Элисон Б. Хаугруд, Юнсянь Жуань, Джозеф Д. Коппок, В. Кит Мискиминс
Исследовательский центр биологии рака, Sanford Research, 2301 E. 60th St North, Sioux Falls, SD 57104, USA
e-mail: [email protected]
Получено: 24 апреля 2014 г.
Принято: 5 сентября 2014 г.
Опубликовано: 12 сентября 2014 г
Аннотация
Уникальный метаболизм клеток рака молочной железы представляет интерес для использования этого явления в терапевтических целях. Метформин, перспективный препарат для лечения рака молочной железы, действует на комплекс I электронно-транспортной цепи, что приводит к накоплению реактивных форм кислорода (ROS), которые в конечном итоге приводят к гибели клеток. Ингибирование комплекса I приводит к выработке лактата — метаболического побочного продукта, который уже в высокой степени вырабатывается перепрограммированными раковыми клетками и ассоциируется с плохим прогнозом. Хотя метформин остается перспективным средством лечения рака, мы искали дополнительный агент, который бы усиливал апоптотические эффекты метформина и одновременно ослаблял выработку лактата, что могло бы привести к значительному повышению эффективности. Дихлорацетат (DCA) — хорошо известный препарат, используемый для лечения молочнокислого ацидоза, который действует через ингибирование киназы пируватдегидрогеназы (PDK), способствуя митохондриальному метаболизму. Наша цель заключалась в изучении синергии и механизмов, с помощью которых эти два препарата убивают клетки рака молочной железы. Клеточные линии были подвергнуты указанным видам лечения и проанализированы на предмет гибели клеток и различных аспектов метаболизма. Гибель клеток и производство ROS анализировали с помощью проточной цитометрии, Вестерн-блот анализа и методов подсчета клеток. Изображения клеток получали с помощью фазово-контрастной микроскопии или конфокальной микроскопии. Метаболизм клеток анализировали с помощью анализатора Seahorse XF24, лактатного анализа и анализа pH. Мы показали, что при совместном использовании DCA и метформина происходит синергетическая индукция апоптоза клеток рака молочной железы. Окислительное повреждение, вызванное метформином, усиливается DCA за счет ингибирования PDK1, что также снижает выработку лактата под действием метформина. Мы продемонстрировали, что DCA и метформин совместно вызывают синергетический каспазозависимый апоптоз, связанный с окислительным повреждением, с одновременным снижением выработки лактата под действием метформина. Инновационные комбинации, такие как метформин и DCA, обещают расширить терапию рака молочной железы.
Ключевые слова: Метформин; дихлорацетат; рак молочной железы; лактат; апоптоз
© Springer Science+Business Media New York 2014
ВВЕДЕНИЕ
Метаболизм раковых клеток превращается в перспективную область для разработки новых терапевтических подходов. По сравнению с нормальными клетками, из которых они получены, раковые клетки метаболически перепрограммированы, они преимущественно используют гликолиз даже в условиях достаточного количества кислорода, это явление известно как эффект Варбурга [1]. Чтобы компенсировать потерю АТФ в результате преимущественного гликолиза (по сравнению с продвижением через окислительное фосфорилирование), раковые клетки повышают регуляцию генов, кодирующих транспортеры глюкозы и гликолитические ферменты, такие как киназа пируватдегидрогеназы (PDK) и лактатдегидрогеназа (LDH). Такая высокая скорость поглощения глюкозы и измененный метаболизм не только обеспечивают АТФ и позволяют клеткам выживать в гипоксических условиях; они также обеспечивают биосинтетические строительные блоки, такие как промежуточные продукты и субстраты для производства аминокислот, NADPH и рибозо-5-фосфата, которые необходимы для синтеза нуклеотидов, белков и мембран, необходимых в быстро делящихся клетках. Это также означает, что митохондриальный цикл ТСА генерирует меньший процент АТФ, что позволяет использовать цитрат в биосинтезе жирных кислот и липидов для производства новых мембран [2]. Большая часть лактата, образующегося в результате гликолиза, поглощается окружающими клетками с последующей рециркуляцией для поддержания роста опухоли и устойчивости к апоптотическим механизмам гибели клеток [2, 3]. Лактат, вырабатываемый опухолями, может нарушать метаболизм Т-клеток и антигенную презентацию дендритных клеток, что приводит к иммунному уклонению опухоли [4, 5]. Таким образом, высокий уровень использования глюкозы и гликолиза дает раковым клеткам многочисленные преимущества. Однако уникальный метаболический профиль раковых клеток можно использовать и в терапевтических целях. В данном исследовании мы изучили активность и взаимодействие двух препаратов, направленных на метаболизм, — метформина и дихлорацетата (DCA) — для определения их влияния на рост и выживание клеток рака молочной железы.
Метформин гидрохлорид (1,1-диметилбигуанид гидрохлорид) — это пероральный препарат, широко используемый для лечения диабета второго типа. Исследования показали, что у пациентов с диабетом, принимающих метформин, заболеваемость раком и связанная с ним смертность ниже, чем у пациентов, не принимающих метформин. В одном из таких исследований пациенты с диабетом, страдающие раком молочной железы и принимающие метформин, значительно лучше отвечали на неоадъювантную химиотерапию по сравнению с пациентами, не принимающими метформин [6-8]. Исследования in vitro показали, что метформин подавляет рост многих типов раковых клеток, включая клетки рака молочной железы, рака толстой кишки, рака простаты, рака яичников и глиомы [9-12]. Известно, что метформин активирует AMP-активированную протеинкиназу (AMPK), что приводит к ингибированию синтеза белка и роста клеток [13]. Однако одной активации AMPK недостаточно, чтобы привести к апоптотической гибели клеток [14]. Исследования показали, что метформин накапливается в митохондриях и мягко ингибирует комплекс I электронно-транспортной цепи, что происходит до активации AMPK [15-18]. Поскольку комплекс I ингибирован, затрудненное прохождение электронов приводит к образованию супероксида в митохондриальном матриксе, повреждая митохондриальные белки, липиды и нуклеиновые кислоты. В исследованиях, в которых было показано, что метформин способствует гибели клеток, основным путем является апоптоз [10, 12, 19]. Ранее мы показали, что метформин вызывает как каспазозависимую, так и поли(АДФ-рибоза) полимеразную (PARP) гибель клеток в большинстве клеточных линий рака молочной железы, при этом он не является цитотоксичным для нетрансформированных эпителиальных клеток молочной железы [20]. Поли(АДФ-рибоза) полимераза-зависимая гибель клеток была связана с серьезными изменениями формы и функции митохондрий, что позволило сделать вывод о том, что повреждение митохондрий в раковых клетках является ключевым медиатором метформин-индуцированной гибели клеток. Основываясь на этих наблюдениях, мы предположили, что соединения, способствующие окислительному метаболизму митохондрий, будут усиливать вызванное метформином повреждение митохондрий и взаимодействовать с метформином в уничтожении раковых клеток. Поскольку лечение метформином также способствует выработке лактата [21], такое соединение в идеале должно бороться с этим эффектом.
Дихлорацетат также является перорально доступным препаратом с хорошо изученной фармакокинетикой и был протестирован для лечения молочнокислого ацидоза (потенциального побочного эффекта метформина) и митохондриальной недостаточности [27]. Дихлорацетат является ингибитором PDK, который фосфорилирует пируватдегидрогеназу (PDH), делая ее неактивной [23]. Пируватдегидрогеназа — фермент, катализирующий превращение пирувата в ацетил-КоА для вступления в митохондриальный цикл трикарбоновых кислот (ТКК) и окислительного фосфорилирования. В раковых клетках активность PDK часто повышена, действуя как привратник, снижающий поток пирувата из цитоплазмы в метаболизм митохондрий. Считается, что это является важным компонентом метаболического перепрограммирования в раковых клетках, что приводит к снижению окисления глюкозы и выработки лактата [24-26]. Ингибируя PDK, DCA повышает активность PDH, позволяя пирувату вступать в цикл TCA, а не преобразовываться в лактат и выделяться [27].
В данном исследовании мы изучили противоопухолевую активность и взаимодействие двух препаратов, направленных на метаболизм, — метформина и DCA. Мы показали, что DCA усиливает цитотоксичность метформина для клеток рака молочной железы через механизм, включающий окислительное повреждение, одновременно снижая производство лактата метформином, что потенциально обеспечивает двойное терапевтическое преимущество.
Методы
Химикаты и реагенты
Следующие химикаты, реагенты и наборы были приобретены в Sigma-Aldrich, если не указано иное: метформин (1,1-диметилбигуанид), дихлорацетат натрия, 0.4 % раствор трипанового синего, монтажная среда Vectashield для флуоресценции, содержащая 4,6 диамидино-2-фенилиндол (DAPI) (Vector Laboratories), Lactate Assay Kit (Eton Biosciences), ингибитор каспазы OPH-109 (MP Biomedicals), Coomassie Brilliant Blue R250 (Bio-Rad Laboratories), параформальдегид, SYTOX® Green (Life Technologies), Triton X-100 (Eastman) и ингибитор PARP II INH2BP (Epigentek).
Культура клеток
Клеточные линии рака молочной железы человека MCF7 и T47D и эпителиальные клетки молочной железы человека MCF10A были приобретены у ATCC. Клеточная линия карциномы молочной железы мыши 66CL4 была предоставлена доктором Фредом Миллером (Karmanos Cancer Institute, Детройт, MI). После получения клеточных линий клетки немедленно культивировали и размножали для приготовления замороженных ампульных запасов. Клетки пассировали не более 2-3 месяцев, прежде чем создавать новые культуры из замороженных ампул ранних пассажей. Клеточные линии регулярно проверялись на заражение микоплазмами и были подтверждены лабораториями IDEXX RADIL как свободные от микоплазм. Клетки поддерживали в модифицированной среде Дульбекко (DMEM) с 10 % фетальной бычьей сыворотки, 100 Ед/мл пенициллина и 100 мкг/мл стрептомицина. Клетки инкубировали в увлажненномCO2-инкубаторе при 37 °C.
Анализ исключения трипанового синего
Клеточные линии MCF7 и T47D были высеяны на 35-миллиметровую посуду. Клетки обрабатывали метформином и DCA в указанных концентрациях или транспортным средством. После указанного периода времени среда была собрана, чтобы сохранить все плавающие мертвые клетки. Прикрепленные клетки промывали фосфатно-буферным солевым раствором (PBS), который объединяли с собранной средой. Затем клетки собирали трипсинизацией и добавляли в пул. Затем раствор трипанового синего использовался для окрашивания мертвых клеток 1:1, и клетки подсчитывались с помощью гемацитометра.
Вестерн-блоттинг
Клетки высевали на 35-миллиметровую посуду. После обработки в течение указанного периода времени клетки собирали в среду для сбора живых и мертвых клеток. Клетки гранулировали, промывали PBS и повторно гранулировали. Затем клеточные гранулы лизировали путем добавления 1× додецилсульфата натрия (SDS) буфера для образцов [2,5 мМ Трис-HCl (pH 6,8), 2,5 % SDS, 100 мМ дитиотреитола, 10 % глицерина, 0,025 % бромфенолового синего]. Равные количества белка разделяли на 8,5 % SDS-полиакриламидном геле. Белки переносили на мембраны Immobilon P (Millipore) с помощью аппарата Bio-Rad Trans-blot с использованием буфера для переноса [48 мМ Трис-HCl, 39 мМ глицин]. Мембраны погружали в 5 % обезжиренное сухое молоко в Трис-буферном солевом растворе, содержащем Tween 20 (TBS-T) [10 мМ Трис-HCl (pH 7,5), 150 мМ NaCl, 0,1 % Tween-20] с указанным антителом либо на 3 ч при комнатной температуре, либо на 4 °C на ночь. После тщательной промывки в TBS-T наносили соответствующее вторичное антитело, конъюгированное с пероксидазой хрена (HRP). Мембрану снова промывали, после чего белки выявляли с помощью хемилюминесцентного субстрата Super Signal West Pico (Pierce Biochemical). Для выделения митохондрий (рис. 3г) клетки MCF7 выращивали до 95% конфлюентности на 150-миллиметровой посуде. Набор Mitochondrial Isolation Kit for Cultured Cells (Pierce) был использован для выделения митохондрий в соответствии с протоколом производителя. Изображения всех блотов были получены с помощью системы визуализации UVP. Были использованы следующие антитела: Pyruvate dehydrogenase kinase1 (Abcam), PARP (Cell Signaling), анти-4-гидроксиноненаль (Millipore), субъединица комплекса I NDUFB8 (Mitosciences), GAPDH (Ambion), PDH (Abcam) и Phospho-PDHE1 alpha (Calbiochem).
Конфокальная микроскопия
Клетки высевали на стеклянные покровные стекла в 35-миллиметровую посуду и обрабатывали, как указано. Клетки промывали в PBS, фиксировали 4 % параформальдегидом в течение 10 мин, монтировали в среде Vectashield с DAPI на стандартные предметные стекла микроскопа и затем наблюдали на конфокальном микроскопе Olympus FV1000 при 100×. Создание клеточных линий MCF7, стабильно экспрессирующих pAcGFP1-Mito (Clontech; плазмида, кодирующая митохондриальные целевые флуоресцентные белки), было описано ранее [20].
Анализ лактата и рН
Клетки MCF7 и 66CL4 в 35-миллиметровой посуде выращивали до 80 % конфлюентности и обрабатывали, как указано, в среде, не содержащей фенолового красного и карбонатов. Клетки обрабатывали в течение 4-6 ч. Среду из посуды собирали и измеряли концентрацию лактата с помощью коммерчески доступного набора L-Lactate Assay Kit (Eton Bioscience). Для клеточной линии 66Cl4 клетки подсчитывали с помощью гемацитометра для нормализации уровня лактата. Для измерения рН среды использовали рН-метр.
Анализ колониеобразования
Клетки MCF7 высевали из расчета 500 клеток на 60-миллиметровое блюдо. На следующий день клетки обрабатывали, как указано. После 12 дней инкубации клетки промывали PBS и фиксировали 70 % этанолом в течение 5 мин. Затем колонии окрашивали Coomassie Blue [40 % метанол, 12 % ледяная уксусная кислота, 0,24 % Coomassie Blue], промывали, получали изображения и проводили количественную оценку с помощью AlphaImager System и соответствующего программного обеспечения для анализа изображений (AlphaInnotech, Santa Clara, CA).
Проточная цитометрия
Клетки MCF7 обрабатывали, как указано. Затем клетки инкубировали с MitoSOX (5 мкМ) в течение 15 мин, промывали, трипсинизировали и добавляли к клеткам 1 мл 10% FBS DMEM. Уровень MitoSOX в равном количестве клеток для каждого условия обработки определяли методом проточной цитометрии на цитометре Accuri C6.
Ингибирование PDK1 с помощью siRNA
On-TARGET Plus Smartpool siRNA, нацеленные на PDK1, и нецелевые siRNA были приобретены у Thermo Scientific. клетки 66CL4 (250 000/диск) высевали на 35-миллиметровую посуду. На следующий день клетки трансфецировали siRNAs с помощью Dharmafect (Thermo Scientific) в соответствии с протоколом производителя. Трансфекцию повторяли на следующий день. На следующий день клетки промывали и обрабатывали, как указано, в бескарбонатной среде DMEM с 10 % FBS. Через 4 ч клетки подсчитывали с помощью гемацитометра, а среду собирали для измерения pH и уровня лактата. Клетки собирали для анализа уровня PDK1 методом вестерн-блоттинга.
Анализ реактивных веществ тиобарбитуровой кислоты (TBARS)
Клетки MCF7 выращивали до 100 % конфлюентности на 150-миллиметровой посуде. Клетки обрабатывали, как указано, в течение 24 ч метформином (8 мМ) или DCA (5 мМ). Клетки собирали в PBS, сонировали и определяли относительное количество белка с помощью анализа Брэдфорда. Лизаты обрабатывали с помощью набора Quantichrom™ TBARS Assay Kit и считывали на планшетном ридере SpectraMax M5 (λ ex/em = 560 нм/585 нм).
SYTOX® green assay
Клетки MCF7 и T47D были помещены в 96-луночные планшеты и выращены до 90% конфлюентности. Клетки обрабатывали, как указано, затем добавляли краситель нуклеиновых кислот SYTOX® Green (10 мкМ) и инкубировали в течение 20 мин. перед считыванием на флуоресцентном планшетном ридере при λ ex/em = 485/535 нм с отсечкой 515 нм. Затем клетки пермеабилизировали Тритоном Х-100 (0,4 %) в течение 30 мин, и проводили повторное считывание для определения общего уровня окрашивания ДНК — суррогата общего числа клеток. Значения индекса комбинации определялись с помощью программного обеспечения CalcuSyn.
Скорость потребления кислорода
Скорость потребления кислорода (СПК) измеряли с помощью анализатора Seahorse XF24 в соответствии с инструкциями производителя (Seahorse Bioscience, North Billerica, MA, США). Клетки T47D высевали в количестве 40 000 клеток/лунку в луночные планшеты XF24. На следующий день клетки промывали и уравновешивали безбуферной средой в течение 30 мин при 37 °C в инкубаторебез CO2 перед переносом в анализатор XF24. Было получено начальное измерение OCR, после чего в лунки через инъекционные порты добавляли DCA до конечной концентрации 5 мМ. OCR измеряли каждые 30 минут.
Статистический анализ
Сравнение двух групп проводили с помощью непарного t-теста с поправкой Уэлча, созданной в программе Graph Pad Prism (La Jolla, CA). Значения P меньше или равные 0,05 считались значимыми.
Результаты
DCA и метформин синергично индуцируют апоптоз в клетках рака молочной железы
Ранее мы показали, что метформин вызывает два различных типа гибели клеток после 3 дней лечения в большинстве клеточных линий рака молочной железы, при этом он не является цитотоксичным для нормальных эпителиальных клеток молочной железы [20]. Индукция клеточной смерти в клетках рака молочной железы была тесно связана с изменением структуры митохондрий, что позволяет предположить, что метформин, известный ингибитор комплекса I митохондриальной электронно-транспортной цепи, убивает раковые клетки путем изменения функции митохондрий. Это позволило нам предположить, что увеличение транспорта электронов в митохондриях усилит гибель раковых клеток под действием метформина. Поскольку DCA способствует митохондриальному окислению пирувата, мы сначала изучили, может ли DCA усилить цитотоксичность клеточных линий рака молочной железы, вызванную метформином.
Клетки рака молочной железы MCF7 и T47D обрабатывали метформином, DCA или их комбинацией. Концентрация метформина составляла 8 мМ, что представляет собой физиологически значимую дозу метформина, как показано в работе Owen et al. [17]. Что касается DCA, то в сыворотке крови пациентов могут быть достигнуты низкие миллимолярные концентрации, поэтому диапазон 0,5-5 мМ является клинически значимым [27-29] и использовался в данных экспериментах. Количество живых и мертвых клеток определяли методом исключения трипанового синего (рис. 1а). Гибель клеток измеряли в клетках MCF7 (рис. 1а слева), обработанных в течение 2 дней, и клетках T47D (рис. 1асправа), обработанных в течение 4 дней. В обеих клеточных линиях метформин индуцировал гибель клеток, как это наблюдалось в предыдущих экспериментах [20]. Однако гибель клеток значительно увеличивалась при одновременном лечении DCA и метформином, в то время как только DCA оказывал минимальный цитотоксический эффект (рис. 1а). Когда нетрансформированные эпителиальные клетки молочной железы MCF10A подвергались той же обработке, гибели клеток не наблюдалось, как показано на рис. 1б. Для дальнейшего изучения этого наблюдения гибели клеток в раковых клетках были проведены титрование дозы и анализ цитотоксичности SYTOX Green (рис. 1c). Комбинация препаратов была проверена на синергизм по методу Чоу [30] с использованием программного обеспечения CalcuSYN, которое генерирует индекс комбинации (CI) препаратов; синергизм проявляется, если CI меньше 1. Клетки MCF7 (рис. 1c слева) имели CI 0,00047 при лечении в течение двух дней комбинацией DCA и метформина. КИ клеток T47D составил 9,762e-006 после 4 дней лечения (рис. 1c справа). Эти значения указывают на сильную синергическую цитотоксичность комбинации DCA и метформина в этих клеточных линиях рака молочной железы.
Далее наблюдаемые цитотоксические эффекты DCA (2,5 мМ) и метформина (1 мМ) были подтверждены с помощью анализа на образование колоний. При этих концентрациях оба препарата по отдельности уменьшали размер колоний, но существенно не снижали их количество (рис. 1d, e). Однако при совместном применении двух препаратов наблюдалось значительное снижение числа колоний (рис. 1e). Эти наблюдения показывают, что DCA усиливает цитотоксичность клеток рака молочной железы, вызванную метформином, по сравнению с одним из препаратов.
DCA способствует окислительному фосфорилированию через ингибирование PDK1 и ослабляет вызванное метформином производство лактата
Чтобы подтвердить, что наблюдаемые эффекты DCA связаны с ингибированием ожидаемой мишени, PDK1, мы показали, что DCA действительно снижает уровень фосфорилированной PDH по сравнению с контролем в клетках MCF7 и MCF10A с помощью вестерн-блота (рис. 2a). Стимуляция пируватдегидрогеназы после обработки DCA, как ожидается, усилит окислительный метаболизм пирувата. Этот эффект был изучен с помощью анализатора Seahorse XF24. Измеряли OCR контрольных и обработанных ДКА клеток MCF7 (рис. 2б). Клетки, обработанные дихлорацетатом, имели значительно более высокий показатель OCR по сравнению с контрольными клетками.
Метформин, являясь мягким ингибитором комплекса I и усилителем активности AMPK, как ожидается, будет стимулировать гликолитическую скорость и увеличивать продукцию лактата [11,14,15,17]. Напротив, DCA, как ожидается, перенаправит пируват в митохондриальный метаболизм за счет выработки лактата. Чтобы проверить это, измеряли pH и уровень лактата в среде культивируемых клеток MCF7 после обработки метформином, DCA или обоими препаратами. Клетки, обработанные метформином, вырабатывали значительно больше лактата по сравнению с необработанным контролем. Только дихлорацетат оказывал незначительный эффект, но значительно снижал выработку лактата, вызванную метформином. PH среды соответствовал наблюдаемым изменениям в уровнях лактата (рис. 2c). То есть метформин значительно снижал pH среды, и этот эффект в значительной степени устранялся DCA.
Для дальнейшего обоснования роли PDK в производстве лактата и ингибировании вступления пирувата в цикл ТСА, PDK был нокаутирован с помощью siRNA в клетках 66CL4. Эта клеточная линия получена из карциномы молочной железы мыши и может быть полезна для переноса этих экспериментов в доклинические условия в будущих исследованиях. Клетки были трансфицированы либо siRNA, нацеленной на PDK1 (PDK1siRNA), либо не нацеленной контрольной siRNA (C. siRNA). Уровень PDK1 определяли с помощью вестерн-блота, и клетки, трансфицированные PDK1 siRNA, имели заметно более низкий уровень PDK1 по сравнению с клетками, трансфицированными C. siRNA (рис. 2d). SiRNA-опосредованная репрессия PDK1 уменьшила выработку лактата и увеличила pH среды, что соответствует известным функциям PDK1 (рис. 2e, f). Нокдаун PDK1 эффективно снижал вызванные метформином изменения лактата и pH среды. Дихлорацетат еще больше снижал лактат и повышал рН в клетках, трансфицированных PDK1 siRNA. Способность ДХА отменять действие метформина на лактат и рН была несколько ослаблена, когда PDK1 был нокаутирован, что еще раз говорит о том, что ДХА оказывает свое действие через PDK1. Таким образом, можно сделать вывод, что потеря PDK1 путем трансфекции siRNA или ингибирования DCA предотвращает выработку лактата, вызванную метформином.
DCA в сочетании с метформином усиливает окислительное повреждение и последующую каспаза-зависимую гибель клеток
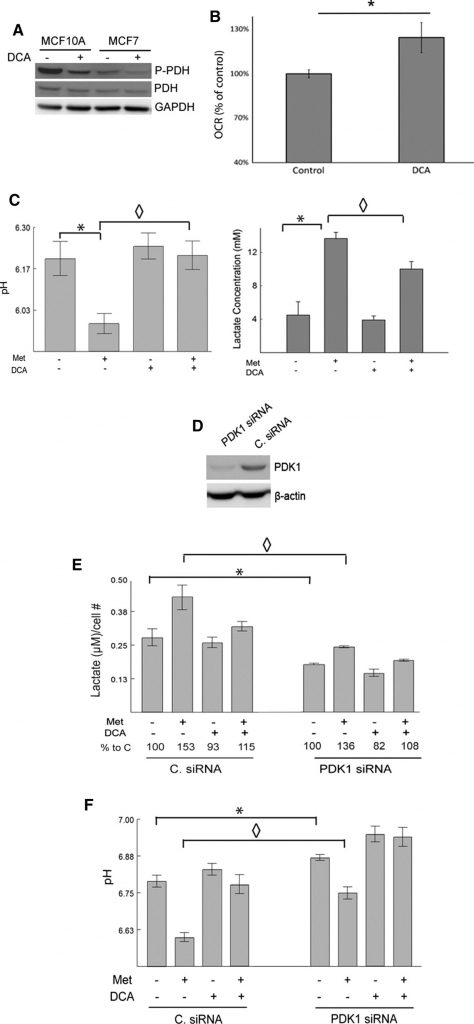
Метформин ингибирует комплекс I электронно-транспортной цепи, препятствуя потоку электронов и приводя к образованию супероксида в матриксе митохондрий. Поскольку DCA способствует вступлению пирувата в цикл ТСА, мы предположили, что DCA может усиливать продукцию ROS как потенциальный механизм цитотоксического синергизма с метформином. MitoSOX, специфический индикатор продукции супероксида митохондриями, окрашивал клетки, которые анализировались методом проточной цитометрии. После 1 ч лечения ни метформин (красная линия, рис. 3а, левая панель), ни DCA (не показано) не увеличивали продукцию супероксида митохондриями. Однако комбинированное лечение DCA и метформином привело к правому сдвигу интенсивности флуоресценции, что указывает на увеличение продукции супероксида. Через 3 ч лечения клетки, обработанные метформином, начали продуцировать повышенные уровни митохондриального супероксида метформина (красная линия, рис. 3а, правая панель) по сравнению с необработанными клетками (черная линия). Комбинация DCA и метформина показала еще более высокие уровни окрашивания супероксида. Эти результаты показывают, что метформин вмешивается в транспорт электронов в комплексе I, что приводит к увеличению продукции супероксида, и что DCA потенцирует эффект метформина.
Накопление супероксида и других ROS может привести к повреждению клеточных компонентов, включая белки, мембраны и ДНК. Окислительное повреждение ДНК может привести к двунитевым разрывам и фосфорилированию гистона H2AX (p-H2AX), который является полезным суррогатным маркером такого повреждения ДНК [31]. Поэтому для оценки повреждения ДНК, связанного с продукцией ROS в ответ на лечение метформином и DCA, можно проанализировать уровень фосфорилированного H2AX. Эпителиальные клетки молочной железы человека MCF10A и клеточные линии рака молочной железы MCF7 и 66CL4 были проанализированы методом вестерн-блоттинга после лечения DCA, метформином или их комбинацией в течение 24 ч (рис. 3б). В клетках MCF10A ни при одном из методов лечения не наблюдалось обнаруживаемого количества p-H2AX. В клетках MCF7 p-H2AX присутствовал в необработанных клетках и значительно увеличивался под действием комбинации метформина и DCA, но не под действием одного из препаратов. В клетках 66CL4 p-H2AX наблюдался только в клетках, обработанных DCA плюс метформин. Эти данные свидетельствуют о том, что сочетание ДКА и метформина усиливает окислительное повреждение ДНК.
В качестве второго уровня доказательств мы измерили перекисное окисление липидов, которое происходит, когда свободные радикалы повреждают липиды в клеточных мембранах. Для оценки степени окисления липидов после лечения ДКА и метформином использовались два отдельных метода. Во-первых, анализ TBARS, который измеряет побочные продукты перекисного окисления липидов и используется для оценки степени окислительного повреждения клеточных мембран, вызванного ROS. TBARS увеличился в клетках MCF7, обработанных либо метформином, либо только DCA, и наибольшее увеличение наблюдалось при совместном применении обоих препаратов (рис. 3c). Второй метод, использованный для оценки окисления липидов, — вестерн-блоттинг белковых аддуктов 4-гидроксиноненола (4-HNE), который образуется в результате окисления ненасыщенных жирных кислот в клеточных мембранах и образует ковалентные аддукты с белками, что свидетельствует об окислительном повреждении клеточных мембран. Митохондриальные экстракты клеток MCF7, обработанные DCA, метформином или обоими препаратами в течение 24 ч, были проанализированы на уровень белковых аддуктов 4-HNE (рис. 3d). Повышение уровня митохондриальных аддуктов 4-HNE наблюдалось только в клетках, обработанных комбинацией метформина и DCA. Это подтверждает полученные ранее результаты о том, что комбинация этих препаратов вызывает повышенное производство ROS.
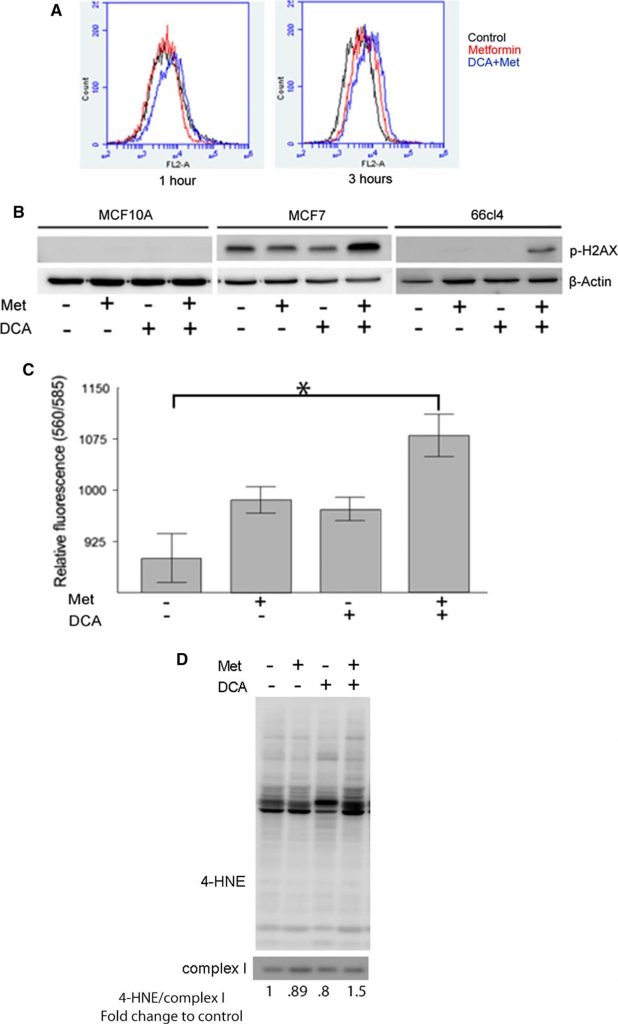
Наши предыдущие данные показывают, что в клетках, чувствительных к метформину, образуются большие вакуоли, которые, как было установлено с помощью визуализации GFP-связанных митохондрий и электронной микроскопии, являются структурно измененными митохондриями. Эти измененные митохондрии, по-видимому, конкретно связаны с PARP-зависимым путем клеточной смерти, который задерживается во времени по сравнению с апоптотической клеточной смертью [20]. Поскольку DCA синергирует с метформином в гибели клеток, было исследовано влияние DCA на структуру митохондрий. Была создана стабильная линия клеток MCF7, экспрессирующая зеленый флуоресцентный белок, нацеленный на митохондрии (pAcGFP1-Mito). Клетки обрабатывали в течение одного дня перед визуализацией с помощью фазового контраста и конфокальной микроскопии (рис. 4). В клетках, обработанных только метформином, наблюдались крупные митохондрии, как было показано ранее [20]. Напротив, клетки, обработанные DCA, имели более мелкие и многочисленные митохондрии. Дихлорацетат также предотвратил увеличение митохондрий, вызванное метформином. По фазовому контрасту увеличенные митохондрии были видны в клетках, обработанных метформином, и отсутствовали в клетках, обработанных комбинацией. Также была исследована вторая клеточная линия T47D, чувствительная к метформину. Опять же, увеличенные митохондрии наблюдались при лечении метформином, в то время как при комбинированном лечении они отсутствовали.
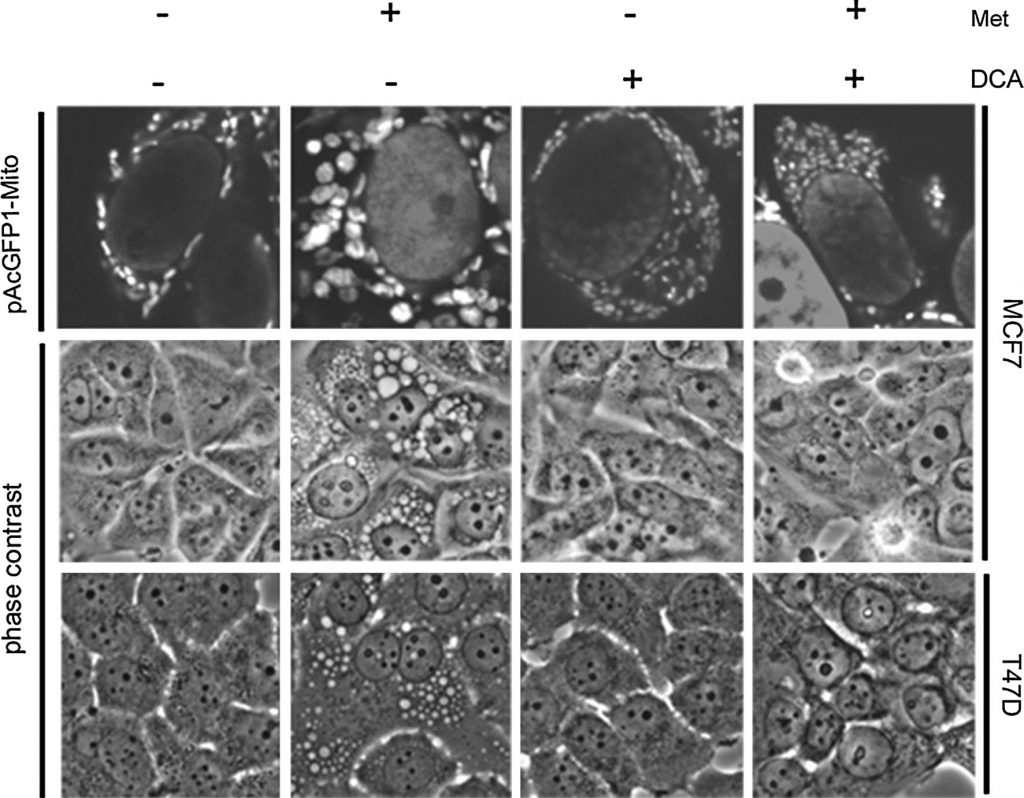
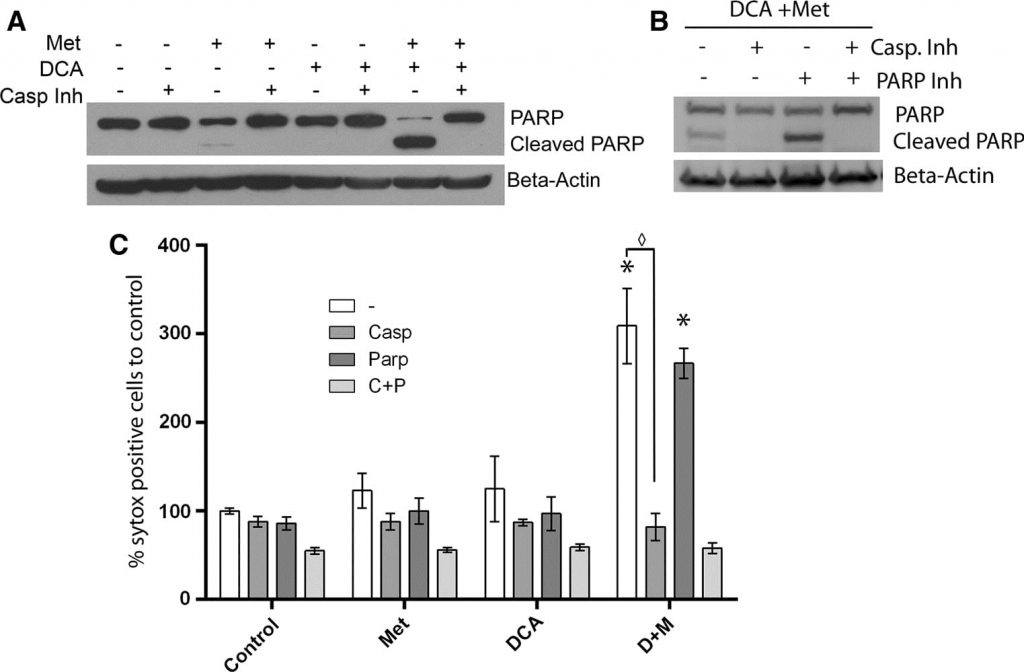
В нашей предыдущей работе мы обнаружили, что увеличение митохондрий коррелирует с PARP-зависимой гибелью клеток, которая была отсрочена во времени по сравнению с апоптотической гибелью клеток. Это побудило нас изучить особенности механизма клеточной гибели, индуцированной DCA в сочетании с метформином [20], поскольку увеличения митохондрий не наблюдается. Чтобы охарактеризовать гибель клеток, вызванную совместной обработкой метформином и DCA, клетки MCF7 обрабатывали необратимым ингибитором каспаз широкого спектра действия (Q-Val-Asp-OPh) вместе с DCA, метформином или обоими препаратами в течение одного дня. Вестерн-блоттинг (рис. 5a) выявил значительное увеличение расщепленного PARP и снижение интактного PARP в клетках, обработанных совместно DCA и метформином. Добавление ингибитора каспазы полностью блокировало расщепление PARP. Подобный эффект ранее наблюдался в клетках, обработанных только метформином в течение 2,5 дней [20]. Однако в однодневной временной точке один лишь метформин вызывал лишь незначительное увеличение расщепленного PARP. Таким образом, эти данные указывают на то, что каспазозависимый апоптоз ускоряется при добавлении DCA к метформину в этих клетках рака молочной железы. Для дальнейшего исследования того, происходит ли PARP-зависимая смерть при совместном лечении DCA и метформина, также использовался ингибитор PARP. Через два дня после лечения был проведен анализ SYTOX Green (рис. 5c) для количественной оценки гибели клеток, чтобы определить, имеет ли ингибитор PARP аддитивный эффект по отношению к ингибитору каспазы. Ингибитор PARP не оказывал защитного эффекта ни сам по себе, ни в сочетании с ингибитором каспазы. Вестерн-блот подтвердил этот вывод (рис. 5б) и показал, что ингибитор PARP не предотвращал расщепление PARP. Поскольку увеличение митохондрий связано с PARP-зависимой гибелью клеток в клетках, обработанных метформином, отсутствие увеличения митохондрий при добавлении DCA предполагает, что апоптоз происходит исключительно каспазозависимым образом.
Обсуждение
Метформин — давно известный препарат, используемый для лечения диабета второго типа. Аналогичным образом, DCA был изучен в клинических испытаниях для лечения молочнокислого ацидоза и митохондриальной недостаточности. Ранее было показано, что метформин замедляет пролиферацию и способствует гибели многих линий раковых клеток путем апоптоза [9, 10, 12, 32]. Метформин концентрируется в митохондриях, где он ингибирует комплекс I электронно-транспортной цепи, вызывая аномальный поток электронов к кислороду, производя супероксид в митохондриальном матриксе [22, рис. 3]. Производство реактивных форм кислорода является проблематичным для клеток рака молочной железы, поскольку многие из них имеют пониженную экспрессию супероксиддисмутазы марганца (MnSOD), которая обычно устраняет митохондриальный супероксид [33]. Хотя проапоптотическое и ингибирующее рост действие метформина на рак заслуживает внимания, наше наблюдение, что метформин усиливает гликолиз, о чем свидетельствует повышенное производство лактата in vitro [21], может ограничить его эффективность в качестве сольного агента. Поскольку высокий уровень лактата был описан как благоприятный для опухоли и ассоциируется с плохим клиническим исходом [26, 34-38], мы попытались ослабить индуцированное метформином производство лактата как средство потенциального повышения терапевтической эффективности. В этом исследовании мы обнаружили, что DCA помогает ослабить выработку лактата, индуцированную метформином, и что DCA достигает этого путем ингибирования PDK1.
Наши данные показывают, что DCA и метформин синергично вызывают апоптоз с большей скоростью, чем лечение только метформином. Наши данные также показывают, что DCA отвлекает пируват для использования в окислительном фосфорилировании, что приводит к накоплению ROS в контексте ингибирования комплекса I метформином. Мы пришли к выводу, что это увеличение производства ROS приводит к ускорению темпов апоптоза.
В нашей предыдущей работе мы показали, что метформин вызывает не только каспазозависимую апоптотическую гибель клеток, но и более позднюю PARP-зависимую гибель клеток, которая была выявлена при использовании панкаспазного ингибитора [20]. Эта форма клеточной гибели была связана с морфологическими изменениями в митохондриях. В данном исследовании DCA предотвратил морфологические изменения митохондрий, вызванные метформином, в то время как совместное лечение также привело к более высокой скорости гибели клеток. Кроме того, ингибирование пан-каспазы почти полностью предотвратило гибель клеток и устранило расщепление PARP, связанное с совместным лечением метформином и DCA. Все вместе это указывает на то, что механизм гибели клеток, запускаемый совместным лечением, — это каспазозависимый апоптоз, и апоптоз ускоряется по сравнению с метформином. Возможно, что этот апоптотический процесс слишком катастрофичен и не требует или не дает времени для увеличения митохондрий, связанного с PARP-зависимой гибелью клеток, вызванной только метформином.
Во время подготовки этой рукописи было опубликовано исследование Choi и Lim [39], в котором изучалась совместная обработка DCA и метформина в раковых клетках. Наши результаты подтверждают и значительно расширяют выводы этого исследования. В то время как исследование Choi и Lim было почти полностью сосредоточено на клетках HeLa, наше исследование демонстрирует эффективность комбинации DCA и метформина на нескольких клеточных линиях рака молочной железы. Важно отметить, что мы также показали, что эта комбинация препаратов не убивает нетрансформированные эпителиальные клетки молочной железы, MCF10A, в тех же условиях, в которых препараты убивают раковые клетки. Мы провели тщательное титрование, которое продемонстрировало очень сильный синергизм препаратов в индуцировании клеточной смерти в клетках рака молочной железы. Эти результаты показывают, что DCA будет эффективен при концентрациях, которые могут быть достигнуты терапевтически у людей [27, 29], и при гораздо более низких уровнях, чем использовались в экспериментах в исследовании Choi и Lim (10-20 мМ) [39]. В идеале максимальная доза DCA, используемая для лечения раковых клеток, должна быть достаточно низкой, чтобы избежать неблагоприятных эффектов, таких как нейропатия. В исследовании Michelakis и др. достигнутый уровень DCA в сыворотке крови составил 0,5 мМ у пациентов с глиобластомой, принимавших 6,25 мг/кг перорально дважды в день [28]. Более высокие дозы, такие как 25 мг/кг, связаны с дозозависимой нейропатией. Следовательно, клинически значимые концентрации, скорее всего, будут находиться в диапазоне 0,5-5 мМ.
Как представленные здесь результаты, так и результаты Choi и Lim [39] показали, что гибель клеток, вызванная комбинацией DCA и метформина, связана с окислительным стрессом. Далее мы показали, что это связано с индуцированной метформином продукцией супероксида митохондриями. Дихлорацетат еще больше увеличивает производство супероксида митохондриями в присутствии метформина, что приводит к окислительному повреждению митохондрий, клеточных липидов и ДНК. Новые комбинации, такие как метформин и DCA, обещают расширить терапию рака молочной железы.
Благодарности
Эта статья была поддержана грантами Susan G. Komen for the Cure (KG100497) и Национального института рака Национальных институтов здоровья (1R01CA180033). Проточная цитометрия и центры визуализации были поддержаны грантами COBRE P20GM103548 и P20GM103620 от Национального института общих медицинских наук при Национальном институте здоровья.
Конфликт интересов
Авторы заявляют, что у них нет конфликта интересов.
Этические стандарты
Авторы заявляют, что эксперименты соответствуют действующему законодательству страны, в которой они проводились.
ССЫЛКИ
1 Warburg O (1956) On the origin of cancer cells. Science 123(80):309-314. doi:10.1016/S0306-9877(96)90136-X
2 DeBerardinis RJ, Lum JJ, Hatzivassiliou G, Thompson CB (2008) The biology of cancer: metabolic reprogramming fuels cell growth and proliferation. Cell Metab 7:11-20. doi:10.1016/j.cmet.2007.10.002
3 Zamzami N, Kroemer G (2001) The mitochondrion in apoptosis : how Pandora’ s box opens. Nat Rev Mol Cell Biol 2:67-71. doi:10.1038/35048073
4 Gottfried E, Kunz-Schughart LA, Ebner S et al (2006) Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Кровь 107:2013-2021. doi:10.1182/blood-2005-05-1795
5 Fischer K, Hoffmann P, Voelkl S et al (2007) Inhibitory effect of tumor cell-derived lactic acid on human T cells. Кровь 109:3812-3819. doi:10.1182/blood-2006-07-035972
6 Jiralerspong S, Palla SL, Giordano SH et al (2009) Метформин и патологические полные ответы на неоадъювантную химиотерапию у больных раком молочной железы с диабетом. J Clin Oncol 27:3297-3302. doi:10.1200/JCO.2009.19.6410
7 Evans JMM, Donnelly LA, Emslie-Smith AM et al (2005) Metformin and reduced risk of cancer in diabetic patients. BMJ 330:1303-1304. doi:10.1136/bmj.38393.572188.EB
8 Libby G, Donnelly LA, Donnan PT et al (2009) New users of Metformin are at low risk of incident cancer. Diabetes Care 32:1620-1625. doi:10.2337/dc08-2175
9 Zakikhani M, Dowling R, Fantus IG et al (2006) Метформин является AMP-киназозависимым ингибитором роста клеток рака молочной железы. Cancer Res 66:10269-10273. doi:10.1158/0008-5472.CAN-06-1500
10 Isakovic A, Harhaji L, Stevanovic D et al (2007) Dual antiglioma action of metformin: cell cycle arrest and mitochondria-dependent apoptosis. Cell Mol Life Sci 64:1290-1302. doi:10.1007/s00018-007-7080-4
11 Hadad SM, Appleyard V, Thompson AM (2008) Therapeutic metformin/AMPK activation promotes the angiogenic phenotype in the ERα negative MDA-MB-435 breast cancer model. Breast Cancer Res Treat 114:391. doi:10.1007/s10549-008-0016-3
12 Buzzai M, Jones RG, Amaravadi RK et al (2007) Системное лечение противодиабетическим препаратом метформином избирательно ухудшает рост p53-дефицитных опухолевых клеток. Cancer Res 67:6745-6752. doi:10.1158/0008-5472.CAN-06-4447
13 Sarbassov DD, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17:596-603. doi:10.1016/j.ceb.2005.09.009
14 Zhuang Y, Miskimins WK (2008) Арест клеточного цикла в клетках рака молочной железы, обработанных метформином, включает активацию AMPK, даунрегуляцию циклина D1 и требует p27Kip1 или p21Cip1. J Mol Signal 3:1-11. doi:10.1186/1750-2187-3-18
15 Hinke SA, Martens GA, Cai Y et al (2007) Methyl succinate antagonises biguanide-induced AMPK-activation and death of pancreatic beta-cells through restoration of mitochondrial electron transfer. Br J Pharmacol 150:1031-1043. doi:10.1038/sj.bjp.0707189
16 Zou M-H, Kirkpatrick SS, Davis BJ et al (2004) Активация AMP-активируемой протеинкиназы противодиабетическим препаратом метформином in vivo. Роль митохондриальных реактивных видов азота. J Biol Chem 279:43940-43951. doi:10.1074/jbc.M404421200
17 Owen MR, Doran E, Halestrap AP (2000) Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J 614:607-614. doi:10.1042/0264-6021:3480607
18 Carvalho C, Correia S, Santos MS et al (2008) Metformin promotes isolated rat liver mitochondria impairment. Mol Cell Biochem 308:75-83. doi:10.1007/s11010-007-9614-3
19d Kefas BA, Cai Y, Kerckhofs K et al (2004) Metformin-induced stimulation of AMP-activated protein kinase in beta-cells impairs their glucose responsiveness and can lead to apoptosis. Biochem Pharmacol 68:409-416. doi:10.1016/j.bcp.2004.04.003
20 Zhuang Y, Miskimins WK (2012) Метформин вызывает как каспазозависимую, так и поли(ADP-рибоза) полимеразозависимую клеточную смерть в клетках рака молочной железы. Mol Cancer Res 9:603-615. doi:10.1158/1541-7786.MCR-10-0343.Metformin
21 Bailey CJ, Wilcock C, Day C (1992) Effect of metformin on glucose metabolism in the splanchnic bed. Br J Pharmacol 105:1009-1013
22StacpoolePW, Lorenz AC, Thomas RG, Harman EM (1988) Dichloroacetate in the treatment of lactic acidosis. Ann Intern Med 108:58-63. doi:10.1056/NEJM198308183090702
23 Whitehouse BSUE, Cooper RH, Randle PJ (1974) Mechanism of activation of pyruvate dehydrogenase by dichloroacetate and other halogenated carboxylic acids. Biochem J 141:761-774. doi:10.1056/NEJM198308183090702
24 Hussien R, Brooks GA (2011) Экспрессия изоформ лактаттранспортера и лактатдегидрогеназы в митохондриях и плазматической мембране в клеточных линиях рака молочной железы. Physiol Genomics 43:255-264. doi:10.1152/physiolgenomics.00177.2010
25 Kim J, Tchernyshyov I, Semenza GL, Dang CV (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 3:177-185. doi:10.1016/j.cmet.2006.02.002
26 Wigfield SM, Winter SC, Giatromanolaki A et al (2008) PDK-1 регулирует производство лактата при гипоксии и связан с плохим прогнозом при сквамозном раке головы и шеи. Br J Cancer 98:1975-1984. doi:10.1038/sj.bjc.6604356
27 Stacpoole PW, Kurtz TL, Han Z, Langaee T (2008) Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev 60:1478-1487. doi:10.1016/j.addr.2008.02.014
28 Michelakis ED, Sutendra G, Dromparis P et al (2010) Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2:31ra34. doi:10.1126/scitranslmed.3000677
29 Mori M, Yamagata T, Goto T et al (2004) Лечение дихлорацетатом митохондриальной цитопатии: долгосрочные эффекты в MELAS. Brain Develop 26:453-458. doi:10.1016/j.braindev.2003.12.009
30 Chou T (2010) drug combination studies and their synergy quantification using the Chou-Talalay Method drug combination studies and their synergy quantification using the Chou-Talalay Method. Cancer Res 70:440-446. doi:10.1158/0008-5472.CAN-09-1947
31 Mah L, Karagiannis TC (2010) γH2AX: чувствительный молекулярный маркер повреждения и восстановления ДНК. Лейкемия 24:679-686. doi:10.1038/leu.2010.6
32 Alimova IN, Liu B, Fan Z et al (2009) Метформин ингибирует рост клеток рака молочной железы, образование колоний и индуцирует остановку клеточного цикла in vitro. Cell Cycle 8:909-915. doi:7933 [pii]
33 Soini Y, Vakkala M, Kahlos K et al (2001) Экспрессия MnSOD реже встречается в опухолевых клетках инвазивных карцином молочной железы, чем в карциномах in situ или неопластических эпителиальных клетках молочной железы. J Pathol 195:156-162. doi:10.1002/path.946
34 Walenta S, Wetterling M, Lehrke M et al. (2000) High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers high lactate levels predictibility of metastases, tumor recurrence, and restricted patient survival in human cervical. Cancer Res 60:916-921
35 Leek R, Harris AL (2009) Экспрессия лактатдегидрогеназы 5 в плоскоклеточном раке головы и шеи связана с прогнозом после радикальной или послеоперационной лучевой терапии. Oncology 77:285-292. doi:10.1159/000259260
36 Isidoro A, Casado E, Redondo A et al (2005) Breast carcinomas fulfill the Warburg hypothesis and provide metabolic markers of cancer prognosis. Carcinogenesis 26:2095-2104. doi:10.1093/carcin/bgi188
37 Isidoro A, Martínez M, Fernández PL et al (2004) Изменение биоэнергетического фенотипа митохондрий является отличительной чертой рака молочной железы, желудка, легких и пищевода. Biochem J 378:17-20. doi:10.1042/BJ20031541
38 Walenta S, Schroeder T (2004) Лактат в солидных злокачественных опухолях: потенциальная основа метаболической классификации в клинической онкологии. Curr Med Chem 11:2195-2204
39 Choi YW, Lim IK (2014) Sensitization of metformin-cytotoxicity by dichloroacetate through reprogramming glucose metabolism in cancer cells. Cancer Lett 346:300-308. doi:10.1016/j.canlet.2014.01.015
Связанный контент: