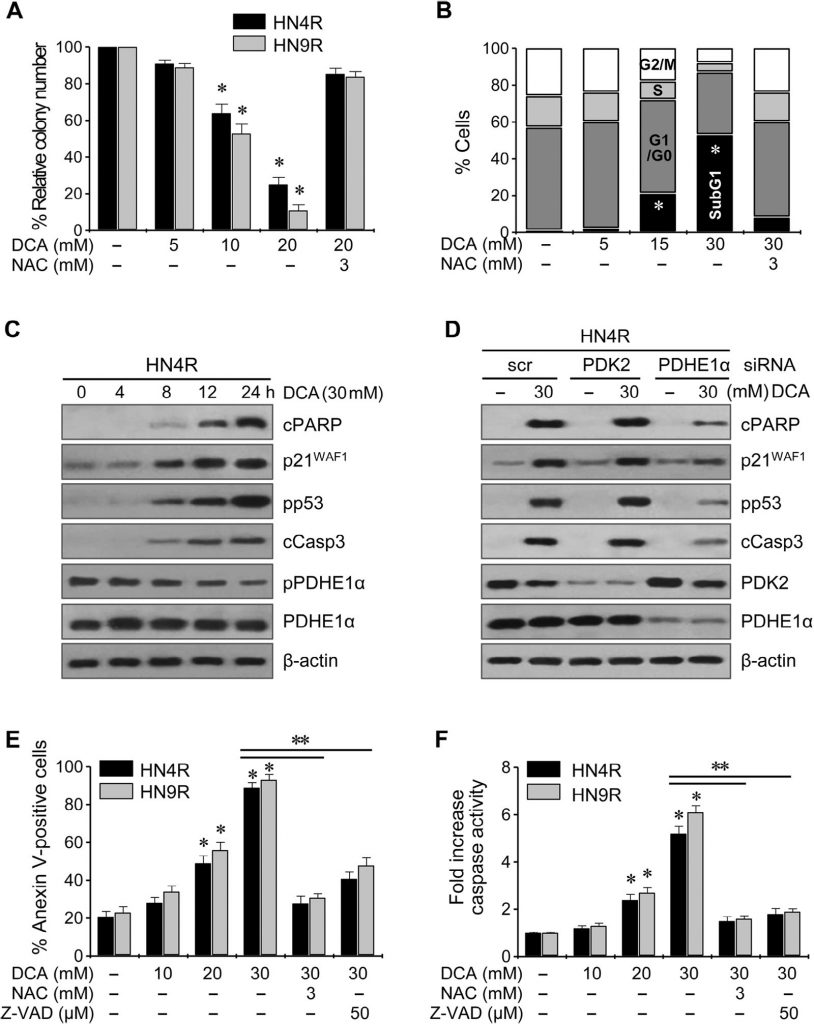
Клоногенный анализ привел к заметному снижению числа колоний HN4-cisR и HN9-cisR при воздействии ДКА (P < 0,01) (рис. 4A ). Проточная цитометрия с окрашиванием йодистым пропидием показала значительное изменение клеточного цикла в клетках HNC: вогонин увеличил популяцию апоптотических клеток sub-G1 (P < 0,05), и этот эффект блокировался при совместном воздействии с NAC (рис. 4B ). Вестерн-блот анализ показал, что DCA снижал pPDHE1α, но увеличивал cPARP, p21, фосфо-p53 и расщепленную каспазу-3 (cCasp3) в зависимости от времени (рис. 4C ). Активация PDHE1α путем нокдауна PDK2 в клетках AMC-HN4-cisR не привела к значительному увеличению проапоптотических белков cPARP и cCasp3 (рис. 4D ); однако нокдаун PDHE1α снизил экспрессию p21 и фосфорилирование p53. Проточная цитометрия с использованием йодистого пропидия и окрашивания аннексином-V показала эффективную индукцию апоптоза и гибели клеток в цисплатин-устойчивых клетках HNC под действием DCA; эффект снижался при совместном воздействии DCA и антиоксиданта NAC или пан-каспазного ингибитора Z-VAD-fmk (рис. 4E). Это было подтверждено измерением активности каспазы, которая значительно увеличивалась под действием ДКА в зависимости от концентрации (рис. 4F).
