Хелена Популо1,2, Регина Калдас1,2,3, Хосе Мануэль Лопес1,2,4,5, Джоана Пардал5, Вальдемар Максимо1,2,4 и Паула Соарес†,2,4
† Институт исследований и инноваций в области здравоохранения (Instituto de Investigacao e Inovacao em Saude), Университет Порту, Порту, Португалия
1 Институт молекулярной патологии и иммунологии Университета Порту (IPATIMUP), Университет Порту, Порту, Португалия
Тел: +22 557 0700; Факс: +22 557 0799; E-mail: [email protected]
2 Институт исследований и инноваций в области здравоохранения (Instituto de Investigacao e Inovac¸aoem Saude), Университет Порту, Порту, Португалия
3 Медицинский факультет, Университет Порту, Порту, Португалия
4 Кафедра патологии и онкологии, Медицинский факультет, Университет Порту, Порту, Португалия
Служба анатомической патологии больницы Сан-Жоао, Порту, Португалия
Опубликовано онлайн: 14 мая 2015 г
Аннотация
Цель: Мы задались целью проверить, есть ли основания считать дихлорацетат (ДХА), который ингибирует киназу пируватдегидрогеназы (PDK) и обращает метаболический сдвиг раковых клеток от гликолиза к окислительному фосфорилированию, перспективным препаратом для терапии больных кожной меланомой (КМ).
Дизайн и методы исследования: Мы оценили профиль экспрессии PDK 1, 2 и 3 в серии образцов меланомы, чтобы проверить, экспрессируют ли опухоли меланомы мишени DCA, коррелирует ли эта экспрессия с активацией важных сигнальных каскадов для меланомагенеза, а также с прогнозом пациентов с меланомой. Мы также установили чувствительность клеточных линий меланомы к лечению DCA, оценив их метаболические изменения, пролиферацию и выживаемость.
Результаты: Мы обнаружили, что обе изоформы PDK 1 и 2 сверхэкспрессированы в КМ по сравнению с невусами, причем эта экспрессия связана с экспрессией эффекторов пути mTOR и не зависит от мутационного статуса BRAF. Клеточные линии меланомы, обработанные DCA, показали изменение метаболизма, то есть снижение потребления глюкозы и выработки лактата, снижение пролиферации, увеличение апоптоза и снижение активации пути mTOR.
Заключение: Наши результаты показывают, что экспрессия PDK может играть определенную роль в развитии меланомы и что DCA может быть полезен для терапии КМ, отдельно или в комбинации с ингибиторами mTOR.
Ключевые слова: дихлорацетат, меланома, метаболизм, mTOR, киназа пируватдегидрогеназы
© 2015 Informa UK, Ltd. ISSN 1472-8222, e-ISSN 1744-7631
ВВЕДЕНИЕ
Кожная меланома (КМ) — очень агрессивная злокачественная опухоль, и, несмотря на то, что она является наименее распространенным видом рака кожи, на нее приходится большинство смертей от рака кожи. Поскольку заболеваемость КМ растет, в настоящее время она является наиболее вероятным инвазивным раком, развивающимся в возрасте до 50 лет у мужчин [1,2]. Воздействие ультрафиолетового излучения считается основным фактором риска меланомагенеза [2]. КМ можно классифицировать по различным гистологическим подтипам, наиболее распространенной является поверхностная распространяющаяся меланома (ПРМ), за которой следуют узловая меланома (УМ), меланома lentigo maligna (МММ) и акральная лентигинозная меланома (АЛМ). SSM и NM возникают на коже с периодическим воздействием солнца, в то время как LMM возникает на хронически поврежденной солнцем коже, а ALM ограничена кожей без воздействия солнца. Эта гистологическая классификация не имеет прогностического значения [3,4]. При стадировании КМ учитывается толщина опухоли, изъязвление, частота митозов, поражение узлов и наличие метастазов [5]. К счастью, большинство случаев РМ диагностируется на ранней стадии, при этом 5-летняя выживаемость достигает 98% [1]. Тем не менее, для пациентов с метастатической меланомой медиана выживаемости составляет всего 8 — 9 месяцев [5].
КМ является очень гетерогенной опухолью, и многие клеточные сигнальные пути дерегулируются в меланомагенезе [6,7]. Путь MAPK конститутивно активирован в большинстве РМ. Мутации NRAS были зарегистрированы в 10 — 20% РМ, наиболее распространенной является NRASQ61K/R. Мутации BRAF, в частности мутация BRAFV600E, были выявлены в 40-60% случаев. Наша группа наблюдала ассоциацию между наличием мутаций BRAF и недавно описанными мутациями промотора TERT в КМ [8]. Мутации BRAFV600E и, в меньшей степени, NRASQ61K/R также были выявлены почти в 80% невусов (доброкачественных меланоцитарных образований, которые в 25% случаев считаются предшественниками КМ), что указывает на то, что активация MAPK-пути может быть необходимой, но недостаточной для развития меланомы [7,9,10]. Фактически, BRAFV600E связан с онкоген-индуцированным старением и поэтому может привести во многих невусах к состоянию остановки роста [11]. Активация пути PI3K-AKTmTOR может преодолеть этот фенотип старения и ускорить прогрессию опухоли [12]. Наша группа подтвердила важность PI3K-AKT-mTOR пути в агрессивности КМ, связав его активацию с наличием мутаций BRAF и худшими прогностическими характеристиками. Действительно, сверхэкспрессия эффекторов пути mTOR была связана с более высокой частотой митозов, более высоким уровнем Кларка, увеличенной толщиной опухоли и наличием изъязвления кожи [13].
До 2011 года варианты лечения пациентов с распространенным КМ в основном основывались на традиционной химиотерапии, которая имела низкую частоту ответов и незначительное влияние на общую выживаемость пациентов (ОВ). С тех пор было одобрено пять новых препаратов для пациентов с КМ IV стадии [14]. Ипилимумаб, анти-CTLA4 антитело, показал заметное улучшение OS пациентов, но его неблагоприятные эффекты не позволили использовать его для всех пациентов [15]. Вемурафениб и дабрафениб, селективные ингибиторы BRAFV600E, и траметиниб, ингибитор MEK, воздействуют на MAPK-путь и, как сообщается, позволяют достичь частоты ответов более 50%, что даже лучше, чем у ипилимумаба. Однако их улучшение OS не превышает 7-8 месяцев из-за развития резистентности [16-18]. Недавно другой иммунный модулятор, пембролизумаб, антитело против рецептора запрограммированной смерти-1 (PD-1), был одобрен для лечения пациентов с неоперабельной или метастатической меланомой и прогрессированием заболевания после приема ипилимумаба или, в случае положительной мутации BRAFV600 , ингибитора BRAF [19]. В настоящее время проводятся клинические испытания препаратов, направленных на путь PI3K-AKT-mTOR [4,14], однако требуются новые терапевтические подходы.
Метаболизм раковых клеток отличается от метаболизма нормальных клеток. В нормальных клетках, в зависимости от наличия O2, глюкоза может быть частично метаболизирована до лактата посредством гликолиза в гипоксических условиях, или полностью окислена доCO2 в присутствии O2, посредством митохондриального окислительного фосфорилирования, более эффективного энергетического процесса. Раковые клетки, с другой стороны, метаболизируют большую часть глюкозы до лактата, независимо от наличия O2, так называемый эффект Варбурга [20,21]. Приобретение гликолитического фенотипа раковыми клетками до конца не изучено, однако признана существенная роль транскрипционного фактора гипоксия-индуцибельного фактора 1a (HIF1-α) [22]. Известно, что белок HIF1-α стабилизируется при гипоксии, но онкогенные пути, такие как MAPK и mTOR, также опосредуют его активацию при раке (рис. 1) [22,23]. Убедительные доказательства указывают на то, что РМ приобретает этот гликолитический фенотип, что может быть подтверждено при сканировании пациентов с помощью ФДГПЭТ [24-27]. Фактически, HIF1-α, по-видимому, сверхэкспрессируется в КМ, поскольку кожа является умеренно-гипоксической средой, а производство меланина косвенно стимулирует экспрессию HIF1-α путем производства ROS [28-30]. Тем не менее, цитоплазматический гликолиз должен быть отсоединен от митохондриального окислительного фосфорилирования, чтобы большая часть пирувата могла быть преобразована в лактат. Этот последний процесс управляется киназой пируватдегидрогеназы (PDK), ферментом, регулируемым HIF1-α (рис. 1) [31,32].
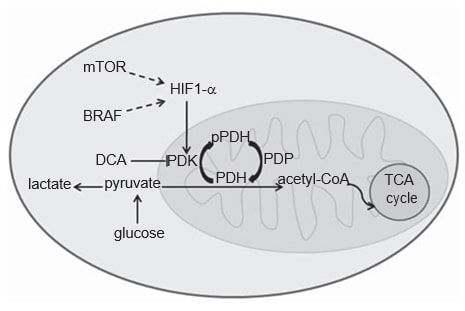
PDK является элементом митохондриального пируватдегидрогеназного комплекса (PDC). В состав PDC входит фермент пируватдегидрогеназа (PDH) и его регуляторные белки: PDK, который, фосфорилируя PDH, действует как ингибитор, и фосфатаза пируватдегидрогеназы, которая активирует PDH путем дефосфорилирования [33]. Дефосфорилированная PDH катализирует окислительное декарбоксилирование пирувата в ацетил-КоА,CO2 и NADH (H+ ), тем самым связывая гликолитический путь с окислительным путем цикла трикарбоновых кислот (Рисунок 1) [33]. Существует четыре изоформы PDK (1, 2, 3, 4), которые различаются по присущей им активности, тканевому распределению и чувствительности к селективному ингибитору — дихлорацетату (DCA) [33-35]. Хотя PDK1 является единственной, способной связываться со всеми сайтами фосфорилирования PDH, PDK3 представляется наиболее активной изоформой [33]. PDK2 наиболее чувствительна к ингибированию DCA и повсеместно экспрессируется в различных тканях человека, в то время как другие более тканеспецифичны [33,34]. PDK4 в основном связан с физиологической метаболической гибкостью и является единственной изоформой, которая не повышается под действием HIF1-α [32,36]. Экспрессия изоформ PDK была оценена в различных типах рака, а именно в плоскоклеточной карциноме головы и шеи (HNSCC) [37], раке толстой кишки [38], почечно-клеточной карциноме [39] и раке желудка [40].
DCA используется для лечения состояний, связанных с молочнокислым ацидозом в условиях митохондриальной дисфункции, таких как врожденные митохондриальные заболевания [35]. В клетках немелкоклеточного рака легких, глиобластомы и рака молочной железы Bonnet и др. сообщили, что DCA вызывает апоптоз и снижает рост клеток, способствуя окислению глюкозы, деполяризации митохондриальной мембраны и выработке ROS [41]. Эти эффекты были воспроизведены в различных моделях рака и, по-видимому, являются избирательными для раковых клеток [35,41,42]. Более того, DCA также способен подавлять ангиогенез через косвенное ингибирование HIF1-α [43]. Все эти данные в сочетании с хорошо известным профилем безопасности у людей доказывают, что DCA является перспективным препаратом для лечения рака. Клиническое исследование по оценке действия DCA у пациентов с глиобластомой уже было проведено
Кожная меланома (КМ) — очень агрессивная злокачественная опухоль, и, несмотря на то, что она является наименее распространенным видом рака кожи, на нее приходится большинство смертей от рака кожи. Поскольку заболеваемость КМ растет, в настоящее время она является наиболее вероятным инвазивным раком, развивающимся в возрасте до 50 лет у мужчин [1,2]. Воздействие ультрафиолетового излучения считается основным фактором риска меланомагенеза [2]. КМ можно классифицировать по различным гистологическим подтипам, наиболее распространенной является поверхностная распространяющаяся меланома (ПРМ), за которой следуют узловая меланома (УМ), меланома lentigo maligna (МММ) и акральная лентигинозная меланома (АЛМ). SSM и NM возникают на коже с периодическим воздействием солнца, в то время как LMM возникает на хронически поврежденной солнцем коже, а ALM ограничена кожей без воздействия солнца. Эта гистологическая классификация не имеет прогностического значения [3,4]. При стадировании КМ учитывается толщина опухоли, изъязвление, частота митозов, поражение узлов и наличие метастазов [5]. К счастью, большинство случаев РМ диагностируется на ранней стадии, при этом 5-летняя выживаемость достигает 98% [1]. Тем не менее, для пациентов с метастатической меланомой медиана выживаемости составляет всего 8 — 9 месяцев [5]. КМ является очень гетерогенной опухолью, и многие клеточные сигнальные пути дерегулируются в меланомагенезе [6,7]. Путь MAPK конститутивно активирован в большинстве РМ. Мутации NRAS были зарегистрированы в 10 — 20% РМ, наиболее распространенной является NRASQ61K/R. Мутации BRAF, в частности мутация BRAFV600E, были выявлены в 40-60% случаев. Наша группа наблюдала ассоциацию между наличием мутаций BRAF и недавно описанными мутациями промотора TERT в КМ [8]. Мутации BRAFV600E и, в меньшей степени, NRASQ61K/R также были выявлены почти в 80% невусов (доброкачественных меланоцитарных образований, которые в 25% случаев считаются предшественниками КМ), что указывает на то, что активация MAPK-пути может быть необходимой, но недостаточной для развития меланомы [7,9,10]. Фактически, BRAFV600E связан с онкоген-индуцированным старением и поэтому может привести во многих невусах к состоянию остановки роста [11]. Активация пути PI3K-AKTmTOR может преодолеть этот фенотип старения и ускорить прогрессию опухоли [12]. Наша группа подтвердила важность PI3K-AKT-mTOR пути в агрессивности КМ, связав его активацию с наличием мутаций BRAF и худшими прогностическими характеристиками. Действительно, сверхэкспрессия эффекторов пути mTOR была связана с более высокой частотой митозов, более высоким уровнем Кларка, увеличенной толщиной опухоли и наличием изъязвления кожи [13].
До 2011 года варианты лечения пациентов с распространенным КМ в основном основывались на традиционной химиотерапии, которая имела низкую частоту ответов и незначительное влияние на общую выживаемость пациентов (ОВ). С тех пор было одобрено пять новых препаратов для пациентов с КМ IV стадии [14]. Ипилимумаб, анти-CTLA4 антитело, показал заметное улучшение OS пациентов, но его неблагоприятные эффекты не позволили использовать его для всех пациентов [15]. Вемурафениб и дабрафениб, селективные ингибиторы BRAFV600E, и траметиниб, ингибитор MEK, воздействуют на MAPK-путь и, как сообщается, позволяют достичь частоты ответов более 50%, что даже лучше, чем у ипилимумаба. Однако их улучшение OS не превышает 7-8 месяцев из-за развития резистентности [16-18]. Недавно другой иммунный модулятор, пембролизумаб, антитело против рецептора запрограммированной смерти-1 (PD-1), был одобрен для лечения пациентов с неоперабельной или метастатической меланомой и прогрессированием заболевания после приема ипилимумаба или, в случае положительной мутации BRAFV600, ингибитора BRAF [19]. В настоящее время проводятся клинические испытания препаратов, направленных на путь PI3K-AKT-mTOR [4,14], однако необходимы новые терапевтические подходы. Метаболизм раковых клеток отличается от метаболизма нормальных клеток. В нормальных клетках, в зависимости от наличия O2, глюкоза может быть частично метаболизирована до лактата посредством гликолиза в гипоксических условиях, или полностью окислена доCO2 в присутствии O2, посредством митохондриального окислительного фосфорилирования, более эффективного энергетического процесса. Раковые клетки, с другой стороны, метаболизируют большую часть глюкозы до лактата, независимо от наличия O2, так называемый эффект Варбурга [20,21]. Приобретение гликолитического фенотипа раковыми клетками до конца не изучено, однако признана существенная роль транскрипционного фактора гипоксия-индуцибельного фактора 1a (HIF1-α) [22]. Известно, что белок HIF1-α стабилизируется при гипоксии, но онкогенные пути, такие как MAPK и mTOR, также опосредуют его активацию при раке (рис. 1) [22,23]. Убедительные доказательства указывают на то, что РМ приобретает этот гликолитический фенотип, что может быть подтверждено при сканировании пациентов с помощью ФДГПЭТ [24-27]. Фактически, HIF1-α, по-видимому, сверхэкспрессируется в КМ, поскольку кожа является умеренно-гипоксической средой, а производство меланина косвенно стимулирует экспрессию HIF1-α путем производства ROS [28-30]. Тем не менее, цитоплазматический гликолиз должен быть отсоединен от митохондриального окислительного фосфорилирования, чтобы большая часть пирувата могла быть преобразована в лактат. Этот последний процесс управляется киназой пируватдегидрогеназы (PDK), ферментом, который регулируется HIF1-α (рис. 1) [31,32]. PDK является элементом митохондриального пируватдегидрогеназного комплекса (PDC). PDC включает в себя фермент пируватдегидрогеназу (PDH) и ее регуляторные белки: PDK, который, фосфорилируя PDH, действует как ингибитор, и фосфатаза пируватдегидрогеназы, которая активирует PDH путем дефосфорилирования [33]. Дефосфорилированная PDH катализирует окислительное декарбоксилирование пирувата в ацетил-КоА,CO2 и NADH (H+ ), тем самым связывая гликолитический путь с окислительным путем цикла трикарбоновых кислот (Рисунок 1) [33]. Существует четыре изоформы PDK (1, 2, 3, 4), которые различаются по присущей им активности, тканевому распределению и чувствительности к селективному ингибитору — дихлорацетату (DCA) [33-35]. Хотя PDK1 является единственной, способной связываться со всеми сайтами фосфорилирования PDH, PDK3 представляется наиболее активной изоформой [33]. PDK2 наиболее чувствительна к ингибированию DCA и повсеместно экспрессируется в различных тканях человека, в то время как другие более тканеспецифичны [33,34]. PDK4 в основном связан с физиологической метаболической гибкостью и является единственной изоформой, которая не повышается под действием HIF1-α [32,36]. Экспрессия изоформ PDK была оценена в различных типах рака, а именно в плоскоклеточной карциноме головы и шеи (HNSCC) [37], раке толстой кишки [38], почечно-клеточной карциноме [39] и раке желудка [40]. DCA используется для лечения состояний, связанных с молочнокислым ацидозом в условиях митохондриальной дисфункции, таких как врожденные митохондриальные заболевания [35]. В клетках немелкоклеточного рака легких, глиобластомы и рака молочной железы Bonnet и др. сообщили, что DCA вызывает апоптоз и снижает рост клеток, способствуя окислению глюкозы, деполяризации митохондриальной мембраны и выработке ROS [41]. Эти эффекты были воспроизведены в различных моделях рака и, по-видимому, являются избирательными для раковых клеток [35,41,42]. Более того, DCA также способен подавлять ангиогенез через косвенное ингибирование HIF1-α [43]. Все эти данные в сочетании с хорошо известным профилем безопасности у людей доказывают, что DCA является перспективным препаратом для лечения рака. Уже было проведено клиническое исследование по оценке действия DCA у пациентов с глиобластомой, в ходе которого было отмечено усиление апоптоза и снижение ангиогенеза, причем у четырех из пяти пациентов были получены обнадеживающие результаты [44].
В настоящем исследовании оценивалась экспрессия изоформ PDK 1 — 3 в КМ и ее связь с экспрессией основных каскадов сигнальных путей меланомагенеза и с прогнозом пациентов с меланомой. Также были проведены исследования in vitro для оценки влияния лечения DCA на метаболизм, пролиферацию и выживаемость клеточных линий меланомы с различными генетическими профилями.
Материалы и методы
Отбор образцов, клинико-патологические и прогностические параметры
Формалин-фиксированные, парафинированные ткани из 120 случаев КМ и 22 меланоцитарных невусов (12 сложных невусов и 10 невусов Шпица) были получены из службы анатомической патологии больницы С. Жоао, Порту, и больницы С. Маркос, Брага. Клинико-патологические (Таблица 1) и последующие данные были получены из историй болезни пациентов и онкологических регистров больницы С. Жоао и больницы С. Маркос, а также из RORENO (онкологический регистр Северного региона). Все случаи были пересмотрены и стадированы в соответствии с 7-м изданием AJCC [5]. Данные последующего наблюдения включают данные о рецидивах и метастазах (DFS; n = 108) и количестве смертей от меланомы (болезнь-специфическая смертность; OS; n = 118). Средняя продолжительность наблюдения за пациентами по DFS составила 51 месяц (SE ± 3,59, диапазон 1-195), а по OS — 55 месяцев (SE ± 3,48, диапазон 1-207). Данная работа была одобрена местным этическим комитетом и соответствовала национальным этическим правилам.
Иммуногистохимический анализ
Окрашивание анализируемых белков проводилось на 3-мкм парафиновых срезах репрезентативных участков опухоли, смонтированных на предметных стеклах, покрытых поли-L-лизином. Срезы депарафинизировали и регидратировали, затем проводили процедуру микроволнового извлечения антигена с помощью 10 мМ цитратного буфера натрия pH 6,0 с 1 мМ ЭДТА pH 9,0 (PDK2) или ЭДТА буфера pH 9,0 (PDK1 и PDK3). Секции инкубировали в течение ночи при 4°C в увлажненной камере с первичными антителами PDK1 (поликлональное, кролик, 1:50), PDK2 (поликлональное, кролик, 1:150), PDK3 (моноклональное, мышь, 1:500), все от Sigma-Aldrich Co. (Сент-Луис, Миссури, США). Детекцию проводили с помощью Envision G/2 System/AP (K5355; Dako, Дания). антищелочной фосфатазы методом щелочной фосфатазы (APAAP), и цвет развивался с постоянным красным хромогеном. Слайды контрастировали гематоксилином, а затем монтировали с помощью водной монтажной среды. Метод APAAP применялся для того, чтобы избежать вмешательства меланиновой пигментации в иммуногистохимический анализ. Ткань желудка человека использовалась в качестве отрицательного (пропуск первичного антитела) и положительного контроля.
Иммуногистохимическая оценка
Три наблюдателя (J.M.L., H.P. и R.C.) оценивали иммунореактивность опухолевых клеток без знания каких-либо клинических данных случаев. Иммунореактивность прилегающей неопухолевой ткани использовалась в качестве внутреннего контроля. Для PDK1 — 3 был определен балл IHC, который является результатом умножения балла интенсивности окрашивания (отрицательный = 0, слабый = 1, умеренный = 2 и сильный = 3) и балла протяженности иммунореактивности опухолевых клеток (0 — 5% = 0, 6 — 25% = 1, 26 — 50% = 2, 51 — 75% = 3, 76 — 100% = 4). Затем оценки IHC классифицировались как отрицательные/низкие (значение оценки £ 4) и умеренные/высокие (значение оценки > 4). Анализ каскадов MAPK и PI3KAKT-mTOR в части серий был проведен ранее [13], и его результаты были использованы в данном исследовании.
Выделение ДНК и анализ мутаций
Выделение ДНК из опухолей размером менее 5 мм проводилось после микродиссекции с помощью системы PALM MicroLaser Systems (PALM, Германия) и с использованием набора Quiamp DNA micro kit (Quiagen, Хильден). В опухолях размером более 5 мм выделение ДНК проводилось путем ручного препарирования 10 мкм цельных участков парафинированной ткани с использованием набора Invisorb spin tissue mini kit (Invitek, Берлин). Фрагменты экзона 15 BRAF и экзона 2 NRAS амплифицировали с помощью полимеразной цепной реакции (ПЦР) с использованием ранее описанных праймеров [45]. Геномную ДНК (25 — 100 нг) амплифицировали методом ПЦР с использованием следующих условий циклирования: 35 с при 94°C, 40 с при 58°C для BRAF и 57°Cдля NRAS, и 45 с при 72°Cв течение 40 циклов.
| Клинико-патологические особенности | |
| Количество случаев (n) | 120 |
| Средний возраст (± SD) | 61.5 ± 17.0 |
| Пол (n [%]) | |
| Женский | 68 (56.7) |
| Мужской | 52 (43.3) |
| Воздействие солнца (n [%]) | |
| Отсутствует | 27 (22.7) |
| Периодическое | 72 (60.5) |
| Хроническое | 20 (16.8) |
| Гистологический подтип (n [%]) | |
| ЛММ | 16 (13.3 |
| АЛМ | 24 (20.0) |
| НМ | 19 (15.8) |
| SSM | 61 (50.8) |
| Медианная толщина (мм) | 3.7 (0 — 70) |
| Эпидермальное изъязвление (n [%]) | |
| Отсутствует | 79 (65.8) |
| Присутствует | 41 (34.2) |
| Количество митозов (n [%]) | |
| < 1/мм2 | 45 (37.5) |
| ≥1/мм2 | 75 (62.5 |
| pT (n [%]) | |
| ≤ pT2 | 62 (51.7) |
| >pT2 | 58 (48.3) |
Все продукты ПЦР были очищены и непосредственно секвенированы на автоматическом секвенаторе ABI Prism 3130 xl (Perkin-Elmer, Foster City, CA) с использованием набора для секвенирования ABI Prism Dye Terminator Cycle (Perkin-Elmer). Реакция секвенирования проводилась в прямом направлении, а в образцах, предположительно несущих мутации, проводилась независимая ПЦР-амплификация в прямом и обратном направлении. Опять же, мутационный анализ BRAF и NRAS в части серий был проведен ранее [13], и его результаты были использованы в данном исследовании.
Клеточные линии и условия культивирования
В данной работе были использованы две клеточные линии: клеточная линия меланомы кожи A375, несущая BRAFV600E, и клеточная линия меланомы кожи Mewo, несущая BRAFwt. Обе клеточные линии были протестированы на наличие микоплазмы.
A375 поддерживали в среде RPMI (Gibco/BRL — Invitrogen), а Mewo — в среде DMEM (Gibco/BRL — Invitrogen). Все среды были дополнены 10% фетальной бычьей сывороткой, 100 U/мл пенициллина и 100 мкг/мл стрептомицина. Клеточные линии поддерживали в увлажненной атмосфере (5%CO2) при 37°C.
Обработка клеточных линий меланомы с помощью DCA
Натрий DCA, приобретенный у Sigma-Aldrich (Сент-Луис, МО, ЕСА), растворяли в dH2O, добавляли в культуральную среду и использовали для обработки через 24 и 48 ч. Клетки меланомы, инкубированные с культуральной средой, дополненной dH2O, служили контролем.
Анализ жизнеспособности клеток
Влияние DCA на рост клеточных линий меланомы анализировали с помощью анализа PrestoBlue (PB). Клетки высевали в 96-луночные планшеты при плотности 5 103 в 200 мкл среды. Через 24 ч среду заменяли на среду, содержащую 5, 20, 40, 60 мМ DCA. Клетки инкубировали в течение 24 и 48 ч, промывали PBS (pH 7,4) и оценивали рост клеток с помощью PB в соответствии с инструкциями производителя. Во время инкубации с клетками реагент PB модифицируется восстановительной средой жизнеспособных клеток и становится высокофлуоресцентным. Флуоресценцию измеряли с помощью микропланшетного ридера (Synergy HT Multi-Mode Microplate Reader, BioTek Instruments Inc., Winooski, VT, USA) при длинах волн возбуждения и испускания 560 и 590 нм, соответственно. Поглощение лунок, содержащих культуральную среду и опухолевые клетки, использовалось в качестве контроля, и каждое экспериментальное условие оценивалось в трех экземплярах и повторялось в двух экземплярах. Сравнивая измеренную флуоресценцию/абсорбцию лунок, содержащих DCA, с измерениями лунок, содержащих необработанные клетки, можно было составить профили доза-ответ и определить значения IC50 (концентрация, подавляющая выживаемость на 50%), используя GraphPadPrism5.0 (GraphPad Software, Inc., La Jolla, CA).
Количественное определение глюкозы и лактата
Для измерения уровня глюкозы и лактата клетки меланомы высевали в 6-луночные планшеты при конечной плотности 2 105 клеток/лунку и инкубировали при 37°C в течение 24 ч. Затем клетки обрабатывали 35 мМ DCA. В качестве контроля клетки инкубировали с транспортным средством (dH2O). После 24 и 48 ч лечения культуральную среду собирали. Уровень глюкозы в культуральной среде определяли количественно с помощью набора Glucose GOD/PAP Kit (Roche Applied Sciences) и вычитали из исходного уровня (0 ч). Лактат определяли аналогичным образом, используя ферментативный колориметрический анализ LO-POD (Spinreact, Sant Esteve de Bas, Испания).
Анализ клеточного цикла и апоптоза
Для анализа профиля клеточного цикла и апоптоза клетки меланомы высевали в 6-луночные планшеты при конечной плотности 1 105 клеток/лунку и инкубировали при 37°C в течение 24 ч. Затем клетки обрабатывали DCA в концентрации 35 мМ в течение 24 и 48 ч лечения. В качестве контроля клетки инкубировали с транспортным средством (dH2O). Для анализа клеточного цикла клетки собирали и фиксировали на ночь в ледяном 70% этаноле. Затем клетки ресуспендировали в PBS с 0,1 мг/мл RNase A и 5 мкг/мл йодистого пропидия перед анализом. Для измерения апоптоза клетки собирали и анализировали уровень апоптоза методом проточной цитометрии с использованием набора Annexin-V FITC Apoptosis Kit (Clontech Laboratories, Inc., Saint-Germainen-Laye, Франция) в соответствии с инструкциями производителя. Анализ содержания клеточной ДНК и экстернализации фосфатидилсерина проводили с помощью проточного цитометра (BD Accuri C6), регистрируя не менее 20 000 событий на образец. Данные анализировали с помощью программного обеспечения FlowJo 7.6.5 (Tree Star, Inc., Ashland, США).
Вестерн-блот анализ и антитела
Клетки лизировали в течение 15 мин при 4°C с использованием RIPA-буфера (1% NP-40 в 150 мМ NaCl, 50 мМ Трис (pH 7,5), 2 мМ ЭДТА), содержащего ингибиторы фосфатаз и протеаз. Количественное определение белков проводили с помощью модифицированного анализа Брэдфорда (Biorad). Образцы белка (50 мкг) разделяли в 10% гелях SDS/ PAGE и наносили на мембрану Hybond ECL (Amersham Biosciences). Мы использовали следующие первичные антитела: PDH и pPDH Ser293, от Abcam; PDK1 (Sigma-Aldrich), PDK2 (Sigma-Aldrich), HIF1-α (Transduction Laboratories) и эффекторы пути mTOR pS6 Ser235/236, s6, p-4EBP1 Thr37/46, 4EBP1, все от Cell Signaling Technology. Вторичные антитела конъюгировали с пероксидазой (Santa Cruz Biotechnology) и визуализировали с помощью раствора для детекции ECL. Мембраны повторно окрашивали козьим поликлональным антиактином (Santa Cruz Biotechnology) для контроля загрузки белка. Все эксперименты и количественные оценки (с использованием программного обеспечения Bio-Rad Quantity One 1-D Analysis (версия 4.6.6)) проводились в трех экземплярах.
Статистический анализ
Статистический анализ проводили с использованием STAT VIEW-J 5.0 (SAS Institute, Inc., Cary, NC). Связь между средним уровнем экспрессии (баллом) маркеров иммуногистохимии и клинико-патологическими параметрами оценивали с помощью ANOVA. При необходимости проводилась коррекция множественных сравнений с помощью post hoc тестов Бонферрони и Тамхана. Корреляцию между показателями иммунореактивности различных маркеров оценивали с помощью точного теста Фишера. Данные экспериментов с клеточными линиями анализировали с помощью непарного t-теста Стьюдента с двумя хвостами. Для оценки данных о выживаемости меланомы использовали метод Каплана-Мейера и лог-ранг тест. Для определения прогностической ценности ковариатов в отношении OS и DFS с помощью регрессионной модели Кокса были проведены унивариантный и многовариантный анализы. OS и DFS рассчитывались с момента постановки диагноза до смерти от заболевания или метастазов, соответственно, или цензурировались на момент последнего наблюдения или смерти, не связанной с заболеванием. Значение p < 0,05 считалось статистически значимым.
Результаты
Экспрессия PDK в КМ и невусах
Как в КМ, так и в невусах изоформы PDK 1, 2 и 3 экспрессировались не только в клетках меланоцитов/меланомы, но и в кератиноцитах, а также в клетках сальных желез и волосяных фолликулов. Уровни экспрессии всех анализируемых изоформ PDK положительно коррелировали между собой, что свидетельствует о сопутствующей экспрессии этих белков (табл. 2).
| Белок | Средний уровень экспрессии (± SD) | Корреляция (значение p) | Корреляция (p значение) | Корреляция (p значение) |
| PDK1 | 4.8 ± 3.5 | < 0.01* | < 0.01§ | |
| ПДК2 | 4.6 ± 3.6 | < 0.01† | ||
| PDK3 | 7.0 ± 3.2 |
*между PDK1 и PDK2; † между PDK2 и PDK3; § между PDK1 и PDK3.
Иммуногистохимическая экспрессия PDK1
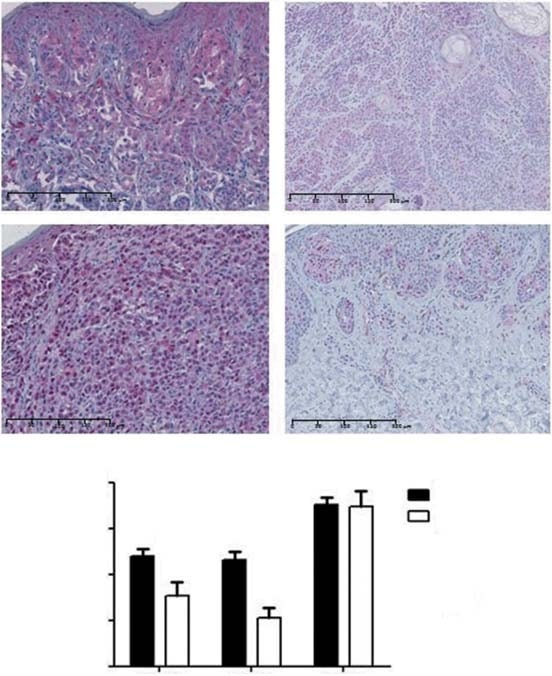
Цитоплазматическое окрашивание PDK1 наблюдалось в 88% КМ и 76% невусов (рис. 2А). Отрицательное/низкое окрашивание наблюдалось в 52 и 76%, а умеренное/высокое — в 48 и 24% меланом и невусов, соответственно. Таким образом, КМ демонстрировали значительно более высокие средние уровни экспрессии PDK1, чем невусы (p = 0,03; Рисунок 2B). Что касается гистологических подтипов КМ, LMM демонстрировали более высокие уровни экспрессии PDK1, чем NM (p = 0,04) и ALM (p = 0,04). Не было обнаружено связи между экспрессией PDK1 и типом солнечного облучения. Что касается прогностических факторов, PDK1 больше экспрессировался в опухолях толщиной £ 1 мм (p = 0,02). Более высокий уровень экспрессии PDK1 был связан с более низкой стадией опухоли (p = 0,04), но не с общей и безболезненной выживаемостью пациентов. Мы также оценили, связана ли экспрессия изоформ PDK с MAPK и PI3K-AKT-mTOR путями. Не было обнаружено связи между экспрессией PDK1 и мутациями BRAF/NRAS или активацией пути MAPK (оцениваемой по экспрессии pERK). Экспрессия PDK1 положительно коррелировала с экспрессией белков каскада PI3K-AKT-mTOR, а именно mTOR и 4EBP1 (p = 0,02 и p = 0,04, соответственно).
Иммуногистохимическая экспрессия PDK2
PDK2 проявлял как ядерную, так и цитоплазматическую экспрессию в 87% КМ и 83% невусов (рис. 2А). Отрицательное/низкое окрашивание наблюдалось в 58 и 89%, а умеренное/высокое — в 42 и 11% меланом и невусов, соответственно. Средний уровень экспрессии PDK2 был выше в КМ по сравнению с невусами (p < 0,01; рис. 2B). Что касается гистологических подтипов КМ, то в SSM уровень экспрессии PDK2 был выше, чем в ALM (p = 0,02). Мы не обнаружили никакой связи между типом солнечного облучения и экспрессией PDK2. На более низких стадиях опухоли уровень PDK2 был значительно выше (p = 0,02), хотя не было обнаружено связи между экспрессией этой изоформы и основными прогностическими факторами КМ, а также с общей и безболезненной выживаемостью пациентов. Экспрессия PDK2 не была связана с мутационным статусом BRAF/NRAS или с активацией MAPK-пути. Была обнаружена значительная положительная корреляция между экспрессией PDK2 и экспрессией AKT из каскада PI3K-AKT-mTOR (p = 0,03).
Иммуногистохимическая экспрессия PDK3
PDK3 проявлял цитоплазматическую экспрессию во всех КМ и невусах. Отрицательное/низкое окрашивание наблюдалось в 28 и 22%, а умеренное/высокое — в 72 и 78% КМ и невусов, соответственно. Уровень экспрессии изоформы PDK3 был одинаковым между КМ и невусами (рис. 2B). При сравнении среднего уровня экспрессии PDK3 между подтипами КМ и типом солнечного облучения существенных различий не было. Опухоли толщиной £ 1 мм и опухоли с более низкой стадией имели более высокий уровень PDK3 (p = 0,01 и p < 0,01, соответственно). Язвенная болезнь, частота митозов, общая и безболезненная выживаемость пациентов не были значительно связаны с экспрессией PDK3. Не было обнаружено связи между экспрессией PDK3 и мутациями BRAF/NRAS, а также активацией каскада MAPK. Что касается пути PI3K-AKT-mTOR, экспрессия PDK3 положительно коррелировала с общей экспрессией AKT, mTOR и 4EBP1 (p < 0,01, p < 0,01 и p = 0,04, соответственно). Ввиду общей экспрессии PDK3 в каждом КМ и невусе, последующий анализ in vitro не включал эту изоформу PDK.
Влияние обработки DCA на жизнеспособность клеточных линий меланомы
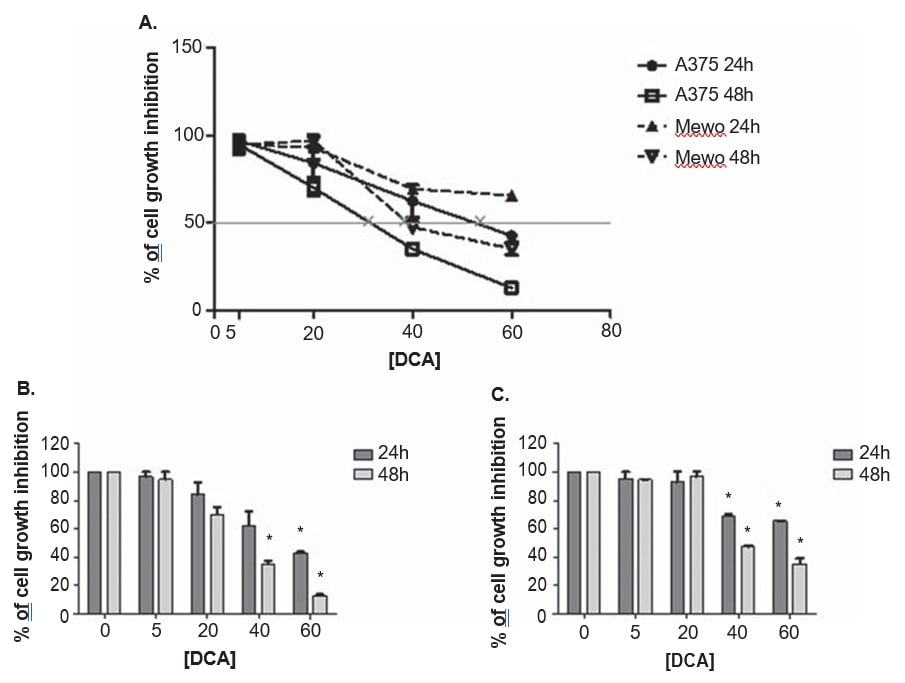
Клеточные линии меланомы A375 и Mewo подвергались воздействию возрастающих концентраций DCA для определения влияния на жизнеспособность клеток с помощью анализа PrestoBlue. ДКА снижал жизнеспособность обеих клеточных линий дозозависимым образом после 24 и 48 часов обработки, хотя A375 был немного более чувствителен к обработке ДКА, с большим ингибированием роста по сравнению с клеточной линией Mewo (Рисунок 3). Влияние DCA на жизнеспособность клеток A375 наблюдалось после обработки 5 мМ, а в линии клеток Mewo было более интенсивным после обработки 20 мМ, со значениями ингибирования от 3 до 87% в A375 и от 5 до 65% в Mewo. Значения IC50 были оценены как 33 ± 5,5 мМ для A375 и 39,9 ± 2,2 мМ для линии клеток Mewo после 48 ч обработки DCA (рис. 3).
Влияние обработки DCA на уровень потребления глюкозы и выработки лактата клеточными линиями меланомы
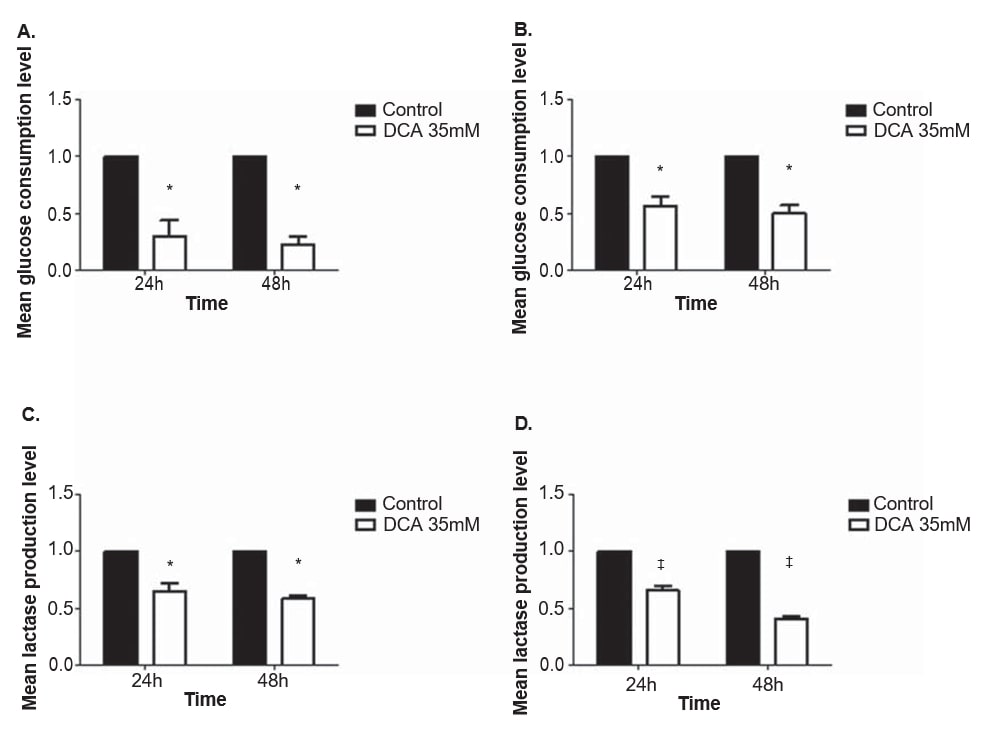
Для оценки влияния DCA на метаболизм клеточных линий меланомы, уровни глюкозы и лактата были определены в культуральной среде после 24 и 48 ч обработки 35 мМ DCA, концентрации, близкой к IC50 для обеих клеточных линий. Потребление глюкозы и выработка лактата снизились в обеих клеточных линиях меланомы после обработки ДКА (рис. 4). Клеточные линии A375 и Mewo продемонстрировали значительное снижение уровня потребления глюкозы через 24 и 48 часов после обработки ДКА (p = 0,01 и p < 0,01, соответственно). Уровень выработки лактата также значительно снизился через 24 и 48 часов в линиях клеток A375 (p = 0,01 и p < 0,01, соответственно) и Mewo (p < 0,01).
Эффект лечения DCA на эффекторы PDK и mTOR пути, а также на экспрессию HIF1-α в клеточных линиях меланомы
Эффективность лечения DCA в ингибировании активности PDK оценивалась путем анализа экспрессии фосфорилированного нижележащего эффектора PDH с помощью вестерн-блота. Через 24 ч после обработки ДКА наблюдалось значительное ингибирование фосфорилирования PDH в обеих клеточных линиях (p < 0,01; рис. 5). Экспрессия PDK1 и PDK2 не изменялась под действием ДКА (рис. 5), как и ожидалось. Также была проведена оценка экспрессии двух нижележащих эффекторов и индикаторов активации пути mTOR — S6 и 4EBP1. Хотя лечение DCA не изменило экспрессию S6 и 4EBP1, уровень экспрессии фосфорилированных форм обоих белков был снижен в A375 (p = 0,03 и не существенно, соответственно) и в Mewo (p < 0,01 и p = 0,01, соответственно; Рисунок 5). Поскольку DCA может влиять на уровень HIF1-α, экспрессия этого белка также была оценена, и после лечения DCA изменений не наблюдалось (рис. 5). Аналогичные результаты были получены через 48 ч после обработки ДКА, который значительно ингибирует фосфорилирование PDH в обеих клеточных линиях (p <0,01). Уровень экспрессии фосфорилированных форм S6 и 4EBP1 был значительно снижен в A375 (p = 0,01 и p = 0,02, соответственно) и в Mewo (p = 0,01 и не значимо, соответственно). Экспрессия PDK1, PDK2 и HIF1-α не была изменена (данные не показаны).
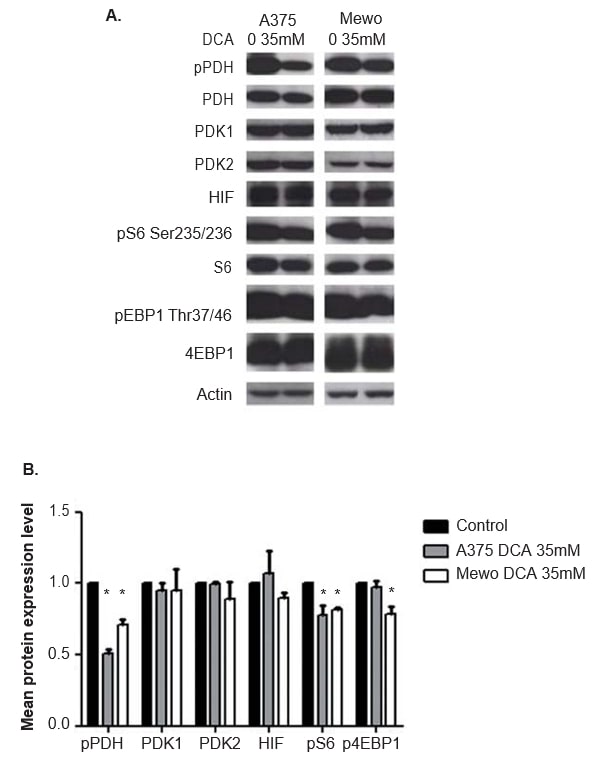
Влияние обработки DCA на клеточный цикл и апоптоз клеточных линий меланомы
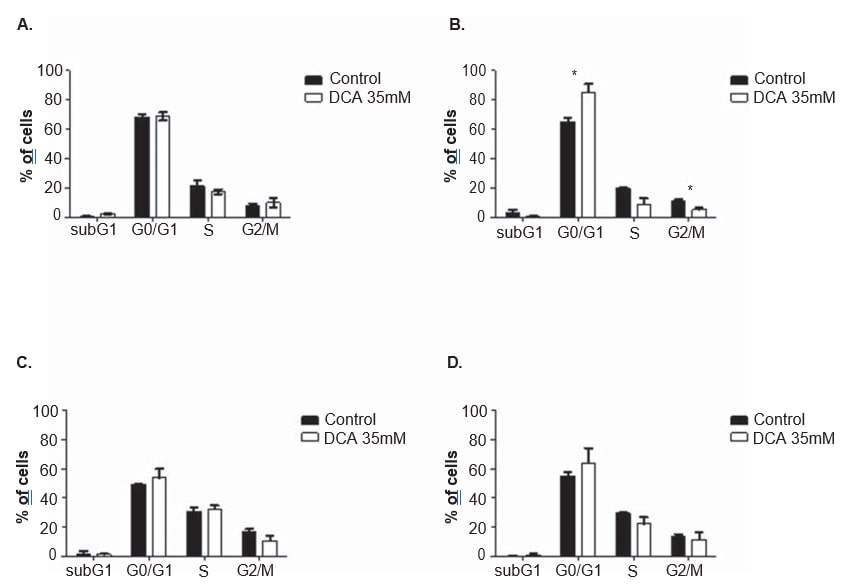
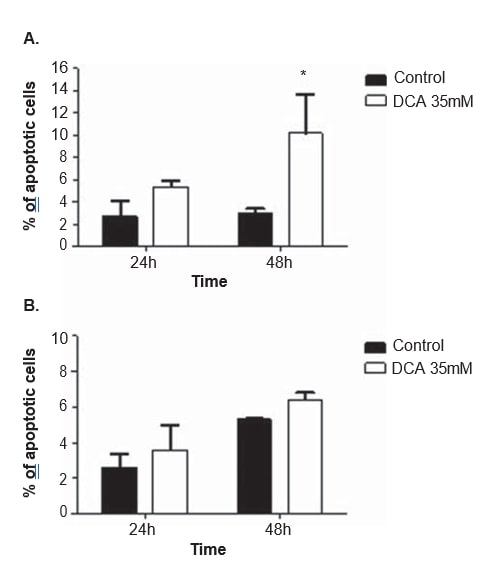
Для выяснения механизма действия DCA, анализ клеточного цикла и измерение апоптоза проводили в обеих клеточных линиях, после 24 и 48 ч обработки 35 мМ DCA. Клеточный цикл анализировали с помощью йодистого пропидия. После 48 ч обработки ДКА клеточная линия A375 показала значительное увеличение процента клеток в фазе G0/G1 клеточного цикла (с 65,0 ± 4,9%, наблюдаемых в необработанных клетках, до 85,2 ± 10,2% в клетках, обработанных ДКА; p = 0,04), значительное снижение G2/M (с 11.3 ± 1,3% в клетках без обработки до 5,2 ± 2,2% в клетках, обработанных ДКА; p = 0,01) и снижение, не достигшее порога статистической значимости, в фазе S (с 19,7 ± 1,8% в клетках без обработки до 9,1 ± 6,7% в клетках, обработанных ДКА; p = 0,06; рис. 6). Такая же тенденция наблюдалась в клеточной линии Mewo через 48 ч после обработки ДКА (увеличение процента клеток в G0/G1 и снижение в S и G2/M), хотя и не достигала статистической значимости (рис. 6). Анализ методом проточной цитометрии проводился после окрашивания аннексином V/пропидиум йодидом. Наблюдалось значительное увеличение количества апоптотических клеток в клеточной линии A375 после 48 ч обработки ДКА (3,0 ± 0,5% в необработанных клетках до 16,9 ± 4,6% в обработанных ДКА клетках; p < 0,01; Рисунок 7). Наблюдалось незначительное увеличение процента апоптотических клеток в клеточной линии A375 через 24 ч и в клеточной линии Mewo через 24 ч и 48 ч после обработки ДКА.
Обсуждение
В этой работе мы впервые установили, что КМ сверхэкспрессирует белки PDK1 и PDK2. PDK являются ключевыми белками, регулируемыми HIF1-α, которые приводят злокачественные клетки в фенотип аэробного гликолиза, являющегося отличительной чертой рака [20,22,31].
Эффект Варбурга считается фактором резистентности к традиционным химиотерапевтическим препаратам [21]. Сверхэкспрессия PDK, обнаруженная в КМ, побудила нас оценить эффект лечения DCA в клеточных линиях меланомы. Мы наблюдали, что лечение DCA снижает потребление глюкозы и производство лактата в клеточных линиях меланомы, что согласуется с эффектом DCA в переходе от аэробного гликолиза к окислительному фосфорилированию, о котором ранее сообщалось в раковых клетках [41]. Этот сдвиг в метаболизме глюкозы, вероятно, является результатом наблюдаемого значительного снижения экспрессии фосфорилированной формы PDH, что также подтверждает эффективность лечения DCA. Следует отметить, что правильным показателем действия ДКА является уровень фосфорилированной PDH, поскольку, ингибируя активность PDK, а не ее экспрессию, ДКА стимулирует дефосфорилирование PDH (рис. 1).
Мы наблюдали дозозависимое снижение жизнеспособности клеток после обработки DCA в обеих клеточных линиях меланомы. Кроме того, в обеих клеточных линиях наблюдалось снижение пролиферации через остановку G0/G1, а также увеличение апоптоза, но статистически значимое только в клеточной линии A375. Тот факт, что концентрация DCA, выбранная в данном исследовании (35 мМ), немного ниже IC50 для клеточной линии Mewo, может объяснить менее выраженный эффект, наблюдаемый в этих клетках.
Связь между DCA и HIF-1a в раке не до конца понятна, поскольку DCA увеличивает потребление кислорода тканями и производство ROS, что должно приводить к повышению или стабилизации HIF-1a, соответственно [46,47]. Напротив, некоторые авторы сообщали, что ДКА приводит к снижению экспрессии HIF1-α [25,48,49]. В нашей работе не наблюдалось изменений в уровне HIF1-α после обработки DCA клеточных линий меланомы, что согласуется с данными Shahrzad и др., которые сообщили, что экспрессия HIF1-α снижается только после обработки DCA в гипоксических условиях [49].
Чтобы преодолеть вызванную мутантом BRAF старение и перейти к более агрессивным фенотипам, КМ, вероятно, активирует другие сигнальные каскады, такие как путь mTOR [12]. Этот путь способствует эффекту Варбурга, повышая активность HIF1-α, что, в свою очередь, может увеличить экспрессию PDK [23,31]. Ранее мы уже сообщали о полной активации этого пути в КМ [13]. В данном исследовании мы наблюдали одновременную экспрессию PDK 1, 2 и 3 и эффекторов пути mTOR в КМ. Экспрессия изоформ PDK в КМ положительно коррелировала с экспрессией mTOR, а также с эффекторами upstream и downstream этого пути: AKT и 4EBP1. Более того, мы наблюдали значительное снижение pS6 и p4EBP1 в клеточных линиях A375 и Mewo после обработки DCA.
Невусы имеют общие генетические изменения с КМ, так как в обоих случаях наблюдается высокая частота мутаций BRAF [10]. Экспрессия PDK, хотя и в гораздо меньшей степени, также наблюдалась в невусах. Мы проанализировали, может ли мутационный статус BRAF и последующая активация ERK быть связана с более высокой экспрессией PDK. Мы не обнаружили никакой связи ни в невусах, ни в КМ. Клеточная линия A375 имеет мутацию BRAFV600E, а клеточная линия Mewo — дикий тип этого гена, и ответ обеих клеточных линий на лечение DCA был сходным, несмотря на различную интенсивность. Более того, уже сообщалось, что мутационный статус BRAF не изменяет чувствительность к лечению DCA [50]. Эти результаты контрастируют с нашими предыдущими результатами по использованию ингибиторов пути mTOR в нескольких клеточных линиях, полученных из меланомы, где мы обнаружили более высокую чувствительность к лечению RAD001 в клеточных линиях КМ, несущих мутацию BRAFV600E [51].
Изоформы PDK экспрессировались в цитоплазме клеток меланомы, что подтверждает известную митохондриальную функцию таких изоформ [33]. Что касается PDK2, то в большинстве случаев КМ наблюдалась ядерная экспрессия. Такое ядерное расположение также было отмечено для PDK1 в HNSCC [37]. Насколько нам известно, роль PDK в ядре еще предстоит выяснить, но недавно было выдвинуто предположение о ядерной функции PDH, эффектора PDK. Сутендра и др. сообщили, что PDH участвует в ядерном образовании ацетил-КоА [52]. В ответ на стимуляцию фактором роста PDH и его фосфатаза транслоцируются из митохондрий, где они ингибируются PDK, в ядро, но ядерное местоположение PDK не было определено [52]. Тем не менее, существуют доказательства того, что некоторые гликолитические ферменты участвуют в других клеточных функциях, помимо гликолиза, а именно в регуляции транскрипции [53]. Необходимы дополнительные исследования, чтобы выяснить, выполняет ли PDK также ядерную функцию.
Сообщалось, что при почечно-клеточной карциноме экспрессия PDK1 снижается по мере прогрессирования опухоли [39]. Соответственно, в нашей работе было обнаружено, что PDK экспрессируется на более низких стадиях опухоли, не влияя на общую и безболезненную выживаемость пациентов с меланомой. В отличие от этого, сверхэкспрессия PDK была определена как маркер опухолевой прогрессии, плохого прогноза и рецидива в HNSCC и раке желудка [37,40,54]. Хотя нашей целью не было изучение прогностической роли PDK в КМ, наши результаты позволяют предположить, что PDK в КМ могут играть более активную роль в развитии меланомы, чем в ее прогрессии. Мы осознаем, что наша серия состоит в основном из первичных КМ и что для прояснения этого вопроса потребуются дальнейшие исследования в прогрессирующей (метастатической) меланоме.
Нашей целью в данном исследовании было изучение биомаркеров для новых терапевтических подходов у пациентов с КМ. Наши результаты свидетельствуют о потенциальной пользе DCA в лечении пациентов с КМ, что согласуется с данными Abildgaard и др., которые сообщили о снижении уровня АТФ и роста меланомы при лечении DCA [50]. Эти авторы также сообщили о синергической комбинации между ДКА и ингибитором BRAF, вемурафенибом [50]. Кроме того, наши результаты показывают одновременную экспрессию эффекторов путей PDK и mTOR в КМ и снижение активности пути mTOR при лечении DCA. Учитывая результаты Хонга и др., которые сообщили о синергетическом эффекте комбинации ДКА с ингибитором S6K1 (эффектор пути mTOR) [55], можно предположить, что синергетический терапевтический эффект может быть достигнут при КМ при использовании комбинации ДКА с прямым ингибитором mTOR.
Заключение
Мы впервые сообщаем о сверхэкспрессии PDK1 и 2 в РМ по сравнению с невусами. Было установлено, что экспрессия PDK связана с экспрессией эффекторов пути mTOR и не связана с мутационным статусом BRAF. Лечение DCA приводит к изменению метаболизма, снижению пролиферации, увеличению апоптоза и снижению активации пути mTOR в клеточных линиях меланомы. Принимая все эти результаты вместе, мы пришли к выводу, что экспрессия PDK может играть роль в развитии меланомы и что ее ингибирование с помощью DCA отдельно или в комбинации с прямыми ингибиторами mTOR может принести пользу пациентам с КМ.
Благодарности
Мы благодарны всем пациентам, принявшим участие в этом исследовании, а также врачам, предоставившим клиническую, патологическую и последующую информацию. Мы благодарим доктора Мадалену Пинто из CEQUIMED, фармацевтического факультета Университета Порту, Португалия, которая любезно предоставила нам клеточную линию меланомы кожи A375, и доктора Марка Мареэля из отделения радиотерапии и ядерной медицины Университетской больницы Гента, Бельгия, который любезно предоставил нам клеточную линию меланомы кожи Mewo. Мы благодарим Габриэлу Альмейду за полезные технические советы по проведению анализа PB. Мы также благодарим профессора Мануэля Собриньо Симоэса за критическое прочтение этой рукописи. H Pópulo и R Caldas внесли равный вклад в эту работу.
Декларация интересов
Данное исследование было поддержано Португальским фондом науки и технологий через грант Post-Doc для HP (Ref.: SFRH/BPD/85249/2012). Дальнейшее финансирование было получено в рамках проекта «Микроокружение, метаболизм и рак», который был частично поддержан Programa Operacional Regional do Norte (ON.2 — O Novo Norte) в рамках Quadro de Referência Estratégico Nacional (QREN) и Fundo Europeu de Desenvolvimento Regional (FEDER). IPATIMUP объединяет исследовательскую группу i3S, которая частично поддерживается FCT, Португальским фондом науки и технологий. Эта работа финансировалась из средств FEDER через Оперативную программу по факторам конкурентоспособности — COMPETE и национальных фондов через FCT, в рамках проектов «PEst-C/SAU/LA0003/2013» Авторы не раскрывают потенциальных конфликтов интересов. Авторы не имеют других соответствующих связей или финансового участия с какой-либо организацией или субъектом, имеющим финансовый интерес или финансовый конфликт с предметом или материалами, обсуждаемыми в рукописи, кроме раскрытых.
ССЫЛКИ
1 Siegel R, Ma J, Zou Z, et al. Статистика рака, 2014. CA Cancer J Clin 2014;64(1):9-29
2 Nikolaou V, Stratigos AJ. Новые тенденции в эпидемиологии меланомы. Br J Dermatol 2014;170(1):11-19
3 Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol 2006;19(Suppl 2):S34-40
4 Populo H, Soares P, Lopes JM. Insights into melanoma: targeting the mTOR pathway for therapeutics. Expert Opin Ther Targets 2012;16(7):689-705
5 Balch CM, Gershenwald JE, Soong SJ, et al. Final version of 2009 AJCC melanoma staging and classification. J Clin Oncol 2009;27(36):6199-206
6 Lopez-Bergami P, Fitchman B, Ronai Z. Understanding signaling cascades in melanoma. Photochem Photobiol 2008;84(2):289-306
7 Bertolotto C. Меланома: от меланоцита до генетических изменений и клинических вариантов. Scientifica 2013; 2013:22
8 Populo H, Boaventura P, Vinagre J, et al. TERT promoter mutations in skin cancer: the effects of sun exposure and X-irradiation. J Invest Dermatol 2014;134(8):2251-7
9 Elder DE. Предшественники меланомы и их мимика: невусы особых участков. Modern pathology Inc 2006;19(Suppl 2):S4-20
10 Kumar R, Angelini S, Snellman E, et al. BRAF мутации являются распространенными соматическими событиями
11 в меланоцитарных невусах. J Invest Dermatol 2004;122(2):342-8 Michaloglou C, Vredeveld LC, Soengas MS, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature 2005;436(7051):720-4
12 Vredeveld LC, Possik PA, Smit MA, et al. Abrogation of BRAFV600E-induced senescence by PI3K pathway activation contributes to melanomagenesis. Genes Dev 2012;26(10):1055-69
13 Populo H, Soares P, Faustino A, et al. Активация пути mTOR в кожной меланоме связана с более плохими характеристиками прогноза. Pigment Cell Melanoma Res 2011;24(1):254-7
14 Olszanski AJ. Текущая и будущая роль таргетной терапии и иммунотерапии в прогрессирующей меланоме. J Manag Care Spec Pharm 2014;20(4):346-56
15 Hodi FS, O’Day SJ, McDermott DF, et al. Улучшение выживаемости с помощью ипилимумаба у пациентов с метастатической меланомой. N Engl J Med 2010;363(8):711-23
16 Chapman PB, Hauschild A, Robert C, et al. Улучшение выживаемости с помощью вемурафениба при меланоме с мутацией BRAF V600E. N Engl J Med 2011;364(26):2507-16
17 Flaherty KT, Robert C, Hersey P, et al. Улучшение выживаемости при ингибировании MEK в меланоме с мутацией BRAF. N Engl J Med 2012;367(2):107-14
18 Ballantyne AD, Garnock-Jones KP. Дабрафениб: первое глобальное одобрение. Drugs 2013;73(12):1367-76
19 Доступно на сайте:http://www.fda.gov/Drugs/InformationOnDrugs/ApprovedDrugs/ucm412861.htm
20 Gillies RJ, Robey I, Gatenby RA. Причины и последствия повышенного метаболизма глюкозы при раковых заболеваниях. J Nucl Med 2008;49(Suppl 2):24S-42S
21 Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis 2013;4:e532
22 Semenza GL. HIF-1 опосредует метаболические реакции на внутриопухолевую гипоксию и онкогенные мутации. J Clin Invest 2013;123(9):3664-71
23 Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer 2008;8(11):851-64
24 Scott DA, Richardson AD, Filipp FV, et al. Comparative metabolic flux profiling of melanoma cell lines: beyond the Warburg effect. J Biol Chem 2011;286(49):42626-34
25 Kluza J, Corazao-Rozas P, Touil Y, et al. Inactivation of the HIF-1alpha/PDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates therapeutic activity of pro-oxidants. Cancer Res 2012;72(19):5035-47
26 Baudy AR, Dogan T, Flores-Mercado JE, et al. FDG-PET является хорошим биомаркером как раннего ответа, так и приобретенной резистентности в мутантных меланомах BRAFV600, леченных вемурафенибом и ингибитором MEK GDC-0973. Исследование EJNMMI 2012;2(1):22
27 Hall A, Meyle KD, Lange MK, et al. Дисфункциональное окислительное фосфорилирование делает клетки злокачественной меланомы зависимыми от гликолиза, управляемого онкогеном (V600E)BRAF. Oncotarget 2013;4(4):584-99
28 Kumar SM, Yu H, Edwards R, et al. Мутант V600E BRAF увеличивает экспрессию гипоксия-индуцибельного фактора-1альфа в меланоме. Cancer Res 2007;67(7):3177-84
29 Kuphal S, Winklmeier A, Warnecke C, et al. Конститутивная активность HIF-1 в злокачественной меланоме. Eur J Cancer 2010;46(6):1159-69
30 Slominski A, Kim TK, Brozyna AA, et al. Роль меланогенеза в регуляции поведения меланомы: Меланогенез приводит к стимуляции экспрессии HIF-1альфа и HIF-зависимых сопутствующих путей. Archives of biochemistry and biophysics 2014;563:79-93
31 Kim JW, Tchernyshyov I, Semenza GL, et al. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006;3(3):177-85
32 Lu CW, Lin SC, Chen KF, et al. Индукция киназы-3 пируватдегидрогеназы гипоксия-индуцибельным фактором-1 способствует метаболическому переключению и лекарственной устойчивости. J Biol Chem 2008;283(42):28106-14
33 Patel MS, Korotchkina LG. Регуляция пируватдегидрогеназного комплекса. Biochem Soc Trans 2006;34(Pt 2):217-22
34 Bowker-Kinley MM, Davis WI, Wu P, et al. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J 1998;329(Pt 1):191-6
35 Papandreou I, Goliasova T, Denko NC. Противораковые препараты, направленные на метаболизм: является ли дихлорацетат новой парадигмой? Int J Cancer 2011;128(5):1001-8
36 Zhang S, Hulver MW, McMillan RP, et al. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Питание и метаболизм 2014;11(1):10
37 Wigfield SM, Winter SC, Giatromanolaki A, et al. PDK-1 регулирует производство лактата в условиях гипоксии и ассоциируется с плохим прогнозом при сквамозном раке головы и шеи. Br J Cancer 2008;98(12):1975-84
38 Lu CW, Lin SC, Chien CW, et al. Сверхэкспрессия киназы 3 пируватдегидрогеназы увеличивает лекарственную устойчивость и ранний рецидив при раке толстой кишки. Am J Pathol 2011;179(3):1405-14
39 Baumunk D, Reichelt U, Hildebrandt J, et al. Expression parameters of the metabolic pathway genes pyruvate dehydrogenase kinase-1 (PDK-1) and DJ-1/PARK7 in renal cell carcinoma (RCC). World J Urol 2013;31(5):1191-6
40 Hur H, Xuan Y, Kim YB, et al. Экспрессия киназы-1 пируватдегидрогеназы при раке желудка как потенциальная терапевтическая мишень. Int J Oncol 2013;42(1):44-54
41 Bonnet S, Archer SL, Allalunis-Turner J, et al. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell 2007;11(1):37-51
42 Kankotia S, Stacpoole PW. Дихлорацетат и рак: Новый дом для сиротского препарата? Biochimica et biophysica acta 2014;1846(2):617-29
43 Sutendra G, Dromparis P, Kinnaird A, et al. Активация митохондрий путем ингибирования PDKII подавляет сигнализацию HIF1a и ангиогенез при раке. Oncogene 2013;32(13):1638-50
44 Michelakis ED, Sutendra G, Dromparis P, et al. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med 2010;2(31):31ra34
45 Castro P, Rebocho AP, Soares RJ, et al. Перестройка PAX8-PPARgamma часто обнаруживается в фолликулярном варианте папиллярной карциномы щитовидной железы. J Clin Endocrinol Metab 2006;91(1):213-20
46 Sun W, Zhou S, Chang SS, et al. Митохондриальные мутации способствуют накоплению HIF1alpha через увеличение реактивных видов кислорода и up-regulated pyruvate dehydrogenase kinase 2 в плоскоклеточной карциноме головы и шеи. Clin Cancer Res 2009;15(2):476-84
47 Cairns RA, Bennewith KL, Graves EE, et al. Фармакологически увеличенная гипоксия опухоли может быть измерена с помощью позитронно-эмиссионной томографии с 18F-фторазомицином арабинозидом и усиливает ответ опухоли на гипоксический цитотоксин PR-104. Clin Cancer Res 2009;15(23):7170-4
48 Sun RC, Board PG, Blackburn AC. Таргетинг метаболизма с помощью триоксида мышьяка и дихлорацетата в клетках рака молочной железы. Mol Cancer 2011;10:142
49 Shahrzad S, Lacombe K, Adamcic U, et al. Дихлорацетат натрия (DCA) снижает апоптоз при гипоксии колоректальных опухолей. Cancer Lett 2010;297(1):75-83
50 Abildgaard C, Dahl C, Basse AL, et al. Биоэнергетическая модуляция с помощью дихлорацетата уменьшает рост клеток меланомы и потенцирует их ответ на ингибирование BRAFV600E. J Transl Med 2014;12:247
51 Populo H, Tavares S, Faustino A, et al. Мутации GNAQ и BRAF показывают дифференциальную активацию пути mTOR в трансформированных клетках человека. Peer J 2013;1:e104
52 Sutendra G, Kinnaird A, Dromparis P, et al. Ядерный пируватдегидрогеназный комплекс важен для образования ацетил-КоА и ацетилирования гистонов. Cell 2014;158(1):84-97
53 Kim JW, Dang CV. Многогранные роли гликолитических ферментов. Trends Biochem Sci 2005;30(3):142-50
54 Xuan Y, Hur H, Ham IH, et al. Dichloroacetate attenuates hypoxia-induced resistance to 5-fluorouracil in gastric cancer through the regulation of glucose metabolism. Exp Cell Res 2014;321(2):219-30
55 Hong SE, Shin KS, Lee YH, et al. Inhibition of S6K1 enhances dichloroacetate-induced cell death. J Cancer Res Clin Oncol 2014.