Санг Хёк Ву1,*, Сунг Кым Со1,*, Юнхва Пак1,2, Ын-Кю Ким3, Мин-Ки Сон4, Хён-Ах Ким4, Джи-Ёнг Сонг1, Санг-Гу Хванг1, Джин Кён Ли5, Ву Чул Но4, Ин-Чул Парк1
1Отделениерадиационных исследований рака, Корейский институт радиологических и медицинских наук, Новон-гу, Сеул, 01812, Республика Корея
2Школа наук о жизни и биотехнологий, Корейский университет, Сонбук-гу, Сеул, 02841, Республика Корея
3Отделение хирургии, Центр рака молочной железы, Больница Бунданг Сеульского национального университета, Медицинский колледж Сеульского национального университета, Bundang-gu, Seongnam, 13620, Republic of Korea
4Отделение хирургии, Больница Корейского онкологического центра, Корейский институт радиологических и медицинских наук, Nowon-gu, Seoul, 01812, Republic of Korea
5Кирамский радиационный биобанк, Корейский институт радиологических и медицинских наук, Nowon-gu, Seoul, 01812, Republic of Korea
авторы внесли равный вклад в эту работу
Переписка с: Ин-Чул Парк, e-mail: [email protected]
Ключевые слова: тамоксифен, рак молочной железы, дихлорацетат, рецептор эпидермального фактора роста, киназа пируватдегидрогеназы
Получено: 18 мая 2016 г.
Принято: 22 июля 2016 г.
Опубликовано онлайн: 01 августа 2016 г
Аннотация
Метаболическое перепрограммирование в раковых клетках недавно было признано важнейшей отличительной чертой неоплазии. В этом контексте метаболические изменения представляют собой привлекательную терапевтическую мишень, и в доклинических исследованиях были получены обнадеживающие результаты применения препаратов, направленных на различные метаболические процессы. Недавно несколько исследований показали, что дихлорацетат (ДХА), специфический ингибитор киназы пируватдегидрогеназы, может быть потенциальным противораковым препаратом при большом количестве разнообразных опухолей. Однако точный механизм его действия до конца не изучен, что важно для использования ДКА в лечении рака. В настоящем исследовании мы обнаружили, что DCA сенсибилизировал клетки рака молочной железы MCF7 к клеточной гибели, вызванной тамоксифеном, путем снижения экспрессии рецептора эпидермального фактора роста (EGFR). Снижение экспрессии EGFR было вызвано деградацией белка. Более того, митоген-активированная протеинкиназа p38 играла важную роль в деградации EGFR, вызванной DCA/тамоксифеном. Наконец, DCA также способствовал сравнимой с тамоксифен-индуцированной гибели клеток в устойчивых к тамоксифену клетках MCF7, которые были созданы в результате длительного лечения тамоксифеном. В целом, наши результаты свидетельствуют о том, что DCA является привлекательным потенциальным препаратом, который сенсибилизирует клетки к клеточной гибели, вызванной тамоксифеном, и преодолевает резистентность к тамоксифену посредством даунрегуляции экспрессии EGFR в клетках рака молочной железы.
ВВЕДЕНИЕ
Пролиферирующие раковые клетки имеют значительно отличающиеся метаболические требования по сравнению с большинством нормальных дифференцированных клеток. Например, для поддержания быстрого клеточного роста и пролиферации раковые клетки дифференцированно изменяют метаболические потоки по сравнению с окружающей тканью, чтобы обеспечить достаточное количество биоэнергетики и биосинтетических промежуточных продуктов. Хорошо известное явление, наблюдаемое в большинстве раковых клеток, — это переход к аэробному гликолизу, независимо от поступления кислорода, что называется «эффектом Варбурга», при котором пируват напрямую превращается в молочную кислоту вместо того, чтобы вступить в цикл лимонной кислоты [1]. Поскольку все раковые клетки зависят от такого изменения метаболизма, эти измененные пути представляют собой привлекательные терапевтические мишени [2]. Были предприняты усилия по воздействию на перепрограммированный метаболизм отдельно или в сочетании с химиотерапией рака как в доклинических, так и в клинических исследованиях [3]. Интересно, что это специфическое для рака метаболическое ремоделирование обращается вспять под действием дихлорацетата (ДХА), малой молекулы, нацеленной на митохондрии, которая может проникать в большинство тканей после перорального приема [4]. Он специфически ингибирует киназу пируватдегидрогеназы (PDK), члена семейства киназ, что приводит к реактивации пируватдегидрогеназы (PDH), ключевого фермента, который смещает поток пирувата в митохондрии для продвижения окисления глюкозы вместо гликолиза [4]. Хотя DCA недавно был оценен в нескольких доклинических испытаниях против рака [5], реакция раковых клеток на лечение DCA, которая определяет, принесет ли DCA клиническую пользу при лечении рака, до конца не выяснена.
Более 70% раковых опухолей молочной железы экспрессируют рецептор эстрогена (ER) и зависят от эстрогена для стимулирования роста и прогрессии опухоли [6]. Таким образом, эндокринная терапия должна рассматриваться как дополнение к хирургическому вмешательству у большинства пациентов, поскольку она вызывает ремиссию опухоли и обеспечивает постоянный клинический эффект. Антиэстрогенный препарат тамоксифен является наиболее часто используемым средством лечения больных с ER-положительным раком молочной железы как на ранних, так и на поздних/метастатических стадиях [7]. В качестве адъювантной терапии при раннем раке молочной железы тамоксифен улучшает общую выживаемость, и считается, что его широкое применение внесло значительный вклад в снижение смертности от рака молочной железы, наблюдаемое в последнее десятилетие [8]. Несмотря на очевидные преимущества лечения тамоксифеном при раке молочной железы, почти все пациентки с метастатической болезнью и до 25% пациенток, получающих адъювантный тамоксифен, в конечном итоге рецидивируют и умирают от болезни [9, 10]. Поэтому биологические механизмы, лежащие в основе внутренней (de novo) и приобретенной резистентности к тамоксифену, имеют важное клиническое значение. Лучшее понимание этих механизмов может предложить новые стратегии преодоления резистентности к тамоксифену и улучшить лечение рака молочной железы.
В настоящем исследовании мы продемонстрировали, что экспрессия EGFR в клетках рака молочной железы снижается при лечении ДКА. Комбинация DCA и тамоксифена еще больше снизила уровень EGFR. Мы показали, что DCA усиливает цитотоксичность тамоксифена для клеток рака молочной железы за счет подавления экспрессии EGFR. Кроме того, DCA повышал чувствительность устойчивых к тамоксифену клеток MCF7 к тамоксифену за счет снижения уровня EFGR. Эти результаты свидетельствуют о потенциальном использовании DCA в лечении рака молочной железы путем ослабления сигнального пути EGFR.
Результаты
Ингибирование PDK снижает уровень EGFR и усиливает клеточную гибель, вызванную тамоксифеном, в клетках рака молочной железы
Недавние исследования показали, что воздействие на PDK с помощью ингибитора PDK, такого как DCA, сдвигает метаболизм раковых клеток с гликолиза на окислительное фосфорилирование путем дефосфорилирования митохондриальной пируватдегидрогеназы [4, 11]. Чтобы выяснить, как сигнальные пути факторов роста и киназ в сочетании с PDK регулируют эффект Варбурга при раке молочной железы, мы изучили уровни экспрессии нескольких рецепторов факторов роста в клетках MCF7 с нокдауном PDK. Интересно, что истощение PDK путем обработки siRNA привело к снижению EGFR, в отличие от отсутствия или незначительного влияния на другие рецепторы факторов роста (рис. 1A). Четыре изофермента PDK (PDK1, PDK2, PDK3, PDK4) были идентифицированы в тканях млекопитающих [12]. Чтобы подтвердить даунрегуляцию EGFR при обработке siPDK, мы исследовали экспрессию EGFR в клетках, обработанных siRNA против каждой изоформы PDK. Каждая siRNA отменяла только экспрессию целевой PDK, однако все они вызывали даунрегуляцию EGFR, что говорит о том, что PDK может не регулировать экспрессию EGFR изоформно-специфическим образом (рис. 1B). В соответствии с этими результатами, DCA также снижал уровень EGFR в дозозависимой манере (Рисунок 1С). Когда мы проанализировали концентрацию лактата в культуральной среде, не было обнаружено значительного изменения концентрации лактата при лечении тамоксифеном/DCA или siRNA против EGFR, даже если она снижалась при лечении DCA (Дополнительный рисунок S1). Таким образом, мы предполагаем, что даунрегуляция EGFR может быть не связана с метаболическим сдвигом в клетках рака молочной железы, вызванным DCA. Поскольку активация сигнального пути EGFR способствует резистентности к тамоксифену [13], мы проверили, сенсибилизирует ли клетки к тамоксифену снижение уровня EGFR путем ингибирования PDK. Как показано на рисунке 1D, совместная обработка тамоксифеном и DCA привела к заметному снижению жизнеспособности клеток. Совместная обработка в течение 72 ч снизила жизнеспособность клеток до менее чем 30% от контрольной (рис. 1E). Затем оценивали гибель клеток в клетках, подвергнутых совместной обработке, с помощью окрашивания Annexin V/PI. Через 48 часов после лечения комбинация тамоксифена и DCA вызвала гибель 30% клеток, по сравнению с 15% или 8% при использовании только тамоксифена или DCA, соответственно (рис. 1F). Ранее мы сообщали, что апоптотическая гибель клеток рака молочной железы вызвана потерей мембранного потенциала митохондрий (ММП) [14]. Таким образом, мы проверили, участвует ли потеря ММП в гибели клеток, вызванной совместным лечением, однако не было обнаружено значительных изменений ММП между необработанными и обработанными тамоксифеном/DCA клетками (Дополнительный рисунок S2).
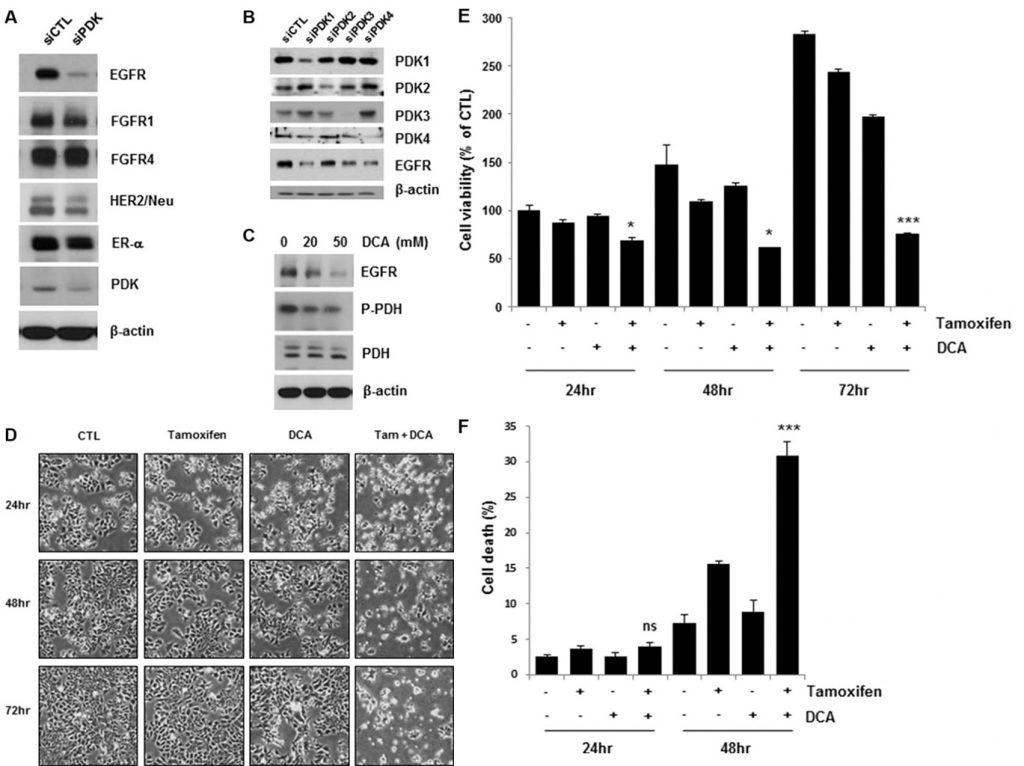
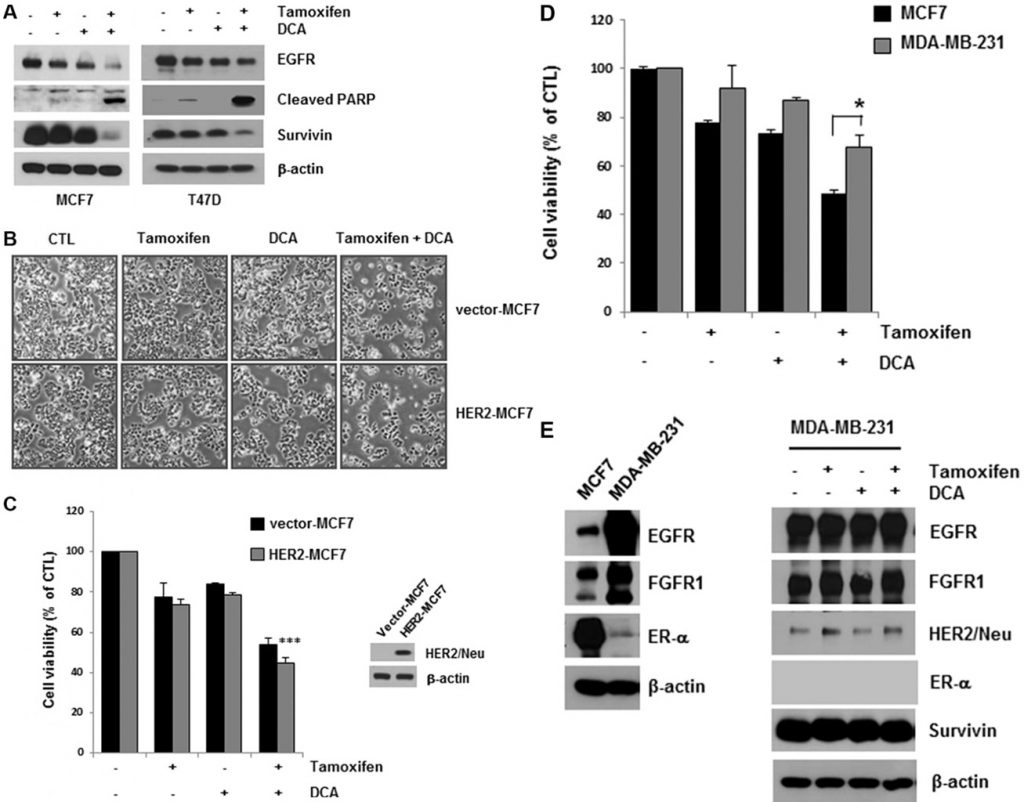