DCA сенсибилизирует цисплатин-устойчивые клетки HNC к цисплатину in vitro и in vivo
Цисплатин (10 мкМ) не вызывал значительной цитотоксичности или экспрессии апоптотических белков в цисплатин-устойчивых линиях HNC HN4-cisR и HN9-cisR по сравнению с родительскими чувствительными к цисплатину линиями HN4 и HN9 (рис.
5A); однако DCA вызывал заметное снижение выживаемости в цисплатин-устойчивых клетках HNC (P < 0,05), которое блокировалось предварительной обработкой NAC. DCA индуцировал экспрессию апоптотических белков и увеличивал цисплатин-индуцированную цитотоксичность и экспрессию апоптотических белков в клетках HN4-cisR (рис.
B). В комбинации DCA увеличивал цитотоксичность цисплатина в клетках HN4-cisR за счет повышения активности каспаз до степени, превышающей сумму эффектов каждого из агентов в отдельности (CI < 1, P < 0,01), которые ослаблялись при глушении гена PDHE1α (рис. 5C). Уровень ΔΨm был выше в клетках HN4-cisR, устойчивых к цисплатину, чем в чувствительных к цисплатину родительских клетках HN4 (ΔΨm, средняя интенсивность флуоресценции [MFI]: 1 ± 0 против 0,54 ± 0,09, P < 0,001) и снижался под действием DCA или комбинации цисплатина и DCA, что ослаблялось глушением гена PDHE1α (рис. 5D).
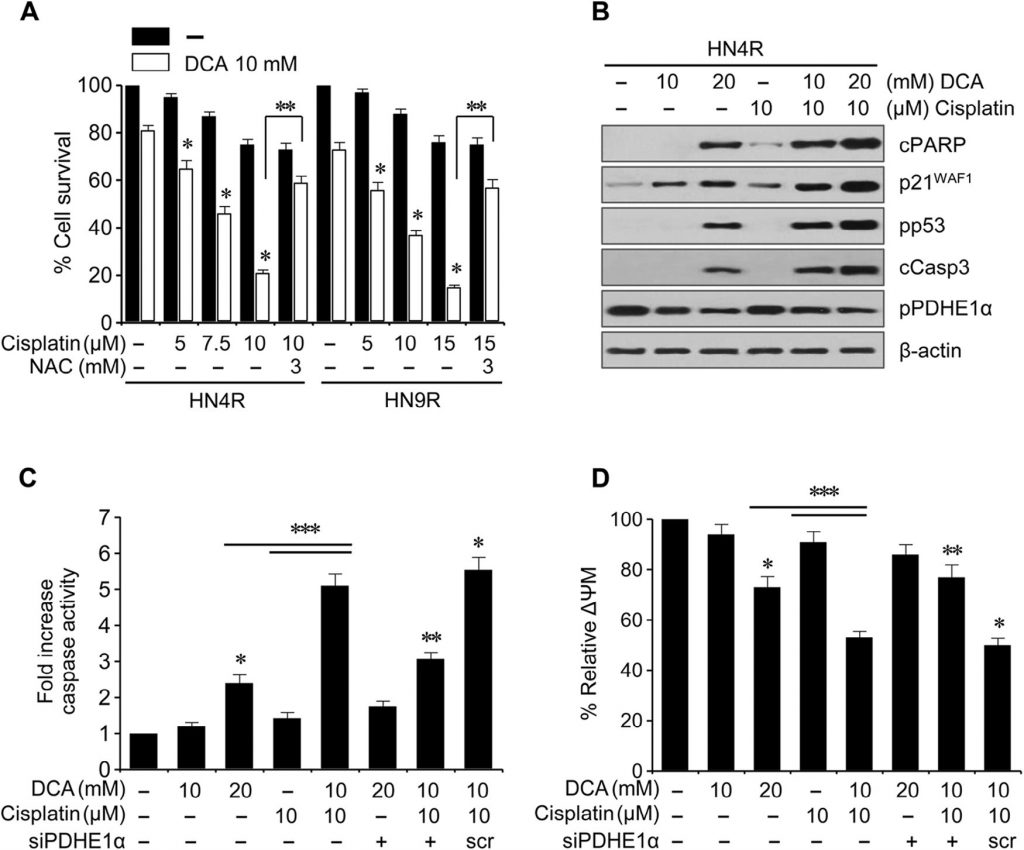
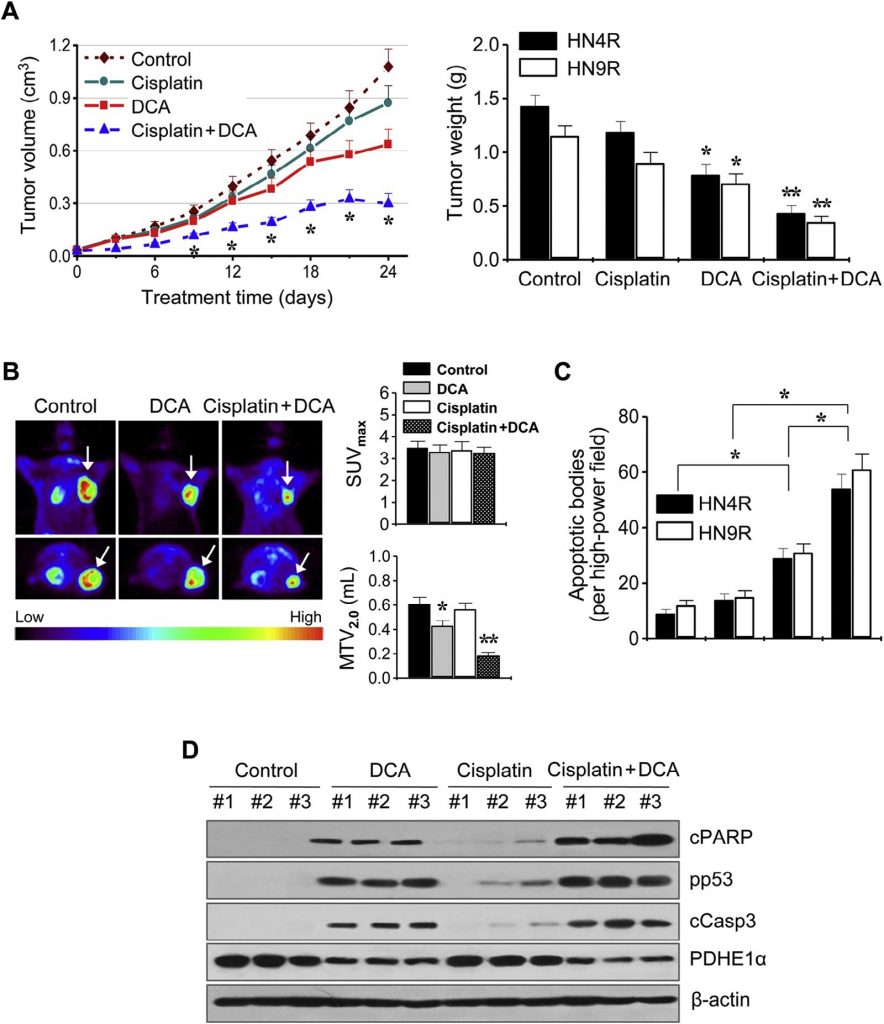
Эти результаты были далее изучены на мышиных моделях опухолевых ксенотрансплантатов in vivo. Атимичные мыши BALB/c, несущие опухоли AMC-HN4-cisR или HN9-cisR, получали DCA, цисплатин, DCA плюс цисплатин или транспортное средство. Комбинация цисплатина и DCA синергетически подавляла рост опухоли (рис. 6A). Визуализация роста опухоли in vivo проводилась с помощью ПЭТ с 18 F-ФДГ на 21 день лечения. Очаговые поглощения 18 F-ФДГ наблюдались в местах имплантации опухоли, где измерялось максимальное стандартизированное поглощение (SUVmax) и метаболический объем опухоли при SUV 2.0 (MTV2.0). SUVmax не отличался между группами лечения (P > 0,1), но MTV2.0 был значительно ниже в группах, получавших DCA и комбинацию, чем в других группах (P < 0,05) (рис. 6B). Анализ апоптоза in situ показал, что TUNEL-положительные апоптотические тельца чаще встречались в опухолях, получавших DCA и цисплатин плюс DCA, чем в опухолях, получавших транспортное средство (P < 0,01) (рис. 6C). Вестерн-блот анализ опухолевых тканей показал, что уровень апоптотических белков увеличился в большей степени в клетках HN4-cisR, обработанных комбинацией цисплатина и DCA, чем в опухолях, обработанных одним агентом (рис. 6D).
Обсуждение
Повышенный гликолиз, который часто встречается при раке, тесно связан с нарушением митохондрий или дефектным окислительным фосфорилированием и способствует терапевтической резистентности [18,19]. Аэробный гликолиз был связан с устойчивостью к химиотерапии [20] или радиотерапии [21]. В настоящем исследовании клеточные линии, устойчивые к цисплатину, показали повышенный гликолиз, что указывает на биохимическую связь между гликолизом и химиорезистентностью. Устойчивые к лекарствам клетки испытывают большую потребность в АТФ, чем нормальные клетки, для поддержания клеточного гомеостаза и активации путей выживания, которые позволяют избежать гибели клеток при генотоксическом стрессе [20]. Устойчивые раковые клетки увеличивают гликолиз для быстрой выработки АТФ, чтобы удовлетворить внутриклеточные потребности. Это было четко обнаружено в наших клетках, устойчивых к цисплатину, по сравнению с родительскими чувствительными клетками, что указывает на то, что лекарственная устойчивость в HNC напрямую связана с увеличением гликолиза. Это означает, что изменение биоэнергетики раковых клеток в сторону увеличения гликолиза связано с химиорезистентностью [14,22]. Таким образом, уровень гликолиза в раковых клетках человека может быть биомаркером химиорезистентности и требует клинического подтверждения в раковых тканях человека.
В настоящем исследовании показано, что DCA смещает генерацию энергии с гликолиза на митохондриальное окисление глюкозы в HNC. Это вызванное DCA изменение метаболического сдвига в сторону гликолиза устраняет пролиферативное преимущество раковых клеток и в конечном итоге приводит к их гибели [23]. DCA, являясь структурным аналогом пирувата, ингибирует PDK и реактивирует PDH, фермент, входящий в комплекс, который преобразует пируват в ацетил-КоА, основной субстрат цикла Кребса [10,23]. Поскольку большинство типов рака создают гипоксическую среду, раковые клетки полагаются на анаэробный гликолиз в качестве основного источника энергии. Активация гипоксия-индуцибельного фактора (HIF) индуцирует митохондриальную PDK [24]. Митохондриальная активность PDH в раке блокируется PDK, что приводит к снижению доступности ацетил-КоА для митохондриального окисления глюкозы [24]. Повышенная экспрессия PDK связана с лекарственной устойчивостью при раке [25,26]. Регуляция PDK2 в результате митохондриальных мутаций и стабилизации HIF1α также наблюдается в клетках HNC [27]. В настоящем исследовании также была подтверждена связь между сверхэкспрессией PDK2 и устойчивостью к цисплатину в клетках HNC. Эти данные указывают на важность PDK как новой молекулярной мишени в терапии рака [10]. Предыдущие данные показали, что генетическое или фармакологическое ингибирование PDK изменяет биоэнергетику рака и восстанавливает митохондрий-зависимый апоптоз в раковых клетках [11,13]. Поэтому ДКК эффективен для преодоления резистентности к лечению в раковых опухолях с агрессивным фенотипом in vitro и in vivo[12-14].
Активация PDH под действием DCA вызывает накопление mROS в раковых клетках. В раковых клетках снижен митохондриальный метаболизм глюкозы, что приводит к снижению активности электронно-транспортной цепи (ЭТЦ) и уменьшению mROS [10], [28]. Митохондриальное ремоделирование ΔΨm гиперполяризации и снижение выработки mROS в раковых клетках приводит к устойчивости к апоптозу при генотоксическом стрессе. В устойчивых раковых клетках с более гликолитическим фенотипом HK2 приводит к транслокации на внешнюю митохондриальную мембрану и увеличению ΔΨm [29]. Ингибирование гликолиза и транслокации HK2 снижает ΔΨm рака и отменяет устойчивость к апоптозу [29]. В настоящем исследовании повышенная экспрессия HK2 и PDK2 и мембранный потенциал митохондрий также были обнаружены в устойчивых к цисплатину раковых клетках. DCA заставляет пируват поступать в митохондрии через активацию PDH, регулятора митохондриального окисления глюкозы, снижает ΔΨm и увеличивает mROS [13]. В настоящем исследовании DCA просто отменил повышенное митохондриальное ремоделирование в клетках HNC, устойчивых к цисплатину, что способствовало гибели раковых клеток. Активация митохондриального окисления глюкозы под действием DCA вызвала mROS и активацию митохондриальной сигнализации, что привело к активации p53 и связанных с ним проапоптотических путей и к гибели устойчивых к химиотерапии раковых клеток. В нашем исследовании фармакологическое ингибирование выработки mROS или каспаза-опосредованного апоптоза ослабляло цитотоксический эффект DCA в клетках HNC, что подтверждает известные механизмы действия препарата.
Доклинические и клинические исследования поддерживают применение ДКА у онкологических больных и у пациентов с молочнокислым ацидозом, связанным с митохондриальными заболеваниями [10,23]. В ряде исследований продемонстрировано цитотоксическое действие DCA, отдельно или в комбинации с другими препаратами, на различные опухоли, полученные из всех трех зародышевых слоев [23]. Предыдущее исследование показало, что глиобластома, один из самых агрессивных видов рака человека, сверхэкспрессирует PDK2 в раковых тканях, взятых у 49 пациентов, и регрессирует у пяти пациентов после лечения ДКА, что доказывает клиническую пользу ДКА в резистентных раковых опухолях [13]. В недавнем исследовании, посвященном HNC, сравнивался эффект DCA на трех клеточных линиях плоскоклеточной карциномы полости рта (OSCC) [15]. Клетки OSCC с дефицитом митохондриального окислительного фосфорилирования (т.е. высоким гликолизом) были более чувствительны к DCA, чем другие [15,30]. Настоящее исследование подтвердило, что клетки HNC с высокими биоэнергетическими изменениями были более чувствительны к DCA. Фактически, поскольку химиорезистентные раковые клетки имеют тенденцию к высокому гликолизу, эти клетки могут быть мишенью для ингибиторов метаболизма [31]. Таким образом, DCA является хорошим кандидатом для лечения раковых клеток с агрессивным фенотипом, включая химиорезистентные клетки HNC. Однако, поскольку потенциальные противораковые эффекты DCA все еще спорны, особенно в опухолях в условиях гипоксии, необходимы дальнейшие доклинические и клинические исследования DCA и рака [32]. Недавний систематический обзор показал, что DCA синергирует со многими стандартными противораковыми препаратами, а клинические испытания на ранней стадии свидетельствуют о том, что хронический DCA безопасен и хорошо переносится при пероральном приеме 12,5 мг/кг дважды в день [23].
Наше исследование показало, что DCA синергирует с цисплатином и таким образом преодолевает устойчивость к цисплатину в клетках HNC. Поскольку цисплатин является химиотерапевтическим препаратом первой линии при НЯК, комбинация цисплатина и DCA может быть эффективной в клинических условиях для снижения токсичности и преодоления устойчивости к раковым препаратам. Предыдущие отчеты показали, что DCA потенцирует цитотоксический эффект цисплатина, вызывая митохондрий-зависимый апоптоз [33,34]. Соединение двух молекул DCA с цисплатином, называемое митаплатином, вызывает селективную гибель раковых клеток [35,36]. Эти результаты свидетельствуют о том, что DCA может иметь дополнительные преимущества в сочетании с существующими противораковыми методами лечения. В нашем исследовании DCA и его комбинация с цисплатином не влияли на SUVmax, но значительно снижали MTV2.0. Это означает, что DCA эффективно подавляет рост опухоли in vivo, несмотря на отсутствие значительного изменения максимального поглощения глюкозы в локализованных опухолью ROI. В настоящем исследовании впервые показано, что DCA восстанавливает цитотоксический эффект цисплатина в лекарственно-устойчивых клетках HNC in vitro и in vivo. DCA вызвал значительное увеличение апоптоза, опосредованного цисплатином, через активацию p53, PARP и каспазы в цисплатин-резистентных клетках HNC. DCA сенсибилизировал устойчивые к цисплатину клетки НЯК, что привело к повышению цитотоксичности и более эффективной терапии агрессивного НЯК. В совокупности эти результаты могут иметь огромное клиническое значение: вызывая гибель резистентных клеток с помощью DCA, можно снизить дозу цисплатина, необходимую в клинических условиях, и тем самым минимизировать потенциальные побочные эффекты цисплатиновой химиотерапии.
В заключение, наши данные свидетельствуют о том, что высокий уровень гликолиза и сверхэкспрессия PDK тесно связаны с устойчивостью к цисплатину в клетках HNC. DCA переключает биоэнергетику клеток НЯК на митохондриальное окислительное фосфорилирование. Это приводит к снижению ΔΨm и увеличению mROS, тем самым сенсибилизируя химиорезистентные клетки НЯК к цисплатину in vitro и in vivo. Эти данные дают основания для дальнейших доклинических и клинических исследований DCA, перспективного противоракового препарата, при НЯК с агрессивным фенотипом. Однако наше заключение следует принимать во внимание с осторожностью при противоположном сценарии. Дефектное митохондриальное окислительное фосфорилирование было связано с чувствительностью раковых клеток к состоянию низкой глюкозы, что может быть использовано в качестве биомаркера для терапии рака [37]. Раковая клетка также приспособлена для обеспечения пролиферации путем поглощения и включения питательных веществ в биомассу, а не для эффективного производства АТФ [38]. Поэтому доклинические и клинические эффекты DCA на HNC и другие виды рака должны быть изучены далее.
Благодарности
Это исследование было поддержано грантом (2015R1A2A1A15054540) Программы фундаментальных научных исследований Национального исследовательского фонда Кореи (NRF), Министерства науки, ИКТ и планирования будущего, и грантом (HI14C23050000) Корейского проекта исследований и разработок в области технологий здравоохранения, Министерства здравоохранения и социального обеспечения, Сеул, Республика Корея (J.-L. Roh).
Конфликт интересов
Авторы заявляют об отсутствии конфликтов интересов.
