Received: 14 сентября 2020 г. Принято: 4 декабря 2020 г. Опубликовано: 9 декабря 2020 г
Аннотация
Митохондриальный метаболизм является привлекательной мишенью для терапии рака. Перепрограммирование метаболических путей может потенциально сенсибилизировать опухоли с ограниченными возможностями лечения, такие как трижды негативный рак молочной железы (TNBC), к химио- и/или радиотерапии. Дихлорацетат (DCA) является специфическим ингибитором киназы пируватдегидрогеназы (PDK), что приводит к усиленному производству реактивных видов кислорода (ROS). ROS являются первичными эффекторными молекулами радиации, и увеличение их количества усиливает радиоответ. В данном исследовании мы оценили влияние DCA и радиотерапии на две клеточные линии TNBC, а именно EMT6 и 4T1, в аэробных и гипоксических условиях. Как и ожидалось, лечение DCA уменьшило фосфорилированную пируватдегидрогеназу (PDH) и снизило скорость внеклеточного окисления (ECAR) и производство лактата. Примечательно, что обработка DCA привела к значительному увеличению продукции ROS (до 15 раз) в гипоксических раковых клетках, но не в аэробных клетках. Последовательно, DCA радиосенсибилизировал гипоксические опухолевые клетки и 3D сфероиды, оставляя неизменной внутреннюю радиочувствительность опухолевых клеток. Наши результаты свидетельствуют о том, что, хотя DCA описывается как препарат, стимулирующий окислительное фосфорилирование (OXPHOS), он также может усиливать гипоксические радиореактивные реакции. Таким образом, данное исследование открывает путь для воздействия на митохондриальный метаболизм гипоксических раковых клеток, в частности, для борьбы с радиорезистентностью.
Рак молочной железы является наиболее распространенным онкологическим заболеванием у женщин во всем мире и ежегодно приводит к 627 000 смертей [1]. За последние десятилетия был достигнут значительный прогресс в лечении рака молочной железы. Однако для пациентов с тройным негативным/базально-подобным раком молочной железы доступны лишь ограниченные методы лечения [2,3,4]. Стандартом лечения рака молочной железы высокого риска является неоадъювантная химиотерапия и хирургическое вмешательство с последующим послеоперационным облучением всей груди/грудной стенки. В настоящее время исследователи фокусируются либо на гипофракционировании адъювантной радиотерапии (исследование FAST-Forward [5], либо на комбинации химиотерапии с предоперационной радиотерапией. Применение предоперационной радиотерапии может привести к улучшению выживаемости без болезни и качества жизни [6,7,8,9,10,11].
Основным эффектом радиации, особенно для излучения с низкой линейной передачей энергии, является индукция реактивных форм кислорода (ROS). Во время радиотерапии ROS образуются в результате радиолиза воды во внеклеточной среде, которые являются токсичными для опухолевых клеток и близлежащих нормальных тканей. Около двух третей радиационно-индуцированных повреждений ДНК объясняются ROS в клетках млекопитающих [12]. Ответ клеток на радиационно-индуцированное повреждение ДНК сильно зависит от присутствия кислорода. Молекулы кислорода действительно могут исправлять повреждения ДНК, вызванные свободными радикалами. Это называется «гипотезой фиксации кислорода» [12,13]. В отсутствие кислорода радикалы ДНК восстанавливаются соединениями, содержащими сульфгидрильные группы, которые восстанавливают ДНК до первоначальной формы. Согласно этой гипотезе, гипоксия, определяемая низким уровнем кислорода в опухоли, является одной из основных причин клинической неудачи радиотерапии [14,15]. Гипоксия является общей характеристикой микроокружения опухоли. ROS и гипоксия — два фактора с противоположным влиянием на радиореактивность опухоли [16]. Общепринятая гипотеза гласила, что в гипоксических областях опухоли возникает меньший окислительный стресс из-за нехватки кислорода — субстрата ROS. Однако последние данные показали, что в гипоксических условиях клетки генерируют больше ROS, в основном через митохондриальный метаболизм [17,18,19,20].
Определяющей отличительной чертой опухолевых клеток является способность изменять свой метаболизм, обеспечивая их энергией и метаболитами, необходимыми для роста и выживания в условиях недостатка питательных веществ и кислорода. Однако в присутствии O2 раковые клетки также адаптируют свой метаболизм к гликолизу, перенаправляя митохондриальное окисление пирувата на производство лактата [21,22]. Этот эффект называют эффектом Варбурга. Последние сообщения показывают, что эффект Варбурга связан с устойчивостью к цитотоксическому стрессу, вызванному химиотерапией или радиотерапией [23,24,25,26,27]. Таким образом, методы лечения, блокирующие или снижающие гликолитический метаболизм, могут повысить чувствительность опухолевых клеток к радиотерапии.
В гипоксических условиях гипоксия-индуцибельный фактор 1-альфа (HIF1α) вызывает увеличение экспрессии киназ пируватдегидрогеназы (PDK1-4) [28]. Эти ферменты отвечают за переключение метаболизма в митохондриях путем регулирования статуса фосфорилирования (т.е. состояния активности) пируватдегидрогеназы (ПДГ), которая является основным белком-привратником между гликолизом и митохондриальным окислительным фосфорилированием (OXPHOS). Дихлорацетат (DCA), низкомолекулярный ингибитор PDK, может обратить эффект Варбурга, активируя PDH и перенаправляя метаболизм пирувата обратно в митохондрии. Ингибирование PDK с помощью DCA используется для лечения молочнокислого ацидоза и наследственных митохондриальных заболеваний [29,30]. В целом, эти наблюдения позволили рассматривать DCA как потенциальный противораковый препарат [30,31].
Было продемонстрировано, что DCA повышает радиочувствительность клеток рака толстой кишки и простаты, а также плоскоклеточной карциномы пищевода и опухолей глиобластомы [32,33,34,35]. Однако аналогичных исследований на клетках рака молочной железы не проводилось. Основной механизм радиосенсибилизации в этих моделях был связан с окислительным стрессом. В то же время, остановка клеточного цикла на фазе G2-M и снижение резервных возможностей митохондрий также способствовали радиосенсибилизирующему эффекту. На основании ранее описанных результатов в настоящее время проводятся два клинических испытания (одно при карциноме головы и шеи и одно при глиобластоме), изучающие противоопухолевые эффекты комбинированного лечения DCA с радиотерапией[36,37]. Все доклинические исследования проводились в аэробных условиях, но нет данных об эффектах лечения DCA в гипоксических условиях. Поэтому в настоящем исследовании мы сначала проверили гипотезу о том, что DCA снижает уровень лактата и переключает метаболизм с гликолитического фенотипа на OXPHOS. Затем мы определили, может ли DCA радиосенсибилизировать гипоксические клетки рака молочной железы, и изучили лежащие в основе этого механизмы. Результаты этого исследования могут иметь важное значение для клинических испытаний, направленных на использование ингибиторов PDK для улучшения радиоответа у больных раком молочной железы.
Результаты
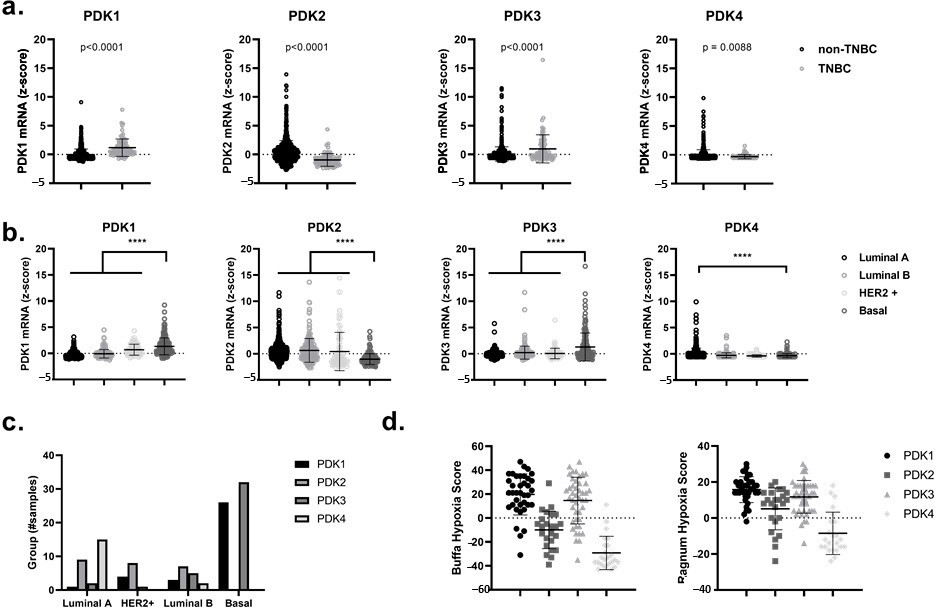
Высокая экспрессия PDK1 и PDK3 при трижды негативном раке молочной железы (TNBC) и базальноподобном раке молочной железы коррелирует с сигнатурой генов, связанных с гипоксией Прежде всего, с помощью онлайнового и общедоступного портала cBioPortal for Cancer Genomics, мы проанализировали уровни мРНК четырех различных изомеров PDK, а именно PDK1, PDK2, PDK3 и PDK4, в данных, полученных от пациентов из набора данных TCGA (PanCancer Atlas and Cell 2015) [38,39]. Мы показали значительное увеличение (p < 0,0001) экспрессии PDK1 и PDK3 в TNBC по сравнению с не-TNBC и в базальноподобных раках молочной железы по сравнению с другими подтипами, такими как люминальный A, люминальный B и HER2-обогащенный рак молочной железы (Рисунок 1a-c). Примечательно, что повышение уровня PDK1 и PDK3 у больных раком молочной железы может коррелировать с более высокими показателями гипоксии по шкалам Ragnum и Buffa [40,41] (рис. 1d). Эти оценки гипоксии основаны на дифференциальной экспрессии специфических генов, связанных с гипоксией, и могут быть свободно доступны через cBioPortal for Cancer Genomics. Наблюдаемое повышение уровня PDK1 и PDK3 в базальноподобных раках молочной железы и их корреляция с гипоксическим фенотипом (который связан с активацией HIF1α-зависимой транскрипционной программы) позволяет предположить, что использование ингибиторов PDK может служить привлекательным терапевтическим методом для целей гипоксической радиосенсибилизации [42,43,44].
Рисунок 1. Повышенная экспрессия мРНК PDK1 и PDK3 коррелирует с генными подписями, связанными с гипоксией, в трижды негативном и базальноподобном раке молочной железы.(a) Экспрессия мРНК PDK1-4 (RNA-seq) в подтипе нетрижды-негативного рака молочной железы (нет-ТНБК) по сравнению со случаями ТНБК.(b) Экспрессия мРНК PDK1-4 (RNA-seq) в различных подтипах рака молочной железы, классифицированных по классификации Pam50 набора данных TCGA (люминальный A, люминальный B, HER2+ и базальный).(c) Количество образцов в наборе данных TCGA, имеющих z-scorez > 2 для PDK1-4 в различных подтипах рака молочной железы, классифицированных по системе классификации Pam50.(d) Оценка гипоксии по шкале Баффа (слева) и оценка гипоксии по шкале Рагнума (справа) коррелирует со сверхэкспрессией(z > 2) мРНК PDK1-4. **** p < 0.0001.
DCA снижал фосфорилированную PDH, уровень внеклеточного лактата и скорость внеклеточного закисления (ECAR) Мы начали наши эксперименты in vitro с проведения анализа жизнеспособности (Рисунок S1a,b), чтобы определить ингибирующие рост свойства DCA. DCA снижал жизнеспособность клеток в дозозависимой манере в раковых клетках EMT6 и 4T1 независимо от статуса O2 (Рисунок S1a). Анализ на пролиферацию также показал, что увеличение концентрации DCA привело к тому, что мы наблюдали переход от задержки роста к цитостатическому и даже цитотоксическому эффекту (Рисунок S1b).
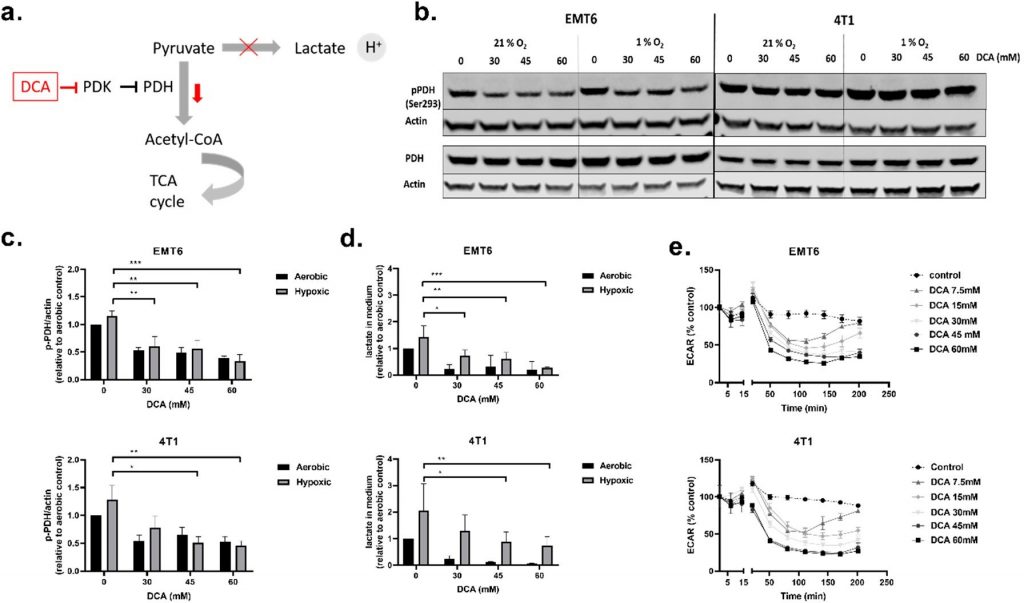
Далее мы исследовали влияние DCA на активность PDK1-4 и метаболизм клеток TNBC. PDH является ключевым белком-привратником гликолиза и митохондриальной OXPHOS, то есть PDH катализирует лимитирующее по скорости декарбоксилирование пирувата в ацетил-КоА. PDH ингибируется путем фосфорилирования (на Ser293) PDK, и это ингибирование может быть отменено путем дефосфорилирования фосфатазой пируватдегидрогеназы (PDP) [45,46](Рисунок 2a). При приемлемых дозах токсичности (30 мМ, 45 мМ и 60 мМ) мы оценили влияние ДКА на активность ПДГ, измерив уровень фосфорилированной ПДГ (p-PDH), лактата во внеклеточной среде и ЭКАР клеток в режиме реального времени (Рисунок 2b-e). Все три дозы DCA снижали количество p-PDH в клетках EMT6 и 4T1 дозозависимым образом как в условиях кислорода, так и в условиях гипоксии. Снижение p-PDH было значительным для всех доз DCA в EMT6, тогда как в 4T1 только 45 мМ и 60 мМ значительно снижали p-PDH в гипоксических условиях (Рисунок 2b,c). Оценивая эффект более низких доз DCA, мы обнаружили, что в клетках EMT6 количество p-PDH начинает снижаться при обработке 3 мМ DCA, а в клетках 4T1 — при обработке 10 мМ DCA (Рисунок S2a,b). Количество лактата в среде было повышено в гипоксических условиях по сравнению с аэробными условиями в обеих клеточных линиях, что подтверждает чистое увеличение гликолитического оборота в клетках, лишенных О2 [47,48]. В соответствии с результатами Вестерн-блота, обработка DCA привела к дозозависимому снижению лактата в среде для обеих клеточных линий (Рисунок 2d). Хотя снижение высвобождения лактата было значительным в аэробных условиях, дозозависимое снижение продукции конечного гликолитического продукта наблюдалось и в условиях гипоксии. Наконец, DCA в дозе 7,5 мМ вызывал зависимое от времени снижение ECAR как в EMT6, так и в 4T1 (Рисунок 2e). Первоначальное снижение ECAR после лечения дозами ниже 30 мМ DCA было компенсировано через 2,5 ч в EMT6. В 4T1 мы наблюдали тот же эффект при дозах ДКА ниже 15 мМ. Эти результаты показывают, что обработка клеточных линий мышиных TNBC с помощью DCA ингибирует фосфорилирование PDH и снижает степень гликолиза как в аэробных, так и в гипоксических условиях.
Рисунок 2. Дихлорацетат (ДХА) снижает фосфорилированную пируватдегидрогеназу (PDH), лактат во внеклеточной среде и ECAR клеток TNBC.(a) Схема, показывающая влияние DCA на киназы пируватдегидрогеназы (PDK) и последующие эффекты.(b) Репрезентативный вестерн-блот p-PDH (Ser293) и общего PDH в клетках 4T1 и EMT6 после обработки DCA (30 мМ, 45 мМ и 60 мМ) в аэробных и гипоксических условиях.(c) Нормализованная относительная экспрессия p-PDH (Ser293) в клетках 4T1 и EMT6 после обработки DCA (30 мМ, 45 мМ и 60 мМ) в аэробных и гипоксических условиях.(d) После обработки DCA с указанными концентрациями в аэробных или гипоксических условиях лактат измеряли с помощью L-лактатного анализа.(e) ECAR клеток EMT6 и 4T1 измеряли с течением времени после введения указанных концентраций DCA с помощью анализатора Seahorse. Скорость внеклеточного закисления (ECAR) выражали в процентах по отношению к контролю. * p < 0,05, ** p < 0,01, *** p < 0,001.
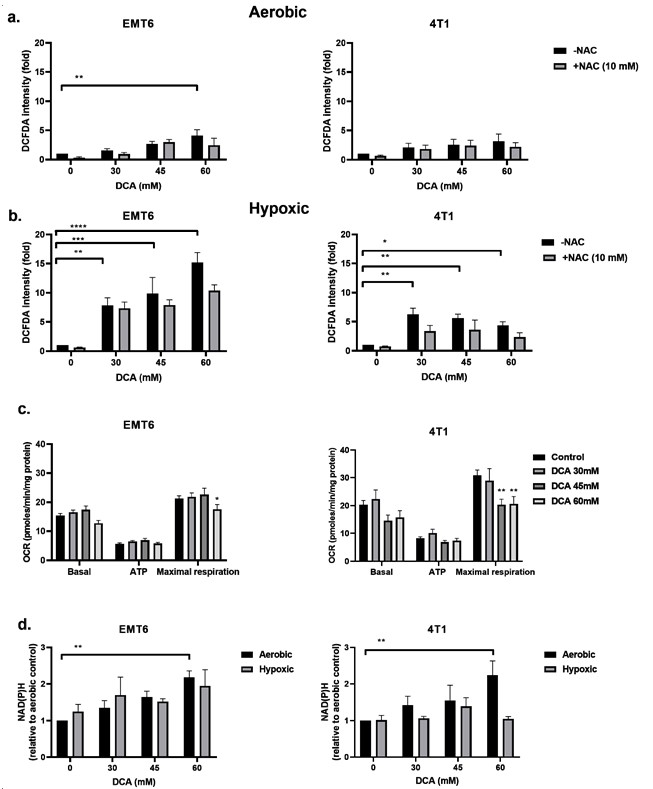
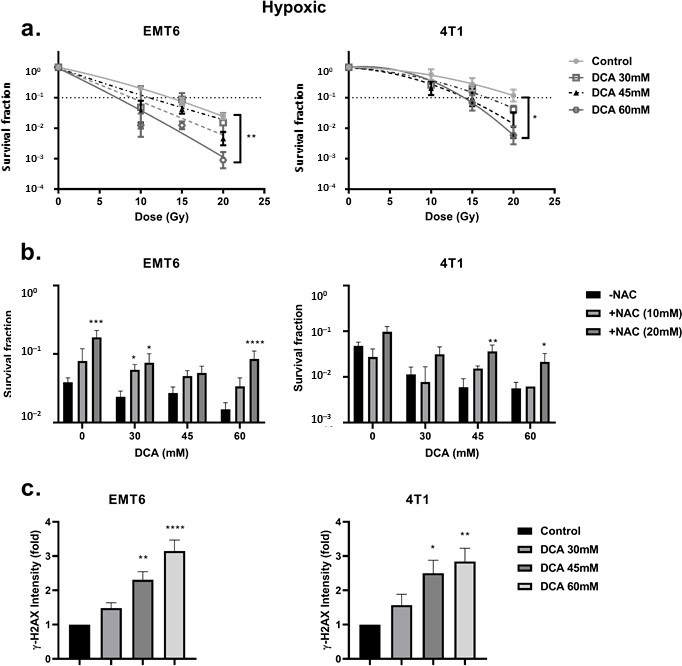
DCA индуцирует продукцию ROS в гипоксических раковых клетках Ингибирование PDK и активация PDH связаны с повышением уровня внутриклеточной ROS [49,50]. ROS имеет ключевое значение в возникновении повреждений ДНК после облучения. Мы исследовали уровень ROS в клетках EMT6 и 4T1 в аэробных и гипоксических условиях с помощью зонда CM-H2DCFDA. Как показано на рисунке 3a,b, DCA вызывал дозозависимое производство ROS в клетках EMT6 и 4T1. В аэробных условиях только самая высокая доза (60 мМ) DCA вызывала значительное увеличение ROS в пять раз по сравнению с контролем в клетках EMT6. В клетках 4T1 в аэробных условиях не было обнаружено значительного повышения уровня ROS. Повышение уровня ROS было частично нейтрализовано добавлением N-ацетил-цистеина (NAC), разрушающего ROS. В гипоксических условиях мы продемонстрировали дозозависимое увеличение ROS до 15 раз в клетках EMT6 и до 5 раз в клетках 4T1; самая низкая доза DCA действительно приводила к значительному увеличению ROS на обоих типах клеток (рис. 3b).
Мы предположили, что повышенная продукция ROS в условиях гипоксии может быть результатом совместного воздействия изменений в электронно-транспортной цепи митохондрий (из-за уменьшения O2 в качестве конечного акцептора электронов) и вынужденного окислительного метаболизма пирувата под действием DCA. Используя анализатор Seahorse, мы обнаружили, что DCA не влияет на базальное дыхание и производство АТФ, но значительно снижает максимальную дыхательную способность в опухолевых клетках EMT6 и 4T1 (Рисунок 3c). Другая возможность заключается в том, что DCA повышает уровень NAD(P)H, что связано с увеличением продукции ROS. Мы увидели, что в аэробных условиях при обработке DCA наблюдалось дозозависимое увеличение NAD(P)H в клетках EMT6 и 4T1 (Рисунок 3д). В гипоксических условиях мы продемонстрировали возможное увеличение NAD(P)H в клетках EMT6, обработанных 60 мМ DCA, но без увеличения в клетках 4T1 (Рисунок3d). Вместе с приведенными выше данными по ROS, эти результаты свидетельствуют о том, что, хотя митохондриальный метаболизм по-прежнему сохраняется, локальное увеличение продукции ROS при воздействии DCA может изменить целостность и функцию митохондрий.
Рисунок 3. DCA индуцирует производство ROS в гипоксических клетках, перегружая митохондриальный метаболизм. Клетки EMT6 и 4T1 обрабатывали DCA в течение ночи при указанных концентрациях, а N-ацетил-цистеин (NAC) (10 мМ) добавляли за 1 ч до и во время обработки. Генерацию ROS измеряли методом проточной цитометрии с использованием зонда CM-H2DCFDA в аэробных(а) и гипоксических(б) условиях.(в) Измерения скорости потребления кислорода (СПК) были получены со временем с помощью анализатора Seahorse с использованием митохондриального стресс-теста. Для получения подробной информации о цепи переноса электронов в митохондриях последовательно добавляли специфические ингибиторы, состоящие из олигомицина, FCCP, ротенона и антимицина А. По этим результатам рассчитывали базальный митохондриальный OCR, ATP-связанный OCR и максимальный OCR.(d) NAD(P)H измеряли методом проточной цитометрии с использованием датчика JZL1707 NAD(P)H в аэробных и гипоксических условиях. * p < 0,05, ** p < 0,01, *** p < 0,001, **** p < 0,0001.
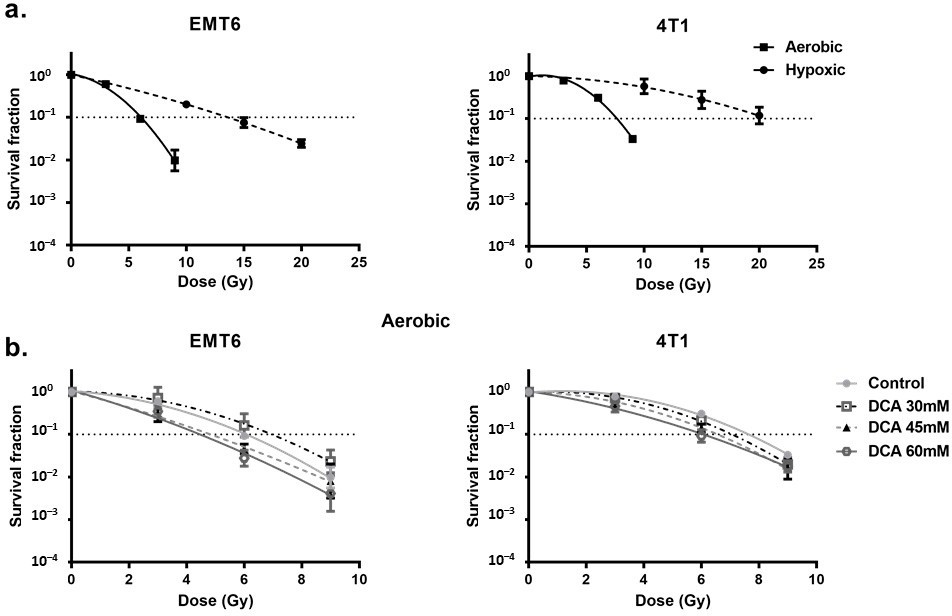
DCA радиосенсибилизирует гипоксические клетки, опосредованно ROS Сначала мы рассмотрели радиорезистентность клеток, вызванную гипоксией. При сравнении гипоксических и аэробных условий мы обнаружили сильно ослабленную радиореактивность: коэффициент усиления кислорода составил 2,7 и 2,3 для опухолевых клеток EMT6 и 4T1, соответственно (рис. 4a). В этих условиях мы наблюдали, что обработка DCA вызывала небольшой эффект внутренней радиосенсибилизации в клетках EMT6, но не в клетках 4T1 (Рисунок 4b). Интересно, что в соответствии с результатами генерации ROS в гипоксических условиях, 60 мМ DCA значительно (p < 0,05) преодолевал гипоксическую радиорезистентность с коэффициентами усиления 2,3 и 1,5 при 60 мМ для опухолевых клеток EMT6 и 4T1, соответственно (Рисунок 5a). Радиосенсибилизирующий эффект был отменен NAC в клетках EMT6 и 4T1 (рис. 5b). Основной причиной радиационно-индуцированной гибели клеток под действием ROS является индукция двунитевых разрывов ДНК (ds-ДНК) [12,51]. Поэтому мы исследовали повреждение ds-ДНК после обработки DCA путем количественной оценки состояния фосфорилирования γH2AX в гипоксических условиях. Как показано на рисунке 5c, ДКА увеличивал образование повреждений ds-ДНК в EMT6 и 4T1 дозозависимым образом.
Рисунок 4. DCA обладает небольшим радиосенсибилизирующим действием на аэробные опухолевые клетки. Клетки EMT6 и 4T1 обрабатывали DCA в течение ночи при указанных концентрациях.(a) Радиочувствительность клеток EMT6 и 4T1 в аэробных или гипоксических условиях.(b) Радиосенсибилизирующий эффект DCA в аэробных условиях. Рисунок 5. DCA радиосенсибилизирует гипоксические опухолевые клетки через регуляцию ROS. Клетки EMT6 и 4T1 обрабатывали DCA в течение ночи в указанных концентрациях.(a) Радиосенсибилизирующий эффект DCA в гипоксических условиях оценивался по анализу образования колоний.(b) Предварительная обработка NAC (10 мМ и 20 мМ) отменяла радиосенсибилизирующий эффект различных концентраций DCA при 15 Гр для EMT6 и 20 Гр для 4T1.(c) Двухцепочечные разрывы ДНК анализировались методом проточной цитометрии с использованием окрашивания gammaH2AX. * p < 0,05, ** p < 0,01, *** p < 0,001, **** p < 0,0001.
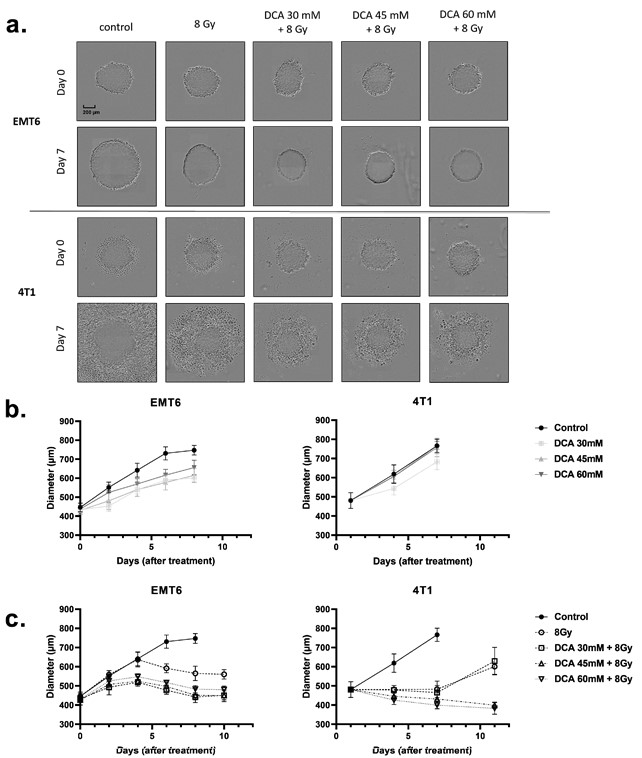
DCA радиосенсибилизирует трехмерные клеточные культуры (сфероиды) Вышеизложенные результаты заставили нас изучить, может ли DCA также улучшить радиочувствительность трехмерных (3D) моделей клеточных культур (рис. 6a-c), которые лучше имитируют физико-химические свойства опухолевой микросреды, включая градиенты кислорода. Используя сфероиды, полученные из культур клеток EMT6 и 4T1, мы измерили рост сфероидов после обработки DCA и радиотерапии (Рисунок 6a). Обработка только DCA привела к небольшой задержке роста клеток EMT6, но не изменила рост сфероидов 4T1 (рис. 6b). Облучение в 8 Гр уменьшило рост опухолевых сфероидов, и этот эффект еще больше усилился при сочетании с обработкой DCA (Рисунок 6a,c). Следует отметить, что в то время как цитотоксический эффект наблюдался в сфероидах 4T1 (о чем свидетельствовали ореолы мертвых клеток), цитостатический эффект наблюдался в сфероидах EMT6 (Рисунок 6a).
Рисунок 6. Лечение DCA и облучение 8 Гр снижает рост опухолевых сфероидов EMT6 и 4T1.(a) Репрезентативные фотографии и(b,c) зависимый от времени рост сфероидов EMT6 и 4T1 при обработке DCA в указанных концентрациях и в сочетании с радиотерапией 8 Гр.
Комбинация DCA и радиотерапии не задерживает рост опухоли in vivo Далее мы проверили, можно ли подтвердить in vitro преимущества комбинации DCA с радиотерапией in vivo (рис. S3a-d). Мыши, которым вводили клетки рака молочной железы EMT6 или 4T1, подвергались однократному (12 Гр или 15 Гр, соответственно) (Рисунок S3a,c) или фракционированному (5*4 Гр или 5*6 Гр, соответственно) облучению (Рисунок S3b,d). Однократные и фракционированные дозы облучения для каждого типа опухоли сходны по биологической эффективной дозе (BED) и различаются в зависимости от присущей радиочувствительности используемых клеточных линий. Важно отметить, что введение DCA как внутривенно, так и внутривенно в течение 10 дней было безопасным и не вызывало заметной токсичности (рис. S4a-d). Только облучение задерживало рост опухоли в EMT6 на семь дней при однократной фракции и на четыре дня при фракционированном облучении (Рисунок S3a,b). В опухолях 4T1 облучение задерживало рост опухоли на пять дней при одиночном облучении и на 10 дней при фракционированном облучении, как и ожидалось (рис. S3c,d). DCA (300 мг/кг), вводимый внутрибрюшинно (ip) или интратуморально (it), не задерживал рост опухоли, как и комбинация DCA с облучением (рис. S3a-d). Далее мы проверили, может ли лечение DCA вызывать гипоксию в опухолях (Рисунок S3e,f). Хотя степень гипоксии, окрашенной пимонидазолом, в опухолях EMT6 не изменилась, в опухолях 4T1 в ответ на ДКА наблюдалась тенденция к уменьшению гипоксии.
Обсуждение
Целью данного исследования было выяснить, может ли воздействие DCA на митохондриальный метаболизм сенсибилизировать клетки TNBC/базальноподобного рака молочной железы к радиотерапии. Большинство TNBC и базальноподобных раков молочной железы являются агрессивными опухолями, для которых возможности лечения ограничены, а прогноз неблагоприятный [2,3,4]. Эти опухоли демонстрируют повышенный гликолитический фенотип, который поддерживает их плохой прогноз и дополнительно коррелирует с радиорезистентностью [52]. В настоящем исследовании мы обнаружили, что уровни мРНК двух из четырех изоформ PDK (PDK1 и PDK3) повышены в подтипах TNBC и базальноподобного рака молочной железы. Сверхэкспрессия PDK была обнаружена во многих образцах опухолей человека [42,53,54,55,56,57,58,59,60,61], а во многих линиях раковых клеток наблюдается значительная сверхэкспрессия изоформ PDK [50,62,63]. Сообщалось, что сверхэкспрессия PDK связана с плохим прогнозом при различных типах опухолей [53,54,55,56,57,58,59,60,61]. На сверхэкспрессию PDK в раковых клетках влияют несколько транскрипционных факторов, таких как HIF1 [28,64]. HIF1 активно подавляет OXPHOS путем трансактивации генов, кодирующих PDK1 и PDK3. PDK, в свою очередь, фосфорилируют и инактивируют PDH [42]. Таким образом, повышение уровня PDK при раке может быть связано как с трансформирующими мутациями, так и с гипоксическим микроокружением опухоли. В соответствии с этими выводами мы наблюдали, что повышение уровня мРНК PDK1 и PDK3 коррелирует с профилями генов, связанных с гипоксией. Таким образом, метаболическое перепрограммирование с помощью ферментов PDK, направленное на переход от гликолиза к OXPHOS, представляется перспективным терапевтическим направлением для лечения рака молочной железы с ограниченными возможностями терапии.
Главный вывод нашего исследования заключается в том, что ингибитор PDK DCA может снижать гликолитическую активность клеток рака молочной железы в присутствии кислорода, а также в условиях гипоксии [29]. Мы обнаружили, что DCA снижает количество фосфорилированной PDH и вызывает дозозависимое снижение уровня внеклеточного лактата и ECAR в аэробных и гипоксических клетках. Далее мы объединили DCA и радиотерапию, исходя из гипотезы, что, обратив гликолитический фенотип и направив больше пирувата на митохондриальное окисление, опухолевые клетки смогут производить больше ROS и стать более чувствительными к облучению. Внутренний радиосенсибилизирующий эффект DCA был зарегистрирован для клеток глиобластомы [34,35], немелкоклеточной карциномы легких (NSCLC) [65,66], колоректальной [35], рака простаты [32] и радиорезистентных клеток медуллобластомы [67]. Предполагаемые механизмы — остановка клеточного цикла в фазе G2-M, создание дополнительных повреждений ДНК и последовательная гибель клеток в ответ на повышенное производство митохондриальных ROS. В настоящем исследовании мы обнаружили, что в то время как минимальный радиосенсибилизирующий эффект наблюдался при самой высокой нетоксичной концентрации DCA в аэробных условиях, DCA сильно радиосенсибилизировал гипоксические клетки рака молочной железы как в 2D, так и в 3D сфероидах. Хотя теоретически ROS связаны с окислительными механизмами, между гипоксией и ROS в опухолях существует партнерство. Гипоксия усиливает генерацию ROS через продление времени жизни семихиноновых радикалов; в свою очередь, ROS помогает опухолевым клеткам адаптироваться к гипоксии через стабилизацию HIF1-α [16,68]. Однако внеклеточные воздействия могут нарушить это партнерство, вызывая чрезмерное производство ROS, что нарушает митохондриальное дыхание и, таким образом, снижает гипоксическую фракцию в опухолях [16,69,70]. В этом контексте триоксид мышьяка подавляет потребление кислорода опухолевыми клетками через увеличение внутриклеточного ROS, что приводит к усилению радиореактивности [71]. Подавление гликолиза, как сообщается, также увеличивает радиоответ. Это можно сделать с помощью ритонавира (ингибитор транспортера глюкозы), 2-дезоксиглюкозы (ингибитор гексокиназы) и лонидамина (ингибитор гексокиназы), которые проходят клинические испытания при различных видах рака[72,73,74,75]. Другая возможность заключается в том, что ROS образуется после лечения DCA из-за индукции NADPH-оксидазы [76]. Однако не было найдено прямых доказательств того, что DCA может стимулировать NADPH-оксидазу [77]. Мы наблюдали дозозависимое увеличение NAD(P)H в аэробных условиях, но не в гипоксических условиях в клетках EMT6 и 4T1. Поэтому мы предположили, что увеличение NAD(P)H и ап-регуляция NADPH-оксидазы играют лишь второстепенную роль в увеличении ROS в гипоксических условиях. По нашему мнению, основной механизм наблюдаемого эффекта радиосенсибилизации, скорее всего, обусловлен многократным увеличением продукции ROS (до 15 раз) после обработки DCA в гипоксических условиях.
В соответствии с литературными данными, мы продемонстрировали необходимость сверхфизиологических концентраций DCA, чтобы вызвать изменение метаболической активности, увеличение образования ROS и радиосенсибилизацию [29]. Наша основная гипотеза заключается в том, что эти эффекты являются следствием ингибирования PDK под действием DCA [78]. Однако концентрации, необходимые для того, чтобы вызвать измеренные результаты, в несколько раз превышают константу ингибирования (Ki) PDK1-4. Следует отметить, что ДКА физиологически существует как анион, относительно непроницаем для мембран, несмотря на свой небольшой размер, и поэтому для его поглощения требуется митохондриальный переносчик пирувата[79,80]. Однако конъюгация DCA с липофильным переносчиком усиливает митохондриальный транспорт. Это снизило значение IC50 ДКА с миллимолярного до низкого микромолярного диапазона, что находится в пределах диапазона Ki PDK1-4 [81]. DCA имитирует эффективное ингибирование PDK1-4 с помощью siRNA, а DCA, добавленный к PDK siRNA, не оказывал дополнительного эффекта [49,50,60,82,83,84,85,86,87,88]. Далее, DCA, подобно малым молекулам, может прямо или косвенно воздействовать на другие клеточные и молекулярные мишени. Недавние исследования показали, что лечение DCA увеличивает концентрацию каждого промежуточного продукта TCA, но не влияет на поглощение глюкозы или гликолиз [89]. Другие исследователи показали, что DCA может увеличить биосинтез КоА de novo. Поскольку высокие концентрации КоА могут быть токсичными для клеток, этот метаболический эффект может быть частично ответственен за токсичность раковых клеток, опосредованную DCA [90]. Недавние исследования выдвинули новую гипотезу, предполагающую, что эффективность DCA против рака может быть обусловлена его способностью антагонизировать ацетат. Высокий уровень ацетата может усиливать синтез ДНК, РНК и белка. Кроме того, он может быть связан с устойчивостью к противораковым препаратам [91]. Наконец, исследователи обнаружили, что DCA может активировать сигнальный путь AMPK, что приводит к каскаду последующих метаболических и противораковых эффектов [92,93]. Тем не менее, наша гипотеза остается в силе: в гипоксических условиях доставка пирувата в митохондрии вызывает повышение уровня ROS, что приводит к радиосенсибилизации раковых клеток. Нам не удалось воспроизвести эти эффекты in vivo, и необходима дальнейшая работа, чтобы определить, как перевести радиосенсибилизирующие эффекты DCA в гипоксические опухолевые компартменты. Вопросы фармакокинетики, связанные с введением DCA in vivo, можно исключить, а использованная доза DCA (150 мг/кг) вполне соответствует дозам, используемым в литературе [29]. Эквивалентная для человека доза ДКА, использованная нами in vivo, составляет 12 мг/кг в день, что вполне соответствует допустимой зоне, используемой в клинических испытаниях. Возможное объяснение неудачных экспериментов in vivo заключается в том, что для оказания радиосенсибилизирующего эффекта in vivo необходима более высокая доза DCA. За последние 30 лет DCA действительно применялся с достаточным успехом в качестве препарата для лечения диабета 2 типа, приобретенной и врожденной гиперлипопротеинемии, ишемии миокарда, приобретенного и врожденного молочнокислого ацидоза, а в последнее время и рака [29,30]. В нескольких испытаниях I/II фазы изучается безопасность DCA и его активность в качестве противоракового агента. DCA быстро всасывается и даже может преодолевать гематоэнцефалический барьер. В двух испытаниях I фазы изучалась безопасность перорального DCA у пациентов с рецидивирующими злокачественными опухолями головного мозга или метастазами в головной мозг от нецентральных раковых опухолей нервной системы [94,95]. Эти исследования показали, что DCA в целом хорошо переносится пациентами. Альтернативное объяснение загадочной разницы между эффектами in vitro и in vivo на самом деле более вероятно связано с изменениями в метаболических фенотипах, когда раковые клетки (эктопически) вводятся in vivo. Действительно, повышение радиочувствительности DCA in vitro достигается в условиях гипоксии и высокого гликолитического метаболизма, так что при лечении DCA наблюдается сдвиг гликолиза в сторону оксофосфорилирования вместе с дополнительным повышением ROS.
Ограниченная степень гипоксии в опухолях in vivo может, таким образом, отражать ограниченную способность DCA вызывать сдвиг, который уже присутствует в хорошо окисляемых опухолях, в значительной степени зависящих от OXPHOS. Необходимы дальнейшие исследования для проверки радиосенсибилизирующего действия DCA в моделях опухолей молочной железы мышей, характеризующихся ограниченным ангиогенезом и повышенной гипоксической фракцией.
В заключение, мы продемонстрировали, что DCA преодолевает гипоксическую радиорезистентность клеток рака молочной железы в 2D и 3D системах, что может быть в первую очередь связано с повышением уровня ROS. Примечательно, что DCA-индуцированный сдвиг метаболизма гликолиза в сторону OXPHOS также создает окислительный стресс в гипоксических условиях. В течение многих лет DCA использовался для лечения метаболических состояний и наследственных митохондриальных заболеваний. В последнее десятилетие DCA был в значительной степени перепрофилирован в противораковый препарат с многообещающими доклиническими данными, сообщениями о случаях и клиническими испытаниями, как описано ранее. Текущие доклинические результаты показывают, что необходимо дальнейшее изучение противоракового потенциала DCA, учитывая, что гипоксические опухолевые клетки не щадит ингибитор PDK, который может вызывать смертельный окислительный стресс, в частности, в сочетании с радиотерапией.
Материалы и методы
Когортный анализ рака молочной железы TCGA Профили экспрессии мРНК PDK1-4 (RNA Seq V2 RSEM или log RNA Seq V2 RSEM) были запрошены с сайта cBioPortal в виде преобразованных по z-score данных [38,39]. Запрашиваемые данные оценивались по 1084 общедоступным случаям рака молочной железы из атласа TCGA PanCancer Atlas и по 817 случаям рака молочной железы из базы данных TCGA Cell 2015. Для набора данных TCGA Cell 2015 анализ PDK1-4 проводился путем сравнения трижды-отрицательной субпопуляции с остальными случаями рака молочной железы. Тройной негативный рак молочной железы определялся «отрицательным» статусом по результатам иммуногистохимии генов рецептора эстрогена (ER), рецептора прогестерона (PR) и рецептора эпидермального фактора роста человека 2 (HER2) (всего 83 случая). В базе данных TCGA PanCancer Atlas был проведен анализ экспрессии PDK1-4 по различным категориям Pam50. Pam50 — это 50-генная сигнатура, которая классифицирует рак молочной железы на пять молекулярно присущих подтипов: Люминальный A, Люминальный B, обогащенный HER2, базальноподобный и нормальный. Экспрессия мРНК рака молочной железы TCGA и клинические данные анализировались непосредственно на сайте cBioPortal или загружались для дальнейшего анализа. Анализ оценки гипоксии по шкале Баффа и Рагнума для образцов, в которых экспрессия PDK1-4 имела z-score выше 2, проводился непосредственно на сайте cBioPortal.
Клеточные линии и химические вещества Клеточная линия мышиной аденокарциномы молочной железы EMT6 была любезно предоставлена Эдит Лорд (Университет Рочестера, Онкологический центр, Нью-Йорк), а клетки 4T1 были получены из American Type Culture Collection. Все эксперименты проводили в среде Roswell Park Memorial Institute 1640 (Thermo Fisher Scientific, Waltham, MA, США), дополненной 10% фетальной бычьей сывороткой (Greiner Bio-One, Kremsmünster, Австрия). Для всех обработок клеток использовался буфер HEPES. Химические вещества были получены от Sigma-Aldrich (Sigma-Aldrich, Сент-Луис, МО, США), если не указано иное
Обработка EMT6 и 4T1 выращивали до конфлюентности и обрабатывали DCA в течение 16 ч в указанных концентрациях. N-ацетил цистеин (NAC) добавляли в концентрации 10 мМ или 20 мМ к культурам за 1 ч до и во время обработки ДКА. После этого культуры использовали для дальнейшего анализа, как описано ниже. Обработку проводили либо в аэробных, либо в гипоксических условиях. Гипоксия индуцировалась путем инкубации в сбалансированном по азоту/углекислоте газе, содержащем 1% кислорода [96]
МТТ-анализ Цитотоксичность DCA оценивали с помощью МТТ-анализа, как описано в другом месте <a href=»#97″>[97,</a></sup><a href=»#98″><sup>98]</sup>.</a> Вкратце, клетки выращивали в 96-луночных планшетах и обрабатывали указанными концентрациями. После обработки среду аспирировали, добавляли 50 мкл реагента МТТ (5 мг/мл) на 1,5 ч. Затем добавляли 200 мкл растворителя МТТ (19:1 ДМСО/HCL) и перемешивали, чтобы растворить кристаллы формазана, образовавшиеся внутри клеток. Абсорбцию измеряли при длине волны 540 нм с помощью спектрофотометра (Bio-Rad Laboratories, Hercules, CA, USA). Жизнеспособность клеток определяли путем нормализации обработанных клеток к необработанным контрольным клеткам.</p>
<p><strong>Кинетический анализ роста<br></strong>Влияние DCA на пролиферацию клеток EMT6 и 4T1 оценивали с помощью кинетического анализа роста. Клетки выращивали до конфлюентности в 96-луночных культуральных планшетах и обрабатывали DCA в указанных концентрациях в 6 повторах. Фотомикрофотографии делались каждые два часа с помощью прибора Incucyte live cell imager (Essen Biosciences, Ньюарк, Великобритания), а конфлюентность культур измерялась с помощью программного обеспечения Incucyte (Incucyte ZOOM 2018A, Essen Biosciences) в течение 80 ч в культуре.</p>
<p><strong>Вестерн-блот<br></strong>Вестерн-блот анализ проводили как описано ранее <sup><a href=»#99″>[99]</a></sup>. Вкратце, клетки лизировали в 1% буфере тритон-Х, дополненном ингибитором фосфатазы (P5726), ингибитором протеазы (P8340) и трифторацетатом леупептина (L2023). Лизаты центрифугировали, и концентрацию белка определяли с помощью Bio-Rad DC protein assay (Bio-Rad 500-0116). Эквивалентные количества белка загружали на 12% разрешающий акриламидный гель. Перенос белка проводили в течение ночи при 4 °C с использованием нитроцеллюлозной мембраны (0,45 мкМ, Thermo 88018, Thermo Fisher Scientific). Мембраны блокировали 5% BSA в TBS и промывали (TBST). Блокированные мембраны были помечены первичными антителами в течение ночи при 4 °C. Первичные антитела маркировали вторичными антителами ближнего инфракрасного диапазона (IRDyes 680 RD или 800 CW, LI-COR Biosciences, Lincoln, NE, USA), детектировали и количественно определяли с помощью Odyssey Fc Imaging System (LI-COR Biosciences). Первичными антителами были: фосфо-PDH (ABS204, MERCK, Дармштадт, Германия), общий-PDH (C54G1, cell signaling), антибета ACTIN (A1978) и антиальфа TUBULIN (T9026)</p>
<p><strong>Анализ лактата<br></strong>После лечения проводился анализ L-лактата в соответствии с инструкциями производителя. Вкратце, супернатант клеток был взят и внесен в мастер-реакционную смесь. После этого проводилась инкубация в течение 30 минут при комнатной температуре в темноте. Затем измеряли абсорбцию при 570 нм и рассчитывали количество L-лактата в среде. Кроме того, клетки лизировали, и общее количество белков исследовали с помощью BCA protein assay (23227, Thermo Fisher); этот шаг выполнялся для нормализации значений лактата к количеству белков в клетке.</p>
<p><strong>Seahorse Metabolic Profiling<br></strong>Скорость потребления кислорода (OCR) и скорость внеклеточного окисления (ECAR) определяли с помощью анализатора Seahorse XF96 (Agilent Technologies, Santa Clara, CA, USA), как сообщалось ранее <sup><a href=»#100″>[100]</a></sup>. Вкратце, 1,5 × 10<sup>5</sup> клеток высевали в 96-луночные планшеты. Затем клетки обрабатывали DCA, либо на ночь (для измерения OCR), либо в течение 3 ч (во время работы Seahorse, для ECAR). Клетки уравновешивали в небуферной среде Dulbecco’s Modified Eagle Medium (DMEM) с 2 мМ глютамина и 10 мМ глюкозы при 37 °C в инкубаторе без CO<sub>2</sub> и затем измеряли с помощью анализатора Seahorse. Для получения подробной информации о цепи переноса электронов в митохондриях последовательно добавляли специфические ингибиторы, состоящие из олигомицина, FCCP, ротенона и антимицина А. Показатель ECAR был нормализован к базальному уровню.</p>
<p><strong>Продукция ROS<br></strong>Внутриклеточный уровень ROS определяли с помощью 5-(6)-хлорметил-2′,7′-дихлордигидро-флуоресцеин диацетата (CM-H<sub>2</sub>DCFDA), чувствительного к окислению флуоресцентного зонда (Abcam, Кембридж, Великобритания), как описано ранее <sup><a href=»#97″>[97]</a></sup>. Вкратце, после обработки клетки окрашивали 5 мкМ CM-H2DCFDA при 37 °C в течение 30 мин. Среднюю интенсивность флуоресценции измеряли на проточном цитометре FACSCanto (BD Bioscience, Франклин Лейкс, штат Нью-Джерси, США) и анализировали с помощью программного обеспечения Flowjo (BD Bioscience)</p>
<p><strong>Измерение NAD(P)H<br></strong>Внутриклеточный уровень NAD(P)H определяли с помощью набора для проточного цитометрического анализа Cell Meter<sup>TM</sup> внутриклеточного NADH/NADPH (AAT BioQuest, Sunnyvale, CA, США), в соответствии с инструкциями производителя. Вкратце, после обработки клетки окрашивали датчиком JZLA707 NAD(P)H при 37 °C в течение 45 мин. Среднюю интенсивность флуоресценции измеряли на проточном цитометре FACSCanto (BD Bioscience) и анализировали с помощью программного обеспечения Flowjo (BD Bioscience).</p>
<p><strong>Радиация и клоногенный анализ<br></strong>После лечения клетки облучали в указанных дозах на 6 MV Linac (Varian Truebeam STx, Palo Alto, CA, USA; BrainLAB AG, Feldkirchen, Germany) и высевали в 6-луночные планшеты для образования колоний. Перед посевом клетки подсчитывали и нормировали к контрольным условиям. Через 7-12 дней культуры фиксировали кристаллическим фиолетовым и подсчитывали колонии (>50 клеток). Фракции выживания (ФВ) подгоняли под линейно-квадратичную модель с помощью программного обеспечения GraphPad Prism 8 (GraphPad Prism Software Inc, Сан-Диего, Калифорния, США). Радиосенсибилизация оценивалась на уровне 0,1 выживших фракций.</p>
<p><strong>Трехмерные (3D) клеточные культуры (сфероиды)<br></strong>Сфероиды готовили с клетками EMT6 и 4T1 путем высева 4000 клеток/лунку в 96-луночный планшет с ультранизким прикреплением (Corning, Corning, NY, USA). DCA добавляли в среду, когда диаметр сфероидов составлял около 500 мкм. После этого сфероиды облучали при 8 Гр, а затем промывали свежей средой, которую обновляли каждые 3 дня. Рост сфероидов отслеживался с помощью системы IncuCyte Live Cell Imaging System (Essen Bioscience) в течение 10 дней.</p>
<p><strong>Модель опухоли у мышей</br></strong> Опухолевые клетки 4T1 и EMT6 (0,5 × 10<sup>6</sup>) инокулировали в левую заднюю конечность сингенных мышей Balb/c (самки, возраст 7-9 недель; Charles River Laboratories, L’Arbresle Cedex, Франция). Когда опухоли достигали приблизительно 150 мм<sup>3</sup>, мышей рандомизировали и в течение 10 дней подряд лечили DCA 300 мг/кг (внутрибрюшинно или интратуморально). Мышей облучали однократной дозой 12 Гр (опухоли EMT6) или 15 Гр (опухоли 4T1) или схемой фракционированного облучения 5*4 Гр (опухоли EMT6) и 5*6 Гр (опухоли 4T1), когда опухоли достигали приблизительно 150 мм3. Облучение проводилось на установке Linac 6 MV (Varian Truebeam STx). В течение всего эксперимента опухоли измеряли электронным штангенциркулем, а объем опухоли рассчитывали по формуле: Объем = (Длина × Ширина<sup>2</sup>) × 0,5. Эксперименты были одобрены Этическим комитетом по использованию лабораторных животных Врийе Университета Брюсселя (этическое досье №: 16-552-2 (18/4/2017) и 18-552-2 (1/6/2018) </p>
<p><strong>Окрашивание пимонидазолом срезов опухоли<br></strong> Опухоли инокулировали, как описано в части 4.13. После лечения, пимонидазол (60 мг/кг; Hypoxyprobe) вводили i.v. в хвостовую вену. Опухоли вырезали через 1,5 часа, взвешивали, замораживали и хранили в пластиковых флаконах при -80 °C. Затем срезы опухолей (5 мкм) иммуноокрашивали с помощью анти-Pimo кроличьего антитела (Hypoxyprobe, Burlington, MA, USA), которое окрашивали антикроличьим FITC антителом (Abcam). Слайды опухоли монтировали монтажной жидкостью (монтажная среда DAKO, Agilent), смешанной с dapi (Sigma-Aldrich), и покрывали покровным стеклом. Изображения получали с помощью флуоресцентной конфокальной микроскопии (EVOS FL, Thermo Fisher) и анализировали с помощью ImageJ.</p>
<p><strong>Статистика</strong><br>Все анализы проводились с использованием GraphPad Prism 8.4.3. Данные выражены как среднее ± SEM по крайней мере трех независимых экспериментов, если не указано иное. Для статистического анализа использовались непарный <em>t</em>-тест, односторонний ANOVA с последующим тестом множественного сравнения Даннетта и двусторонний ANOVA с тестом множественного сравнения Даннетта, Сидака или Тьюки: * <em>p</em> < 0,05, ** <em>p</em> < 0,01, *** <em>p</em> < 0,001, **** <em>p</em> < 0,0001.</p>
<h2>Дополнительные материалы</h2>
<p>Дополнительные материалы можно найти на сайте https://www.mdpi.com/1422-0067/21/24/9367/s1.</p>
<h2>Авторские материалы</h2>
<p>Концептуализация, S.d.M., I.D. и H.J.; курирование данных, S.d.M., C.C., K.L.L. и H.V.; исследование, S.d.M.; методология, T.G.; контроль, I.D., H.J., О.Ф. и М.Д.Р.; визуализация, С.д.М.; написание первоначального проекта, С.д.М.; написание и редактирование, И.Д., К.К., Х.В., М.В.Д.Г., Л.К., О.Ф. и М.Д.Р. Все авторы обсудили результаты и внесли свой вклад в окончательную рукопись. Все авторы прочитали и согласились с опубликованной версией рукописи.</p>
<h2>Финансирование</h2>
<p>Эта работа была поддержана стратегической исследовательской программой «Общественная польза от безмаркерной стереотаксической радиотерапии тела: статистическая поддержка на основе количественной визуализации» (Zwaartepunt, SRP 53, 2019-2024) исследовательского совета Врийского университета Брюсселя.</p>
<h2>Благодарности</h2>
<p>Авторы благодарят Валерия Веровского за ценные и конструктивные предложения.</p>
<h2>Конфликты интересов</h2>
<p>Авторы заявляют об отсутствии потенциальных конфликтов интересов.</p>
<h2>Примечание издателя:</h2>
<p>MDPI сохраняет нейтралитет в отношении юрисдикционных претензий в опубликованных картах и институциональной принадлежности.</p>
<h2>Аббревиатуры</h2>
<figure class=»wp-block-table»><table><tbody><tr><td>DCA</td><td>Дихлорацетат</td></tr><tr><td>ECAR</td><td>Скорость внеклеточного закисления</td></tr><tr><td>NAC</td><td>N-ацетил-..цистеин</td></tr><tr><tr><td>OCR</td><td>Скорость потребления кислорода</td></tr><tr><td>OXPHOS</td><td>Окислительное фосфорилирование</td></tr><tr><td>PDH</td><td>Пируватдегидрогеназа</td></tr><tr><td>PDK</td><td>Пируватдегидрогеназа</td></tr><tr><td>Pyruvate киназа дегидрогеназы</td></tr><tr><td>PDP</td><td>фосфатаза пируватдегидрогеназы</td></tr><tr><td>ROS</td><td>Reactive oxygen species</td></tr><tr><td>TBNC</td><td>Triple-негативный рак молочной железы</td></tr></tbody></table></figure>
<h2>REFERENCES</h2>
<span id=»1″ class=»referencess blue-text»>1</span> Всемирная организация здравоохранения. Рак молочной железы. Доступно онлайн: https://www.who.int/cancer/prevention/ diagnosis-screening/breast-cancer/en/ (дата обращения: 10 сентября 2020 г.).
<br><span id=»2″ class=»referencess blue-text»>2</span> Bianchini, G.; Balko, J.M.; Mayer, I.A.; Sanders, M.E.; Gianni, L. Triple-negative breast cancer: Вызовы и возможности гетерогенного заболевания. Nat. Rev. Clin. Oncol. 2016, 13, 674-690. [CrossRef] [PubMed]
<br><span id=»3″ class=»referencess blue-text»>3</span> Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular subtypes and local-regional control of breast cancer. Surg. Онкол. Clin. N. Am. 2018, 27, 95-120. [CrossRef] (PubMed)
<br><span id=»4″ class=»referencess blue-text»>4</span> Prat, A.; Pineda, E.; Adamo, B.; Galván, P.; Fernández, A.; Gaba, L.; Díez, M.; Viladot, M.; Arance, A.; Muñoz, M. Clinical implications of the intrinsic molecular subtypes of breast cancer. Breast 2015, 24 (Suppl. 2), S26-S35. [CrossRef] [PubMed]
<br><span id=»5″ class=»referencess blue-text»>5</span> Brunt, A.M.; Haviland, J.S.; Wheatley, D.A.; Sydenham, M.A.; Alhasso, A.; Bloomfield, D.J.; Chan, C.; Churn, M.; Cleator, S.; Coles, C.E.; et al. Hypofractionated breast radiotherapy for 1 week versus 3 weeks (FAST-Forward): 5-летняя эффективность и поздние эффекты нормальной ткани результаты многоцентрового рандомизированного испытания 3-й фазы, не имеющего противопоказаний. Lancet 2020, 395, 1613-1626. [CrossRef]
<br><span id=»6″ class=»referencess blue-text»>6</span> Poleszczuk, J.; Luddy, K.; Chen, L.; Lee, J.K.; Harrison, L.B.; Czerniecki, B.J.; Soliman, H.; Enderling, H. Neoadjuvant radiotherapy of early-stage breast cancer and long-term disease-free survival. Breast Cancer Res. 2017, 19, 1-7. [CrossRef]
<br><span id=»7″ class=»referencess blue-text»>7</span> Lightowlers, S.V.; Boersma, L.J.; Fourquet, A.; Kirova, Y.M.; Offersen, B.V.; Poortmans, P.; Scholten, A.N.; Somaiah, N.; Coles, C.E. Preoperative breast radiation therapy: Показания и перспективы. Eur. J. Cancer 2017, 82, 184-192. [CrossRef]
<br><span id=»8″ class=»referencess blue-text»>8</span> Palta, M.; Yoo, S.; Adamson, J.D.; Prosnitz, L.R.; Horton, J.K. Preoperative single fraction partial breast radiotherapy for early-stage breast cancer. Int. J. Radiat. Oncol. Biol. Phys. 2012, 82, 37-42. [CrossRef]
<br><span id=»9″ class=»referencess blue-text»>9</span> Horton, J.K.; Blitzblau, R.C.; Yoo, S.; Geradts, J.; Chang, Z.; Baker, J.A.; Georgiade, G.S.; Chen, W.; Siamakpour-Reihani, S.; Wang, C.; et al. Preoperative Single-Fraction Partial Breast Radiation Therapy: Novel Phase 1, Dose-Escalation Protocol with Radiation Response Biomarkers. Int. J. Radiat. Oncol. Biol. Phys. 2015, 92, 846-855. [CrossRef]
<br><span id=»10″ class=»referencess blue-text»>10</span> Roth, S.L.; Audretsch, W.; Bojar, H.; Lang, I.; Willers, R.; Budach, W. Retrospective study of neoadjuvant versus adjuvant radiochemotherapy in locally advanced noninflammatory breast cancer: Преимущество выживаемости в категории cT2 при неоадъювантной радиохимиотерапии. Strahlentherapie und Onkologie 2010, 186, 299-306. [CrossRef]
<br><span id=»11″ class=»referencess blue-text»>11</span> Пред- или послеоперационная ускоренная радиотерапия. Доступно онлайн: https://ClinicalTrials.gov/show/ NCT03783364 (дата обращения: 27 августа 2020 г.).
<br><span id=»12″ class=»referencess blue-text»>12</span> Hall, E.J.; Giaccia, A.J. Radiobiology for the Radiologist; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2012; Том 7.
<br><span id=»13″ class=»referencess blue-text»>13</span> De Ridder, M.; Tournel, K.; Van Nieuwenhove, Y.; Engels, B.; Hoorens, A.; Everaert, H.; De Beeck, B.O.; Vinh-Hung, V.; De Grève, J.; Delvaux, G.; et al. Phase II study of preoperative helical tomotherapy for rectal cancer. Int. J. Radiat. Oncol. Biol. Phys. 2008, 70, 728-734. [CrossRef]
<br><span id=»14″ class=»referencess blue-text»>14</span> Gray, L.H.; Conger, A.D.; Ebert, M.; Hornsey, S.; Scott, O.C. The concentration of oxygen dissolved in tissues at the time of irradiation as a factor in radiotherapy. Br. J. Radiol. 1953, 26, 638-648. [CrossRef] [PubMed]
<br><span id=»15″ class=»referencess blue-text»>15</span> Brown, J.M.; Wilson, W.R. Exploiting tumour hypoxia in cancer treatment. Nat. Rev. Cancer 2004, 4, 437-447. [CrossRef] [PubMed]
<br><span id=»16″ class=»referencess blue-text»>16</span> Wang, H.; Jiang, H.; Van De Gucht, M.; De Ridder, M. Hypoxic Radioresistance: Can ROS Be the Key to Overcome It? Cancers 2019, 11, 112. [CrossRef] [PubMed]
<br><span id=»17″ class=»referencess blue-text»>17</span> Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807-819. [CrossRef]
<br><span id=»18″ class=»referencess blue-text»>18</span> Chandel, N.S.; McClintock, D.S.; Feliciano, C.E.; Wood, T.M.; Melendez, J.A.; Rodriguez, A.M.; Schumacker, P.T. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: A mechanism of O2 sensing. J. Biol. Chem. 2000, 275. [CrossRef]
<br><span id=»19″ class=»referencess blue-text»>19</span> Azimi, I.; Petersen, R.M.; Thompson, E.W.; Roberts-Thomson, S.J.; Monteith, G.R. Hypoxia-induced reactive oxygen species mediate N-cadherin and SERPINE1 expression, EGFR signalling and motility in MDA-MB-468 breast cancer cells. Sci. Rep. 2017, 7, 1-11. [CrossRef]
<br><span id=»20″ class=»referencess blue-text»>20</span> Johnson, M.K.; Vathanayagam, R.R.; Wang, E.S. Hypoxia-Associated Effects on Reactive Oxygen Species Generation by Human Acute Myeloid Leukemia Cells. Blood 2011, 118, 4998. [CrossRef]
<br><span id=»21″ class=»referencess blue-text»>21</span> Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646-674. [CrossRef]
<br><span id=»22″ class=»referencess blue-text»>22</span> Warburg, O. On the origin of cancer cells. Science 1956, 123, 309-314. [CrossRef]
<br><span id=»23″ class=»referencess blue-text»>23</span> Pitroda, S.P.; Wakim, B.T.; Sood, R.F.; Beveridge, M.G.; Beckett, M.A.; MacDermed, D.M.; Weichselbaum, R.R.; Khodarev, N.N. STAT1-dependent expression of energy metabolic pathways links tumour growth and radioresistance to the Warburg effect. BMC Med. 2009, 7, 68. [CrossRef]
<br><span id=»24″ class=»referencess blue-text»>24</span> Song, K.; Li, M.; Xu, X.; Xuan, L.I.; Huang, G.; Liu, Q. Resistance to chemotherapy is associated with altered glucose metabolism in acute myeloid leukemia. Oncol. Lett. 2016, 12, 334-342. [CrossRef]
<br><span id=»25″ class=»referencess blue-text»>25</span> Zhou, Y.; Tozzi, F.; Chen, J.; Fan, F.; Xia, L.; Wang, J.; Gao, G.; Zhang, A.; Xia, X.; Brasher, H.; et al. Intracellular ATP levels are a pivotal determinant of chemoresistance in colon cancer cells. Cancer Res. 2012, 72, 304-314. [CrossRef] [PubMed]
<br><span id=»26″ class=»referencess blue-text»>26</span> Shimura, T.; Noma, N.; Sano, Y.; Ochiai, Y.; Oikawa, T.; Fukumoto, M.; Kunugita, N. AKT-mediated enhanced aerobic glycolysis causes acquired radioresistance by human tumor cells. Radiother. Oncol. 2014, 112, 302-307. [CrossRef] [PubMed]
<br><span id=»27″ class=»referencess blue-text»>27</span> Sattler, U.G.; Meyer, S.S.; Quennet, V.; Hoerner, C.; Knoerzer, H.; Fabian, C.; Yaromina, A.; Zips, D.; Walenta, S.; Baumann, M.; et al. Glycolytic metabolism and tumour response to fractionated irradiation. Radiother. Oncol. 2010, 94, 102-109. [CrossRef] [PubMed]
<br><span id=»28″ class=»referencess blue-text»>28</span> Kim, J.W.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated Expression of Pyruvate Dehydrogenase Kinase: Метаболический переключатель, необходимый для клеточной адаптации к гипоксии. Cell Metab. 2006, 3, 177-185. [CrossRef] [PubMed]
<br><span id=»29″ class=»referencess blue-text»>29</span> Kankotia, S.; Stacpoole, P.W. Dichloroacetate and cancer: Новый дом для сиротского препарата? Biochim. Biophys. Acta 2014, 1846, 617-629. [CrossRef]
<br><span id=»30″ class=»referencess blue-text»>30</span> James, M.O.; Jahn, S.C.; Zhong, G.; Smeltz, M.G.; Hu, Z.; Stacpoole, P.W. Therapeutic applications of dichloroacetate and the role of glutathione transferase zeta-1. Pharmacol. Ther. 2017, 170, 166-180. [CrossRef] <br><span id=»31″ class=»referencess blue-text»>31</span> Tataranni, T.; Piccoli, C. Dichloroacetate (DCA) and Cancer: Обзор в направлении клинического применения. Oxidative Med. Cell. Longev. 2019, 2019, 1-14. [CrossRef]
<br><span id=»32″ class=»referencess blue-text»>32</span> Cao, W.; Yacoub, S.; Shiverick, K.T.; Namiki, K.; Sakai, Y.; Porvasnik, S.; Urbanek, C.; Rosser, C.J. Dichloroacetate (DCA) sensitizes both wild-type and over expressing Bcl-2 prostate cancer cells in vitro to radiation. Prostate 2008, 68, 1223-1231. [CrossRef]
<br><span id=»33″ class=»referencess blue-text»>33</span> Dong, G.; Chen, Q.; Jiang, F.; Yu, D.; Mao, Q.; Xia, W.; Shi, R.; Wang, J.; Xu, L. Diisopropylamine Dichloroacetate Enhances Radiosensitization in Esophageal Squamous Cell Carcinoma by Increasing Mitochondria-Derived Reactive Oxygen Species Levels. Oncotarget 2016, 7, 68170-68178. [CrossRef]
<br><span id=»34″ class=»referencess blue-text»>34</span> Shen, H.; Hau, E.; Joshi, S.; Dilda, P.J.; McDonald, K.L. Sensitization of Glioblastoma Cells to Irradiation by Modulating the Glucose Metabolism. Mol. Cancer Ther. 2015, 14, 1794-1804. [CrossRef] [PubMed]
<br><span id=»35″ class=»referencess blue-text»>35</span> Zwicker, F.; Kirsner, A.; Peschke, P.; Roeder, F.; Debus, J.; Huber, P.E.; Weber, K.J. Dichloroacetate induces tumor-specific radiosensitivity in vitro but attenuates radiation-induced tumor growth delay in vivo. Strahlentherapie und Onkologie 2013, 189, 684-692. [CrossRef] [PubMed]
<br><span id=»36″ class=»referencess blue-text»>36</span> Комбинация радиотерапии и темозоломида с дихлорацетатом у пациентов с недавно диагностированной глиобластомой. Доступно онлайн: https://ClinicalTrials.gov/show/NCT00703859 (дата обращения: 13 июля 2020 г.).
<br><span id=»37″ class=»referencess blue-text»>37</span> Исследование DCA (дихлорацетат) в комбинации с цисплатином и окончательным облучением при карциноме головы и шеи. Доступно онлайн: https://ClinicalTrials.gov/show/NCT01386632 (дата обращения: 13 июля 2020 г.).
<br><span id=»38″ class=»referencess blue-text»>38</span> Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: Открытая платформа для изучения многомерных данных геномики рака. Cancer Discov. 2012, 2, 401-404. [CrossRef] [PubMed]
<br><span id=»39″ class=»referencess blue-text»>39</span> Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6. [CrossRef] [PubMed]
<br><span id=»40″ class=»referencess blue-text»>40</span> Ragnum, H.B.; Vlatkovic, L.; Lie, A.K.; Axcrona, K.; Julin, C.H.; Frikstad, K.M.; Hole, K.H.; Seierstad, T.; Lyng, H. The tumour hypoxia marker pimonidazole reflects a transcriptional program associated with aggressive prostate cancer. Br. J. Cancer 2015, 112, 382-390. [CrossRef]
<br><span id=»41″ class=»referencess blue-text»>41</span> Buffa, F.M.; Harris, A.L.; West, C.M.; Miller, C.J. Large meta-analysis of multiple cancers reveals a common, compact and highly prognostic hypoxia metagene. Br. J. Cancer 2010, 102, 428-435. [CrossRef]
<br><span id=»42″ class=»referencess blue-text»>42</span> Zhang, W.; Zhang, S.L.; Hu, X.; Tam, K.Y. Targeting Tumor Metabolism for Cancer Treatment: Является ли киназа пируватдегидрогеназы (PDKs) жизнеспособной противораковой мишенью? Int. J. Biol. Sci. 2015, 11, 1390-1400. [CrossRef]
<br><span id=»43″ class=»referencess blue-text»>43</span> Tang, L.; Wei, F.; Wu, Y.; He, Y.; Shi, L.; Xiong, F.; Gong, Z.; Guo, C.; Li, X.; Deng, H.; et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J. Exp. Clin. Cancer Res. 2018, 37, 1-15. [CrossRef]
<br><span id=»44″ class=»referencess blue-text»>44</span> Stacpoole, P.W. Therapeutic Targeting of the Pyruvate Dehydrogenase Complex/Pyruvate Dehydrogenase Kinase (PDC/PDK) Axis in Cancer. J. Natl. Cancer Inst. 2017, 109. [CrossRef]
<br><span id=»45″ class=»referencess blue-text»>45</span> Saunier, E.; Benelli, C.; Bortoli, S. The Pyruvate Dehydrogenase Complex in Cancer: Старый метаболический привратник, регулируемый новыми путями и фармакологическими агентами. Int. J. Cancer 2016, 138, 809-817. [CrossRef]
<br><span id=»46″ class=»referencess blue-text»>46</span> Колобова Е., Туганова А., Булатников И., Попов К.М. Регуляция активности пируватдегидрогеназы через фосфорилирование по нескольким сайтам. Биохим. J. 2001, 358, 69-77. [CrossRef] [PubMed]
<br><span id=»47″ class=»referencess blue-text»>47</span> Корбе, К.; Пинто, А.; Мартерус, Р.; де Хесус, Ж.П.С.; Поле, Ф.; Ферон, О. Ацидоз приводит к перепрограммированию метаболизма жирных кислот в раковых клетках через изменения в митохондриальном и гистоновом ацетилировании. Cell Metab. 2016, 24, 311-323. [CrossRef]
<br><span id=»48″ class=»referencess blue-text»>48</span> Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding theWarburg effect: Метаболические требования клеточной пролиферации. Science 2009, 324, 1029-1033. [CrossRef] [PubMed]
<br><span id=»49″ class=»referencess blue-text»>49</span> Bonnet, S.; Archer, S.L.; Allalunis-Turner, J.; Haromy, A.; Beaulieu, C.; Thompson, R.; Lee, C.T.; Lopaschuk, G.D.; Puttagunta, L.; Bonnet, S.; et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007, 11, 37-51. [CrossRef] [PubMed]
<br><span id=»50″ class=»referencess blue-text»>50</span> Woolbright, B.L.; Choudhary, D.; Mikhalyuk, A.; Trammel, C.; Shanmugam, S.; Abbott, E.; Pilbeam, C.C.; Taylor, J.A. The Role of Pyruvate Dehydrogenase Kinase-4 (PDK4) in Bladder Cancer and Chemoresistance. Mol. Cancer Ther. 2018, 17, 2004-2012. [CrossRef] [PubMed]
<br><span id=»51″ class=»referencess blue-text»>51</span> Okamoto, S.; Narita, T.; Sasanuma, H.; Takeda, S.; Masunaga, S.-I.; Bessho, T.; Tano, K. Impact of DNA repair pathways on the cytotoxicity of piperlongumine in chicken DT40 cell-lines. Genes Cancer 2014, 5, 285-292. [CrossRef] [PubMed]
<br><span id=»52″ class=»referencess blue-text»>52</span> Sun, X.; Wang, M.; Yu, X.; Guo, J.; Sun, T.; Li, X.; Yao, L.; Dong, H.; Xu, Y. Metabolic Reprogramming in Triple-Negative Breast Cancer. Front. Oncol. 2020, 10, 428. [CrossRef]
<br><span id=»53″ class=»referencess blue-text»>53</span> Lu, C.-W.; Lin, S.-C.; Chien, C.-W.; Lin, S.-C.; Lee, C.-T.; Lin, B.-W.; Lee, J.-C.; Tsai, S.-J. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am. J. Pathol. 2011, 179, 1405-1414. [CrossRef]
<br><span id=»54″ class=»referencess blue-text»>54</span> Jha, M.K.; Suk, K. Pyruvate dehydrogenase kinase as a potential therapeutic target for malignant gliomas. Brain Tumor Res. Treat. 2013, 1, 57-63. [CrossRef]
<br><span id=»55″ class=»referencess blue-text»>55</span> Blouin, J.M.; Penot, G.; Collinet, M.; Nacfer, M.; Forest, C.; Laurent-Puig, P.; Coumoul, X.; Barouki, R.; Benelli, C.; Bortoli, S. Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int. J. Cancer 2011, 128, 2591-2601. [CrossRef]
<br><span id=»56″ class=»referencess blue-text»>56</span> Hur, H.; Xuan, Y.; Kim, Y.B.; Lee, G.; Shim, W.; Yun, J.; Ham, I.H.; Han, S.U. Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int. J. Oncol. 2013, 42, 44-54. [CrossRef] [PubMed]
<br><span id=»57″ class=»referencess blue-text»>57</span> Wigfield, S.M.; Winter, S.C.; Giatromanolaki, A.; Taylor, J.; Koukourakis, M.L.; Harris, A.L. PDK-1 регулирует производство лактата при гипоксии и связан с плохим прогнозом при сквамозном раке головы и шеи. Br. J. Cancer 2008, 98, 1975-1984. [CrossRef] [PubMed]
<br><span id=»58″ class=»referencess blue-text»>58</span> Sun, W.; Zhou, S.; Chang, S.S.; McFate, T.; Verma, A.; Califano, J.A. Mitochondrial mutations contrib to HIF1alpha accumulation through increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma. Клин. Cancer Res. 2009, 15, 476-484. [CrossRef] [PubMed]
<br><span id=»59″ class=»referencess blue-text»>59</span> Shen, Y.C.; Ou, D.L.; Hsu, C.; Lin, K.L.; Chang, C.Y.; Lin, C.Y.; Liu, S.H.; Cheng, A.L. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br. J. Cancer 2013, 108, 72-81. [CrossRef] [PubMed]
<br><span id=»60″ class=»referencess blue-text»>60</span> Fujiwara, S.; Kawano, Y.; Yuki, H.; Okuno, Y.; Nosaka, K.; Mitsuya, H.; Hata, H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br. J. Cancer 2013, 108, 170-178. [CrossRef]
<br><span id=»61″ class=»referencess blue-text»>61</span> Guda, M.R.; Asuthkar, S.; Labak, C.M.; Tsung, A.J.; Alexandrov, I.; Mackenzie, M.J.; Prasad, D.V.; Velpula, K.K. Targeting PDK4 inhibits breast cancer metabolism. Am. J. Cancer Res. 2018, 8, 1725-1738.
<br><span id=»62″ class=»referencess blue-text»>62</span> Roh, J.L.; Park, J.Y.; Kim, E.H.; Jang, H.J.; Kwon, M. Activation of mitochondrial oxidation by PDK2 inhibition reverses cisplatin resistance in head and neck cancer. Cancer Lett. 2016, 371, 20-29. [CrossRef]
<br><span id=»63″ class=»referencess blue-text»>63</span> Dupuy, F.; Tabariès, S.; Andrzejewski, S.; Dong, Z.; Blagih, J.; Annis, M.G.; Omeroglu, A.; Gao, D.; Leung, S.; Amir, E.; et al. PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab. 2015, 22, 577-589. [CrossRef]
<br><span id=»64″ class=»referencess blue-text»>64</span> McFate, T.; Mohyeldin, A.; Lu, H.; Thakar, J.; Henriques, J.; Halim, N.D.; Wu, H.; Schell, M.J.; Tsang, T.M.; Teahan, O.; et al. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J. Biol. Chem. 2008, 283, 22700-22708. [CrossRef]
<br><span id=»65″ class=»referencess blue-text»>65</span> Shavit, R.; Ilouze, M.; Feinberg, T.; Lawrence, Y.R.; Tzur, Y.; Peled, N. Mitochondrial induction as a potential radio-sensitizer in lung cancer cells-A short report. Cell. Oncol. 2015, 38, 247-252. [CrossRef]
<br><span id=»66″ class=»referencess blue-text»>66</span> Allen, K.T.; Chin-Sinex, H.; DeLuca, T.; Pomerening, J.; Sherer, J.; Watkins, J.; Foley, J.; Jesseph, J.; Mendonca, M. Dichloroacetate alters Warburg metabolism, inhibits cell growth, and increases the X-ray sensitivity of human A549 and H1299 NSC lung cancer cells. Free Radic. Biol. Med. 2015, 89, 263-273. [CrossRef] [PubMed]
<br><span id=»67″ class=»referencess blue-text»>67</span> Sun, L.; Moritake, T.; Ito, K.; Matsumoto, Y.; Yasui, H.; Nakagawa, H.; Hirayama, A.; Inanami, O.; Tsuboi, K. Metabolic analysis of radioresistant medulloblastoma stem-like clones and potential therapeutic targets. PLoS ONE 2017, 12, e0176162. [CrossRef] (PubMed)
<br><span id=»68″ class=»referencess blue-text»>68</span> Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Инициаторы, усилители или ахиллесова пята? Nat. Rev. Cancer 2014, 14, 709-721. [CrossRef] [PubMed]
<br><span id=»69″ class=»referencess blue-text»>69</span> Feng, H.; Wang, J.; Chen, W.; Shan, B.; Guo, Y.; Xu, J.; Wang, L.; Guo, P.; Zhang, Y. Hypoxia-induced autophagy as an additional mechanism in human osteosarcoma radioresistance. J. Bone Oncol. 2016, 5, 67-73. [CrossRef]
<br><span id=»70″ class=»referencess blue-text»>70</span> Samanta, D.; Park, Y.; Andrabi, S.A.; Shelton, L.M.; Gilkes, D.M.; Semenza, G.L. PHGDH Expression Is Required for Mitochondrial Redox Homeostasis, Breast Cancer Stem Cell Maintenance, and Lung Metastasis. Cancer Res. 2016, 76, 4430-4442. [CrossRef]
<br><span id=»71″ class=»referencess blue-text»>71</span> Diepart, C.; Karroum, O.; Magat, J.; Feron, O.; Verrax, J.; Calderon, P.B.; Grégoire, V.; Leveque, P.; Stockis, J.; Dauguet, N.; et al. Лечение триоксидом мышьяка снижает скорость потребления кислорода опухолевыми клетками и радиосенсибилизирует солидные опухоли. Cancer Res. 2012, 72, 482-490. [CrossRef]
<br><span id=»72″ class=»referencess blue-text»>72</span> Maggiorella, L.; Wen, B.; Frascogna, V.; Opolon, P.; Bourhis, J.; Deutsch, E. Combined radiation sensitizing and anti-angiogenic effects of ionizing radiation and the protease inhibitor ritonavir in a head and neck carcinoma model. Anticancer Res. 2005, 25, 4357-4362.
<br><span id=»73″ class=»referencess blue-text»>73</span> Dwarakanath, B.S. Цитотоксичность, радиосенсибилизация и химиосенсибилизация опухолевых клеток 2-дезокси-D-глюкозой in vitro. J. Cancer Res. Ther. 2009, 5 (Suppl. 1), 27-31. [CrossRef]
<br><span id=»74″ class=»referencess blue-text»>74</span> Kim, J.H.; Kim, S.H.; He, S.Q.; Alfieri, A.A.; Young, C.W. Potentiation of radiation effects on multicellular tumor spheroids (MTS) of HeLa cells by lonidamine. Int. J. Radiat. Oncol. Biol. Phys. 1989, 16, 1277-1280. [CrossRef]
<br><span id=»75″ class=»referencess blue-text»>75</span> Nath, K.; Guo, L.; Nancolas, B.; Nelson, D.S.; Shestov, A.A.; Lee, S.C.; Roman, J.; Zhou, R.; Leeper, D.B.; Halestrap, A.P.; et al. Mechanism of antineoplastic activity of lonidamine. Biochim. Biophys Acta 2016, 1866, 151-162. [CrossRef]
<br><span id=»76″ class=»referencess blue-text»>76</span> Blackburn, A.C.; Matthaei, K.I.; Lim, C.; Taylor, M.C.; Cappello, J.Y.; Hayes, J.D.; Anders, M.W.; Board, P.G. Deficiency of glutathione transferase zeta causes oxidative stress and activation of antioxidant response pathways. Мол. Pharmacol. 2006, 69, 650-657. [CrossRef] [PubMed]
<br><span id=»77″ class=»referencess blue-text»>77</span> Theodoratos, A.; Tu, W.J.; Cappello, J.; Blackburn, A.C.; Matthaei, K.; Board, P.G. Phenylalanine-induced leucopenia in genetic and dichloroacetic acid generated deficiency of glutathione transferase Zeta. Биохим. Pharmacol. 2009, 77, 1358-1363. [CrossRef] [PubMed]
<br><span id=»78″ class=»referencess blue-text»>78</span> Michelakis, E.D.; Webster, L.; Mackey, J.R. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br. J. Cancer 2008, 99, 989-994. [CrossRef] [PubMed]
<br><span id=»79″ class=»referencess blue-text»>79</span> Stacpoole, P.W. The pharmacology of dichloroacetate. Metabolism 1989, 38, 1124-1144. [CrossRef]
<br><span id=»80″ class=»referencess blue-text»>80</span> Halestrap, A.P. Митохондриальный переносчик пирувата. Кинетика и специфичность для субстратов и ингибиторов. Биохим. J. 1975, 148, 85-96. [CrossRef]
<br><span id=»81″ class=»referencess blue-text»>81</span> Pathak, R.K.; Marrache, S.; Harn, D.A.; Dhar, S. Mito-DCA: молекулярный каркас, нацеленный на митохондрии, для эффективной доставки метаболического модулятора дихлорацетата. ACS Chem. Biol. 2014, 9, 1178-1187. [CrossRef]
<br><span id=»82″ class=»referencess blue-text»>82</span> Gang, B.P.; Dilda, P.J.; Hogg, P.J.; Blackburn, A.C. Targeting of two aspects of metabolism in breast cancer treatment. Cancer Biol. Ther. 2014, 15, 1533-1541. [CrossRef]
<br><span id=»83″ class=»referencess blue-text»>83</span> Sutendra, G.; Dromparis, P.; Kinnaird, A.; Stenson, T.H.; Haromy, A.; Parker, J.M.R.; McMurtry, M.S.; Michelakis, E.D. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene 2013, 32, 1638-1650. [CrossRef]
<br><span id=»84″ class=»referencess blue-text»>84</span> Xuan, Y.; Hur, H.; Ham, I.H.; Yun, J.; Lee, J.Y.; Shim, W.; Kim, Y.B.; Lee, G.; Han, S.U.; Cho, Y.K. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp. Cell Res. 2014, 321, 219-230. [CrossRef]
<br><span id=»85″ class=»referencess blue-text»>85</span> Cesi, G.; Walbrecq, G.; Zimmer, A.; Kreis, S.; Haan, C. ROS production induced by BRAF inhibitor treatment rewires metabolic processes affecting cell growth of melanoma cells. Mol. Cancer 2017, 16, 1-16. [CrossRef]
<br><span id=»86″ class=»referencess blue-text»>86</span> Kluza, J.; Corazao-Rozas, P.; Touil, Y.; Jendoubi, M.; Maire, C.; Guerreschi, P.; Jonneaux, A.; Ballot, C.; Balayssac, S.; Valable, S.; et al. Инактивация сигнальной оси HIF-1α/PDK3 стимулирует меланому к митохондриальному окислительному метаболизму и потенцирует терапевтическую активность про-оксидантов. Cancer Res. 2012, 72, 5035-5047. [CrossRef] [PubMed]
<br><span id=»87″ class=»referencess blue-text»>87</span> Sun, H.; Zhu, A.; Zhou, X.; Wang, F. Suppression of pyruvate dehydrogenase kinase-2 re-sensitizes paclitaxel-resistant human lung cancer cells to paclitaxel. Oncotarget 2017, 8, 52642-52650. [CrossRef] (PubMed)
<br><span id=»88″ class=»referencess blue-text»>88</span> Velpula, K.K.; Bhasin, A.; Asuthkar, S.; Tsung, A.J. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013, 73, 7277-7289. [CrossRef] [PubMed]
<br><span id=»89″ class=»referencess blue-text»>89</span> Zhang, W.; Hu, X.; Zhou, W.; Tam, K.Y. Liquid Chromatography-Tandem Mass Spectrometry Method Revealed that Lung Cancer Cells Exhibited Distinct Metabolite Profiles upon the Treatment with Different Pyruvate Dehydrogenase Kinase Inhibitors. J. Proteome Res. 2018, 17, 3012-3021. [CrossRef]
<br><span id=»90″ class=»referencess blue-text»>90</span> Dubuis, S.; Ortmayr, K.; Zampieri, M. A framework for large-scale metabolome drug profiling links coenzyme A metabolism to the toxicity of anti-cancer drug dichloroacetate. Commun. Biol. 2018, 1, 1-11. [CrossRef]
<br><span id=»91″ class=»referencess blue-text»>91</span> El Sayed, S.M.; Baghdadi, H.; Ahmed, N.S.; Almaramhy, H.H.; Mahmoud, A.A.; El-Sawy, S.A.; Ayat, M.; Elshazley, M.; Abdel-Aziz, W.; Abdel-Latif, H.M.; et al. Дихлорацетат — антиметаболит, который антагонизирует ацетат и лишает раковые клетки его преимуществ: Новая медицинская гипотеза, основанная на доказательствах. Med. Hypotheses 2019, 122, 206-209. [CrossRef]
<br><span id=»92″ class=»referencess blue-text»>92</span> Li, X.; Liu, J.; Hu, H.; Lu, S.; Lu, Q.; Quan, N.; Rousselle, T.; Patel, M.S.; Li, J. Dichloroacetate Ameliorates Cardiac Dysfunction Caused by Ischemic Insults Through AMPK Signal Pathway-Not Only Shifts Metabolism. Toxicol. Sci. 2019, 167, 604-617. [CrossRef]
<br><span id=»93″ class=»referencess blue-text»>93</span> Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for cancer prevention and treatment. Oncotarget 2015, 6, 7365-7378. [CrossRef]
<br><span id=»94″ class=»referencess blue-text»>94</span> Dunbar, E.M.; Coats, B.S.; Shroads, A.L.; Langaee, T.; Lew, A.; Forder, J.R.; Shuster, J.J.; Wagner, D.A.; Stacpoole, P.W. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Investig. New Drugs 2014, 32, 452-464. [CrossRef]
<br><span id=»95″ class=»referencess blue-text»>95</span> Michelakis, E.D.; Sutendra, G.; Dromparis, P.; Webster, L.; Haromy, A.; Niven, E.; Maguire, C.; Gammer, T.L.; Mackey, J.R.; Fulton, D.; et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci. Transl. Med. 2010, 2. [CrossRef]
<br><span id=»96″ class=»referencess blue-text»>96</span> De Ridder, M.; Verovski, V.N.; Chiavaroli, C.; Berge, D.L.V.D.; Monsaert, C.; Law, K.; Storme, G.A. The radiosensitizing effect of immunoadjuvant OM-174 requires cooperation between immune and tumor cells through interferon-gamma and inducible nitric oxide synthase. Int. J. Radiat. Oncol. Biol. Phys. 2006, 66, 1473-1480. [CrossRef] [PubMed]
<br><span id=»97″ class=»referencess blue-text»>97</span> Wang, H.; Bouzakoura, S.; de Mey, S.; Jiang, H.; Law, K.; Dufait, I.; Corbet, C.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Ауранофин радиосенсибилизирует опухолевые клетки путем воздействия на тиоредоксин-редуктазу и вызванное этим перепроизводство реактивных форм кислорода. Oncotarget 2017, 8, 35728-35742. [CrossRef] [PubMed]
<br><span id=»98″ class=»referencess blue-text»>98</span> de Mey, S.; Jiang, H.; Corbet, C.; Wang, H.; Dufait, I.; Law, K.; Bastien, E.; Verovski, V.; Gevaert, T.; Feron, O.; et al. Antidiabetic Biguanides Radiosensitize Hypoxic Colorectal Cancer Cells Through a Decrease in Oxygen Consumption. Front. Pharmacol. 2018, 9, 1073. [CrossRef] [PubMed]
<br><span id=»99″ class=»referencess blue-text»>99</span> Noeparast, A.; Teugels, E.; Giron, P.; Verschelden, G.; De Brakeleer, S.; Decoster, L.; De Grève, J. Non-V600 BRAF mutations recurrently found in lung cancer predict sensitivity to the combination of Trametinib and Dabrafenib. Oncotarget 2016, 8, 60094-60108. [CrossRef]
<br><span id=»100″ class=»referencess blue-text»>100</span> Polet, F.; Corbet, C.; Pinto, A.; Rubio, L.I.; Martherus, R.; Bol, V.; Drozak, X.; Grégoire, V.; Riant, O.; Feron, O. Снижение доступности серина дополняет ингибирование метаболизма глутамина для блокирования роста лейкозных клеток. Oncotarget 2015, 7, 1765-1776. [CrossRef]
<p></p>
<p>Сопутствующий контент:</p>
<figure class=»wp-block-embed is-type-wp-embed is-provider-dca-guide wp-block-embed-dca-guide»><div class=»wp-block-embed__wrapper»>