ДКА плюс тамоксифен еще больше снижали уровень EGFR в клетках MCF7 и T47D по сравнению с одним ДКА (рис. 2А). Гибель клеток, вызванная совместным лечением, была подтверждена путем обнаружения расщепления PARP, маркера апоптоза (рис. 2А). Сурвивин является антиапоптотической молекулой, а также мишенью ER [15]. Совместное лечение также снижало уровень сурвивина, который может опосредовать апоптоз в клетках (Рисунок 2А). Хотя лечение тамоксифеном несколько снизило уровень EGFR в клетках MCF7 и T47D, значительного увеличения гибели клеток в них не наблюдалось, что говорит о том, что для выживания клеток рака молочной железы необходим критический уровень EGFR (Рисунок 2А).
Данные, полученные на клеточных линиях, показали, что сверхэкспрессия HER2 может способствовать приобретенной резистентности к эндокринной терапии [13]. Чтобы определить, влияет ли сверхэкспрессия HER2 на цитотоксичность тамоксифена и DCA, мы исследовали жизнеспособность клеток MCF7 (HER2-MCF7), сверхэкспрессирующих HER2, после лечения тамоксифеном и DCA. Результаты показали, что тамоксифен и DCA значительно снизили жизнеспособность клеток даже в клетках HER2-MCF7 (рис. 2B и 2C), что говорит о том, что DCA может усилить индуцированную тамоксифеном гибель клеток в клетках рака молочной железы, экспрессирующих HER2. Далее мы оценили ингибирующий рост эффект совместного лечения на клеточной линии трижды-отрицательного рака молочной железы MDA-MB-231. Как показано на рисунке 2D, клетки MDA-MB-231 были менее чувствительны к тамоксифену и DCA, чем клетки MCF7. Поскольку даунрегуляция EGFR наблюдалась в ER-положительных клетках, мы изучили влияние тамоксифена и DCA на уровень EGFR в клетках MDA-MB-231. EGFR был высоко экспрессирован в клетках MDA-MB-231 по сравнению с клетками MCF7, и его уровень не был значительно снижен тамоксифеном и DCA (Рисунок 2E). Далее мы изучили цитотоксичность тамоксифена и DCA в неопухолевых иммортализованных эпителиальных клетках молочной железы линии MCF10A. Интересно, что экспрессия EGFR в клетках MCF10A была сопоставима с таковой в клетках MDA-MB-231, и после обработки тамоксифеном и DCA в клетках MCF10A не наблюдалось ни даунрегуляции EGFR, ни гибели клеток (Дополнительный рисунок S3). Эти результаты показывают, что антипролиферативный эффект тамоксифена и DCA в клетках рака молочной железы зависит от даунрегуляции EGFR.
Совместное лечение тамоксифеном и DCA вызывает p38 MAPK-опосредованную деградацию EGFR
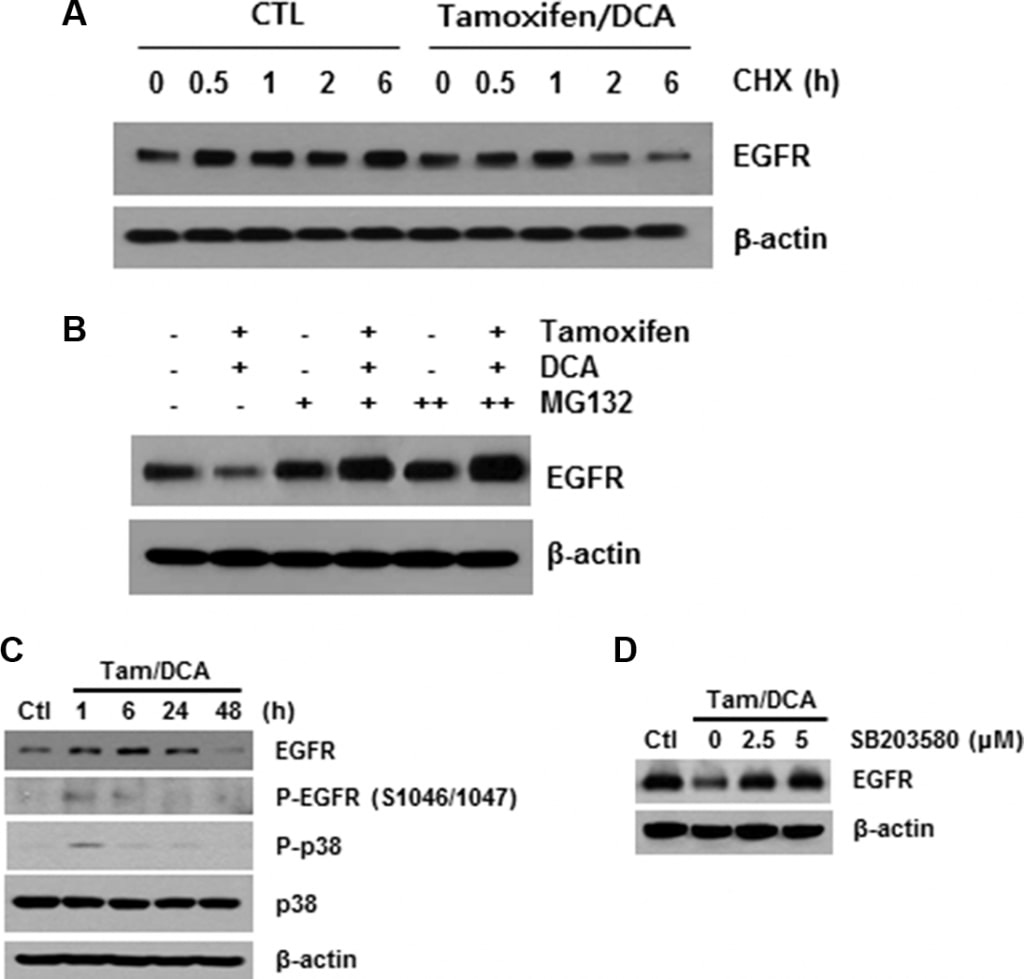
Как описано выше, связывание лиганда вызывает быстрое автофосфорилирование, что приводит к удалению EGFR с клеточной поверхности путем эндоцитоза в ранний эндосомальный компартмент [16]. Поэтому далее мы исследовали роль модификации рецептора в тамоксифен/DCA-опосредованной даунрегуляции EGFR. После блокирования синтеза белка с помощью циклогексимида мы обнаружили, что стабильность EGFR была значительно нарушена в клетках, обработанных тамоксифеном/DCA, по сравнению с контролем (Рисунок 3A). Затем мы оценили влияние MG132, ингибитора протеасомы, на деградацию EGFR, вызванную тамоксифеном/DCA. Лечение MG132 восстановило экспрессию EGFR в клетках, обработанных тамоксифеном/DCA, дозозависимым образом (Рисунок 3B).
Фосфорилирование EGFR по остаткам серина и треонина представляет собой механизм ослабления активности EGFR, и среди них участки фосфорилирования серина 1046/1047 (Ser 1046/7) необходимы для десенситизации EGFR [17]. Недавно стало известно, что p38 митоген-активируемая протеинкиназа (MAPK) индуцирует фосфорилирование EGFR на Ser 1046/7, что приводит к его деградации в раковых клетках [18]. Поэтому мы изучили влияние тамоксифена и DCA на фосфорилирование p38 MAPK в клетках MCF7. p38 MAPK был значительно фосфорилирован в течение 1 ч, и фосфорилирование сохранялось в течение 24 ч после обработки тамоксифеном и DCA (рис. 3C). Более того, деградация EGFR, вызванная совместным лечением, значительно подавлялась, когда клетки предварительно обрабатывались специфическим ингибитором p38 MAPK, SB203580 (Рисунок 3D), что указывает на то, что активация p38 MAPK играет роль в вызванной тамоксифеном/DCA даунрегуляции EGFR в клетках MCF7.
Ингибиторы EGFR усиливают клеточную гибель, вызванную тамоксифеном
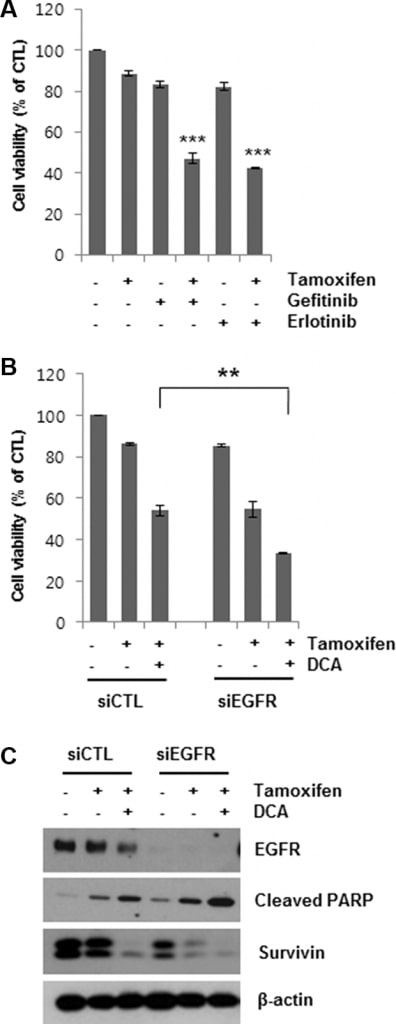
Показав, что DCA-опосредованная деградация EGFR может усиливать клеточную гибель, вызванную тамоксифеном, в клетках MCF7, мы затем определили, усиливает ли ингибирование EGFR клеточную гибель в клетках, обработанных тамоксифеном. Гефитиниб и эрлотиниб являются селективными обратимыми ингибиторами связывания тирозинкиназы EGFR с АТФ [19, 20]. Совместная обработка клеток MCF7 5 мкМ гефитиниба или эрлотиниба в течение 48 ч заметно увеличивала гибель клеток, вызванную тамоксифеном (Рисунок 4A). Аналогично, нокдаун EGFR с помощью siEGFR усиливал клеточную гибель, вызванную тамоксифеном (Рисунок 4B). Снижение жизнеспособности клеток при комбинированном лечении siEGFR и тамоксифена было сравнимо с таковым при лечении DCA и тамоксифена. Более того, лечение siEGFR еще больше снизило уровень EGFR в клетках, обработанных тамоксифеном/DCA, что привело к увеличению апоптотической гибели клеток наряду с понижением уровня сурвивина по сравнению с si-контролем (Рисунок 4C). Эти результаты подтверждают вывод о том, что DCA сенсибилизирует клетки рака молочной железы к тамоксифену через снижение уровня EGFR.
экспрессия c-myc и Nanog связана с цитотоксическим действием DCA в клетках MCF7, обработанных тамоксифеном
Активация EGFR в раке, как было показано, увеличивает различные транскрипционные факторы, которые могут влиять на тип и продолжительность EGFR-сигнализации [21]. Среди них Nanog и c-myc играют плейотропную роль в опухолевом генезе, включая устойчивость к стандартной терапии при карциноме молочной железы [22, 23]. Для дальнейшего изучения противоопухолевой активности, опосредованной даунрегуляцией EGFR в клетках рака молочной железы, мы исследовали экспрессию c-myc и Nanog в клетках MCF7 после котерапии тамоксифеном и DCA. Уровни c-myc и Nanog были значительно снижены в клетках, подвергнутых котерапии тамоксифеном и DCA (рис. 5A). Аналогично, экспрессия обоих белков была подавлена тамоксифеном в комбинации с гефитинибом или эрлотинибом (Рисунок 5B), что указывает на то, что экспрессия EGFR необходима для поддержания экспрессии c-myc и Nanog в клетках MCF7, обработанных тамоксифеном. Тем не менее, siEGFR практически не влиял на экспрессию c-myc и Nanog, что указывает на то, что даунрегуляция EGFR необходима, но недостаточна для снижения экспрессии этих белков (Дополнительный рисунок S4). Чтобы определить, вовлечена ли сниженная экспрессия этих двух белков в цитотоксическое действие тамоксифена и DCA в клетках MCF7, мы проверили действие siRNAs против c-myc и Nanog в клетках, подвергшихся воздействию тамоксифена/DCA. Нокдаун c-myc и Nanog значительно повышал чувствительность клеток к тамоксифену/DCA по сравнению с контролем (Рисунок 5C). И наоборот, сверхэкспрессия FLAG-c-myc и FLAG-Nanog путем трансфекции клеток векторами FLAG-c-myc и FLAG-Nanog значительно защищала клетки от цитотоксичности, вызванной тамоксифеном/DCA (Рисунок 5D). В целом, наши данные свидетельствуют о том, что снижение уровня EGFR комбинацией тамоксифена и DCA может вызывать гибель клеток MCF7 частично через ингибирование экспрессии c-myc и Nanog. Известно, что гены самообновления, такие как c-myc и Nanog, связаны со свойствами раковых стволовых клеток 24. Чтобы проверить, подавляет ли совместное лечение тамоксифеном и DCA стволоподобные клетки рака молочной железы, мы провели анализ проточной цитометрии для оценки доли субпопуляции стволоподобных клеток в клетках MCF7 на основе экспрессии CD44. После совместного лечения популяция клеток MCF7 с высоким уровнем CD44 снизилась с 42,5% до 19,9% (рис. 5E). Этот результат позволил предположить, что DCA плюс тамоксифен могут подавлять способность раковых стволоподобных клеток в клетках рака молочной железы.

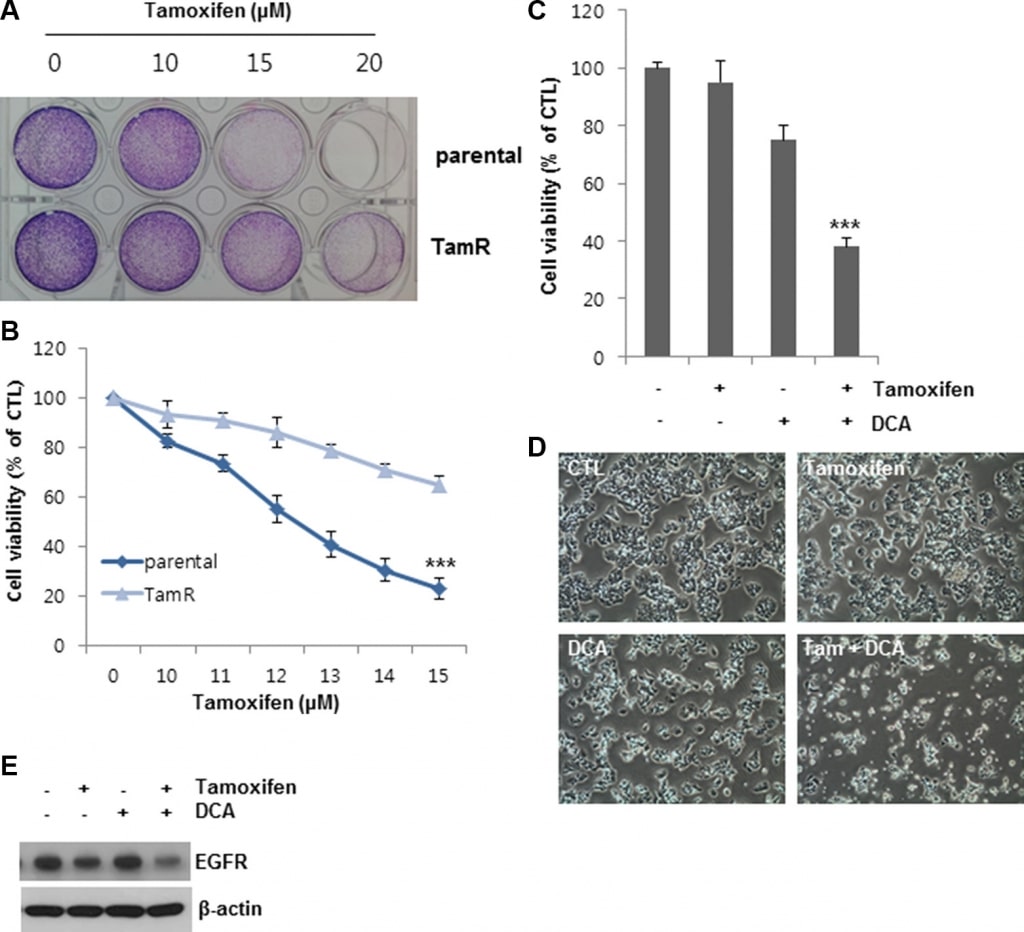
Комбинированное лечение DCA и тамоксифеном может преодолеть устойчивость к тамоксифену в клетках рака молочной железы
Для дальнейшего подтверждения наблюдаемого влияния DCA на вызванную тамоксифеном гибель клеток, мы создали тамоксифенрезистентные (TamR) клетки MCF7 путем лечения тамоксифеном в течение длительного периода времени. По сравнению с необработанными клетками, жизнеспособность клеток MCF7 и TamR MCF7 составила 20 и 60%, соответственно, после обработки 13 мкМ тамоксифена, что указывает на то, что клетки TamR MCF7 были менее чувствительны к той же концентрации тамоксифена, чем родительские клетки MCF7 (Рисунок 6A и 6B). Только DCA ингибировал рост клеток TamR MCF7 примерно на 25% по сравнению с контролем (Рисунок 6C и 6D). В отличие от этого, совместная обработка DCA с тамоксифеном подавляла рост клеток более чем на 60% по сравнению с контролем (Рисунок 6C и 6D). Уровень EGFR в клетках TamR также был снижен в результате совместного лечения (Рисунок 6E). Эти результаты свидетельствуют о том, что DCA может преодолеть устойчивость к тамоксифену в клетках рака молочной железы путем снижения уровня EGFR на белковом уровне.
Обсуждение
По мере углубления понимания метаболического фенотипа опухолевых клеток, в качестве новой противораковой стратегии было предложено воздействовать на метаболические различия между опухолевыми и нормальными клетками. Несмотря на увеличение числа потенциальных противораковых препаратов, направленных на метаболические процессы раковых клеток, для успешного воздействия на метаболизм раковых клеток необходимо выяснить, как клетки реагируют на эти препараты. Здесь мы продемонстрировали, что DCA ингибирует не только активность PDK, но и экспрессию EGFR в клетках рака молочной железы. Совместное лечение DCA и тамоксифеном вызвало протеасомно-зависимую деградацию белков EGFR в клетках рака молочной железы через активацию p38 MAPK. Наши результаты показывают, что снижение уровня EGFR играет ключевую роль в апоптотической гибели клеток, индуцированной комбинацией DCA и тамоксифена. Насколько нам известно, это первое сообщение, показывающее, что DCA сенсибилизирует клетки рака молочной железы к клеточной гибели, вызванной тамоксифеном, через даунгрегуляцию EGFR.
В ER-положительных клетках рака молочной железы, которые развили эндокринную резистентность, экспрессия ER может быть непосредственно подавлена усиленной сигнализацией рецепторов факторов роста из-за сверхэкспрессии EGFR и HER2, которые впоследствии активируют MAPK и подавляют транскрипцию ER [25]. Кроме того, резистентность к тамоксифену в клетках рака молочной железы была связана со сверхэкспрессией EGFR и высоким уровнем фосфорилированной внеклеточной активированной киназы 1/2 [2]. Таким образом, стратегия сочетания ингибитора EGFR с эндокринным препаратом представляет достаточный интерес и заслуживает дальнейшего изучения для терапии рака молочной железы. В данном исследовании мы обнаружили, что снижение уровня EGFR в клетках рака молочной железы, обработанных DCA, повышает чувствительность клеток к тамоксифену. Даунрегуляция EGFR при совместном лечении DCA с тамоксифеном была подтверждена в нескольких ER-положительных клеточных линиях рака молочной железы, включая MCF7, T47D и BT474 (рис. 2A и не показано). В дополнение к HER2-амплифицированной клеточной линии рака молочной железы BT474, клетки HER2-MCF7 также показали даунрегуляцию EGFR под действием DCA/тамоксифена, что позволяет предположить, что комбинированное лечение будет применимо к HER2-положительному раку молочной железы. К сожалению, клеточная линия трижды негативного рака молочной железы MDA-MB-231, которая, как известно, сверхэкспрессирует EGFR [26], показала рефрактерный фенотип, а также отсутствие явного изменения уровня EGFR в ответ на совместное лечение. Одна из возможностей заключается в том, что усиление EGFR в клетках MDA-MB-231 перекрывает его даунрегуляцию при совместном лечении DCA и тамоксифена. Другое объяснение заключается в том, что сигнальный(ые) путь(и), опосредующий(ие) даунрегуляцию EGFR, блокируется(ются) неизвестным механизмом в клетках MDA-MB-231. Для выяснения основного механизма поддержания экспрессии EGFR в ответ на совместное лечение ДКА и тамоксифеном в ER-отрицательных клетках рака молочной железы потребуются дальнейшие исследования.
Известны четыре изоформы PDK, каждая из которых активна в ответ на различные внутриклеточные и внеклеточные условия. PDK1 активируется гипоксией [27]; PDK2 активируется продуктами PDH — ацетил-КоА и NADH [28]; PDK3 активируется АТФ [29]; PDK4 транскрипционно регулируется гормональными сигналами [30]. PDK2 обладает наибольшей активностью фосфорилирования пируватдегидрогеназного комплекса, за ним следуют PDK4, PDK1 и PDK3 [12]. PDK2 наиболее восприимчив к ингибированию DCA из-за его повсеместной экспрессии [31]. Хотя мы обнаружили экспрессию четырех изоформ в клетках MCF7 (Рисунок 1С), функциональные различия между изоформами PDK в раке молочной железы все еще остаются неясными. Представляет интерес дальнейшее изучение того, приводят ли сигнальные пути, подавляющие определенную изоформу PDK, к даунрегуляции EGFR. Известно, что PDK функционирует в митохондриях, и несколько исследований показали, что EGFR также транслоцируется в митохондрии после стимуляции EGF [32, 33]. Кроме того, было высказано предположение, что взаимодействие EGFR с PDK в митохондриальном матриксе играет важную роль в EGFR-индуцированном росте опухоли в глиобластоме [34]. Здесь мы обнаружили, что блокада активности PDK с помощью ингибитора или сайленсинга снижает общий клеточный уровень EGFR в клетках рака молочной железы, обработанных тамоксифеном. Необходимы дальнейшие исследования, чтобы определить, подвергается ли только митохондриально-ассоциированный EGFR деградации при совместной обработке DCA и тамоксифеном в клетках рака молочной железы. В целом, воздействие EGF на клетки вызывает быстрое аутофосфорилирование, включая тирозин (Tyr) 1045, который обеспечивает место стыковки для убиквитин-лигазы c-Cbl, что приводит к убиквитинированию EGFR и удалению EGFR путем эндоцитоза с поверхности клетки в ранний эндосомальный компартмент [35]. Однако совместное лечение DCA и тамоксифеном не повлияло на фосфорилирование EGFR по Tyr 1045 (данные не показаны). Вместо этого мы наблюдали фосфорилирование EGFR по Ser 1046/7 под действием DCA и тамоксифена, которое блокировалось специфическим ингибитором p38 MAPK, SB203580. Предполагается, что p38 MAPK играет ключевую роль в интернализации и деградации EGFR [18], однако для выяснения того, как фосфорилирование EGFR по сериновым остаткам вызывает его даунрегуляцию, а также для идентификации медиатора верхнего уровня активации p38 MAPK после лечения ДКА/тамоксифеном, необходимы дальнейшие исследования.
Nanog и c-myc были идентифицированы как транскрипционные факторы, и их роль в поддержании самообновления в эмбриональных стволовых клетках была установлена в предыдущих исследованиях [22, 36]. Недавно предложенный механизм внутренней резистентности к эндокринной терапии предполагает существование специализированного подмножества раковых клеток, называемых опухоль-инициирующими клетками (ОИК) [37]. TICs обладают способностью к самообновлению и образованию новых опухолей, состоящих полностью из клонированных типов клеток, присутствующих в родительской опухоли. Здесь мы продемонстрировали, что DCA снижает экспрессию c-myc и Nanog в клетках MCF7, обработанных тамоксифеном. Более того, экспрессия этих белков была связана с цитотоксическим эффектом комбинированного лечения DCA и тамоксифена. Таким образом, DCA может быть использован для расширения терапевтического потенциала тамоксифена путем подавления развития внутренней резистентности в терапии рака молочной железы.
В заключение, наши результаты показали, что DCA сенсибилизировал ER-положительные клетки рака молочной железы к тамоксифену путем снижения уровня EGFR. Лечение DCA и тамоксифеном подавляло экспрессию генов самообновления и выживаемость устойчивых к тамоксифену клеток MCF7. Таким образом, мы предполагаем, что DCA может быть эффективным терапевтическим агентом для лечения рака молочной железы, устойчивого к тамоксифену. Более того, стратегии комбинирования с DCA могут быть полезны для повышения эффективности лечения другими цитотоксическими химиотерапевтическими препаратами или таргетной терапией. В будущем необходимо провести дальнейшие эксперименты, включая исследования на животных и клинические испытания.
Материалы и методы
Культура клеток и реактивы
Клетки рака молочной железы человека MCF7, T47D и MDA-MB-231 были приобретены в American Type Culture Collection (Rockville, MD, USA) и выращивались в рекомендованной среде роста (Invitrogen, Carlsbad, CA, USA). HER2-экспрессирующие MCF7 (HER2-MCF7) и контрольные векторные клетки (vector-MCF7) были любезно предоставлены доктором Инчолом Шином (Университет Ханьянг, Сеул, Корея). Устойчивые к тамоксифену (TamR) клетки MCF7 были получены путем культивирования клеток MCF7 в присутствии 10 мкМ тамоксифена в течение более 6 месяцев. В качестве контроля родительские клетки культивировали в течение такого же времени в обычной среде. После установления устойчивости клетки пассировали не более 3 месяцев. Для экспериментов, включающих лечение тамоксифеном, клетки регулярно культивировали в среде Дульбекко, модифицированной Орла, не содержащей фенолового красного, плюс 10% фетальной бычьей сыворотки, очищенной от древесного угля, непосредственно перед началом лечения. Антитела против EGFR, фосфо-EGFR (S1046/1047), FGFR1, FGFR4, PDK1, PDH, survivin, cleaved PARP, p38, phospho-p38 и Nanog были приобретены у Cell Signaling Technology (Danvers, MA, USA). Антитела против HER2/Neu, ER-α и c-myc, siRNAs, нацеленные на EGFR, Nanog, PDK4 и c-myc, и отрицательный контроль (скремблированные) siRNAs были приобретены у Santa Cruz Biotechnology (Dallas, TX, USA). Антитела против PDK2 и PDK3 были приобретены у Thermo Fisher Scientific (Waltham, MA, США). Антитело против β-актина, антитело против FLAG, тамоксифен, DCA, циклогексимид и MG132 были приобретены у компании Sigma-Aldrich (Сент-Луис, МО, США). Антитело phospho-PDH (S293) и SB203580 были получены от BD Biosciences Pharmingen (San Diego, CA, USA), а гефитиниб и эрлотиниб — от Selleck Chemicals (London, ON, Canada).
Трансфекции и лечение
Клетки трансфецировали целевой siRNA (50 нМ) с помощью Lipofectamine RNAiMAX (Invitrogen), как описано производителем. Клетки трансфецировали 1 мкг FLAG-c-myc pcDNA 3.1 и FLAG-Nanog-pcDNA 3.1 с помощью Lipofectamine 2000, как описано производителем. Через 6 ч клетки обрабатывали тамоксифеном и/или DCA в течение 24-48 ч, а затем анализировали, как описано в другом месте.
Измерение жизнеспособности клеток
Жизнеспособность клеток определяли путем измерения митохондриального превращения 3-(4,5-диметилтиазолил2)-2,5-дифенилтетразолия бромида (МТТ) в окрашенный продукт. Клетки обрабатывали, как указано, и среду заменяли бессывороточной средой, содержащей 1 мМ МТТ. После 2 ч инкубации при 37°C клетки солюбилизировали в ДМСО. Количество формазана, превращенной формы МТТ, определяли путем измерения абсорбции при 595 нм. Оценка апоптоза
Апоптоз определяли путем анализа флуоресцентно-активированной сортировки клеток с использованием набора для определения апоптоза Annexin V-FITC (BioVision, Milpitas, CA, США) в соответствии с указаниями производителя. Вкратце, после обработки клетки обрабатывали трипсином, а затем ресуспендировали в буфере для связывания (10 мМ HEPES/NaOH, pH 7,4, 140 мМ NaCl, 2,5 мМ CaCl2 ), включающем Annexin V-FITC и йодистый пропидий. После инкубации в течение 15 мин флуоресценцию клеток анализировали методом проточной цитометрии. Гибель клеток измеряли как процент клеток в аннексин V и PI положительной популяции.
Вестерн-блоттинг
Клетки собирали и лизировали в буфере RIPA (50 мМ Трис-HCl pH 7,5, 150 мМ NaCl, 1% Nonidet P40, 0,5% дезоксихолата натрия и 0,1% SDS), дополненном коктейлем ингибиторов протеаз/фосфатаз (Roche, Мангейм, Германия). Равные количества белков (20-50 мкг) разделяли методом SDS-PAGE и переносили на нитроцеллюлозную мембрану. Мембраны блокировали путем инкубации с 5% обезжиренным молоком в трисбуферном физиологическом растворе в течение 1 ч, а затем инкубировали в течение ночи с соответствующими первичными антителами. Мембраны инкубировали с HRP-конъюгированным вторичным антителом в течение 1 ч. Иммунореактивные белки визуализировали с помощью усиленных хемилюминесцентных реагентов (Amersham Biosciences, Литтл Чалфонт, Великобритания).
Выявление CD44-позитивной популяции клеток
Клетки окрашивали антителами в разведении 1∶100 в PBS в течение 15 минут. Использовали следующие антитела; FITC-CD44 и FITC-конъюгированные мышиные IgG изотипные контрольные антитела были получены от BD Biosciences Pharmingen. Меченые клетки анализировали на проточной цитометрии. Популяцию CD44-позитивных клеток определяли по интенсивности FITC.
Статистический анализ
Все представленные данные являются репрезентативными как минимум для двух отдельных экспериментов. Сравнение между группами анализировали с помощью t-теста Стьюдента. Звездочки (***p < 0,001, **p < 0,01, *p < 0,05) указывают на статистическую значимость.
Благодарности и финансирование
Данное исследование было поддержано программой фундаментальных научных исследований Национального исследовательского фонда Кореи (NRF), финансируемой Министерством науки, ИКТ и планирования будущего (№ 1711031812, 1711023318 и 1711031800).
Конфликты интересов
Авторы заявляют об отсутствии конфликтов интересов.
ССЫЛКИ
1 1. Vander Heiden MG, Cantley LC, Thompson CB. Понимание эффекта Варбурга: метаболические требования клеточной пролиферации. Science. 2009; 324:1029-33.
2ZhaoY, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013; 4:e532.
3 Vander Heiden MG. Targeting cancer metabolism: a therapeutic window opens. Nat Rev Drug Discov. 2011; 10:671-84.
4 Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007; 11: 37-51.
5 Kankotia S, Stacpoole PW. Дихлорацетат и рак: новый дом для сиротского препарата? Biochim Biophys Acta. 2014; 1846:617-29.
6 Osborne CK. Тамоксифен в лечении рака молочной железы. N Engl J Med. 1998 Nov 26; 339:1609-18.
7 Johnston SR. Новые стратегии при эстроген-рецептор-положительном раке молочной железы. Clin Cancer Res. 2010 Apr 1; 16:1979-87. doi: 10.1158/1078-0432.CCR-09-1823.
8 Пето Р, Борехам Дж, Кларк М, Дэвис С, Берал В. Смертность от рака молочной железы в Великобритании и США снизилась на 25% в 2000 году в возрасте 20-69 лет. Lancet. 2000; 355:1822.
9 Совместная группа исследователей ранних стадий рака молочной железы (EBCTCG). Влияние химиотерапии и гормональной терапии при раннем раке молочной железы на рецидивы и 15-летнюю выживаемость: обзор рандомизированных исследований. Lancet. 2005; 365:1687-717.
10 Early Breast Cancer Trialists’ Collaborative Group (EBCTCG), Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, Dowsett M, Ingle J, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011; 378:771-84.
11 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Комбинированное воздействие на PDK1 и EGFR вызывает регрессию глиобластомы, обращая вспять эффект Варбурга. Cancer Res. 2013; 73:7277-89.
12 Saunier E, Benelli C, Bortoli S. The pyruvate dehydrogenase complex in cancer: Старый метаболический привратник, регулируемый новыми путями и фармакологическими агентами. Int J Cancer. 2016; 138:809-17.
13 Osborne CK, Schiff R. Механизмы эндокринной резистентности при раке молочной железы. Annu Rev Med. 2011; 62:233-47.
14YunSM, Woo SH, Oh ST, Hong SE, Choe TB, Ye SK, Kim EK, Seong MK, Kim HA, Noh WC, Lee JK, Jin HO, Lee YH, et al. Melatonin enhances arsenic trioxide-induced cell death through sustained upregulation of Redd1 expression in breast cancer cells. Mol Cell Endocrinol. 2016; 422:64-73.
15 Zhu J, Lu X, Hua KQ, Sun H, Yu YH, Feng YJ. Рецептор эстрогена α опосредует усиление 17β-эстрадиолом подвижности клеток рака яичников через ап-регуляцию экспрессии сурвивина. Arch Gynecol Obstet. 2012; 286: 729-37.
16 Arteaga CL. Зависимость рецептора эпидермального фактора роста в опухолях человека: больше, чем просто экспрессия? Онколог. 2002; 7:31-9.
17 Sorkin A, Goh LK. Эндоцитоз и внутриклеточное перемещение ErbBs. Exp Cell Res. 2009; 315:683-96.
18 Adachi S, Shimizu M, Shirakami Y, Yamauchi J, Natsume H, Matsushima-Nishiwaki R, To S, Weinstein IB, Moriwaki H, Kozawa O. (-)-Epigallocatechin gallate downregulates EGF receptor through phosphorylation at Ser1046/1047 by p38 MAPK in colon cancer cells. Канцерогенез. 2009; 30:1544-52.
19CiardielloF, Caputo R, Bianco R, Damiano V, Pomatico G, De Placido S, Bianco AR, Tortora G. Противоопухолевый эффект и потенцирование активности цитотоксических препаратов в раковых клетках человека ZD-1839 (Iressa), селективным ингибитором тирозинкиназы рецептора эпидермального фактора роста. Clin Cancer Res. 2000; 6:2053-63.
20 Pollack VA, Savage DM, Baker DA, Tsaparikos KE, Sloan DE, Moyer JD, Barbacci EG, Pustilnik LR, Smolarek TA, Davis JA, Vaidya MP, Arnold LD, Doty JL, et al. Inhibition of epidermal growth factor receptorassociated tyrosine phosphorylation in human carcinomas with CP-358,774: dynamics of receptor in situ and antitumor effects in athymic mice. J Pharmacol Exp Ther. 1999; 291:739-48.
21 Normanno N, De Luca A, Bianco C, Strizzi L, Mancino M, Maiello MR, Carotenuto A, De Feo G, Caponigro F, Salomon DS. Сигнализация рецептора эпидермального фактора роста (EGFR) в раке. Gene. 2006; 366:2-16.
22 Iv Santaliz-Ruiz LE, Xie X, Old M, Teknos TN, Pan Q. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer. 2014; 135:2741-8.
23 Chen Y, Olopade OI. MYC в прогрессии опухолей молочной железы. Expert Rev Anticancer Ther. 2008; 8: 1689-98.
24 Hou ZJ, Luo X, Zhang W, Peng F, Cui B, Wu SJ, Zheng FM, Xu J, Xu LZ, Long ZJ, Wang XT, Li GH, Wan XY, et al. Flubendazole, одобренный FDA антгельминтик, нацелен на стволовые клетки рака молочной железы. Oncotarget. 2015; 6:6326-40. doi:10.18632/oncotarget.3436.
25 Creighton CJ, Hilger AM, Murthy S, Rae JM, Chinnaiyan AM, El-Ashry D. Activation of mitogenactivated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res. 2006; 66:3903-11.
26 Biswas DK, Cruz AP, Gansberger E, Pardee AB. Индуцированная эпидермальным фактором роста активация ядерного фактора каппа В: Основной путь прогрессии клеточного цикла в эстрогенрецептор-негативных клетках рака молочной железы. Proc Natl Acad Sci U S A. 2000; 97:8542-7.
27 Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF1-опосредованная экспрессия киназы пируватдегидрогеназы: метаболический переключатель, необходимый для клеточной адаптации к гипоксии. Cell Metab. 2006; 3:177-85.
28 Hiromasa Y, Hu L, Roche TE. Лиганд-индуцированные эффекты на изоформу 2 киназы пируватдегидрогеназы. J Biol Chem. 2006; 281:12568-79.
29 Kato M, Chuang JL, Tso SC, Wynn RM, Chuang DT. Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. EMBO J. 2005; 24:1763-74.
30 Kwon HS, Huang B, Unterman TG, Harris RA. Протеинкиназа B-альфа ингибирует индукцию гена пируватдегидрогеназы-4 человека дексаметазоном через инактивацию факторов транскрипции FOXO. Diabetes. 2004; 53:899-910.
31 Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Доказательства существования тканеспецифической регуляции комплекса пируватдегидрогеназы млекопитающих. Biochem J. 1998; 329 :191-6.
32 Dasari VR, Velpula KK, Alapati K, Gujrati M, Tsung AJ. Стволовые клетки пуповинной крови ингибируют транслокацию рецептора эпидермального фактора роста в митохондрии при глиобластоме. PLoS One. 2012; 7:e31884.
33 Demory ML, Boerner JL, Davidson R, Faust W, Miyake T, Lee I, Hüttemann M, Douglas R, Haddad G, Parsons SJ. Транслокация рецептора эпидермального фактора роста в митохондрии: регуляция и эффект. J Biol Chem. 2009; 284:36592-604.
34 Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Комбинированное воздействие на PDK1 и EGFR вызывает регрессию глиобластомы, обращая вспять эффект Варбурга. Cancer Res. 2013; 73:7277-89.
35 Massie C, Mills IG. The developing role of receptors and adaptors. Nat Rev Cancer. 2006; 6:403-9.
36 Chappell J, Dalton S. Roles for MYC in the establishment and maintenance of pluripotency. Cold Spring Harb Perspect Med. 2013;3:a014381.
37 Wei W, Lewis MT. Идентификация и нацеливание на клетки, инициирующие опухоль, в лечении рака молочной железы. Endocr Relat Cancer. 2015; 22:R135-55.