ССЫЛКИ
1 А. Джемаль, Ф. Брей, М.М. Центр, Ж. Ферлей, Э. Уорд, Д. Форман, Глобальная статистика рака, CA Cancer J. Clin, Vol. 61, 2011, 69-90
2 Р.И. Хаддад, Д.М. Шин, Последние достижения в области рака головы и шеи, N. Engl. J. Med, Vol. 359, 2008, 1143-1154
3 E.B. Lamont, E.E. Vokes, Химиотерапия в лечении плоскоклеточной карциномы головы и шеи, Lancet Oncol, Vol. 2, 2001, 261-269
4 F. Petrelli, A. Coinu, V. Riboldi, K. Borgonovo, M. Ghilardi, M. Cabiddu, Concomitant platinum-based chemotherapy or cetuximab with radiotherapy for locally advanced head and neck cancer: a systematic review and meta-analysis of published studies, Oral Oncol, Vol. 50, 2014, 1041-1048
5 Д. Ханахан, Р.А. Вайнберг, Основные признаки рака: следующее поколение, Cell, Vol. 144, 2011, 646-674
6 J.R. Cantor, D.M. Sabatini, Метаболизм раковых клеток: одна отличительная черта, много лиц, Cancer Discov, Vol. 2, 2012, 881-898
7 Y. Zhao, E.B. Butler, M. Tan, Targeting cellular metabolism to improve cancer therapeutics, Cell Death Dis, Vol. 4, 2013, e532
8 R.A. Cairns, I.S. Harris, T.W. Mak, Regulation of cancer cell metabolism, Nat. Rev. Cancer, Vol. 11, 2011, 85-95
9 R.H. Xu, H. Pelicano, Y. Zhou, J.S. Carew, L. Feng, K.N. Bhalla, Inhibition of glycolysis in cancer cells: a novel strategy to overcome drug resistance associated with mitochondrial respiratory defect and hypoxia, Cancer Res, Vol. 65, 2005, 613-621
10 G. Sutendra, E.D. Michelakis, Pyruvate dehydrogenase kinase as a novel therapeutic target in oncology, Front. Oncol, Vol. 3, 2013, 38
11 G. Sutendra, P. Dromparis, A. Kinnaird, T.H. Stenson, A. Haromy, J.M. Parker, Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer, Oncogene, Vol. 32, 2013, 1638-1650
12 R.C. Sun, M. Fadia, J.E. Dahlstrom, C.R. Parish, P.G. Board, A.C. Blackburn, Reversal of the glycolytic phenotype by dichloroacetate inhibits metastatic breast cancer cell growth in vitro and in vivo, Breast Cancer Res. Treat, Vol. 120, 2010, 253-260
13 E.D. Michelakis, G. Sutendra, P. Dromparis, L. Webster, A. Haromy, E. Niven, Metabolic modulation of glioblastoma with dichloroacetate, Sci. Transl. Med, Vol. 2, 2010, 31ra34
14 Y.C. Shen, D.L. Ou, C. Hsu, K.L. Lin, C.Y. Chang, C.Y. Lin, Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma, Br. J. Cancer, Vol. 108, 2013, 72-81
15 V. Ruggieri, F. Agriesti, R. Scrima, I. Laurenzana, D. Perrone, T. Tataranni, Dichloroacetate, a selective mitochondria-targeting drug for oral squamous cell carcinoma: a metabolic perspective of treatment, Oncotarget, Vol. 6, 2015, 1217-1230
16 M. Nakamura, K. Nakatani, K. Uzawa, K. Ono, H. Uesugi, K. Ogawara, Establishment and characterization of a cisplatin-resistant oral squamous cell carcinoma cell line, H-1R, Oncol. Rep, Vol. 14, 2005, 1281-1286
17 T.C. Chou, Исследования комбинации лекарств и количественная оценка их синергизма с помощью метода Chou-Talalay, Cancer Res, Vol. 70, 2010, 440-446
18 Н. Гуарагнелла, С. Джаннаттасио, Л. Моро, Митохондриальная дисфункция в химиорезистентности рака, Биохим. Pharmacol, Vol. 92, 2014, 62-72
19 S. Ganapathy-Kanniappan, J.F. Geschwind, Tumor glycolysis as a target for cancer therapy: progress and prospects, Mol. Cancer, Vol. 12, 2013, 152
20 Y. Zhou, F. Tozzi, J. Chen, F. Fan, L. Xia, J. Wang, Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells, Cancer Res, Vol. 72, 2012, 304-314
21 S.P. Pitroda, B.T. Wakim, R.F. Sood, M.G. Beveridge, M.A. Beckett, D.M. MacDermed, STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect, BMC Med, Vol. 7, 2009, 68
22 B. Bhattacharya, S.H. Low, C. Soh, N. Kamal Mustapa, M. Beloueche-Babari, K.X. Koh, Increased drug resistance is associated with reduced glucose levels and an enhanced glycolysis phenotype, Br. J. Pharmacol, Vol. 171, 2014, 3255-3267
23 S. Kankotia, P.W. Stacpoole, Biochim. Biophys. Acta, Vol. 1846, 2014, 617-629
24 J.W. Kim, I. Tchernyshyov, G.L. Semenza, C.V. Dang, HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia, Cell Metab, Vol. 3, 2006, 177-185
25 Y. Sun, A. Daemen, G. Hatzivassiliou, D. Arnott, C. Wilson, G. Zhuang, Metabolic and transcriptional profiling reveals pyruvate dehydrogenase kinase 4 as a mediator of epithelial-mesenchymal transition and drug resistance in tumor cells, Cancer Metab, Vol. 2, 2014, 20
26 C.W. Lu, S.C. Lin, C.W. Chien, S.C. Lin, C.T. Lee, B.W. Lin, Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer, Am. J. Pathol, Vol. 179, 2011, 1405-1414
27 W. Sun, S. Zhou, S.S. Chang, T. McFate, A. Verma, J.A. Califano, Mitochondrial mutations contrib to HIF1alpha accumulation through increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma, Clin. Cancer Res, Vol. 15, 2009, 476-484
28 S. Bonnet, S.L. Archer, J. Allalunis-Turner, A. Haromy, C. Beaulieu, R. Thompson, Ось каналов подавляется при раке, и ее нормализация способствует апоптозу и подавляет рост рака, Cancer Cell, Vol. 11, 2007, 37-51
29 J.G. Pastorino, J.B. Hoek, N. Shulga, Активация гликоген-синтазы киназы 3бета нарушает связывание гексокиназы II с митохондриями путем фосфорилирования вольтаж-зависимого анионного канала и потенцирует цитотоксичность, вызванную химиотерапией, Cancer Res, Vol. 65, 2005, 10545-10554
30 L.H. Stockwin, S.X. Yu, S. Borgel, C. Hancock, T.L. Wolfe, L.R. Phillips, Sodium dichloroacetate selectively target cells with defects in the mitochondrial ETC, Int. J. Cancer, Vol. 127, 2010, 2510-2519
31 E.J. Sullivan, M. Kurtoglu, R. Brenneman, H. Liu, T.J. Lampidis, Targeting cisplatin-resistant human tumor cells with metabolic inhibitors, Cancer Chemother. Pharmacol, Vol. 73, 2014, 417-427
32 S. Shahrzad, K. Lacombe, U. Adamcic, K. Minhas, B.L. Coomber, Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia, Cancer Lett, Vol. 297, 2010, 75-83
33 Д. Хеше, С. Хугестраат, К. Бракманн, У. Карст, Я. Боос, К. Ланверс-Камински, Метаболически направленная терапия дихлорацетатом побеждает цитотоксичность стандартных противораковых препаратов, Cancer Chemother. Pharmacol, Vol. 67, 2011, 647-655
34 J. Xie, B.S. Wang, D.H. Yu, Q. Lu, J. Ma, H. Qi, Дихлорацетат смещает метаболизм с гликолиза на окисление глюкозы и демонстрирует синергетическое ингибирование роста с цисплатином в клетках HeLa, Int. J. Oncol, Vol. 38, 2011, 409-417
35 S. Dhar, S.J. Lippard, Mitaplatin, мощное соединение цисплатина и сиротского препарата дихлорацетата, Proc. Natl. Acad. Sci. U.S.A., Vol. 106, 2009, 22199-22204
36 X. Xue, S. You, Q. Zhang, Y. Wu, G.Z. Zou, P.C. Wang, Митаплатин повышает чувствительность опухолевых клеток к цисплатину, вызывая дисфункцию митохондрий, Mol. Pharm, Vol. 9, 2012, 634-644
37 K. Birsoy, R. Possemato, F.K. Lorbeer, E.C. Bayraktar, P. Thiru, B. Yucel, Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides, Nature, Vol. 508, 2014, 108-112
38 M.G. Vander Heiden, L.C. Cantley, C.B. Thompson, Understanding the Warburg effect: the metabolic requirements of cell
Связанный контент: